

Abstract
In the world of science, in particular the section concerning the field of chemistry, when the results encountered during the experiment do not meet our expectations, our shrewdness may play an important role to open up new unexplored fields that could be much more interesting than what we were seeking. In those cases, our research undergoes an unforeseen shift, delivering novel and challenging results that may altogether alter our point of view and our future work. We have then struck serendipity. Specifically, in our investigation linked to palladacycles we have found that the new trends in their reactivity, as well as in their structure, have been, in many cases, related to this experience, broadening our research scope within this field. Herein, we describe our most relevant findings, which have shed new light upon the reactivity of palladacycles, thus opening new routes that lead to novel unexpected structures.
Keywords: palladacycles, Schiff base, serendipity, thiosemicarbazone, translational chemistry
1. Introduction
In the field of chemistry, or of any other type of scientific research, when what we expect to find is altogether different from what we produce, we come about unimaginable occurrences: our findings yielding the most remarkable and striking, yet not sought, results when researching for something dissimilar. We have then encountered serendipity. In the words of Louis Pasteur “fortune favors the prepared mind”. Progress in basic, applied, as well as translational chemistry can benefit from serendipity. Specifically, in our investigation related to cyclometallated complexes1 and, in particular, to palladacycles,2 we found new trends in their reactivity and novel structural aspects that, in many cases, have been related to this experience, broadening our research scope within this field. The term cyclopalladation was coined by Trofimenko1i to describe the reactions of palladium complexes in which an organic ligand undergoes intramolecular metalation with the formation of a chelate ring comprising a σ metal–carbon bond and a dative bond between the metal and a donor atom from the ligand (Scheme 1).
Scheme 1.
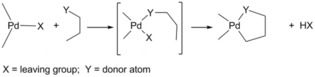
The cyclopalladation reaction.
Herein, we give an account of some of our most relevant findings, including the following aspects, to name but a few, such as palladium penta‐coordination, with structures borderline between trigonal bipyramidal and square pyramidal geometries; catalytic contributions that shed new light upon cross‐coupling, comprising carbon‐carbon formation; and metalloligands, that give way to new homo‐ and heteronuclear species.
2. Pentacoordinated Palladacycles
Palladium(II) essentially displays a square‐planar environment in most of its compounds, whether organometallic or not. Thus, it is also the geometry of choice for palladacycles inclusive of the tertiary mono‐ or diphosphine derivatives. In view of the limited four‐coordination in palladacycles, we reasoned that reaction of the chloro‐ or bromo‐bridged dinuclear species with tridentate phosphines would produce complexes with an uncoordinated phosphorus donor, with the other two bonded to palladium in a chelating mode. At variance, bonding of the phosphorus donors to the metal center in a phoshpine–P,P,P fashion would cleave the palladium–nitrogen bond, leaving an uncoordinated nitrogen atom; in either case, further coordination of the nitrogen or phosphorus atoms should be possible to render bimetallic complexes. However, to our surprise, neither of the two situations happened and, instead, a pseudo‐pentacoordinated palladacycle was produced. Treatment of 1 with triphos (Ph2PCH2CH2)2PPh (Scheme 2) does not give any of the two aforementioned expected compounds, 2 and 3, but rather complex 4 is the resulting species.3
Scheme 2.
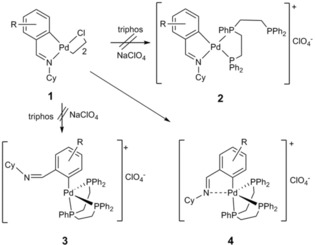
Reactivity of palladacycle 1 towards triphos. Adapted from Ref. 9. Copyright 2001 American Chemical Society.
The 31P‐{1H} NMR spectrum of 4 displays two signals at (δ85.3 ppm, t, 1P) and at (δ42.6 ppm, d, 2P), which concur with a) two phosphorus atoms in a trans geometry; b) the remaining phosphorus atoms almost trans to the metallated phenyl carbon. Fluctuation of the υ(C=N) mode to smaller wavenumbers4 and displacement of the HC=N resonance to higher frequency in the 1H NMR spectrum5 is indicative of some degree of bonding between palladium and the C=N moiety; the spectroscopic results suggest a close to square‐pyramid pattern in solution, with the apical vertex hosting the nitrogen donor. Running the NMR solution spectrum at low temperature (213 K) gave no appreciable modifications. On the contrary, solid‐state NMR for the 31P spectrum shows rather striking differences when compared to the spectrum performed in solution: two signals at (δ75.1 ppm, s, 1P) and at (δ44.6 ppm, dd, 2P) were in agreement with a trigonal‐bipyramid disposition for the metal center, with the former resonance ascribed to the 31P nucleus trans to the phenyl carbon atom at the apical vertex, and the latter assignable to the remaining phosphorus donors in the [N,P,P] plane. The 2 J(PP) coupling constants are 27 Hz (solution spectrum) and 300 Hz [2 J(P1P2) solid state spectrum]. Therefore, the outline of the spectrum in solution, a triplet and a doublet, differs quite significantly from that in the solid state, a singlet and a doublet of doublets, as also does the coupling constant, which greatly increases by approximately 270 Hz, as a consequence of the structural changes mentioned earlier. Five‐coordinate palladacycles have been suggested and reported previously, assuming weak Pd⋅⋅⋅N bonding based on Pd−N bond lengths ranging from 2.710(6) to 2.805(5) Å;6 a length as short as 2.576(4) Å has been published as well.7 Nevertheless, as far as we are aware, the Pd–N distance reported herein for 4 (Figure 1) 2.359(4) Å is, to date, the shortest Pd–N distance reported for this type of species, and quite near to the Pd–N distance in a true trigonal bipyramidal PdII complex of 2.23(2) Å,8 displaying a square‐pyramid setup.
