

Abstract
The synthesis of densely functionalized azetidinesin a highly stereocontrolled manner is challenging, but interest in the bioactivities of these small heterocycles has stimulated methods for their preparation. We recently reported a one-carbon ring expansion of bicyclic methylene aziridines under dirhodium catalysis capable of delivering enantioenriched azetidines. This work explores this ring expansion using computational and experimental studies. DFT computations indicate that the reaction proceeds through formation of an aziridinium ylide, which is precisely poised for concerted, asynchronous ring-opening/closing to deliver the azetidines in a [2,3]-Stevens-type rearrangement. The concerted nature of this rearrangement is responsible for the stereospecificity of the reaction, where axial chirality from the initial allene substrate is transferred to the azetidine product with complete fidelity. The computed mechanistic pathway highlights the key roles of the olefin and the rigid structure of the methylene aziridine in differentiating our observed ring expansion from competing cheletropic elimination pathways noted with ylides derived from typical aziridines.
Keywords: azetidine; methylene aziridines; [2,3]-Stevens rearrangement; aziridinium ylides
Graphical Abstract
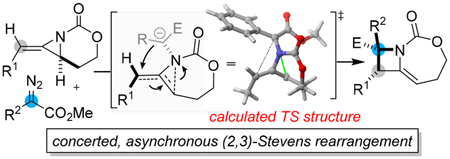
INTRODUCTION
Substituted azetidines occur in several natural products and medicinal chemistry libraries generated through diversity-oriented synthesis (Scheme 1A).1 However, methods to access highly functionalized, enantioenriched azetidines that are not comprised of β-lactam cores are surprisingly rare. The Schomaker group recently reported an efficient ring expansion of carbamate-derived bicyclic methylene aziridines (MAs) to methylene azetidines using substituted α-diazoacetates under dirhodium catalysis (Scheme 1B).2 This transformation was synthetically impressive in its ability to efficiently set adjacent functionalized stereocenters, as well as forge new C−C and C− N bonds. The transformation displayed an unexpected ability to transfer chirality from the initial allene substrate to the azetidine products and represents a rare example of a successful catalytic, intermolecular one-carbon ring expansion reaction involving a three-membered nitrogen heterocycle and a metal-supported carbene.3,4 In this paper, we explore the mechanistic details of this unusual ring expansion using both experimental and computational studies and provide important insight that may expand the scope and utility of these types of useful ring expansions.
Scheme 1.

Bioactive Azetidines and Stereocontrolled Synthesis via Aziridine Ring Expansion
The formation and controlled reactivity of onium ylides, including ammonium, sulfonium, and oxonium ylides, continue to be a powerful approach for transforming simple precursors into stereochemically rich, densely functionalized products, often with high levels of diastereo- and enantiocontrol.5 For example, onium ylides are key intermediates in 1,3-dipolar cycloadditions,6 1,2- and 2,3-rearrangements,7 1,2-ring closures, and various multicomponent reactions, among other useful transformations.8 Catalytic reactions of heteroatoms with metal-supported carbenes are a subset of this class of reactions that provide a convenient entry point to onium ylides.5e,9,10 Insight into the factors responsible for the reactivity and stereocontrol in these systems has been critical for advancing the scope and versatility of these classes of reactions.
Despite their potential utility, onium ylides derived from aziridines and epoxides have seen limited use in synthesis due to their propensity to undergo retro [2 + 1] cycloadditions. In 1972, Watanabe disclosed the first attempted ring expansion of an aziridine to an azetidine through in situ formation of an aziridinium ylide using a copper-bound carbene (Scheme 2).11 Instead of the productive C−C bond formation that was noted with azetidine precursors, the utilization of aziridine substrates led to cheletropic extrusion to form ethylene and the α-imino ester. In 2004, Rowlands attempted to bypass this cheletropic extrusion process through an intramolecular [2,3]-Stevens rearrangement of a vinyl aziridine.12 Even after extensive optimization of the substrate and the reaction conditions, the highest yield of the substituted piperidine that could be obtained was 21%. The low yield was attributed to the dependence of the rearrangement on the configuration of the nitrogen lone pair; that is, effective ring expansion required a syn orientation between the nitrogen lone pair and the alkene upon formation of the ylide. If this condition was not met, decomposition of the aziridinium ylide intermediate occurred through a [1,5]-hydrogen shift.
Scheme 2.

