

Abstract
Signal-transducer-and-activator-of-transcription-3 (STAT3) is a central regulator of immune homeostasis. STAT3 levels are strictly controlled and STAT3 impairment contributes to several diseases including the monogenic autosomal-dominant hyper-IgE syndrome (AD-HIES). We investigated patients of four consanguineous families with an autosomal-recessive disorder resembling the phenotype of AD-HIES, with symptoms of immunodeficiency, recurrent infections, skeletal abnormalities, and elevated IgE. Patients presented with reduced STAT3 expression and diminished Th17 cell numbers, in absence of STAT3 mutations. We identified homozygous nonsense mutations in ZNF341, encoding a zinc-finger transcription factor. Wild-type ZNF341 bound to and activated the STAT3 promoter, whereas the mutant variants showed impaired transcriptional activation, partly due to nuclear translocation failure. In summary, nonsense mutations in ZNF341 account for the STAT3-like phenotype in four autosomal-recessive kindreds. Thus, ZNF341 is a previously unrecognized regulator of immune homeostasis.
One Sentence Summary
Homozygous nonsense mutations in ZNF341 impair its ability to transcriptionally enhance STAT3 expression and thereby cause immunodeficiency.
INTRODUCTION
Immune homeostasis in humans is important to avoid the two extremes of immunodeficiency and autoimmunity/autoinflammation. Signal-transducer-and-activator-of-transcription-3 (STAT3) is an immune rheostat that prevents such diseases by regulating the innate and adaptive immune system (1). Th17 CD4+ T cell differentiation and IL-17 production are dependent on precisely balanced STAT3 activity (2–6), and germline and somatic mutations in STAT3 have been associated with multiple immune disorders and cancer, respectively (7). For instance, heterozygous germline gain-of-function mutations lead to lymphoproliferation and juvenile-onset autoimmunity (8, 9), whereas heterozygous loss-of-function (LOF) mutations in STAT3 cause an autosomal-dominant (AD) immunodeficiency known as hyper-IgE syndrome (HIES, OMIM #147060 and #243700) (10). STAT3-LOF mutations have been shown to exert a dominant-negative effect impairing antibacterial and antifungal host defense and resulting in multisystem disorder also affecting the skeleton, dentition and connective tissue (11, 12). Patients present with the clinical triad of recurrent pneumonia, eczema with cold staphylococcal skin abscesses, and elevated serum IgE levels (11). STAT3-LOF mutations represent the underlying genetic defect in ~75% of sporadic and AD-HIES patients (MIM: 102582), whereas biallelic DOCK8 mutations account for disease in ~80% of patients with the autosomal-recessive (AR) form of HIES (MIM: 611432, (13, 14)). Additionally, mutations in PGM3 (MIM: 172100, (15, 16)) have been described in AR-HIES. At least one of these AR immunodeficiency syndromes also involve dysregulated STAT3 function since the lack of DOCK8 results in reduced STAT3 activation (17, 18).
However, regulatory mechanisms of the STAT3 equilibrium are complex and not fully understood. Regulation at protein level includes phosphorylations and interaction with other STAT family members (19). In addition, epigenetic regulation by HMGB1 (20) or ZNF382 (21) and transcriptional regulation of STAT3 through STAT3 homodimers and other yet unidentified transcription factors have been proposed (22).
Here, we report that ZNF341, a previously uncharacterized C2H2-zinc finger transcription factor, is mutated in families with recurrent bacterial and fungal infections. Two distinct homozygous nonsense mutations in exons 6 and 8 of ZNF341 segregate with a phenotype resembling HIES in four consanguineous families with AR inheritance. We describe ZNF341 as a positive regulator of STAT3 expression and report the clinical and laboratory phenotype of individuals lacking ZNF341.
RESULTS
STAT3 HIES-like phenotype with autosomal-recessive inheritance identified in four consanguineous families
We performed mutational analyses to identify the genetic defects in four consanguineous HIES-families with AR inheritance, in which mutations in known HIES genes had previously been excluded. The clinical triad of HIES consisting of recurrent pneumonias, eczema with cold skin abscesses, and elevated serum IgE levels was present in all three affected individuals (A.II.1, A.II.2 and A.II.3) of the consanguineous Family A (for representative pictures of clinical findings see Fig. 1; pedigree of Family A in Fig. 2A). Additionally, they showed skeletal/connective tissue abnormalities and formation of bronchiectasis and pneumatoceles (Fig. 1D) characteristic for STAT3-HIES and also suffered from recurrent candidiasis. Affected members of Families B, C and D (Fig. 2B–D) presented with a milder phenotype initially diagnosed as atopic dermatitis, with characteristic HIES symptoms occurring later in life. The three Israeli families (Families A, B and C) are descendants from soldiers who lived in Sudan in the 19th century. Family D is of Turkish origin. Increased susceptibility to viral infections, typical in DOCK8-deficient AR-HIES, was not observed in any of the patients.
Fig. 1. Representative clinical and radiological manifestations in patients with homozygous ZNF341 nonsense mutations.
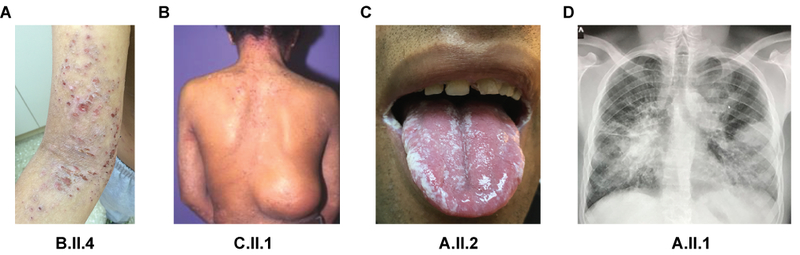
(A) Severe eczema in patient B.II.4 on the upper arm and cubital. (B) Patient C.II.1 with eczema on the neck and a cold skin abscess in the lumbar region missing the typical inflammatory sign of rubor, calor, dolor. (C) Oral thrush due to Candida in patient A.II.2. (D) Chest radiograph of patient A.II.1 showing bilateral pneumonia with positive air bronchogram, bronchiectasis, and pneumatoceles.
Fig. 2. Homozygous nonsense mutations in ZNF341 cause HIES with reduced Th17 cell numbers in patient PBMCs.
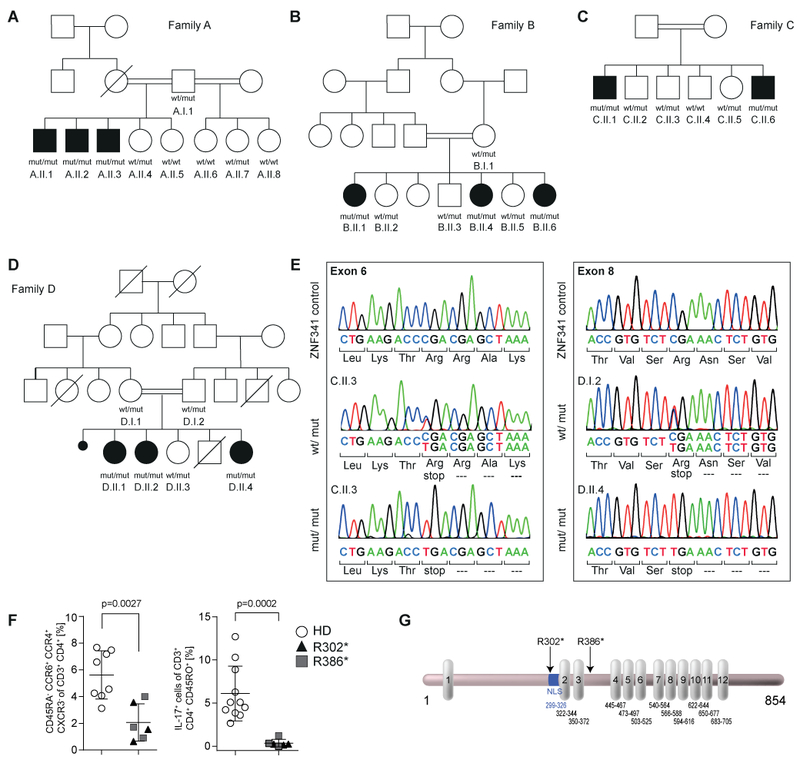
(A-D) Pedigrees and genotypes with the nonsense mutated (mut) alleles g.32345116C>T (c.904C>T; p.Arg302*) for Families A-C and g.32349795C>T (c.1156C>T; p.Arg386*) for Family D. Heterozygous carriers are unaffected. Wt, wild-type. Circles, female; squares, male; filled symbols, affected individuals with HIES; open symbols, healthy members; slash, deceased individual; double horizontal lines, consanguinity; black dot, miscarriage. (E) Both mutations predict premature termination of translation. (F) Flow cytometry of PBMCs demonstrate reduced Th17 cell counts on the basis of CD45RA−CCR6+CCR4+CXCR3− of CD3+CD4+ in patients (n=6; triangles, Family A; squares, Family D) compared to healthy donor controls (HD; n=8; open circles) (left). In contrast to controls (n=11), patient PBMCs (n=6) fail to differentiate into IL17+ cells (CD3+CD4+CD45RO+) upon in vitro stimulation (d4) with Th17 polarizing cytokines IL-1β and IL-6 plus T cell activation/expansion (right). Significance was determined using Mann-Whitney test. (G) ZNF341 is a 854 amino acids “zinc-finger-only” transcription factor with twelve C2H2 motifs (vertical boxes). R302* and R386* (arrows) delete zinc fingers 2–12 and 4–12, respectively. A putative nuclear localization sequence (NLS; blue) is retained in the R386* mutant. Numbers indicate amino acid positions in NP 001269862.