Figure 1.
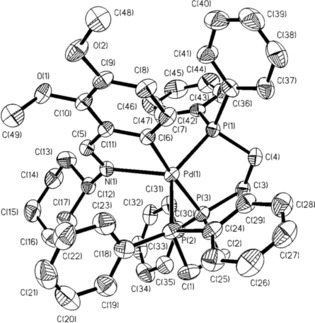
Molecular structure for compound 4. Reproduced from Ref. 3. Copyright 1999 American Chemical Society.
We then extended our research, following this pathway, and found that a rather large array of analogous species could be prepared with different types of nitrogen‐bearing donors, such as bidentate imines, aliphatic nitrogen atoms, and imidazole derivatives (Scheme 3) to give compounds 5 (Figure 2),9 6 (Figure 3),10 and 7 (Figure 4).11 In the case of compound 5, an even yet shorter Pd–N distance was found of 2.338 Å, which remains today as such.9
Scheme 3.
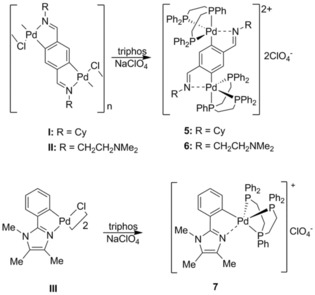
Preparation of compounds 5,9 6,10 and 7.11 Adapted from Ref. 9. Copyright 2001 American Chemical Society.
Figure 2.
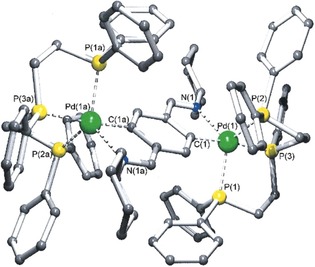
Molecular structure for compound 5. Adapted from Ref. 9. Copyright 2001 American Chemical Society.
Figure 3.
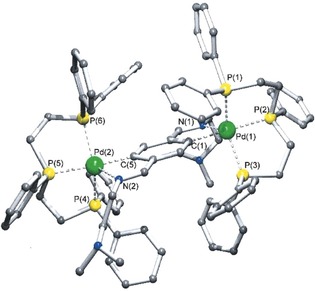
Molecular structure for compound 6. Adapted from Ref. 10. Copyright 2000 Elsevier B.V.
Figure 4.
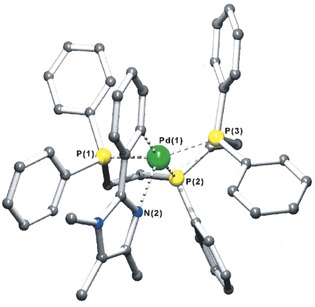
Molecular structure for compound 7. Adapted from Ref. 11. Copyright 2000 Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim.
3. Thiosemicarbazone [C,N,S] Palladacycles and Bidentate [P,S] Metalloligands
Thiosemicarbazones produce four‐metal compounds with two well‐defined bonds between palladium and sulfur, that is, Schelating–Pd and Sbridging–Pd, fastening firmly to the metal as tridentate [C,N,S]. Compared to tridentate [C,N,N] and [C,N,O] analogues, the softer sulfur donor in the terms of Pearson's concept12 strengthens the bond to palladium. Thus, reaction with tertiary diphosphanes yields species with the metal bonded solely to one phosphorus donor, the firmness of the Schelating–Pd linkage impeding the diphosphane from binding to the metal center in a chelate pattern.13, 14 We then hypothesized that the resulting compounds could make use of the non‐coordinated phosphorus lone pair to link to a following metal center, rendering species with a bridging diphosphane and producing compounds analogous to bimetallic thiosemicarbazone complexes,13 in which bonding of the S atom to Pd as Sbridging–Pd is absent.
Notwithstanding, our initial results showed this not to be the issue, as the sulfur donor also displayed coordinative capability by performing as tridentate, thus giving rise to an “unprecedented class of bidentate P,S metalloligands” and conclusive proof of the behavior of these novel palladacycles could be well established (Scheme 4).15 Although the initial attempts were successfully carried out with Ph2PCH2PPh2 (dppm), subsequent research yielded bimetallics with alternative diphosphines such as Ph2P(C=CH2)PPh2 and Ph2PC(CH2)2PPh2, 16 (Figure 5).16 With Ph2Ppy, bidentate [N,S] metalloligands were made, also yielding bimetallic complexes, 17 (Figure 6).17
Scheme 4.
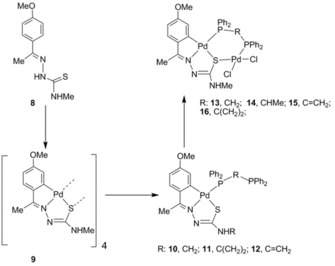
Reaction route leading to bidentate [P,S] metalloligands and bimetallic palladacycles. Adapted from Ref. 15. Copyright 2003 American Chemical Society.
Figure 5.
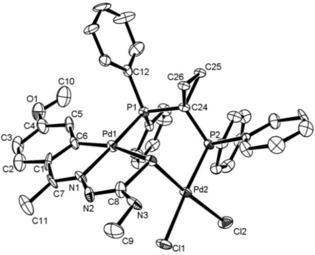
Structure of the bimetallic compound 16. Reproduced from Ref. 16. Copyright 2010 American Chemical Society.
Figure 6.