Reactivity of Typical Aziridinium Ylides
RESULTS AND DISCUSSION
On the basis of our understanding of the unusual features of carbamate-derived methylene aziridines13 and the literature of rhodium-bound carbenes,14 we initially proposed that ring expansion proceeds through formation of an aziridinium ylide, 4.2, followed by a stepwise ring-opening, ring-closing sequence to deliver the functionalized azetidine 4.5 (Scheme 3, red). Despite the precedent for the formation of this type of aziridinium ylide, the difference in the observed reactivity of our system, compared to that of the Watanabe and Rowlands systems, suggested that a better understanding of the factors responsible for controlling the ring expansion was required. Such insight could have broad implications for controlling the reactivity of aziridinium ylides and significantly expand the utility and substrate scope of these types of ring expansions. With the goal of ascertaining the most likely mechanistic pathway, detailing the factors responsible for successful ring expansion, and increasing the reactivity profile of our methylene aziridines, we initiated a more rigorous mechanistic study of this unusual reaction.2
Scheme 3.

Potential Mechanisms for Aziridine Ring Expansion
In addition to our initial proposed mechanism (Scheme 3a red, 4.1−4.5), several other pathways were considered, as highlighted in Scheme 3. In contrast to the stepwise ring-opening/closing path, we recognized the possibility that a concerted [2,3]-type rearrangement proceeding through transition state 4.6 could, in principle, deliver azetidine 4.5 and account for the enantioretention observed previously (Scheme 3b, orange). Ylide 4.2 could diverge to a pathway invoking a cheletropic extrusion similar to that observed by Watanabe and would generate both allene and imine functionality in 4.8, which could be envisaged to undergo a [2 + 2] cycloaddition to furnish 4.5 (Scheme 3c, green). However, no product from cycloaddition with the proximal allene double bond to give 4.9 was noted, seeming to argue against this pathway. Finally, dinuclear Rh catalysts are well-known to promote alkene cyclopropanation reactions; such a scenario applied to 4.1 would lead to the azaspiropentane intermediate 4.10 (Scheme 3d, blue) that could undergo rearrangement to yield 4.5.
Studies sought to answer the following questions: (a) What is the full reaction pathway, as supported by both computational and experimental evidence? (b) What is the source of the high diastereoselectivity and enantioretention that is observed in this ring expansion process? and (c) What specific features enable this rare transformation and how can a better understanding of these factors be utilized to expand the scope of similar reactions?
Previous mechanistic studies of the ring expansion concluded that a radical pathway was not likely as both inter- and intramolecular radical traps, in the form of added TEMPO and a vinyl cyclopropane substrate, respectively, did not affect the observed reactivity.2 Control reactions demonstrated the necessity of forming the rhodium-bound carbene prior to ring expansion. Due to the constraints associated with carrying out kinetic analyses on reactions that require slow addition, we felt that density functional theory (DFT) calculations would be an excellent tool to help elucidate the reaction pathway, with experimental evidence serving as a control on the calculations.
DFT calculations were performed at the dispersion-corrected SMD(CH2Cl2)-B3LYP-D3/def2-SVP level (see Computational Details in the Supporting Information) beginning from the methyl-substituted MA 1a and dirhodium-bound carbene 2a-Rh2, derived from phenyl diazoacetate (Figure 1).15,16 Formation of aziridinium ylide INT1 proceeded through transition state TS1 with a low barrier of 3.2 kcal mol−1 in a moderately exergonic transformation (ΔGR = −7.6 kcal mol−1). In contrast, the potentially competing cyclopropanation pathway had a notably higher transition state energy of 11.3 kcal mol−1 (TS1′), supporting the literature consensus that ylide formation generally outcompetes cyclopropanation.5a Taken together, these results argue against the pathway shown in Scheme 3d, which can be safely ruled out.
Figure 1.