Reduced Th17 CD4+ T cell numbers in patients with STAT3 HIES-like phenotype
Immunophenotyping revealed normal CD19 lymphocyte counts, but an increased percentage of naïve B cells (IgD+CD27−), and reduced memory B cells (CD27+) in the affected individuals. All memory B cell subpopulations including IgG+, IgA+ and IgM+ were significantly reduced (fig. S1A). Patients had normal counts for naïve and memory CD4+ and CD8+ T cells, and for the CD4+ subsets Th1, Th1*, and Th2 (fig. S1B). Patients however presented a significantly reduced percentage of Th17 CD4+ T cells, a key feature of STAT3-HIES (Fig. 2F, left panel). This coincided with reduced expression of CCR6, which is STAT3-dependent (23), in memory CD4 T helper cells (fig. S1B, lower right panel). In addition, peripheral blood mononuclear cells (PBMCs) derived from patients failed to differentiate into IL-17 producing CD4+ T cells (Fig. 2F, right panel, fig. S1C) and showed reduced numbers of IL-22+ T cells (fig. S1D). Detailed case reports, clinical findings and extended immune phenotyping can be found in the Supplementary material (supplements S1 and S2, table S1 and fig. S1). Taken together, these findings support the hypothesis that patients with ZNF341 mutations have a previously unrecognized autosomal recessive immunodeficiency that clinically resembles autosomal dominant HIES due to mutations in STAT3.
Homozygous nonsense mutations in ZNF341 causing the disease phenotype in four HIES families
Genetic defects in STAT3 itself were excluded by sequencing of the exons, cDNA, and the genomic promoter region (supplement S3). We therefore performed genetic linkage analysis of Family A and subsequent whole exome sequencing (WES) on two patients and one healthy sibling of Family A (supplement S4). We identified a homozygous nonsense mutation in exon 6 of ZNF341 (Chr20:32345116C>T; GRCh37; c.904C>T; p.Arg302*, R302* for isoform1; RefSeq NM_001282933.1), which was also present in all patients from Family B and C (Fig. 2A–C + E). At that time, the mutation was absent from dbSNP, but it was later listed as rs746141726 with an allele frequency of 0.0017% and observed only in the heterozygous state. By targeted next-generation sequencing, we identified a second homozygous ZNF341 nonsense mutation in exon 8 (Chr20:32349795C>T; GRCh37; c.1156C>T; p.Arg386*, R386* for isoform1) in Family D, which segregated with the disease status (Fig. 2D + E).
ZNF341 comprises 15 exons and encodes for three isoforms (RefSeq NM_001282933.1; NM_032819.4; NM_001282935.1; three additional non-coding variants are listed in Ensembl, table S2). Expression of mRNA was confirmed in several cell lines and PBMCs (supplement S5 and fig. S2). We focused on the longest isoform 1, a 854 amino acid protein with twelve C2H2 zinc finger domains but no other conserved domain (Fig. 2G) and with putative transcription factor activity (UniProt: Q9BYN7–1 and GO annotation). Both identified mutations predict premature termination of translation, deleting eleven (p.Arg302*; 31.1 kDa) or nine (p.Arg386*; 40.4 kDa) C2H2 zinc finger domains, respectively. As expected, full length ZNF341 isoform 1 was absent in patient-derived (B.II.4) EBV transformed B cell lines (Fig. 3A, left panel). Although the patients presented with high IgE levels, a review of the atopic-dermatitis-meta-analysis of the EAGLE consortium of 10,788 atopic dermatitis cases and 30,047 controls (24) did not reveal any significantly associated variant after Bonferroni correction within locus 20q11.22 (ZNF341 ± 200kb) (fig. S3). In addition, this locus was not reported in a published GWAS meta-analysis of allergic sensitization in 11,903 affected cases and 19,976 controls (25), and no associated variants within this locus are listed in the current version of the NHGRI GWAS Catalog (www.ebi.ac.uk/gwas)(26) for atopy-related traits such as asthma, rhinitis, atopic dermatitis, and allergic sensitization.
Fig. 3. Reduced STAT3 expression in patient-derived cells.
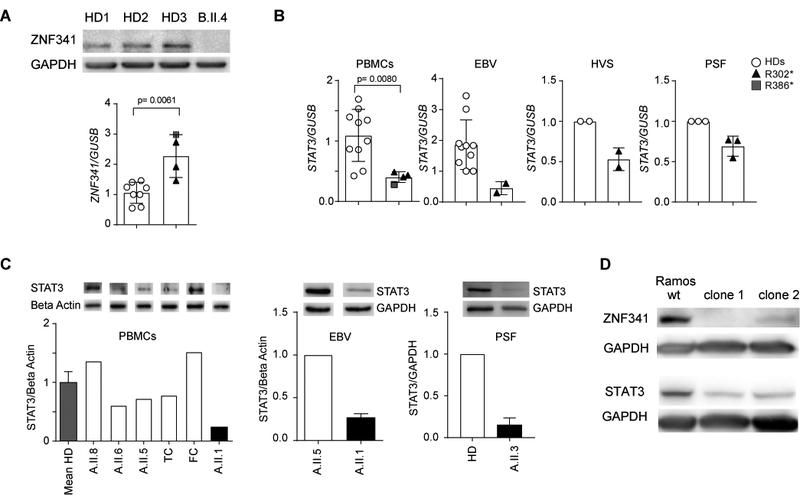
(A) ZNF341 isoform 1 is undetectable in EBV cell lysates from patient B.II.4 (upper panel). Slightly increased relative ZNF341 mRNA expression in patients´ PBMCs (lower panel; patients n=4; HD, n=8). Data from independent experiments were normalized to mean of relative expression in controls. (B) Reduced STAT3 mRNA expression in patient-derived cells. For PBMCs, data from independent experiments were normalized to mean of relative expression in controls (patients, n=4; HD, n=10). EBV cell lines: combined data from two independent experiments were normalized to relative expression of one control (patient A.II.1; HD, n=5). HVS transformed T cell line or primary skin fibroblasts (PSF) of patient A.II.3 compared to healthy donor (mean values and SD of two (HVS) or three (PSF) independent experiments). (C) Western blot and quantitative densitometry demonstrate reduced STAT3 expression in patient-derived PBMCs, EBV cells, and PSF. Beta Actin and GAPDH were used as loading controls. TC, travel control; FC, freezing control. (D) ZNF341 knockout in Ramos cells by using CRISPR/Cas9 technology showed reduced STAT3 protein expression in clone 1 and 2 in comparison to wt Ramos cells. GAPDH was used as loading control.
Reduced STAT3 mRNA and protein in patients with homozygous ZNF341 nonsense mutations
To identify potential target genes of the ZNF341 transcription factor, we compared the transcriptomes of PBMCs derived from patient A.II.1 and his healthy sister A.II.5 (table S3), and confirmed the observations by RT-qPCR for additional individuals. ZNF341 mRNA expression was variable and slightly increased in patients (A.II.1, A.II.2, A.II.3, D.II.4), compared to healthy controls (Fig. 3A, right panel). However, STAT3 mRNA expression was significantly reduced in PBMCs of all affected individuals (A.II.1, A.II.2, A.II.3 and D.II.4) compared to healthy controls (Fig. 3B, left panel).
These findings suggest that the STAT3-like HIES phenotype caused by ZNF341 mutations could be associated with insufficient STAT3 expression, presenting a previously unrecognized pathogenesis in addition to the well-described dominant-negative effect usually associated with STAT3 mutations. Reduced STAT3 mRNA expression was also observed in an EBV-transformed B cell line of patient A.II.1, in a HVS-transformed T cell line of patient A.II.3, and in primary skin fibroblasts (PSF) of patient A.II.3 (Fig. 3B). Moreover, STAT3 protein expression was reduced in ZNF341-mutant cells (patients’ PBMCs, EBV-transformed B cells, and PSF) down to 16–28% of wild-type levels (Fig. 3C). Along the same lines, knockout of ZNF341 in Ramos B cells by CRISPR/Cas9 technology, showed reduced STAT3 protein expression (Fig. 3D). Furthermore, STAT3 Y705-phosphorylation was markedly impaired in PBMCs from patients with R302* and R386* mutations, respectively, following stimulation with IL-6 (Fig. 4A), or with IFN-α in EBV cells from patient A.II.1 (Fig. 4B, left panel). Impaired STAT3 Y705-phosphorylation upon stimulation was even more obvious in EBV cell lines from two patients (B.II.1 and B.II.4) (Fig.4B right panel), indicating that reduced total STAT3 levels consequently lead to overall reduced phospho-STAT3 levels. As we had excluded genetic defects in STAT3 itself, we conclude that the mutations in ZNF341 account for the HIES-phenotype due to the incapability of mutant cells to increase STAT3 protein expression and STAT3 phosphorylation above basal levels, leading to an imbalanced STAT3/phospho-STAT3 ratio in affected individuals.
Fig. 4. Patients´ primary T cells and EBV transformed B cell lines showed reduced Y705-phosphorylation of STAT3.