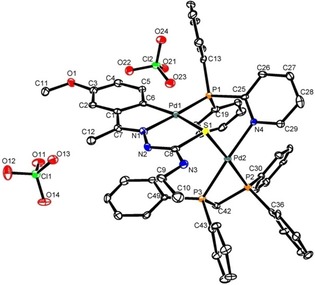
Structure of the bimetallic compound 17. Reproduced from Ref. 17. Copyright 2014 American Chemical Society.
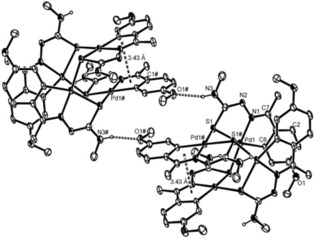
The thiosemicarbazone tetranuclear palladacycles may also form polymers through hydrogen bonds. As an example we have encountered such arrangement in the complex [Pd{4‐MeOC6H3C(Me)=NN=C(S)NHMe}]4 18, (Figure 7).16 The structure of the individual molecules contains the metallated fragments arranged as two sets, mutually at 90°, of nearly co‐planar anti‐parallel pairs separated by about 3.6 Å. Hydrogen bonds and intramolecular π–π interactions between the parallel metallacycles are responsible for the unprecedented beautiful lattice it displays (Figure 8).16
Figure 7.
Structure for the tetranuclear compound 18. Reproduced from Ref. 16. Copyright 2010 American Chemical Society.
Figure 8.
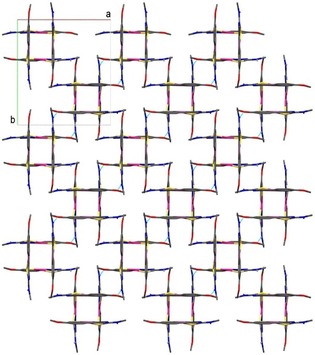
Structure for complex 18 observed on the ab plane. Reproduced from Ref. 16. Copyright 2010 American Chemical Society.
Up to this point, our previous studies related to thiosemicarbazone palladacycles showed that treatment of the ligand with an appropriate palladium salt gave tetranuclear species, always with two perpendicular pairs of parallel metallated systems, set on a central Pd4S4 eight‐membered ring.13, 18 Where bonding of the metal to the phenyl carbon is absent and the metal remains linked only to the nitrogen and sulfur donors, the tetranuclear arrangement of the molecule is preserved.19 Therefore, our outlook for the progress into further visible features related to the chemical behavior of these species was to investigate alternative structural characteristics originated by modifications in the architecture of the organic moiety and of its reaction pattern on treatment with the metal substrate; the resulting tailor‐made route would then be the changes made in the central sulfur–palladium core.
We speculated that reshaping the size and the flexibility of the five‐membered metallated ring, [Pd−C=C−C=N], should vary the angle between the palladium coordination and the metallated phenyl ring planes, in addition to readjusting the angles among the palladium coordination planes, therefore hampering the establishment of the tetrametallic arrangement and generating variation in the atom count and in the spatial arrangement of the PdnSn core. To attain such a situation, the metal should bind to a non‐phenyl carbon, for instance a CH3− carbon atom. The ligand of choice for this purpose was the thiosemicarbazone 2,5‐Me2C6H3C(H)=NN(H)C(=S)−NHEt and attempts were conducted to produce metallation of the methyl group to produce a more flexible six‐membered metallated ring, hoping that the steric hindrance of the 5‐methyl would sufficiently block binding of the metal to the C6 atom (C7 in the molecular structure).
Once more, the resulting Pd3S3 complex (Figures 9 and 10) did not meet with our expectations and a unique trinuclear structure was formed, containing a central Pd3S3 ring of interspersed palladium and sulfur atoms that are combined as two staggered Pd3 and S3 quasi‐parallel triangles at 175.45°.20
Figure 9.
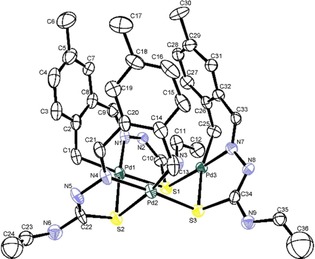
Structure for the Pd3S3 complex. Reproduced with permission from Ref. 20. Copyright 2009 Elsevier.
Figure 10.
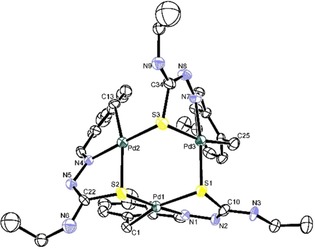
Structure for the Pd3S3 complex along the c axis. Reproduced with permission from Ref. 20. Copyright 2009 Elsevier.
4. Palladacycles as Catalysts
Reactions catalyzed by palladium typify one of the most treasured tools in organic synthesis, spanning now over nearly forty years.21 One of the most influential and widely used modus operandi for attaining carbon–carbon bond formation is the Suzuki–Miyaura cross‐coupling reaction.22 Palladacycles23 have materialized as a preeminent collection of catalysts, partially owing to their water and air stability, with the first phosphapalladacycles being highlighted by Herrmann et al.24 We were interested in considering the demeanor of four‐palladium thiosemicarbazone complexes, deploying a boronic function on the metallated ring, spurred by our previous results with thiosemicarbazone palladacycles. We initiated our research by searching for novel cyclopalladated compounds supported by Suzuki couplings, for which we chose the treatment of palladacycles bearing a boronic acid functionality with aryl halides, for which thiosemicarbazone palladacycles were selected, because the latter palladacycles themselves are rather insoluble in the more usual organic solvents as well as in water. We were able to make complex 22 either directly from the tetranuclear cluster or via triphenylphosphine derivative 21, in both cases under catalytic conditions (Scheme 5).25
Scheme 5.