Computed reaction profile for the process involving methyl-substituted MA 1a and dirhodium-bound carbene 2a-Rh2. Relative free energies (ΔG, computed at 298.15 K and 1 M) and bond distances are given in kcal/mol and angstroms, respectively. All data have been computed at the SMD(CH2Cl2)-B3LYP-D3/def2-SVP level. Values within parentheses were computed at the SMD(CH2Cl2)-B3LYP-D3/def2-TZVPP//SMD(CH2Cl2)-B3LYP-D3/def2-SVP level.16
Dissecting the fate of rhodium-bound aziridinium ylide INT1 proved to be nontrivial. Attempted ring-opening of the strained, allylic C−N bond of INT1 via TS2 was 12.5 kcal mol−1 uphill in energy. However, dissociation of the dirhodium center from the rhodium-bound ylide to produce ylide INT2, followed by a similar ring-opening via TS3, was found to be favored over the process involving TS2 (9.2 kcal mol−1 from INT1). Despite the energetic cost associated with the initial dissociation of the transition metal fragment (relaxed scans performed at different Rh···C distances starting from INT1 indicates that this dissociation is essentially a barrierless process), this metal-free ylide route is believed to be the dominant pathway. In addition, this early dissociation of the dirhodium catalyst is consistent with studies on the fates of rhodium and copper ylides in O−H insertion chemistry.17
In contrast to our previous proposed stepwise pathway from the ylide INT1 to the product 3aa, TS3 suggests an asynchronous, concerted ring-opening, ring-closing cascade to deliver azetidine 3aa, formally considered a [2,3]-Stevens-type rearrangement.18 Zwitterion INT3 could not be identified as a minimum on the potential energy surface. Indeed, intrinsic reaction calculations (IRCs) starting from TS3 demonstrated that the final closing of the C−C bond to form the azetidine exists on a plateau-like energy pathway, where ring closure only begins to occur following completion of the C−N bond rupture and initial planarization of the pseudoallyl cation (Figure 2). The transformation from the ylide INT2 to 3aa merits further discussion. The relief of methylene aziridine ring strain drives the reactivity captured in this final step, which is highly exergonic and releases nearly 60 kcal mol−1 of energy, despite forming congested adjacent stereocenters. Additionally, the low barrier associated with accessing TS3 reflects the lability of the strained aziridinium C−N bond. Despite the exergonicity and near-barrierless nature of TS3, the C−N bond length in TS3 (2.332 Å) resembled the analogous bond length in the product azetidine 3aa (2.507 Å) more closely than that of the ylide INT2 (1.577 Å), highlighting that some degree of positioning for ring closure occurs following aziridinium opening. Interestingly, the distance between the two reactive carbon atoms remains nearly unchanged, 3.512 Å in INT2 and 3.241 Å in TS3, consistent with the IRC depiction of C−N bond breakage, followed by pseudoallyl cation planarization and the final C−C bond formation. In comparison to Singleton’s work on the transition state of acyclic [2,3]-Stevens rearrangements,18 the longer C−C bond distance in TS3 appears to highlight how the constrained structure of the MA can allow for a looser rearrangement transition state.
Figure 2.

Computed intrinsic reaction coordinate at the reference SCM(CH2Cl2)-B3LYP-D3/def2-SVP level for the transformation of TS3 into 3aa.
The concerted nature of TS3 has important repercussions for the diastereoselectivity of the ring expansion reaction. Figure 1 shows calculations of the energies for the full pathways leading to both possible diastereomeric azetidine products 3aa and 3aa-iso. The lower barrier of TS3 compared to TS1 indicated that ylide formation is slow relative to the rearrangement step. As such, ylide formation operates under kinetic rather than thermodynamic control. Our computations (see the SI for further details) suggest that there is no interconversion between INT2 and INT2-iso through rotation about the N−C bond as the rotation barrier was much higher (ΔG‡ = 24.7 kcal mol−1) than that for the process involving TS3. Thus, the stereochemical outcome of the reaction is set during the initial formation of the aziridinium ylide, with the diastereoselectivity of the final ring closure originating from the facial differentiation of the rhodium-bound ylide in its approach to the methylene aziridine. The 3.5 kcal mol−1 difference noted between TS1 and TS1-iso (Figure 1) is in part due to the occurrence of stabilizing noncovalent interactions (NCIs) in TS1 that are not present in TS1-iso. These weak but significant interactions can be readily visualized by means of the NCIPLOT method.19 As clearly shown in Figure 3a, there are two genuine stabilizing NCIs (green surfaces) in TS1, namely, the n···π* interaction involving the carbamate moiety and the carbene ester and the CH···π interaction involving the alkene CH and the carbene phenyl group. Obviously, these interactions cannot be accommodated in TS1-iso, as shown in Figure 3b, which is translated into the lower stability computed for this saddle point.
Figure 3.

Contour plots of the reduced density gradient isosurfaces (density cutoff of 0.03 au) for TS1 (a) and TS1-iso (b). The green surfaces indicate attractive NCIs.