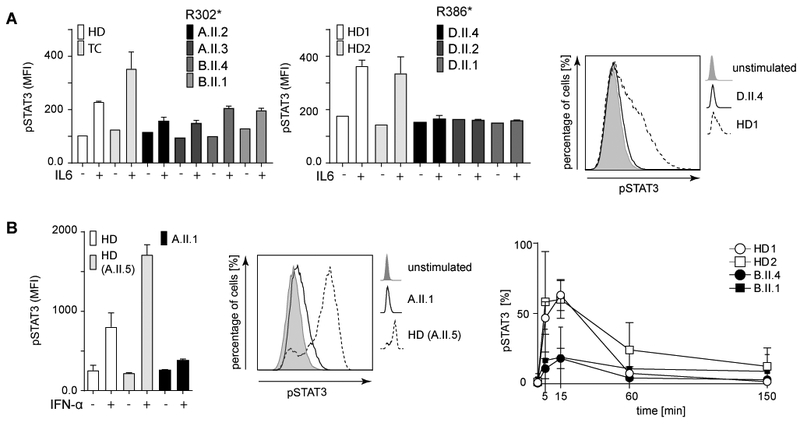
(A) Impaired IL-6 induced Y705-phosphorylation of STAT3 in patients´ PBMCs (gate CD3+). (B) Reduced phospho-STAT3 in IFN-α treated EBV cells from patients with R302* mutations (left). Bar graphs show SD of duplicates; MFI, mean fluorescence intensity. Representative histograms (middle) demonstrating reduced p-STAT3 in patients (solid line) compared to controls (dotted line). Shaded area, unstimulated cells. Only marginal transient increase of p-STAT3 in patients 15 min post stimulation (right). Baseline p-STAT3 levels are reached within 150 min post stimulation. Mean values from independent experiments (HD1, 2 and B.II.4 (n≥3); B.II.1 (n=2) are shown.
Binding of ZNF341 to the STAT3 promotor and subsequent activation of transcription
We next confirmed the transcriptional activation of the endogenous STAT3 promoter upon transient overexpression of GFP-fused ZNF341 in HEK293T cells (fig. S4). In contrast to the R302*-mutant, wild-type-ZNF341 caused a two-fold increase of STAT3 mRNA expression (fig. S4A). Using fluorescence-based reporter assays with synthetic promoters, composed of a genomic STAT3 fragment (−535/−33 relative to the transcription start) fused to the CMV minimal promoter, we observed an average 2.9 -fold increase of reporter expression over basal activity upon co-transfection with wild-type-ZNF341, whereas only marginal activation (average 1.4-fold) was observed with the R302* mutant ZNF341 after 48 hours (Fig. 5A+B). Increased reporter activity (average 2.6-fold) was also observed with the overexpressed ZNF341 variant R386* (which yielded much higher expression levels than the wild-type protein (fig. S4C)), suggesting that residual transcription factor activity is retained (Fig. 5A+B). Similar results were obtained with longer (−997/−33) and shorter (−476/−33) STAT3 promoter fragments and at variable time points (fig. S5).
Fig. 5. ZNF341 binds to the STAT3 promoter.
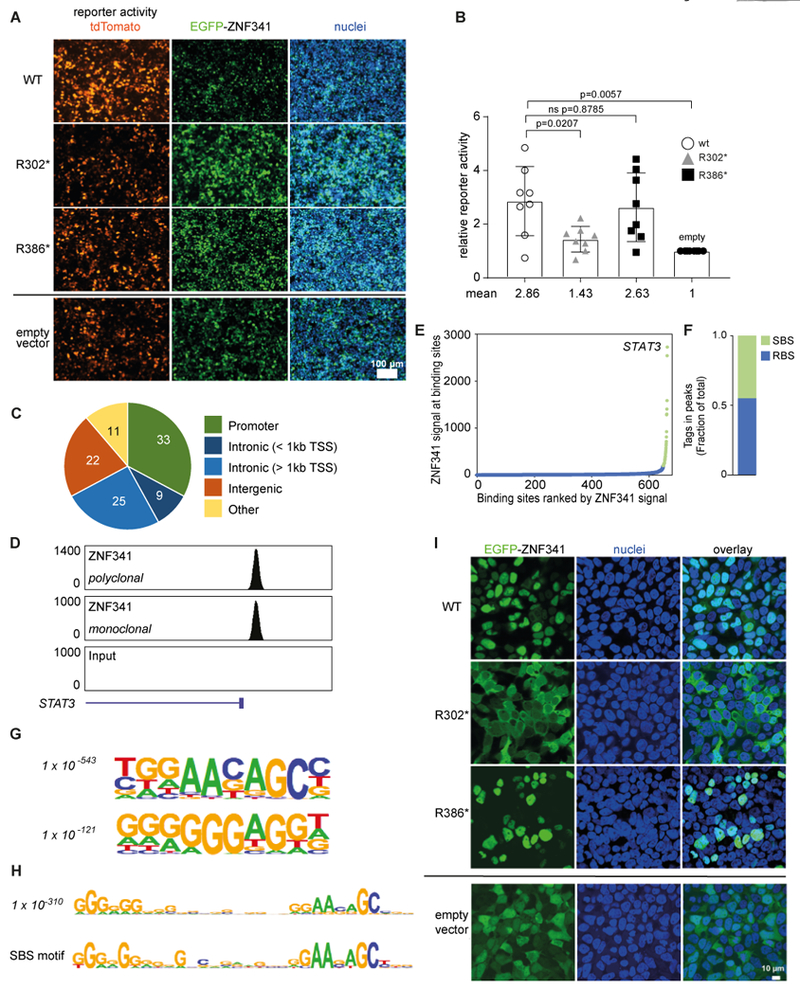
(A) Activation of a synthetic STAT3 promoter (with the −535/−33 upstream genomic sequence fused to CMV minimal promoter) driving a red fluorescence (tdTomato) reporter upon co-transfection with EGFP-tagged wildtype ZNF341 in HEK293T cells (48h). Scale bar 100μM. (B) Relative reporter activity in two independent experiments in quadruplets. Significance and p-values were determined with Mann-Whitney test. (C) ChIP-Seq analysis of ZNF341, performed with distinct antibodies, on EBV-transformed B cells reveal 1658 ZNF341 binding sites across various genomic regions and (D) show high tag densities on the STAT3 promoter region (Chr17: 40,530,000–40,545,000, hg19 build; normalized tags). (E) Distribution of ZNF341 ChIP-Seq signal across the 1658 binding sites. Green, 36 super-binding sites (SBS) with >200 normalized tags; blue, remaining binding sites (RBS). (F) Normalized tags in SBS and RBS as a fraction of total. (G) Cis-regulatory sequences associated with ZNF341 occupancy. P values (italics) reflect the significance of motif occurrence. (H) A 30-nt cis-regulatory sequence associated with ZNF341 occupancy (P value as above) and with ZNF341 occupancy in SBS. (I) Representative confocal images of transfected HEK293T (48h) showing nuclear localization of EGFP-tagged wild-type ZNF341 and R386*, whereas R302* remains cytoplasmic. Scale bar, 10 μM.
To determine whether ZNF341 directly regulates STAT3 by binding to its promoter, we searched for ZNF341 binding sites by ChIP-Seq. ChIP-Seq data, obtained with two different antibodies recognizing ZNF341, were highly correlated (fig. S6A). We identified 1,658 high-confidence ZNF341 binding sites genome-wide, with a high proportion being located at promoters or within short distance to promoters (< 1 kb) (Fig. 5C). The STAT3 promoter displayed high ZNF341 occupancy (Fig. 5D), which was confirmed by ChIP (fig. S6B). This was not observed in patient-derived cells, highlighting the specificity of the antibody as well (fig. S6C). Thirty-six binding sites, termed super-binding sites, were characterized by high level of ZNF341 occupancy (Fig. 5E) and accounted for approximately half of ZNF341 occupancy (as determined by normalized tag count) (Fig. 5F). Expression of the genes associated with the top two binding sites (STAT3 and KAT6A) was decreased in patient-derived PBMCs compared to a healthy control, further supporting the role of ZNF341 as a transcriptional activator (table S3). To identify the sequence recognized by ZNF341, we performed de novo motif analysis. A 10-nt motif was highly enriched at ZNF341 binding sites, as well as a 10-nt G-rich motif (Fig. 5G). When searching for longer motifs, we identified a 30-nt sequence, which contained both 10-nt motifs (Fig. 5H, top panel), located at position −217/−187 in the STAT3 promoter region (relative to the transcription start). This motif could be further refined in the super-binding sites (Fig. 5H, lower panel), suggesting that ZNF341 utilizes several zinc fingers for DNA binding, whereas the remaining zinc fingers may contribute to preferential binding. This might explain why half of ZNF341 occupancy occurs at only 36 preferential binding sites. Thus, ZNF341 has the potential to recognize a highly specific sequence and therefore to regulate a limited number of genes, including STAT3.