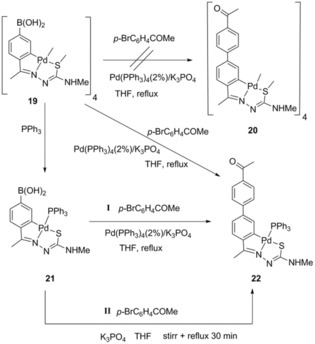
Preparation of compound 22. With Pd(PPh3)4 (I); in the absence of an external catalyst (II). Reproduced from Ref. 25. Copyright 2016 The Royal Society of Chemistry.
We also observed, however, that 22 could be prepared directly from 21 in the absence of catalyst, which was clearly a novelty to the Suzuki–Miyaura cross‐coupling reaction. This was made possible by close inspection of the thin layer chromatography plates, which allowed us to tentatively propose a plausible PdII/PdIV[26] mechanism, which could be in agreement with our discovery (Scheme 6). The structure of complex 22 derived from autocatalysis is depicted in Figure 11, together with the calculated structure according to DFT analysis, which nearly matches the experimental data. Also, because the role of the catalyst is assumed by the palladium reagent itself, the aryl feedstock needed to produce the coupling is set on the metal coordination sphere, from which the final product is discarded; the process is self‐consuming making recovery of the catalyst needless. We conclude by saying that, in the absence of a traditional palladium catalyst, the reaction of a palladacycle functionalized by a boronic acid with an appropriate reagent progresses into the corresponding cross‐coupling product, being yet another example of serendipity.
Scheme 6.
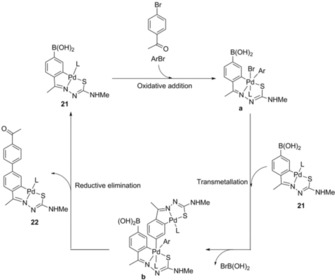
Proposed PdII/PdIV mechanism (L=PPh3). Reproduced from Ref. 25. Copyright 2016 The Royal Society of Chemistry.
Figure 11.
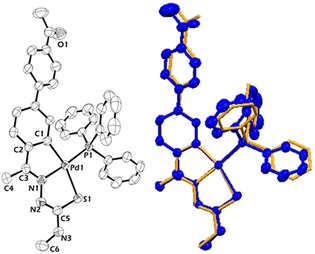
Structure of complex 22 (left). The DFT optimized (orange) and crystal (blue) structures are compared on the right. Reproduced from Ref. 25. Copyright 2016 The Royal Society of Chemistry.
5. Crown Ether Palladacycles
The ability of crown ethers as complexation agents for selectively binding to alkali metal and alkaline‐earth metal cations, as well as to other ionic species, constitutes one of their most prominent properties; the differing dimensions of the crown ether pocket can be modulated to bind selectively with a given cation.27 Hence, they have shown a wide variety of applications related to sensors, membrane ion transport, and potential anticancer species, among others.28 Palladacycles may also furnish a new and attractive itinerary for the synthesis of metalloligands containing a crown ether ring system, adequate for further coordination to metal cations, giving way to complexes with a transition metal/main group metal combination. In accordance with the ring size, sodium29 and silver30 cations may be accommodated in 15‐crown‐5 and in 18‐crown‐6 rings, respectively. The potassium cation is amongst those that show the best affinity for crown ether rings and for which the most studies have been carried out. Its size makes it most suitable for 18‐crown‐6 rings; for the smaller 15‐crown‐5 ether, a sandwich‐type geometry has been suggested for complexation of the cation by two 15‐crown‐5 ether moieties.31, 32, 33 We anticipated that tetranuclear thiosemicarbazone palladacycles could offer an advantageous solution towards the isolation of the aforesaid potassium coordination with 15‐crown‐5 moieties on the four metallated phenyl rings; however, to no avail. Then, our ensuing hypothesis was that adequately separating the two pairs of parallel metallated moieties should produce the needed arrangement, giving molecules that are appropriate for sandwiched potassium coordination through several reaction steps. But, again, this was not the case, and what we found was that the desired compound could be made in a one‐pot reaction to give coordinated potassium in small crown ether rings (Scheme 7).34
Scheme 7.
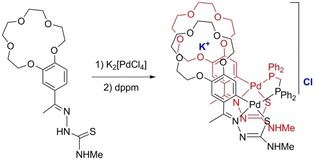
Reaction leading to the synthesis of the sandwiched potassium crown ether complex. Reproduced with permission from Ref. 34. Copyright 2017 Wiley‐VCH Verlag GmbH & Co. KGaA.
This process culminates in the isolation of a unique dinuclear phosphine‐bridged thiosemicarbazone palladacycle containing 15‐crown‐5 ether rings on the metallated phenyls, capable of coordinating to the K+ cation by sandwiching it between the two ether rings, which results in a trinuclear mixed main group–transition metal complex. This result represents a further enrichment in the chemistry of palladacycles as well as in the coordination chemistry of crown ethers. Figure 12 shows the crystal structure for the compound. Figure 13 depicts a view along the b axis together with a space‐filling model (partially).
Figure 12.
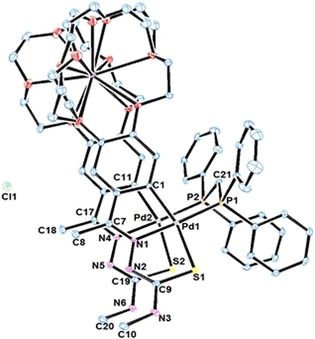
Thermal ellipsoid plot for the trinuclear crown ether complex. Reproduced with permission from Ref. 34. Copyright 2017 Wiley‐VCH Verlag GmbH & Co. KGaA.