Additional evidence for the selectivity of the ylide formation controlling the overall diastereoselectivity of the reaction and the key role of the NCIs can be found from the lower experimental dr values observed for substrates such as methyl or styrenyl α-diazoacetates (7:1 and 4:1, respectively, vs 19:1 for the Ph-substituted counterpart).7 These particular systems lack the phenyl group responsible for the stabilizing CH···π interaction in the corresponding TS1 saddle points, which is reflected in the lower dr values observed experimentally.
As noted earlier, we previously discovered that subjecting enantioenriched methylene aziridines to the reaction conditions transferred chirality to the product azetidine (Scheme 4a). As an additional check of our computations, the absolute configuration of the enantioenriched chlorinated derivative 3bb was determined by single-crystal X-ray diffraction, which indicated that the precursor (S)-1b delivers the (S,S)-3bb azetidine, a result consistent with our computations (see the SI for full details).
Scheme 4.

Stereochemical Probes for the Ring Expansion of MAs to Methylene Azetidines
The transfer of chirality observed in Scheme 4a agrees with our proposed concerted ring-opening, ring-closing pathway proceeding through TS3. The concavity of the methylene aziridine (S)-1b, combined with the barrierless ring-closing process suggested by computations, necessitates that ring closure occur from the back side of (S)-1.20 We were curious whether this observation would hold true with other carbene precursors; thus, we separately subjected both the symmetric iodonium ylide 2c and the phenyl diazonitrile 2d to the reaction conditions with enantioenriched 1b (Scheme 4b). In both cases, enantioretention in the azetidine products 3bc and 3bd was observed. Although it is possible that reactions of these carbene precursors do not proceed through the same concerted pathway, these results strongly support the stereo-specificity of the rearrangement. We found that the TS3-E directly evolves to the final azetidine 3bc′, a situation similar to that found for the analogous phenyl-substituted INT2 in Figure 1 (see Figure 4). The corresponding intrinsic reaction coordination is included in the Supporting Information as Figure S9.
Figure 4.

Computed reaction profile for the INT2-E → 3bc′ transformation. See Figure 1 for additional caption details.
With an understanding of the reaction pathway and the origin of the observed diastereoselectivity and enantioretention, we sought to understand the factors differentiating the fate of our methylene aziridinium ylides, as compared to the simpler aziridinium ylides described by Watanabe and Rowlands.11,12 Thus, saturated bicyclic carbamate aziridines 5a and 5b were synthesized from aziridination of the corresponding homoallylic carbamates and exposed to the ring expansion conditions (Scheme 5).
Scheme 5.

Fate of Bicyclic Aziridinium Ylides
The trans isomer 5a showed no conversion, even under forcing conditions. In contrast, the cis isomer 5b yielded the imine alkene 5c in moderate yield as the sole product of the reaction, highlighting that ready access to the nitrogen lone pair is necessary for reaction with the metal-supported carbene. The product 5c results from the same stereospecific cheletropic extrusion noted by Watanabe as only the cis-alkene isomer was observed. DFT calculations (Figure 5) indicate that the cheletropic extrusion path involving 5b can proceed either directly from the rhodium ylide INT1-b via TS4-b or from the corresponding nonmetallic ylide INT2-b via TS5-b, with feasible activation barriers of 9.9 and 7.2 kcal mol−1, respectively. This is a significantly higher barrier as compared to TS3 (Figure 1) and suggests that the presence of the olefin in the MA is the key difference between the retro [2 + 1] and the ring expansion fates of the aziridinium ylide. Although the computed barrier for the analogous chelotropic extrusion from alkene ylide INT2 is lower (ΔG‡ = 5.1 kcal mol−1), it is still higher than that computed for the process involving TS3, which makes this alternative pathway kinetically unfavored.
Figure 5.

Computed chelotropic extrusion from the aziridinium ylide formed in the reaction of 5b and dirhodium-bound carbene 2a-Rh2. See Figure 1 for additional caption details.