Aberrant cytoplasmic localization of nuclear ZNF341 caused by the R302* mutation
We further characterized the molecular defects of the ZNF341 nonsense mutations by transient overexpression in HEK293T cells (fig. S4). The wild-type ZNF341 achieved moderate protein levels (regardless of whether fused to EGFP or not) whereas both truncated proteins (R302* and R386*) were expressed at robust levels (fig. S4B+C; for expression levels see also fig. S7C+D). Confocal microscopy (Fig. 5I) of EGFP-fused constructs showed that the wild-type ZNF341 localized to the nucleus, as predicted by the protein atlas. In contrast, truncated EGFP-ZNF341-R302* remained in the cytoplasm, indicating that the nuclear localization sequence (NLS) was deleted. Thus, the inability of the R302* mutant to contribute to transcriptional activation is most likely due to the failure of nuclear translocation and accordingly we did not detect any binding of ZNF341 variant R302* to chromatin (fig. S6C, S7A+B). Surprisingly, the truncated mutant EGFP-ZNF341-R386* retained its ability to localize to the nucleus (Fig. 5I). Thus, a potential NLS, which is predicted to reside between K299 and Y326 (http://nls-mapper.iab.keio.ac.jp/cgi-bin/NLS_Mapper_form.cgi), is sufficient for nuclear translocation of ZNF341-R386*. Nevertheless, the ZNF341-R386* variant showed reduced binding to chromatin in an overexpression system (fig. S7A+B). Because ZNF341-R386* lacks nine of the twelve zinc finger domains, residual transcription factor activity may occur only upon massive overexpression in vitro. The clinical phenotype however, suggests that the three remaining zinc fingers are insufficient for ZNF341-R386* binding to and activation of target promoters at physiological levels.
DISCUSSION
JAK-STAT signaling pathways have emerged as critical rheostats for the maintenance of cellular homeostasis, relevant to many human diseases (27–29). Impaired JAK-STAT signaling either causes immunodeficiency if decreased (30–32) or lymphoproliferative disorders, if increased (8, 9, 33). Hence, the regulation of the amount of STAT (and JAK) signaling molecules and their phosphorylation status seem to determine the outcome of cellular signaling events. For example LOF mutations in STAT1 lead to susceptibility to viral and mycobacterial infections (34, 35), whereas GOF mutations in the very same gene lead to a polarization of T lymphocytes away from the Th17 CD4+ T cell lineage towards the Th1 lineage, leading to an increased susceptibility to recurrent fungal infections (36–38) .
Furthermore, decreased STAT3 signaling, due to dominant-negative mutations (10, 12), leads to immunodeficiency characterized by the loss of Th17 CD4+ T cells (3–5). In contrast, somatic GOF mutations in STAT3 lead to large-granular-lymphocytosis (39), and germline GOF mutations in STAT3 lead to a complex immune-dysregulation syndrome (8, 9). Therefore, the mechanisms controlling these cellular regulators (i.e., the transcription factors of the JAK and STAT molecules) become of central importance in human disease. JAK- and STAT inhibitors have already been used to treat lymphoma/cancer, autoimmune diseases such as rheumatoid arthritis (40), and impaired infection control (41, 42).
Here, we describe a novel transcription factor regulating transcription of STAT3: ZNF341. As in HIES patients with STAT3 LOF mutations, the impairment of ZNF341 signaling also led to the defect of naïve T cells to differentiate into Th17 cells (and hence IL17 production), a process critically dependent on STAT3. Thus, we discovered that ZNF341 transcriptionally regulates STAT3 expression, at least its increase above a certain basal level of transcription, but other aspects of the underlying mechanism of impaired JAK-STAT signaling due to homozygous nonsense mutations in ZNF341 still need to be elucidated. Although our patients presented with high IgE levels, to date, ZNF341 has not been associated with atopy related traits such as asthma, rhinitis, atopic dermatitis, and allergic sensitization (43–45). As ZNF341 comprises 12 zinc fingers, and as one zinc finger shows preference to a specific nucleotide triplet (46), ZNF341 has the potential to recognize a very specific sequence and regulate a limited number of genes, which is supported by the super-binding sites and motif analysis. In this regard, KAT6A, the gene whose promoter has the second top-binding site by ChIP, was down-regulated in our transcriptome analysis (table S3). Interestingly, mutations in KAT6A were recently identified in patients presenting with neurodevelopmental disorders (47). Lower KAT6A expression may contribute to the intellectual disabilities observed in our patients. Our data show that ZNF341 acts as a transcriptional activator for cytokine-mediated STAT3 expression. Zinc finger transcription factors lacking other functional domains have been described to recruit either transcriptional activators or repressors or both (48). Overexpressed ZNF341 has been described to bind to PAF1, which plays a role in transcriptional elongation (48), supporting a role for ZNF341 as a transcriptional activator.
In summary, the novel transcription factor ZNF341 is a positive regulator of STAT3 expression. Homozygous nonsense mutations in ZNF341 lead to insufficient STAT3 levels, which prevent Th17 cell differentiation and cause HIES-like phenotypes with recurrent infections. Thus, the well-known HIES phenotype is not only associated with well-described dominant-negative STAT3 mutations, but can also result from insufficient levels of otherwise normal STAT3.
Our observations also have pivotal importance regarding future intervention strategies in STAT3-dependent HIES. Since HIES can be caused by STAT3 insufficiency, gene therapeutic attempts aiming at the inactivation of the mutant allele to eliminate dominant-negative STAT3 mutations, should be carefully considered. On the other hand, treating STAT3-insufficient cells with recombinant ZNF341 might improve infection control. An interesting aspect in future research will be the link between ZNF341 and the maintenance of normal IgE levels, particularly in the context of allergy. Our patients with ZNF341 deficiency had highly elevated serum IgE levels. Hence, augmentation of ZNF341 function may normalize IgE production, possibly by interfering with Th2 cell subset differentiation.
MATERIALS AND METHODS
Experimental Design
The aim of this study was to characterize the underlying genetic defect and pathomechanism in a consanguineous AR-HIES family without mutations in known HIES genes. After the identification of ZNF341 (encoding a transcription factor regulating STAT3) as the disease-causing gene in Family A, additional families with unexplained AR-HIES were tested for ZNF341 mutations.
Patients and Controls
The study was conducted under protocols for human subjects. Samples were collected with the written consent of all study participants and/or their parental guardians after formal ethical approval by the local ethics committees at the University of Freiburg (ethics protocol numbers 239/99_120733 and 302/13), the Rambam Medical Center, the Children´s Medical Center of Israel, Uludag University Medical Faculty, and collaborating institutions. Healthy family members were sequenced for the ZNF341 mutation and wt/wt family members were included into the healthy donor (HD) control group.
Sample preparation
Human PBMCs were isolated by Ficoll density gradient centrifugation and either immediately used or frozen and stored in liquid nitrogen. Epstein-Barr virus (EBV) transformed lymphoblastic B cell lines and Herpesvirus saimiri immortalized T- cell lines (HVS-T) were generated from PBMCs by standard methods. Primary skin fibroblasts (PSF) were generated from a skin biopsy of patient A.II.3 by standard methods and healthy donor PSF were kindly provided by AG Finkenzeller.
Flow cytometry
Immunophenotyping of PBMCs was performed by staining for various cell surface markers. A list of the fluorochrome-conjugated antibodies and the applied gating strategy can be found in supplement S7.
STAT3 phosphorylation
For analysis of STAT3 expression and STAT3 phosphorylation, PBMCs were used either unstimulated or stimulated with human recombinant IL-6 (0.5μg/ml, PeproTech, Hamburg, Germany) for 15 min at 37°C. EBV-transformed B cell lines were stimulated with IFN-α (0.5μg/ml, PeproTech, Hamburg, Germany) for 5, 15, 60 and 150 min at 37°C. Cells were fixed (Lyse/Fix buffer, BD Biosciences) and permeabilized (Fix/Perm III buffer, BD Biosciences) according to the manufacturer΄s instructions. Cells were stained with anti-STAT3-FITC (clone #232209; R&D Systems Wiesbaden-Nordenstadt, Germany), anti CD19 BV421 (clone HIB19, Biolegend), or with phospho-specific PE-coupled anti-STAT3 (pY705) antibodies (clone 4/P-STAT3, BD Biosciences), CD3-FITC (clone SK7, BD Bioscience). Fixable viability dye eFluor 506 (eBioscience, Frankfurt, Germany) was used according to the manufacturer΄s instructions.
In vitro Th17 cell differentiation
For in vitro differentiation assays, freshly purified PBMCs were stimulated with anti-CD2, anti-CD3 and anti-CD28 coated beads, using a T cell activation/expansion kit according to the manufacturer΄s instructions (Miltenyi Biotec, Bergisch Gladbach, Germany), in combination with IL-1ß (10ng/ml) and IL-6 (50ng/ml) or TGF-beta (5ng/ml) and IL-21 (25ng/ml) for four days. Prior to and after induction, cells were stimulated for 4 hours with 50ng/ml PMA and 1μg/ml ionomycin (both from Sigma) in the presence of 5μg/ml Brefeldin A (BD Biosciences). Cells were stained for surface markers with anti-CD4 PercP-Cy5.5 (clone RPTA-T4, BD Biosciences), anti-CD45RO PE-Cy7 (clone UCHL1, ebiosciences) and anti-CD3 APC H7 (clone SK7, BD Bioscience), fixed and permeabilized using Cytofix kit (BD Biosciences). For intracellular staining anti-IFN-γ FITC (Clone B27, BD Biosciences) and anti-IL-17 PE (Clone eBio64DEC17, eBioscience) were used. Fixable viability dye eFluor 506 (eBioscience, Frankfurt, Germany) was used according to the manufacturer΄s instructions.
All flow cytometry data was acquired on a FACS-Canto II flow cytometer (BD Biosciences) and analyzed using FlowJo version X analysis software (Treestar, Ashland, OR).