Figure 13.
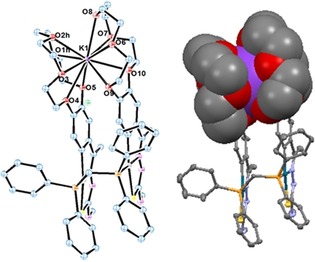
Thermal ellipsoid plot for the trinuclear crown ether complex along the b axis. Space‐filling model for the sandwich coordination. Reproduced with permission from Ref. 34. Copyright 2017 Wiley‐VCH Verlag GmbH & Co. KGaA.
6. Conclusions
The results shown herein put forward how serendipity influences basic chemical research. Often, the deviation of the results found with respect to those proposed initially changes our expectations. This is not at all discouraging, as it allows us to find novel structural aspects of which we would otherwise not be aware, as well as striking reactivity trends that, more often than not, shed new light on the chemical behavior of the species under study. It is necessary to be sharp enough to realize that a disappointing result is actually not only something new, but also groundbreaking; a failed finding should never be ruled out. As Reedjik stated at the 34th International Conference on Coordination Chemistry: “Don't throw it in the bin; serendipity may calling at your door”.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Adolfo Fernández Figueiras graduated in Chemistry in 2012 from the University of Santiago de Compostela (Spain) and is now a Ph.D. candidate in the Chemical Science and Technology Program at the Organometallic and Macrocyclic Chemistry Group, Faculty of Chemistry of the University of Santiago de Compostela (Spain). His scientific interests are focused on cyclometalation complexes derived from iminophosphoranes with palladium and platinum and their applications in catalysis.

Biographical Information
Fátima Lucio‐Martínez graduated in Chemistry in 2012 from the University of Santiago de Compostela (Spain) and is now a Ph.D. candidate in the Chemical Science and Technology Program at the Organometallic and Macrocyclic Chemistry Group, Faculty of Chemistry of the University of Santiago de Compostela (Spain). Her scientific interests are focused on cyclometalation complexes bearing a crown ether in the structure with palladium and platinum and DFT calculations.

Biographical Information
Paula Munín Cruz graduated in Chemistry in 2013 from the University of Santiago de Compostela (Spain) and is now a Ph.D. candidate in the Chemical Science and Technology Program at the Organometallic and Macrocyclic Chemistry Group, Faculty of Chemistry of the University of Santiago de Compostela (Spain). Her scientific interests are focused on cyclometalation complexes and the study of the properties of their derivatives bearing ferrocene phosphine.

Biographical Information
Juan M. Ortigueira graduated in Chemistry from the Universidad de Santiago de Compostela, Galicia (Spain), where he received his Ph.D. degree in Chemistry in 1994. After a stay at the University of Durham (UK) with Professor Melvin Kilner, where he dealt with the chemistry of manganese and rhenium coordination compounds, he was appointed Assitant Professor of Inorganic Chemistry at the Facultad de Ciencias (USC, Campus de Lugo). In 2001, he was appointed Senior Lecturer in Inorganic Chemistry at the USC, where he is currently based. His research interests ctural features with the cyclometallated compounds and supramolecular chemistry.

Biographical Information
M. Teresa Pereira graduated in Chemistry from the Universidad de Santiago de Compostela, Galicia (Spain), where she received her Ph.D. degree in Chemistry in 1986. She was appointed Assitant Professor of Inorganic Chemistry at the Facultad de Química (USC). She completed postdoctoral research at the University of Strasbourg (France) with Dr. Michel Pfeffer, where her research involved palladium organometallic chemisty. She was appointed Lecturer in Inorganic Chemistry at the USC, and later Full Professor in 2008. Her research deals with the chemistry of palladacyles and platinacycles, palladium bimetallics and C−H activation.

Biographical Information
Paula Polo finished her degree in Chemistry in 2010 and received her Ph.D. degree in Chemistry in 2016 from the University of Santiago de Compostela (Spain). Since 2011, she has been a Fellow Researcher at the Faculty of Chemistry at the University of Santiago de Compostela, being part of the Macrocyclic and Organometallic Chemistry research group. Her scientific interests include organometallic compounds, catalytic applications and green chemistry.

Biographical Information
Francisco Reigosa obtained his degree in Chemistry in 2015 from the University of Santiago de Compostela (Spain); he has a MSc from the same university and is now a Ph.D. candidate in the Chemical Science and Technology Program at the Organometallic and Macrocyclic Chemistry Group, Faculty of Chemistry of the University of Santiago de Compostela (Spain). His scientific experience is focused on cyclometalation compounds derived from thiosemicarbazones with palladium, cross‐coupling reactions and DFT calculations.

Biographical Information
José M. Vila graduated in Chemistry from the Universidad Autónoma de Madrid (Spain) and received his Ph.D. degree in Chemistry in 1985 from the Universidad de Santiago de Compostela, Galicia (Spain). He was appointed Assitant Professor of Inorganic Chemistry at the Facultad de Química (USC). He completed postdoctoral research at the University of Leeds (UK) with Professor Bernard L. Shaw in the field of phosphine chemistry with transition metals.He was appointed Lecturer in Inorganic Chemistry at the USC, and later Full Professor in 2007. His research deals with the chemistry of palladium and platinum metallacycles, metalloligands, crown ether palladacycles and palladium catalysts.

Acknowledgements
We thank the Xunta de Galica (Galicia, Spain) under the Grupos de Referencia Competitiva Programme (Project GRC2015/009). F.L.‐M. and F. R. thank the Spanish Ministry of Education (grant FPU13/05014 and FPU 15/07145).