We propose that the alkene in the MA substrate, compared to a typical aziridine, alters two key parameters that combine to lower the energy barrier for ring expansion, as opposed to cheletropic extrusion. The additional alkene in INT2 increases the ring strain of the methylene aziridine by roughly 4.5 kcal mol−1 (see section VII in the Supporting Information for the ring strain calculations); this serves to differentiate the bond strengths of the two aziridine C−N bonds in INT2, effectively biasing which C−N bond breaks first. Indeed, the computed NBO-Wiberg bond indices (WBIs) nicely agree with this hypothesis. Thus, whereas rather similar N−C WBIs were computed for INT2-b (0.78 and 0.79, respectively), the corresponding N−C bond strengths in the alkene-substituted counterpart INT2 are markedly different (0.82 and 0.75, respectively). Therefore, in contrast to saturated bicylic aziridine INT2-b, where the equivalent C−N bond strengths facilitate concerted extrusion, the discrepancy between the bond strengths of the allylic and vinylic C−N bonds in INT2 favors complete rupture of the allylic C−N bond first, effectively leading to subsequent ring expansion.
CONCLUSIONS
Through evidence obtained through combined computational and experimental studies, we have demonstrated that the ring expansion of MAs with rhodium-bound carbenes proceeds through an aziridinium ylide that undergoes a concerted, near-barrierless [2,3]-Stevens rearrangement to deliver substituted azetidines. The concerted nature of this rearrangement confers stereospecificity to the transformation. In comparison to the corresponding saturated bicyclic carbamate aziridines, the olefin of the MA plays a key role in the success of this chemistry, channeling the aziridinium ylide toward ring expansion processes vs the competing cheletropic extrusions. These studies provide a mechanistic basis for expanding our MA chemistry and point to important factors that must be considered in developing further approaches toward more general heterocyclic ring expansion reactions.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Charlie Fry of the University of Wisconsin−Madison is thanked for his assistance with NMR spectroscopy, while Dr. Martha Vestling of UW−Madison is thanked for her assistance with collecting MS data. J.M.S. thanks the NIH R01 GM11412 and the ACS-PRF No. 53146-ND1. The NMR facilities at UW−Madison are funded by the National Science Foundation (NSF; CHE-9208463, CHE-9629688) and National Institutes of Health (NIH; RR08389–01). The QExactive mass spectrometer was acquired with funds from an NIH-S10 award through the National Institutes of Health (NIH-1S10OD020022–1). I.F. acknowledges financial support from the Spanish MINECO-FEDER (Grants CTQ2016–78205-P and CTQ2016–81797-REDC).
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscatal.8b02206.
Experimental procedures, computational details, and characterization data for all new compounds (PDF) Crystallographic information for (S)-3bb (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).For selected references, see:Lowe JT; Lee MD; Akella LB; Davoine E; Donckele EJ; Durak L; Duvall JR; Gerard B; Holson EB; Joliton A; Kesavan S; Lemercier BC; Liu H; Marie J-C; Mulrooney CA; Muncipinto G; Welzel-O’Shea M; Panko LM; Rowley A; Suh B-C; Thomas M; Wagner FF; Wei J; Foley MA; Marcaurelle LA Synthesis and Profiling of a Diverse Collection of Azetidine-based Scaffolds for the Development of CNS-focused Lead-like Libraries. J. Org. Chem 2012, 77, 7187–7211.Shimokawa J; Harada T; Yokoshima S; Fukuyama T Total Synthesis of Gelsemoxonine. J. Am. Chem. Soc 2011, 133, 17634–17637.Matsuura F; Hamada Y; Shioiri T Total Synthesis of Mugineic acid. Efficient Use of the Phenyl Group as the Carboxyl Synthon. Tetrahedron 1993, 49, 8211–8222.Kato N; Comer E; Sakata-Kato T; Sharma A; Sharma M; Maetani M; Bastien J; Brancucci NM; Bittker JA; Corey V; Clarke D; et al. Diversity-oriented Synthesis Yields Novel Multistage Antimalarial Inhibitors. Nature 2016, 538, 344–349.Takikawa H; Maeda T; Seki M; Koshino H; Mori K Synthesis of Sphingosine Relatives. Part 19. Synthesis of Penaresidin A and B, Azetidine Alkaloids with Actomyosin ATPase-activating Properties. J. Chem. Soc., Perkin Trans. 1 1997, 97–112.Diethelm S; Carreira EM Total Synthesis of (±)-Gelsemoxonine. J. Am. Chem. Soc 2013, 135, 8500–8503.Hamada Y; Shioiri T New Methods and Reagents in Organic Synthesis. 63. Synthesis of Mugineic Acid Through Direct C-acylation Using Diphenyl Phosphorazidate (DPPA). J. Org. Chem 1986, 51, 5489–5490.Bott TM; West FG Preparation and Synthetic Applications of Azetidines. Heterocycles 2012, 84, 223–264.