RNA isolation, cDNA generation, quantitative PCR and cDNA sequencing
Total RNA, either from PBMCs, EBV cell lines, HVS transformed T cell lines, PSF, or HEK293T cells, was isolated with the RNeasy Mini Kit using QIAshredder and column DNA digestion (Qiagen). cDNA was synthesized with QuantiTect reverse transcription kit (Qiagen). qPCR was performed in duplicates or triplicates using SYBR Green reagents (Qiagen or Thermo Scientific) and primers for STAT3, ZNF341 and for the house-keeping gene GUSB. Fluorescence intensities were monitored over 40 cycles on a StepOne real-time PCR system (Applied Biosystems/Thermo Fisher Scientific) and relative mRNA expression was calculated with the 2−ΔΔCt method. The STAT3 cDNA sequences were analyzed using long range PCRs with Q5 High-Fidelity DNA Polymerase (NEB) and Sanger sequencing. The most important primer sequences can be found at the end of Supplemental methods and all primer sequences are available upon request.
Western blotting
Fresh or frozen PBMCs, EBV transformed B cells, PSF, or transfected HEK293T lysates were used for blotting. Membranes were probed with primary antibodies separately for STAT3 (MAB1799; R&D Systems), ZNF341 (polyclonal: Atlas Antibodies HPA024607 or monoclonal: customized antibody), GAPDH (G9295; Sigma), or beta-actin (NB 600–532; Novus Biologicals). A detailed protocol can be found in supplement S7.
CRISPR/Cas9-mediated genome editing
ZNF341 knock-out cells were generated using CRISPR/Cas9 technology. A guide RNAs (gRNA) with high target specificity was selected using the CRISPR design tool from MIT (http://crispr.mit.edu). The gRNA (CGTCGGGTCTTCAGCGTTGC) was targeting a region close to R302 and was cloned in the pX330-U6-Chimeric_BB-CBh-hSpCas9 plasmid, a gift from Feng Zhang (Addgene plasmid #42230). ZNF341−/− clones were generated by single cell sorting of EGFP-positive RAMOS cells co-transfected with pX330-ZNF341-gRNA and a EGFP plasmid (1:10 ratio) in 96-wells plates. After about three weeks, clones were screened by Western Blot analysis.
Expression vectors and fluorescence-based promoter-reporter assay
The cDNAs for isoform 1 of wild-type ZNF341 and mutant ZNF341-R302* were cloned from patient derived samples by RT-PCR. Mutant ZNF341-R386* was generated by site-directed mutagenesis. For transient transfection in HEK293T cells, expression vectors for EGFP-fused or non-fused versions were used (pEGFP-C1 and –N1; Takara/Clontech) with the empty vectors as controls. Cells were transfected with XtremeGeneHP reagent (Roche) and harvested 48 h post transfection for RNA or protein analyses.
For the fluorescence-based promoter-reporter assay, three genomic fragments of the human STAT3 promoter, comprising the upstream sequence from −997 to-33, from −535 to −33 or from −476 to −33 relative to the putative transcription start site (49), were amplified by PCR and fused to a 58 bp CMV minimal promoter (without specific transcription factor binding sites). The synthetic promoter constructs provide a TATA box and an artificial transcription start site and drive the expression of a red fluorescent tdTomato reporter protein (Takara/Clontech). Expression vectors for non-fused or EGFP-fused wild-type ZNF341, mutant ZNF341-R302*, or mutant ZNF341-R386*, respectively, were co-transfected with the reporter into HEK293T cells using XtremeGeneHP (Roche) transfection reagent. The empty vector pEGFP-C1 (without ZNF341 cDNA) was used as control. Reporter assays (n=4) were performed in 48 well plates in quadruplets and evaluated with a Fluorospot reader at variable time points (48h/72h/96h; repeated measurements per plate at two distinct time points each). Reporter constructs containing only the CMV minimal promoter (without STAT3 upstream sequences) and RORC- and IL17A- promoter constructs (both fused to CMVmin) were confirmed to be non-activatable by ZNF341 in independent experiments. Cells were analyzed for green (expression and subcellular localization of GFP-fused ZNF341) and red fluorescence (reporter activity) by conventional epi-fluorescence microscopy on a Zeiss Axiovert 200. Images were processed using the Zen software (Zeiss). Fluorescence intensities were measured on a Fluorospot analyzer (CTL, Bonn Germany) and quantified using ImageJ software.
ChIP and ChIP-Seq
Cells were fixed for 15 minutes in 1% formaldehyde in PBS with 2% FCS. Formaldehyde was quenched for 10 minutes with 0.2M glycine. Cells were washed 2 times with ice-cold PBS. 30μl Protein G Dynabeads (Life Technologies) were blocked with 0.5% BSA (w/v) in PBS. Magnetic beads were bound with 5μg of anti-ZNF341 antibody (polyclonal: Atlas Antibodies HPA024607 or monoclonal: customized antibody) or control rabbit IgG (Santa Cruz Biotechnology sc2027x). Crosslinked cells were lysed in lysis buffer (0.5% NP-40, 10mM HEPES, 85mM KCl, 4ul EDTA). After centrifugation, nuclei were resuspended and sonicated in sonication buffer (50mM Tris-HCl pH8.0, 1% SDS, 10mM EDTA) for 5 cycles at 10 sec each on ice (20W) with 50 sec on ice between cycles. Lysates were cleared by centrifugation and Triton X-100 was added at a final concentration of 1%. Lysates were then incubated overnight at 4°C with the previously prepared magnetic beads. Beads were washed once with RIPA (50mM Tris-HCl pH8.0, 150mM NaCl, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100, 1mM EDTA), once with RIPA 500 (50mM Tris-HCl pH8.0, 500mM NaCl, 0.1% SDS, 0.1% Na-Deoxycholate, 1% Triton X-100, 1mM EDTA), once with LiCl wash (10mM Tris-HCl pH8.0, 250mM LiCl, 0.5% NP-40, 0.5% Na-deoxycholate, 1mM EDTA) and finally twice with TE (10mM Tris pH8.0, 1mM EDTA). Bound complexes were eluted from the beads in elution buffer (10mM Tris-HCl pH8.0, 0.5% SDS, 300mM NaCl, 5mM EDTA) for 30 min at 65 °C with shaking. Crosslinks were reversed overnight at 65°C. RNA and protein were digested in the supernatant using RNase A and Proteinase K. DNA was purified using ChIP DNA clean and concentrator columns (Zymoresearch). ChIP primers are listed in supplement S6. ChIP results are represented as percent of input by dividing the signals obtained by the ChIP by the signals obtained from the input sample. For ChIP-Seq, libraries were prepared with the NEBNext primer set and were size-selected with AMPure XP beads (Beckman Coulter). Libraries were run on Illumina HiSeq 2000. Reads were aligned to hg19 using Bowtie with the parameter –m 1 (http://bowtie-bio.sourceforge.net). Data were analyzed using HOMER (http://homer.salk.edu/homer). To correlate ChIP-Seq data obtained with monoclonal and polyclonal antibodies, peaks were identified using the findPeaks command in the combined tag directory and annotated with each tag directory using the –log option. A Pearson correlation test was applied to identify correlation between the tag counts. High-confidence peaks were identified using the getDifferentialPeaksReplicates.pl command using the ChIP-Seq data obtained with the two antibodies as replicates. Motifs of 8, 10 or 12-nt were identified using the findMotifsGenome.pl command with parameter –cpg in a window of 100 bp (-size 100). Longer motifs were identified by searching for motifs of lengths more than 15-nt and top motif was identified using a length of 30 (-len 30). Optimization of the long motif in the super-binding sites was performed with parameter –opt.
Statistical analysis
Data (not including genetic linkage analysis, transcriptome study and motif analysis) were analyzed with the GraphPad InStat software program, version 6, by using nonparametric Mann-Whitney tests. Differences were considered significant at a P value of less than 0.05.
Detailed protocols for Genetic linkage analysis, Whole exome sequencing (WES), Next Generation Sequencing (NGS), Sanger sequencing, CGH Array, and Transcriptome study can be found in Supplementary material and methods (S6).
Supplementary Material
Case reports for Families A-D.
Defective B cell development and Th17 cell differentiation in HIES patients with ZNF341 nonsense mutations.
Exclusion of mutations in STAT3 in the etiology of the HIES phenotype in Family A.
Identification of ZNF341 as an AR-HIES-associated gene in a linkage region on chromosome 20 by whole exome sequencing.
Expression of various ZNF341 mRNA variants in multiple cell lines and PBMCs.
Supplementary materials and methods
B and T cell subsets in patient-derived and healthy donor PBMCs.
Ubiquitous expression of ZNF341 mRNA in various cell types.
No association of the genomic region 20q11.22 spanning the ZNF341 locus with atopy.
Ectopic expression of ZNF341 variants in HEK293T cells.
Ectopically expressed wild-type ZNF341 binds to the STAT3 promoter leading to transcriptional activation.
Occupancy by ZNF341 across the STAT3 promoter as determined by ChIP.
ZNF341 R386* shows reduced binding to the STAT3 promoter.
FACS gating strategy for in vitro Th17 cell differentiation assay.
FACS gating strategy for Y705-phosphorylation of STAT3.
FACS gating strategy for immune phenotyping of PBMCs.
FACS gating strategy for IL22+ T cells.
Clinical and immunological phenotype of HIES patients with ZNF341 mutations.
ZNF341 encodes three protein coding isoforms.
Transcriptome Analysis of patient A.II.1 with gene-specific fold changes in comparison to healthy sibling A.II.5.