A. Fernández-Figueiras, F. Lucio-Martínez, P. Munín-Cruz, J. M. Ortigueira, P. Polo-Ces, F. Reigosa, M. T. Pereira, J. M. Vila, ChemistryOpen 2018, 7, 754.
Contributor Information
Prof. Dr. M. Teresa Pereira, Email: mteresa.pereira@usc.es.
Prof. Dr. José M. Vila, Email: josemanuel.vila@usc.es.
References
- 1.
- 1a. Espinet P., Esteruelas M. A., Oro L. A., Serrano J. L., Sola E., Coord. Chem. Rev. 1992, 117, 215–274; [Google Scholar]
- 1b. Marcos M., Metallomesogens. Synthesis Properties and Applications, (Ed.: J. L. Serrano), VCH, Weinheim, 1996; [Google Scholar]
- 1c. López-Torres M., Fernández A., Fernández J. J., Castro-Juiz S., Suárez A., Vila J. M., Pereira M. T., Organometallics 2001, 20, 1350–1353; [Google Scholar]
- 1d. Pucci D., Barneiro G., Bellusci A., Crispini A., Ghedini M., J. Organomet. Chem. 2006, 691, 1138–1142; [Google Scholar]
- 1e. Ghedini M., Aiello I., Crispini A., Golemme A., La Deda M., Pucci D., Coord. Chem. Rev. 2006, 250, 1373–1390; [Google Scholar]
- 1f. Lowry M. L. S., Bernhard S., Chem. Eur. J. 2006, 12, 7970–7977; [DOI] [PubMed] [Google Scholar]
- 1g. Omae I., J. Organomet. Chem. 2007, 692, 2608–2632; [Google Scholar]
- 1h. Ma D.-L., Ma V. P.-Y., Chan D. S.-H., Leung K.-H., He H.-Z., Leung C.-H., Coord. Chem. Rev. 2012, 256, 3087–3113; [Google Scholar]
- 1i. Trofimenko S., Inorg. Chem. 1973, 12, 1215–1221. [Google Scholar]
- 2.
- 2a. Omae I., Organometallic Intramolecular-Coordination Compounds, Elsevier Science, Amsterdam-New York, 1986; [Google Scholar]
- 2b. Newkome G. R., Puckett W. E., Gupta W. K., Kiefer G. E., Chem. Rev. 1986, 86, 451–489; [Google Scholar]
- 2c. Ryabov A. D., Chem. Rev. 1990, 90, 403–424; [Google Scholar]
- 2d. Dunina V. V., Gorunova O. N., Russ. Chem. Rev. 2004, 73, 309–350; [Google Scholar]
- 2e. Omae I., Coord. Chem. Rev. 2004, 248, 995–1023; [Google Scholar]
- 2f. Dunina V. V., Gorunova O. N., Russ. Chem. Rev. 2005, 74, 871–913; [Google Scholar]
- 2g. Dupont J., Consorti C. S., Spencer J., Chem. Rev. 2005, 105, 2527–2572; [DOI] [PubMed] [Google Scholar]
- 2h. Vicente J., Saura-Llamas I., Comments Inorg. Chem. 2007, 28, 39–72; [Google Scholar]
- 2i. Djukic J., Hijazi A., Flack H. D., Bernardinelli G., Chem. Soc. Rev. 2007, 37, 406–425; [DOI] [PubMed] [Google Scholar]
- 2j. Dupont J., Pfeffer M., Eds., “Palladacycles”, WILEY-VCH, Weinheim, 2008; [Google Scholar]
- 2k. Albrecht M., Chem. Rev. 2010, 110, 576–623; [DOI] [PubMed] [Google Scholar]
- 2l. Omae I., Cyclometalation Reactions. Five-Membered Ring Products as Universal Reagents, Springer, Japan: 2014. [Google Scholar]
- 3. Vila J. M., Pereira M. T., Ortigueira J. M., Fernández J. J., Fernández A., López-Torres M., Adams H., Organometallics 1999, 18, 5484–5487. [Google Scholar]
- 4. Onoue M., Moritani I., J. Organomet. Chem. 1972, 43, 431–436. [Google Scholar]
- 5. Ustynyuk Y., Chertov V. A., Barinov J. V., J. Organomet. Chem. 1971, 29, C53–C54. [Google Scholar]
- 6.