- (2).Schmid SC; Guzei IA; Schomaker JM A Stereoselective [3+ 1] Ring Expansion for the Synthesis of Highly Substituted Methylene Azetidines. Angew. Chem., Int. Ed 2017, 56, 12229–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).For intermolecular carbene-mediated ring expansion of nitrogen heterocycles, see:Bott TM; Vanecko JA; West FG One-carbon Ring Expansion of Azetidines via Ammonium Ylide [1,2]-shifts: A Simple Route to Substituted Pyrrolidines. J. Org. Chem 2009, 74, 2832–2836.Sharma A; Guénée L; Naubron J-V; Lacour J One-Step Catalytic Asymmetric Synthesis of Configurationally Stable Tröger Bases. Angew. Chem., Int. Ed 2011, 50, 3677–3680.He J; Hamann LG; Davies HML; Beckwith REJ Late-stage C−H Functionalization of Complex Alkaloids and Drug Molecules via Intermolecular Rhodium-Carbenoid Insertion. Nat. Commun 2015, 6, 5943–5961.Vanecko JA; West FG Ring Expansion of Azetidinium Ylides: Rapid Access to the Pyrrolizidine Alkaloids Turneforcidine and Platynecine. Org. Lett 2005, 7, 2949–2952.
- (4).For reviews on aziridinium ions in synthesis, see:Topics in Heterocyclic Chemistry; D’hooghe M, Ha H-J; Eds.; Springer: New York, 2016, Vol. 41, p 49–142.Dolfen J; Yadav NN; De Kimpe N; D’hooghe M; Ha H-J Bicyclic Aziridinium Ions in Azaheterocyclic Chemistry−Preparation and Synthetic Application of 1-Azoniabicyclo[n.1.0]alkanes. Adv. Synth. Catal 2016, 358, 3485–3511.
- (5).For selected leading books and reviews, see:Contemporary Carbene Chemistry; Moss RA, Doyle MP, Ed.; Wiley: Hoboken, NJ, 2013.Ford A; Miel H; Ring A; Slattery C; Maguire AR; McKervey MA Modern Organic Synthesis with α-Diazocarbonyl Compounds. Chem. Rev 2015, 115, 9981–10080.Guo X; Hu W Novel Multicomponent Reactions via Trapping of Protic Onium Ylides with Electrophiles. Acc. Chem. Res 2013, 46, 2427–2440.Padwa A; Hornbuckle SF Ylide Formation from the Reaction of Carbenes and Carbenoids with Heteroatom Lone Pairs. Chem. Rev 1991, 91, 263–309.Zhang D; Hu W Asymmetric Multi component Reactions Based on Trapping of Active Intermediates. Chem. Rec 2017, 17, 739–753.
- (6).For selected references, see:Iuhas PC; Georgescu F; Georgescu E; Draghici C; Caproiu MT Regiochemistry of the 1,3-Dipolar Cycloaddition of Some N-Heterocyclic Cycloimmonium Ylides to Unsymmetrical Alkynes. Rev. Roum. Chim 2002, 47, 333–338.Padwa A; Kassir JM; Semones MA; Weingarten MD A Tandem Cyclization-Onium Ylide Rearrangement-Cycloaddition Sequence for the Synthesis of Benzo-Substituted Cyclopentenones. J. Org. Chem 1995, 60, 53–62.Štetinovă J; Juráśek A; Kovać J; Dandarova M; Safar P Furan-related N-onium Salts as Synthons for Preparation of 3-Furylbenzoindolizines. Collect. Czech. Chem. Commun 1986, 51, 412–418.