Acknowledgements:
We are deeply grateful to all affected individuals, their families and all healthy donor controls who participated in this study. We thank Dörte Thiel, Pavla Mrovecova, Katrin Hübscher, Mary Buchta, Hanna Haberstroh, and Nadine Glaser for their excellent technical assistance. We thank Monika Schmidt and Ingrid Müller-Fleckenstein for generating patients’ T cell lines at the Institute of Clinical and Molecular Virology, University of Erlangen-Nürnberg. We thank Prof. Günter Finkenzeller for kindly providing primary skin fibroblasts of a healthy donor from the Plastic Surgery, Medical Center, University of Freiburg. We thank Dr. Michael Leipold for performing the CGH array (Supplement S3) at the Institute of Human Genetics, University of Freiburg. Sequencing for ChIP-Seq was conducted at the Genomics Core Facility of the European Molecular Biology Laboratories (EMBL). We thank the microarray unit of the DKFZ Genomics and Proteomics Core Facility for providing the Illumina Whole-Genome Expression Beadchips (Transcriptome study) and related services.
Funding: Financial support for this research came from the German Ministry of Education and Research (BMBF, grant # 01E01303, sysINFLAME grants # 01ZX1306F and 01ZX1306A), from the Intramural Research Program of the National Institutes of Health, NLM, from the E-rare program of the European Commission EURO-CMC (01GM1502), from the German Center for Infection Research DZIF: 8000805–3 and TTU IICH 07.801, and from the Deutsche Forschungsgemeinschaft (DFG; SFB1160-IMPATH). The funding organizations had no role in study design, the collection, analysis and interpretation of data, the writing of the report, nor the decision to submit the paper for publication. The authors are responsible for the content of this research. J.H. is a fellow of the MOTI-VATE graduate program of the Medical Faculty of Freiburg.
Footnotes
REFERENCES AND NOTES
- 1.Hillmer EJ, Zhang H, Li HS, Watowich SS, STAT3 signaling in immunity. Cytokine & growth factor reviews 31, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al Khatib S, Keles S, Garcia-Lloret M, Karakoc-Aydiner E, Reisli I, Artac H, Camcioglu Y, Cokugras H, Somer A, Kutukculer N, Yilmaz M, Ikinciogullari A, Yegin O, Yuksek M, Genel F, Kucukosmanoglu E, Baki A, Bahceciler NN, Rambhatla A, Nickerson DW, McGhee S, Barlan IB, Chatila T, Defects along the T(H)17 differentiation pathway underlie genetically distinct forms of the hyper IgE syndrome. The Journal of allergy and clinical immunology 124, 342–348, 348 e341–345 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL, Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. The Journal of experimental medicine 205, 1543–1550 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC, Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. The Journal of experimental medicine 205, 1551–1557 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC, Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452, 773–776 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD, Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. The Journal of allergy and clinical immunology 122, 181–187 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yong PF, Freeman AF, Engelhardt KR, Holland S, Puck JM, Grimbacher B, An update on the hyper-IgE syndromes. Arthritis research & therapy 14, 228 (2012)10.1186/ar4069). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanagan SE, Haapaniemi E, Russell MA, Caswell R, Lango Allen H, De Franco E, McDonald TJ, Rajala H, Ramelius A, Barton J, Heiskanen K, Heiskanen-Kosma T, Kajosaari M, Murphy NP, Milenkovic T, Seppanen M, Lernmark A, Mustjoki S, Otonkoski T, Kere J, Morgan NG, Ellard S, Hattersley AT, Activating germline mutations in STAT3 cause early-onset multi-organ autoimmune disease. Nature genetics 46, 812–814 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Milner JD, Vogel TP, Forbes L, Ma CA, Stray-Pedersen A, Niemela JE, Lyons JJ, Engelhardt KR, Zhang Y, Topcagic N, Roberson ED, Matthews H, Verbsky JW, Dasu T, Vargas-Hernandez A, Varghese N, McClain KL, Karam LB, Nahmod K, Makedonas G, Mace EM, Sorte HS, Perminow G, Rao VK, O’Connell MP, Price S, Su HC, Butrick M, McElwee J, Hughes JD, Willet J, Swan D, Xu Y, Santibanez-Koref M, Slowik V, Dinwiddie DL, Ciaccio CE, Saunders CJ, Septer S, Kingsmore SF, White AJ, Cant AJ, Hambleton S, Cooper MA, Early-onset lymphoproliferation and autoimmunity caused by germline STAT3 gain-of-function mutations. Blood 125, 591–599 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B, STAT3 mutations in the hyper-IgE syndrome. The New England journal of medicine 357, 1608–1619 (2007). [DOI] [PubMed] [Google Scholar]
- 11.Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, Malech HL, Miller JA, O’Connell AC, Puck JM, Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. The New England journal of medicine 340, 692–702 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H, Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448, 1058–1062 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Engelhardt KR, McGhee S, Winkler S, Sassi A, Woellner C, Lopez-Herrera G, Chen A, Kim HS, Lloret MG, Schulze I, Ehl S, Thiel J, Pfeifer D, Veelken H, Niehues T, Siepermann K, Weinspach S, Reisli I, Keles S, Genel F, Kutukculer N, Camcioglu Y, Somer A, Karakoc-Aydiner E, Barlan I, Gennery A, Metin A, Degerliyurt A, Pietrogrande MC, Yeganeh M, Baz Z, Al-Tamemi S, Klein C, Puck JM, Holland SM, McCabe ER, Grimbacher B, Chatila TA, Large deletions and point mutations involving the dedicator of cytokinesis 8 (DOCK8) in the autosomal-recessive form of hyper-IgE syndrome. The Journal of allergy and clinical immunology 124, 1289–1302 e1284 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Davis JC, Lamborn IT, Freeman AF, Jing H, Favreau AJ, Matthews HF, Davis J, Turner ML, Uzel G, Holland SM, Su HC, Combined immunodeficiency associated with DOCK8 mutations. The New England journal of medicine 361, 2046–2055 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sassi A, Lazaroski S, Wu G, Haslam SM, Fliegauf M, Mellouli F, Patiroglu T, Unal E, Ozdemir MA, Jouhadi Z, Khadir K, Ben-Khemis L, Ben-Ali M, Ben-Mustapha I, Borchani L, Pfeifer D, Jakob T, Khemiri M, Asplund AC, Gustafsson MO, Lundin KE, Falk-Sorqvist E, Moens LN, Gungor HE, Engelhardt KR, Dziadzio M, Stauss H, Fleckenstein B, Meier R, Prayitno K, Maul-Pavicic A, Schaffer S, Rakhmanov M, Henneke P, Kraus H, Eibel H, Kolsch U, Nadifi S, Nilsson M, Bejaoui M, Schaffer AA, Smith CI, Dell A, Barbouche MR, Grimbacher B, Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IgE levels. The Journal of allergy and clinical immunology 133, 1410–1419, 1419 e1411–1413 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Yu X, Ichikawa M, Lyons JJ, Datta S, Lamborn IT, Jing H, Kim ES, Biancalana M, Wolfe LA, DiMaggio T, Matthews HF, Kranick SM, Stone KD, Holland SM, Reich DS, Hughes JD, Mehmet H, McElwee J, Freeman AF, Freeze HH, Su HC, Milner JD, Autosomal recessive phosphoglucomutase 3 (PGM3) mutations link glycosylation defects to atopy, immune deficiency, autoimmunity, and neurocognitive impairment. The Journal of allergy and clinical immunology 133, 1400–1409, 1409 e1401–1405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keles S, Charbonnier LM, Kabaleeswaran V, Reisli I, Genel F, Gulez N, Al-Herz W, Ramesh N, Perez-Atayde A, Eeder NK, Kutukculer N, Wu H, Geha RS, Chatila TA, Dedicator of cytokinesis 8 regulates signal transducer and activator of transcription 3 activation and promotes TH17 cell differentiation. The Journal of allergy and clinical immunology, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma CS, Avery DT, Chan A, Batten M, Bustamante J, Boisson-Dupuis S, Arkwright PD, Kreins AY, Averbuch D, Engelhard D, Magdorf K, Kilic SS, Minegishi Y, Nonoyama S, French MA, Choo S, Smart JM, Peake J, Wong M, Gray P, Cook MC, Fulcher DA, Casanova JL, Deenick EK, Tangye SG, Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 119, 3997–4008 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shuai K, Liu B, Regulation of JAK-STAT signalling in the immune system. Nature reviews. Immunology 3, 900–911 (2003). [DOI] [PubMed] [Google Scholar]