- 6a. Granell J., Sales J., Vilarrasa J., Declercq P. G., Germain G., Miravitlles C., Solans X., J. Chem. Soc. Dalton Trans. 1983, 2441–2446; [Google Scholar]
- 6b. Granell J., Sainz D., Sales J., Solans X., Font-Altaba M., J. Chem. Soc. Dalton Trans. 1986, 1785–1790; [Google Scholar]
- 6c. Butler I. R., Kalaji M., Nehrlich L., Hurthouse M., Karaulov A. I., Malik K. M. A., J. Chem. Soc. Chem. Commun. 1995, 459–460. [Google Scholar]
- 7. Vicente J., Arcas A., Bautista D., Jones P. G., Organometallics 1997, 16, 2127–2138. [Google Scholar]
- 8. Cecconi F., Ghilardi C. A., Midollini S., Moneti S., Orlandini A., Scapacci G., J. Chem. Soc. Dalton Trans. 1989, 211–216. [Google Scholar]
- 9. López-Torres M., Fernández A., Fernández J. J., Suárez A., Pereira M. T., Ortigueira J. M., Vila J. M., Adams H., Inorg. Chem. 2001, 40, 4583–4587. [DOI] [PubMed] [Google Scholar]
- 10. Fernández A., Fernández J. J., López-Torres M., Suárez A., Ortigueira J. M., Vila J. M., Adams H., J. Organomet. Chem. 2000, 612, 85–95. [Google Scholar]
- 11. Lousame M., Fernández A., López-Torres M., Vázquez-García D., Vila J. M., Suárez A., Ortigueira J. M., Fernández J. J., Eur. J. Inorg. Chem. 2000, 2055–2062. [Google Scholar]
- 12. Pearson R. G., J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar]
- 13. Vila J. M., Pereira M. T., Fernández A., López-Torres M., Adams H., J. Chem. Soc. Dalton Trans. 1999, 4193–4201. [Google Scholar]
- 14. Amoedo A., Graña M., Martínez J., Pereira M. T., López-Torres M., Fernández A., Fernández J. J., Vila J. M., Eur. J. Inorg. Chem. 2002, 613–620. [Google Scholar]
- 15. Martínez J., Pereira M. T., Buceta I., Alberdi G., Amoedo A., Fernández J. J., López-Torres M., Vila J. M., Organometallics 2003, 22, 5581–5584. [Google Scholar]
- 16. Antelo J. M., Adrio L., Pereira M. T., Ortigueira J. M., Fernández J. J., Vila J. M., Cryst. Growth Des. 2010, 10, 700–708. [Google Scholar]
- 17. Pereira M. T., Antelo J. M., Adrio L., Martínez J., Ortigueira J. M., López-Torres M., Vila J. M., Organometallics 2014, 33, 3265–3274. [Google Scholar]
- 18. Amoedo A., Adrio L., Antelo J. M., Martínez J., Pereira M. T., Fernández A., Vila J. M., Eur. J. Inorg. Chem. 2006, 3016–3021. [Google Scholar]
- 19. Hueso-Ureña F., Illán-Cabeza N. A., Moreno-Carretero M. N., Peñas-Chamorro A. L., Faure R., Inorg. Chem. Commun. 1999, 2, 323. [Google Scholar]
- 20. Adrio L., Antelo J. M., Fernández J. J., Hii K. K. (Mimi), Pereira M. T., Vila J. M., J. Organomet. Chem. 2009, 694, 747–751. [Google Scholar]
- 21.
- 21a. Alonso D. A., Nájera C., Chem. Soc. Rev. 2010, 39, 2891–2902; [DOI] [PubMed] [Google Scholar]
- 21b. Tsuji J., Palladium Reagents and Catalysts: Innovations in Organic Synthesis, Wiley, New York: 1995; [Google Scholar]
- 21c. Negishi E. I., Handbook of Organopalladium Chemistry for Organic Synthesis, Wiley, New York, 2002; [Google Scholar]
- 21d.J. J. Li, G. W. Gribble, Palladium in Heterocyclic Chemistry: A Guide for the Synthetic Chemist, Pergamon, New York, 2000.
- 22.
- 22a. Miyaura N., Suzuki A., J. Chem. Soc. Chem. Commun. 1979, 866–867; [Google Scholar]
- 22b. Suzuki A., Chem. Commun. 2005, 4759–4763. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Bedford R. B., Chem. Commun. 2003, 1787–1796; [PubMed] [Google Scholar]
- 23b. Beletskaya I. P., Cheprakov A. V., J. Organomet. Chem. 2004, 689, 4055–4082; [Google Scholar]
- 23c. Xu L.-M., Li B.-J., Yang Z., Shi Z.-J., Chem. Soc. Rev. 2010, 39, 712–733; [DOI] [PubMed] [Google Scholar]
- 23d. Luo Q.-L., Tan J.-P., Li Z.-F., Qin Y., Ma L., Xiao D.-R., Dalton Trans. 2011, 40, 3601–3609; [DOI] [PubMed] [Google Scholar]
- 23e. Marziale A. N., Jantke D., Faul S. H., Reiner T., Herdtweck E., Eppinger J., Green Chem. 2011, 13, 169–177; [Google Scholar]
- 23f. Rosa G., Rosa D. S., RSC Adv. 2012, 2, 5080–5083; [Google Scholar]
- 23g. Susanto W., Chu C.-Y., Ang W. J., Chou T.-C., Lo L.-C., Lam Y., J. Org. Chem. 2012, 77, 2729–2742; [DOI] [PubMed] [Google Scholar]
- 23h. Wang W.-C., Peng K.-F., Chen M.-T., Chen C.-T., Dalton Trans. 2012, 41, 3022–3029; [DOI] [PubMed] [Google Scholar]
- 23i. Karami K., Ghasemi M., Naeini N. H., Tetrahedron Lett. 2013, 54, 1352–1355; [Google Scholar]
- 23j. Rao G. K., Kumar A., Kumar S., Dupare U. B., Singh A. K., Organometallics 2013, 32, 2452–2458; [Google Scholar]
- 23k. Privér S. H., Bennett M. A., Willis A. C., Pottabathula S., Lakshmi Kantam M., Bhargava S. K., Dalton Trans. 2014, 43, 12000–12012; [DOI] [PubMed] [Google Scholar]
- 23l. Cho H.-J., Jung S., Kong S., Park S.-J., Lee S.-M., Lee Y.-S., Adv. Synth. Catal. 2014, 356, 1056–1064; [Google Scholar]
- 23m. Mamidala R., Mukundam V., Dhanunjayarao K., Venkatasubbaiah K., Dalton Trans. 2015, 44, 5805–5809; [DOI] [PubMed] [Google Scholar]
- 23n. Sharma A. K., Joshi H., Bhaskar R., Kumar S., Singh A. K., Dalton Trans. 2017, 46, 2485–2496; [DOI] [PubMed] [Google Scholar]
- 23o. Firinci R., Emin Günay M., Gökçe A. G., Appl. Organomet. Chem. 2017, 31, 1–10; [Google Scholar]
- 23p. Gorunova O. N., Grishin Y. K., Ilyin M. M., Kochetkov K. A., Churakov A. V., Kuz′mina L. G., Dunina V. V., Russ. Chem. Bull. 2017, 66, 282–292. [Google Scholar]
- 24.