- (7).For selected references, see:Murphy GK; West FG [1,2]-or [2,3]-Rearrangement of Onium Ylides of Allyl and Benzyl Ethers and Sulfides via in Situ-Generated Iodonium Ylides. Org. Lett 2006, 8, 4359–4362.Xu B; Tambar UK Copper-Catalyzed Enantioselective, Diastereoselective, and Regioselective [2,3]-Rearrangements of Iodonium Ylides. Angew. Chem., Int. Ed 2017, 56, 9868–9871.Boyer A Rhodium (II)-Catalyzed Stereocontrolled Synthesis of 2-Tetrasubstituted Saturated Heterocycles from 1-Sulfonyl-1, 2, 3-triazoles. Org. Lett 2014, 16, 5878–5881.West TH; Spoehrle SS; Kasten K; Taylor JE; Smith AD Catalytic Stereoselective [2,3]-Rearrangement Reactions. ACS Catal. 2015, 5, 7446–7479.Jaber DM; Burgin RN; Helper M; Zavalij PY; Doyle MP Competitive [2,3]-and [1,2]-Oxonium Ylide Rearrangements. Concerted or stepwise? Org. Lett 2012, 14, 1676–1679.Hosseini SN; Johnston JR; West FG Evidence for Heterolytic Cleavage of a Cyclic Oxonium Ylide: Implications for the Mechanism of the Stevens [1,2]-Shift. Chem. Commun 2017, 53, 12654–12656.Li Z; Davies HML Enantioselective C−C Bond Formation by Rhodium-Catalyzed Tandem Ylide Formation/[2,3]-Sigmatropic Rearrangement between Donor/Acceptor Carbenoids and Allylic Alcohols. J. Am. Chem. Soc 2010, 132, 396–401.
- (8).For other selected references in the area of onium ylide chemistry, see:Yuan W; Szaboó KJ Rhodium-Catalyzed Oxy-Aminofluorination of Diazoketones with Tetrahydrofurans and NFluorobenzenesulfonimide. ACS Catal. 2016, 6, 6687–6691.Yuan W; Eriksson L; Szabó KJ Rhodium-Catalyzed Geminal Oxyfluorination and Oxytrifluoromethylation of Diazocarbonyl Compounds. Angew. Chem., Int. Ed 2016, 55, 8410–8415.Mai BK; Szabó KJ; F. Himo Mechanisms of Rh-Catalyzed Oxyfluorination and Oxytrifluoromethylation of Diazocarbonyl Compounds with Hypervalent Fluoroiodine. ACS Catal. 2018, 8, 4483–4492.Jing C; Xing D; Gao L; Li J; Hu W Divergent Synthesis of Multisubstituted Tetrahydrofurans and Pyrrolidines via Intramolecular Aldol-type Trapping of Onium Ylide Intermediates. Chem. - Eur. J 2015, 21, 19202–19207.Bach R; Harthong S; Lacour J in Comprehensive Organic Synthesis; Knochel P, Molander GA, Ed.; Elsevier Science: York, 2014; Vol. 3, pp 992–1037.Sweeney JB Sigmatropic Rearrangements of ‘Onium’ Ylids. Chem. Soc. Rev 2009, 38, 1027–1038.Murphy GK; West FG In Molecular Rearrangements in Organic Synthesis, Rojas C, Ed.; John Wiley Sons: Hoboken, NJ, 2015; p 497–538.
- (9).For selected references, see:West FG In Modern Rhodium-catalyzed Organic Reactions; Evans PA, Ed.; Wiley-VCH: Weinheim, 2005; pp 417–431.Doyle MP Catalytic Methods for Metal Carbene Transformations. Chem. Rev 1986, 86, 919–939.
- (10).For transformations of epoxides with carbenes, see:Mack DJ; Batory LA; Njardarson JT Intermolecular Oxonium Ylide Mediated Synthesis of Medium-Sized Oxacycles. Org. Lett 2012, 14, 378–381.Ma X; Pan S; Wang H; Chen W Rhodium-Catalyzed Transannulation of N-Sulfonyl-1,2,3-triazoles and Epoxides: Regioselective Synthesis of Substituted 3,4-Dihydro-2H-1,4-oxazines. Org. Lett 2014, 16, 4554–4557.Quinn KJ; Biddick NA; DeChristopher BA Ring Expansion of trans-Divinyl Ethylene Oxide by Oxonium Ylide [2,3]-Sigmatropic Rearrangement. Tetrahedron Lett. 2006, 47, 7281–7283.