- 20.Xu YJ, Li L, Chen Y, Fu B, Wu DS, Li XL, Zhao XL, Chen FP, Role of HMGB1 in regulation of STAT3 expression in CD4(+) T cells from patients with aGVHD after allogeneic hematopoietic stem cell transplantation. Clinical immunology 161, 278–283 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Cheng Y, Geng H, Cheng SH, Liang P, Bai Y, Li J, Srivastava G, Ng MH, Fukagawa T, Wu X, Chan AT, Tao Q, KRAB zinc finger protein ZNF382 is a proapoptotic tumor suppressor that represses multiple oncogenes and is commonly silenced in multiple carcinomas. Cancer research 70, 6516–6526 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Lin L, Yao Z, Bhuvaneshwar K, Gusev Y, Kallakury B, Yang S, Shetty K, He AR, Transcriptional regulation of STAT3 by SPTBN1 and SMAD3 in HCC through cAMP-response element-binding proteins ATF3 and CREB2. Carcinogenesis 35, 2393–2403 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Kluger MA, Melderis S, Nosko A, Goerke B, Luig M, Meyer MC, Turner JE, Meyer-Schwesinger C, Wegscheid C, Tiegs G, Stahl RA, Panzer U, Steinmetz OM, Treg17 cells are programmed by Stat3 to suppress Th17 responses in systemic lupus. Kidney international 89, 158–166 (2016). [DOI] [PubMed] [Google Scholar]
- 24.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, Curtin JA, Bonnelykke K, Tian C, Takahashi A, Esparza-Gordillo J, Alves AC, Thyssen JP, den Dekker HT, Ferreira MA, Altmaier E, Sleiman PM, Xiao FL, Gonzalez JR, Marenholz I, Kalb B, Pino-Yanes M, Xu CJ, Carstensen L, Groen-Blokhuis MM, Venturini C, Pennell CE, Barton SJ, Levin AM, Curjuric I, Bustamante M, Kreiner-Moller E, Lockett GA, Bacelis J, Bunyavanich S, Myers RA, Matanovic A, Kumar A, Tung JY, Hirota T, Kubo M, McArdle WL, Henderson AJ, Kemp JP, Zheng J, Smith GD, Ruschendorf F, Bauerfeind A, Lee-Kirsch MA, Arnold A, Homuth G, Schmidt CO, Mangold E, Cichon S, Keil T, Rodriguez E, Peters A, Franke A, Lieb W, Novak N, Folster-Holst R, Horikoshi M, Pekkanen J, Sebert S, Husemoen LL, Grarup N, de Jongste JC, Rivadeneira F, Hofman A, Jaddoe VW, Pasmans SG, Elbert NJ, Uitterlinden AG, Marks GB, Thompson PJ, Matheson MC, Robertson CF, Ried JS, Li J, Zuo XB, Zheng XD, Yin XY, Sun LD, McAleer MA, O’Regan GM, Fahy CM, Campbell L, Macek M, Kurek M, Hu D, Eng C, Postma DS, Feenstra B, Geller F, Hottenga JJ, Middeldorp CM, Hysi P, Bataille V, Spector T, Tiesler CM, Thiering E, Pahukasahasram B, Yang JJ, Imboden M, Huntsman S, Vilor-Tejedor N, Relton CL, Myhre R, Nystad W, Custovic A, Weiss ST, Meyers DA, Soderhall C, Melen E, Ober C, Raby BA, Simpson A, Jacobsson B, Holloway JW, Bisgaard H, Sunyer J, Probst-Hensch NM, Williams LK, Godfrey KM, Wang CA, Boomsma DI, Melbye M, Koppelman GH, Jarvis D, McLean WH, Irvine AD, Zhang XJ, Hakonarson H, Gieger C, Burchard EG, Martin NG, Duijts L, Linneberg A, Jarvelin MR, Nothen MM, Lau S, Hubner N, Lee YA, Tamari M, Hinds DA, Glass D, Brown SJ, Heinrich J, Evans DM, Weidinger S, Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nature genetics 47, 1449–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonnelykke K, Matheson MC, Pers TH, Granell R, Strachan DP, Alves AC, Linneberg A, Curtin JA, Warrington NM, Standl M, Kerkhof M, Jonsdottir I, Bukvic BK, Kaakinen M, Sleimann P, Thorleifsson G, Thorsteinsdottir U, Schramm K, Baltic S, Kreiner-Moller E, Simpson A, St Pourcain B, Coin L, Hui J, Walters EH, Tiesler CM, Duffy DL, Jones G, Ring SM, McArdle WL, Price L, Robertson CF, Pekkanen J, Tang CS, Thiering E, Montgomery GW, Hartikainen AL, Dharmage SC, Husemoen LL, Herder C, Kemp JP, Elliot P, James A, Waldenberger M, Abramson MJ, Fairfax BP, Knight JC, Gupta R, Thompson PJ, Holt P, Sly P, Hirschhorn JN, Blekic M, Weidinger S, Hakonarsson H, Stefansson K, Heinrich J, Postma DS, Custovic A, Pennell CE, Jarvelin MR, Koppelman GH, Timpson N, Ferreira MA, Bisgaard H, Henderson AJ, Meta-analysis of genome-wide association studies identifies ten loci influencing allergic sensitization. Nature genetics 45, 902–906 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H, The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research 42, D1001–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Leonard WJ, O’Shea JJ, Jaks and STATs: biological implications. Annual review of immunology 16, 293–322 (1998)10.1146/annurev.immunol.16.1.293). [DOI] [PubMed] [Google Scholar]
- 28.Levy DE, Darnell JE Jr., Stats: transcriptional control and biological impact. Nature reviews. Molecular cell biology 3, 651–662 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Villarino AV, Kanno Y, O’Shea JJ, Mechanisms and consequences of Jak-STAT signaling in the immune system. Nature immunology 18, 374–384 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Casanova JL, Holland SM, Notarangelo LD, Inborn errors of human JAKs and STATs. Immunity 36, 515–528 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ghoreschi K, Laurence A, O’Shea JJ, Janus kinases in immune cell signaling. Immunological reviews 228, 273–287 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vainchenker W, Dusa A, Constantinescu SN, JAKs in pathology: role of Janus kinases in hematopoietic malignancies and immunodeficiencies. Seminars in cell & developmental biology 19, 385–393 (2008). [DOI] [PubMed] [Google Scholar]
- 33.Liu L, Okada S, Kong XF, Kreins AY, Cypowyj S, Abhyankar A, Toubiana J, Itan Y, Audry M, Nitschke P, Masson C, Toth B, Flatot J, Migaud M, Chrabieh M, Kochetkov T, Bolze A, Borghesi A, Toulon A, Hiller J, Eyerich S, Eyerich K, Gulacsy V, Chernyshova L, Chernyshov V, Bondarenko A, Grimaldo RM, Blancas-Galicia L, Beas IM, Roesler J, Magdorf K, Engelhard D, Thumerelle C, Burgel PR, Hoernes M, Drexel B, Seger R, Kusuma T, Jansson AF, Sawalle-Belohradsky J, Belohradsky B, Jouanguy E, Bustamante J, Bue M, Karin N, Wildbaum G, Bodemer C, Lortholary O, Fischer A, Blanche S, Al-Muhsen S, Reichenbach J, Kobayashi M, Rosales FE, Lozano CT, Kilic SS, Oleastro M, Etzioni A, Traidl-Hoffmann C, Renner ED, Abel L, Picard C, Marodi L, Boisson-Dupuis S, Puel A, Casanova JL, Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. The Journal of experimental medicine 208, 1635–1648 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Averbuch D, Chapgier A, Boisson-Dupuis S, Casanova JL, Engelhard D, The clinical spectrum of patients with deficiency of Signal Transducer and Activator of Transcription-1. The Pediatric infectious disease journal 30, 352–355 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Boisson-Dupuis S, Kong XF, Okada S, Cypowyj S, Puel A, Abel L, Casanova JL, Inborn errors of human STAT1: allelic heterogeneity governs the diversity of immunological and infectious phenotypes. Current opinion in immunology 24, 364–378 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Baris S, Alroqi F, Kiykim A, Karakoc-Aydiner E, Ogulur I, Ozen A, Charbonnier LM, Bakir M, Boztug K, Chatila TA, Barlan IB, Severe Early-Onset Combined Immunodeficiency due to Heterozygous Gain-of-Function Mutations in STAT1. Journal of clinical immunology 36, 641–648 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dhalla F, Fox H, Davenport EE, Sadler R, Anzilotti C, van Schouwenburg PA, Ferry B, Chapel H, Knight JC, Patel SY, Chronic mucocutaneous candidiasis: characterization of a family with STAT-1 gain-of-function and development of an ex-vivo assay for Th17 deficiency of diagnostic utility. Clinical and experimental immunology 184, 216–227 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, Ouachee-Chardin M, Fouyssac F, Girisha KM, Etzioni A, Van Montfrans J, Camcioglu Y, Kerns LA, Belohradsky B, Blanche S, Bousfiha A, Rodriguez-Gallego C, Meyts I, Kisand K, Reichenbach J, Renner ED, Rosenzweig S, Grimbacher B, van de Veerdonk FL, Traidl-Hoffmann C, Picard C, Marodi L, Morio T, Kobayashi M, Lilic D, Milner JD, Holland S, Casanova JL, Puel A, S. G.-o.-F. S. G. International, Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood 127, 3154–3164 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Koskela HL, Eldfors S, Ellonen P, van Adrichem AJ, Kuusanmaki H, Andersson EI, Lagstrom S, Clemente MJ, Olson T, Jalkanen SE, Majumder MM, Almusa H, Edgren H, Lepisto M, Mattila P, Guinta K, Koistinen P, Kuittinen T, Penttinen K, Parsons A, Knowles J, Saarela J, Wennerberg K, Kallioniemi O, Porkka K, Loughran TP Jr., Heckman CA, Maciejewski JP, Mustjoki S, Somatic STAT3 mutations in large granular lymphocytic leukemia. The New England journal of medicine 366, 1905–1913 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwartz DM, Bonelli M, Gadina M, O’Shea JJ, Type I/II cytokines, JAKs, and new strategies for treating autoimmune diseases. Nature reviews. Rheumatology 12, 25–36 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higgins E, Al Shehri T, McAleer MA, Conlon N, Feighery C, Lilic D, Irvine AD, Use of ruxolitinib to successfully treat chronic mucocutaneous candidiasis caused by gain-of-function signal transducer and activator of transcription 1 (STAT1) mutation. The Journal of allergy and clinical immunology 135, 551–553 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Mossner R, Diering N, Bader O, Forkel S, Overbeck T, Gross U, Grimbacher B, Schon MP, Buhl T, Ruxolitinib Induces Interleukin 17 and Ameliorates Chronic Mucocutaneous Candidiasis Caused by STAT1 Gain-of-Function Mutation. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America 62, 951–953 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Bonnelykke K, Matheson MC, Pers TH, Granell R, Strachan DP, Alves AC, Linneberg A, Curtin JA, Warrington NM, Standl M, Kerkhof M, Jonsdottir I, Bukvic BK, Kaakinen M, Sleimann P, Thorleifsson G, Thorsteinsdottir U, Schramm K, Baltic S, Kreiner-Moller E, Simpson A, St Pourcain B, Coin L, Hui J, Walters EH, Tiesler CM, Duffy DL, Jones G, Ring SM, McArdle WL, Price L, Robertson CF, Pekkanen J, Tang CS, Thiering E, Montgomery GW, Hartikainen AL, Dharmage SC, Husemoen LL, Herder C, Kemp JP, Elliot P, James A, Waldenberger M, Abramson MJ, Fairfax BP, Knight JC, Gupta R, Thompson PJ, Holt P, Sly P, Hirschhorn JN, Blekic M, Weidinger S, Hakonarsson H, Stefansson K, Heinrich J, Postma DS, Custovic A, Pennell CE, Jarvelin MR, Koppelman GH, Timpson N, Ferreira MA, Bisgaard H, Henderson AJ, Australian Asthma Genetics C, Genetics EA, Lifecourse Epidemiology C, Meta-analysis of genome-wide association studies identifies ten loci influencing allergic sensitization. Nature genetics 45, 902–906 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Paternoster L, Standl M, Waage J, Baurecht H, Hotze M, Strachan DP, Curtin JA, Bonnelykke K, Tian C, Takahashi A, Esparza-Gordillo J, Alves AC, Thyssen JP, den Dekker HT, Ferreira MA, Altmaier E, Sleiman PM, Xiao FL, Gonzalez JR, Marenholz I, Kalb B, Pino-Yanes M, Xu CJ, Carstensen L, Groen-Blokhuis MM, Venturini C, Pennell CE, Barton SJ, Levin AM, Curjuric I, Bustamante M, Kreiner-Moller E, Lockett GA, Bacelis J, Bunyavanich S, Myers RA, Matanovic A, Kumar A, Tung JY, Hirota T, Kubo M, McArdle WL, Henderson AJ, Kemp JP, Zheng J, Smith GD, Ruschendorf F, Bauerfeind A, Lee-Kirsch MA, Arnold A, Homuth G, Schmidt CO, Mangold E, Cichon S, Keil T, Rodriguez E, Peters A, Franke A, Lieb W, Novak N, Folster-Holst R, Horikoshi M, Pekkanen J, Sebert S, Husemoen LL, Grarup N, de Jongste JC, Rivadeneira F, Hofman A, Jaddoe VW, Pasmans SG, Elbert NJ, Uitterlinden AG, Marks GB, Thompson PJ, Matheson MC, Robertson CF, Australian Asthma Genetics C, Ried JS, Li J, Zuo XB, Zheng XD, Yin XY, Sun LD, McAleer MA, O’Regan GM, Fahy CM, Campbell L, Macek M, Kurek M, Hu D, Eng C, Postma DS, Feenstra B, Geller F, Hottenga JJ, Middeldorp CM, Hysi P, Bataille V, Spector T, Tiesler CM, Thiering E, Pahukasahasram B, Yang JJ, Imboden M, Huntsman S, Vilor-Tejedor N, Relton CL, Myhre R, Nystad W, Custovic A, Weiss ST, Meyers DA, Soderhall C, Melen E, Ober C, Raby BA, Simpson A, Jacobsson B, Holloway JW, Bisgaard H, Sunyer J, Probst-Hensch NM, Williams LK, Godfrey KM, Wang CA, Boomsma DI, Melbye M, Koppelman GH, Jarvis D, McLean WH, Irvine AD, Zhang XJ, Hakonarson H, Gieger C, Burchard EG, Martin NG, Duijts L, Linneberg A, Jarvelin MR, Nothen MM, Lau S, Hubner N, Lee YA, Tamari M, Hinds DA, Glass D, Brown SJ, Heinrich J, Evans DM, Weidinger S, Genetics EA, Lifecourse Epidemiology Eczema C, Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nature genetics 47, 1449–1456 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Welter D, MacArthur J, Morales J, Burdett T, Hall P, Junkins H, Klemm A, Flicek P, Manolio T, Hindorff L, Parkinson H, The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic acids research 42, D1001–1006 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klug A, The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annual review of biochemistry 79, 213–231 (2010)10.1146/annurev-biochem-010909-095056). [DOI] [PubMed] [Google Scholar]
- 47.Millan F, Cho MT, Retterer K, Monaghan KG, Bai R, Vitazka P, Everman DB, Smith B, Angle B, Roberts V, Immken L, Nagakura H, DiFazio M, Sherr E, Haverfield E, Friedman B, Telegrafi A, Juusola J, Chung WK, Bale S, Whole exome sequencing reveals de novo pathogenic variants in KAT6A as a cause of a neurodevelopmental disorder. American journal of medical genetics. Part A 170, 1791–1798 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Schmitges FW, Radovani E, Najafabadi HS, Barazandeh M, Campitelli LF, Yin Y, Jolma A, Zhong G, Guo H, Kanagalingam T, Dai WF, Taipale J, Emili A, Greenblatt JF, Hughes TR, Multiparameter functional diversity of human C2H2 zinc finger proteins. Genome research 26, 1742–1752 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kato K, Nomoto M, Izumi H, Ise T, Nakano S, Niho Y, Kohno K, Structure and functional analysis of the human STAT3 gene promoter: alteration of chromatin structure as a possible mechanism for the upregulation in cisplatin-resistant cells. Biochimica et biophysica acta 1493, 91–100 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Grimbacher B, Schaffer AA, Holland SM, Davis J, Gallin JI, Malech HL, Atkinson TP, Belohradsky BH, Buckley RH, Cossu F, Espanol T, Garty BZ, Matamoros N, Myers LA, Nelson RP, Ochs HD, Renner ED, Wellinghausen N, Puck JM, Genetic linkage of hyper-IgE syndrome to chromosome 4. American journal of human genetics 65, 735–744 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finck A, Van der Meer JW, Schaffer AA, Pfannstiel J, Fieschi C, Plebani A, Webster AD, Hammarstrom L, Grimbacher B, Linkage of autosomal-dominant common variable immunodeficiency to chromosome 4q. European journal of human genetics : EJHG 14, 867–875 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Lopez-Herrera G, Tampella G, Pan-Hammarstrom Q, Herholz P, Trujillo-Vargas CM, Phadwal K, Simon AK, Moutschen M, Etzioni A, Mory A, Srugo I, Melamed D, Hultenby K, Liu C, Baronio M, Vitali M, Philippet P, Dideberg V, Aghamohammadi A, Rezaei N, Enright V, Du L, Salzer U, Eibel H, Pfeifer D, Veelken H, Stauss H, Lougaris V, Plebani A, Gertz EM, Schaffer AA, Hammarstrom L, Grimbacher B, Deleterious mutations in LRBA are associated with a syndrome of immune deficiency and autoimmunity. American journal of human genetics 90, 986–1001 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cottingham RW Jr., Idury RM, Schaffer AA, Faster sequential genetic linkage computations. American journal of human genetics 53, 252–263 (1993). [PMC free article] [PubMed] [Google Scholar]
- 54.Lathrop GM, Lalouel JM, Julier C, Ott J, Strategies for multilocus linkage analysis in humans. Proceedings of the National Academy of Sciences of the United States of America 81, 3443–3446 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schaffer AA, Gupta SK, Shriram K, Cottingham RW Jr., Avoiding recomputation in linkage analysis. Human heredity 44, 225–237 (1994). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Case reports for Families A-D.
Defective B cell development and Th17 cell differentiation in HIES patients with ZNF341 nonsense mutations.
Exclusion of mutations in STAT3 in the etiology of the HIES phenotype in Family A.
Identification of ZNF341 as an AR-HIES-associated gene in a linkage region on chromosome 20 by whole exome sequencing.
Expression of various ZNF341 mRNA variants in multiple cell lines and PBMCs.
Supplementary materials and methods
B and T cell subsets in patient-derived and healthy donor PBMCs.
Ubiquitous expression of ZNF341 mRNA in various cell types.
No association of the genomic region 20q11.22 spanning the ZNF341 locus with atopy.
Ectopic expression of ZNF341 variants in HEK293T cells.
Ectopically expressed wild-type ZNF341 binds to the STAT3 promoter leading to transcriptional activation.
Occupancy by ZNF341 across the STAT3 promoter as determined by ChIP.
ZNF341 R386* shows reduced binding to the STAT3 promoter.
FACS gating strategy for in vitro Th17 cell differentiation assay.
FACS gating strategy for Y705-phosphorylation of STAT3.
FACS gating strategy for immune phenotyping of PBMCs.
FACS gating strategy for IL22+ T cells.
Clinical and immunological phenotype of HIES patients with ZNF341 mutations.
ZNF341 encodes three protein coding isoforms.
Transcriptome Analysis of patient A.II.1 with gene-specific fold changes in comparison to healthy sibling A.II.5.