- 24a. Herrmann W. A., Brossmer C., Öfele K., Reisinger C.-P., Priermeier T., Beller M., Fischer H., Angew. Chem. Int. Ed. Engl. 1995, 34, 1844–1848; [Google Scholar]; Angew. Chem. 1995, 107, 1989–1992; [Google Scholar]
- 24b. Beller M., Fischer H., Herrmann W. A., Öfele K., Brossmer C., Angew. Chem. Int. Ed. Engl. 1995, 34, 1848–1849; [Google Scholar]; Angew. Chem. 1995, 107, 1992–1993. [Google Scholar]
- 25. Lucio-Martínez F., Adrio L. A., Polo-Ces P., Ortigueira J. M., Fernández J. J., Adams H., Pereira M. T., Vila J. M., Dalton Trans. 2016, 45, 17598–17601. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Blacque O., Frech C. M., Chem. Eur. J. 2010, 16, 1521–1531; [DOI] [PubMed] [Google Scholar]
- 26b. Dang Y., Qu S., Nelson J. W., Pham H. D., Wang Z. X., Wang X., J. Am. Chem. Soc. 2015, 137, 2006–2014. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Izatt R. M., Bradshaw J. S., Nielsen S. A., Lamb J. D., Christensen J. J., Sen D., Chem. Rev. 1985, 85, 271–339; [Google Scholar]
- 27b. Inoue Y., Gokel G. W., Cation Binding by Macrocycles, Marcel Dekker, New York, 1990. [Google Scholar]
- 28.
- 28a. Alexandratos S. D., Stine C. L., React. Funct. Polym. 2004, 60, 3–16; [Google Scholar]
- 28b. Arias J., Bardají M., Espinet P., Inorg. Chem. 2008, 47, 3559–3567; [DOI] [PubMed] [Google Scholar]
- 28c. Arias J., Bardají M., Espinet P., Inorg. Chem. 2008, 47, 1597–1606; [DOI] [PubMed] [Google Scholar]
- 28d. Preihs C., Magda D., Sessler J., J. Porphyrins Phthalocyanines 2011, 15, 539–546; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28e. Kado S., Takeshima Y., Nakahara Y., Kimura K., J. Inclusion Phenom. Macrocyclic Chem. 2012, 72, 227–232; [Google Scholar]
- 28f. Sun Z., Barboiu M., Legrand Y.-M., Petit E., Rotaru A., Angew. Chem. Int. Ed. 2015, 54, 14473–14477; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 14681–14685; [Google Scholar]
- 28g. Yu H.-R., Hu J.-Q., Lu X.-H., Ju X.-J., Liu Z., Xie R., Wang W., Chu L.-Y., J. Phys. Chem. B 2015, 119, 1696–1705; [DOI] [PubMed] [Google Scholar]
- 28h. Gilles A., Barboiu M., J. Am. Chem. Soc. 2016, 138, 426–432; [DOI] [PubMed] [Google Scholar]
- 28i. Sun Z., Gilles A., Kocsis I., Legrand Y.-M., Petit E., Barboiu M., Chem. Eur. J. 2016, 22, 2158–2164; [DOI] [PubMed] [Google Scholar]
- 28j. Godoy F., Maldonado T., Flores E., Agurto N., González R., Ferraudi G., Lappin G., Appl. Organomet. Chem. 2017, 31, 1–9; [Google Scholar]
- 28k. Yu H.-L., Wang W.-Y., Hong B., Si Y.-L., Ma T.-L., Zheng R., RSC Adv. 2017, 7, 41830–41837; [Google Scholar]
- 28l. Rusinov G. L., Charushin V. N., J. Photochem. Photobiol. A 2018, 351, 16–28; [Google Scholar]
- 28m. Ovchinnikova I. G., Kim G. A., Matochkina E. G., Kodess M. I., Slepukhin P. A., Kovalev I. S., Nosova E. V., Rusinov G. L., Charushin V. N., J. Photochem. Photobiol. A 2018, 351, 16–28. [Google Scholar]
- 29. Castro-Juiz S., Fernández A., López-Torres M., Vázquez-García D., Suárez A. J., Vila J. M., Fernández J. J., Organometallics 2009, 28, 6657–6665. [Google Scholar]
- 30. Vázquez-García D., Fernández A., López-Torres M., Rodríguez A., Varela A., Pereira M. T., Vila J. M., Fernández J. J., Organometallics 2011, 30, 396–404. [Google Scholar]
- 31. Dietrich B., Viout P., Lehn J.-M., Macrocyclic Chemistry-Aspects of Organic and Inorganic Supramolecular Chemistry, VHC, New York, 1993, 174. [Google Scholar]
- 32. Pedersen C. J., J. Am. Chem. Soc. 1970, 92, 386–391. [Google Scholar]
- 33.
- 33a. Patel G., Kumar A., Pal U., Menon S., Chem. Commun. 2009, 1849; [DOI] [PubMed] [Google Scholar]
- 33b. Weißenstein A., Würthner F., Chem. Commun. 2015, 51, 3415–3418. [DOI] [PubMed] [Google Scholar]
- 34. Lucio-Martínez F., Bermúdez B., Ortigueira J. M., Adams H., Fernández A., Pereira M. T., Vila J. M., Chem. Eur. J. 2017, 23, 6255–6258. [DOI] [PubMed] [Google Scholar]