- (11).Hata Y; Watanabe M Fragmentation Reaction of Aziridinium Ylids. Tetrahedron Lett. 1972, 13, 3827–3830. [Google Scholar]
- (12).Rowlands GJ; Kentish Barnes W Studies on the [2,3]-Stevens Rearrangement of Aziridinium Ions. Tetrahedron Lett. 2004, 45, 5347–5350. [Google Scholar]
- (13).For previous work on bicyclic methylene aziridines, see:Boralsky LA; Marston D; Grigg RD; Hershberger JC; Schomaker JM Allene Functionalization via Bicyclic Methylene Aziridines. Org. Lett 2011, 13, 1924–1927.Rigoli JW; Boralsky LA; Hershberger JC; Marston D; Meis AR; Guzei IA; Schomaker JM 1, 4-Diazaspiro[2.2]pentanes as a Flexible Platform for the Synthesis of Diamine-Bearing Stereotriads. J. Org. Chem 2012, 77, 2446–2455.Weatherly CD; Rigoli JW; Schomaker JM Synthesis of 1,3-Diaminated Stereotriads via Rearrangement of 1,4-Diazaspiro[2.2]pentanes. Org. Lett 2012, 14, 1704–1707.Adams CS; Boralsky LA; Guzei IA; Schomaker JM Modular Functionalization of Allenes to Aminated Stereotriads. J. Am. Chem. Soc 2012, 134, 10807–10810.Rigoli JW; Guzei IA; Schomaker JM Aminodiols via Stereocontrolled Oxidation of Methyleneaziridines. Org. Lett 2014, 16, 1696–1699.Adams CS; Grigg RD; Schomaker JM Complete Stereodivergence in the Synthesis of 2-Amino-1,3-diols from Allenes. Chem. Sci 2014, 5, 3046–3056.Burke EG; Schomaker JM Oxidative Allene Amination for the Synthesis of Azetidin-3-ones. Angew. Chem., Int. Ed 2015, 54, 12097–12101.Gerstner NC; Adams CS; Tretbar M; Schomaker JM Stereocontrolled Syntheses of Seven-Membered Carbocycles by Tandem Allene Aziridination/[4 + 3] Reaction. Angew. Chem., Int. Ed 2016, 55, 13240–13243.Adams CS; Weatherly CD; Burke EG; Schomaker JM The Conversion of Allenes to Strained Three-membered Heterocycles. Chem. Soc. Rev 2014, 43, 3136–3163.
- (14).For selected references, see:Davies HML; Morton D Guiding Principles for Site-selective and Stereoselective Intermolecular C−H Functionalization by Donor/Acceptor Rhodium Carbenes. Chem. Soc. Rev 2011, 40, 1857–1869.Kornecki KP; Briones JF; Boyarskikh V; Fullilove F; Autschbach J; Schrote KE; Lancaster KM; Davies HM; Berry JF Direct Spectroscopic Characterization of a Transitory Dirhodium Donor-Acceptor Carbene Complex. Science 2013, 342, 351–354.Werlé C; Goddard R; Philipps P; Fares C; Fürstner A Structures of Reactive Donor/Acceptor and Donor/Donor Rhodium Carbenes in the Solid State and Their Implications for Catalysis. J. Am. Chem. Soc 2016, 138, 3797–3805.
- (15).Nakamura E; Yoshikai N; Yamanaka M Mechanism of C−H Bond Activation/C−C Bond Formation Reaction Between Diazo Compound and Alkane Catalyzed by Dirhodium Tetracarboxylate. J. Am. Chem. Soc 2002, 124, 7181–7192. [DOI] [PubMed] [Google Scholar]
- (16).The energetics obtained from the two basis sets applied (def2-SVP and def2-TZVPP) varies largely, likely originating from the relatively large basis set superposition error by using the def2-SVP basis set.
- (17).Liang Y; Zhou H; Yu Z-X Why Is Copper(I) Complex More Competent Than Dirhodium(II) Complex in Catalytic Asymmetric O−H Insertion Reactions? A Computational Study of the Metal Carbenoid O−H Insertion into Water. J. Am. Chem. Soc 2009, 131, 17783–17785. [DOI] [PubMed] [Google Scholar]
- (18).(a) Biswas B; Collins SC; Singleton DA Dynamics and a Unified Understanding of Competitive [2,3]-and [1,2]-Sigmatropic Rearrangements Based on a Study of Ammonium Ylides. J. Am. Chem. Soc 2014, 136, 3740–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Biswas B; Singleton DA Controlling Selectivity by Controlling the Path of Trajectories. J. Am. Chem. Soc 2015, 137, 14244–14247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Johnson ER; Keinan S; Mori-Sánchez P; Contreras-García J; Cohen AJ; Yang W Revealing Noncovalent Interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).The concavity of the methylene aziridines and the corresponding ylides discussed is reflective of the chirality at nitrogen that is present in these species. To the best of our knowledge, these bicyclic aziridines cannot undergo nitrogen inversion and thus contain nitrogen stereocenters; however, for the sake of simplicity in the text, we refer to the bowl-shaped structure of the MA and resulting ylides synonymously in place of nitrogen chirality.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.