

Abstract
The reproductive endocrine systems are vastly different between males and females. This sexual dimorphism of the endocrine milieu originates from sex-specific differentiation of the somatic cells in the gonads during fetal life. Most gonadal somatic cells arise from the adrenogonadal primordium. After separation of the adrenal and gonadal primordia, the gonadal somatic cells initiate sex-specific differentiation during gonadal sex determination with the specification of the supporting cell lineages: Sertoli cells in the testis vs granulosa cells in the ovary. The supporting cell lineages then facilitate the differentiation of the steroidogenic cell lineages, Leydig cells in the testis and theca cells in the ovary. Proper differentiation of these cell types defines the somatic cell environment that is essential for germ cell development, hormone production, and establishment of the reproductive tracts. Impairment of lineage specification and function of gonadal somatic cells can lead to disorders of sexual development (DSDs) in humans. Human DSDs and processes for gonadal development have been successfully modeled using genetically modified mouse models. In this review, we focus on the fate decision processes from the initial stage of formation of the adrenogonadal primordium in the embryo to the maintenance of the somatic cell identities in the gonads when they become fully differentiated in adulthood.
Essential points
Somatic cells in the fetal gonads lay the foundation for the establishment of sexually dimorphic reproductive systems
Somatic cell progenitors in the gonadal primordium undergo several lineage decisions to form the supporting and steroidogenic cells lineages, which ultimately support germ cell development and reproductive functions
The supporting cell lineage, Sertoli cells in the testis and granulosa cells in the ovary, respectively, arises from the coelomic epithelium of the gonadal primordium; Sertoli cells and granulosa cells differentiate from the common somatic precursors through activation and suppression of mutually antagonistic male and female sex determination pathways
The steroidogenic cell lineage, Leydig cells in the testis and theca cells in the ovary, originates from the coelomic epithelium of the gonadal primordium and the neighboring mesonephros
Steroidogenic cells differentiate under paracrine regulation from the supporting cell lineage in both sexes
Gonadal and adrenal cells originate from a joint primordium, which separates into the two lineages during fetal development
Dimorphism of reproductive organs lays the foundation for sex differences in body size, body composition, metabolism, immune system, brain function, stress responses, and disease susceptibility and presentation (1–5). Sexual dimorphism in most mammals is determined by sex chromosome composition in two ways: directly through innate genetic differences between the XX and XY cells, and indirectly though the establishment of gonadal identity and subsequent sex-specific hormonal milieu (6). The origin of most sexually dimorphic traits in the reproductive system can be traced back to early times in fetal development, when the sexually indifferent fetus begins to develop as male or female during sex determination. The drivers of gonadal sex determination are the somatic cells in the fetal gonad. These specialized somatic cells orchestrate the morphogenetic cascade that leads to the formation of testis or ovary and their distinct endocrine cell types. In this review, we focus on how endocrine cell lineages, Sertoli cells and Leydig cells in the testis and granulosa and theca cells in the ovary, are established during embryogenesis through genetic sex determination and paracrine signaling. We also discuss how insights gained from mouse models advance our understanding of human sex determination and the disorders associated with defects in this process.
Disorders of Sex Development in Humans
When the biological sex of the individual does not match the genetic sex, or falls in between the opposite ends of the sexual spectrum, the individual is considered as having a disorder of sex development (DSD). DSDs are defined as “congenital conditions in which the development of chromosomal, gonadal, or anatomical sex is atypical,” and they can range from complete sex reversal to minor reproductive defects that manifest later in life (7). In 2005, the Chicago Consensus Conference reclassified DSDs based on their etiology (chromosomal abnormality or gonadal/reproductive phenotypes) (8, 9). The overall incidence of DSDs is estimated at 1 in 4500 to 5500 live births (7), with some forms of DSDs considerably more frequent than others. Turner syndrome, or 45,XO, occurs in 1 in 2500 live births, whereas 46,XX ovotesticular disorders are very rare at 1 in 100,000 live births (7). Although many of the common DSD conditions, such as sex chromosome aneuploidies, congenital adrenal hyperplasia, and androgen insensitivity syndrome, are well characterized, most DSD cases are idiopathic, as diagnosis remains challenging. Moreover, mutations in key genes in sexual development often give rise to highly variable clinical phenotypes, as a mutation might cause male infertility or premature menopause in one generation and gonadal dysgenesis in another generation, thereby further increasing the difficulty of diagnosing patients with DSDs (10). It is estimated that only 13% of patients with DSDs receive a definitive clinical and genetic diagnosis (11). Utilizing next-generation sequencing techniques could increase the diagnosis rate to 43% to 60%, depending on the specific disorder, which gives hope for more accurate diagnosis and management of individual patients in the future (11, 12). Cases of human DSDs have led to the discovery of a number of genes that we now know are critical for human sexual development. The functions of these genes have been further characterized using genetically modified mouse models, which provide insights into the complex regulatory network controlling sexual differentiation and the specific roles these genes play in facilitating the establishment of different endocrine cell types in the sex-specific development of the gonads.
Formation of the Gonadal Primordium
The gonadal primordium, which is the somatic cell component that eventually gives rise to the testis or ovary, is established on gestational week (GW) 4 in the human embryo and approximately embryonic day (E) 10 in the mouse (13). The gonadal primordium (also known as the genital ridge) forms on the ventral side of the temporary embryonic kidney (mesonephros) as a result of the thickening and proliferation of the coelomic epithelium. Although the anterior-to-posterior thickening of the coelomic epithelium defines the first physical boundary of the gonadal field (14), cells in the coelomic epithelium already acquire a molecular signature unique to the gonads. This includes expression of the transcription factors GATA binding protein 4 (GATA4), Wilms tumor 1 (WT1), and the nuclear receptor steroidogenic factor 1 (SF1, encoded by Nr5a1) (Figs. 1 and 2) (14–16). In the mouse, Gata4 expression is induced by transcription factors sine oculis homeobox (SIX) 1 and SIX4 in the coelomic epithelium before the initial wave of coelomic epithelial thickening (17). Without Gata4, the thickening of the coelomic epithelium fails to occur, subsequently leading to agenesis of the gonads (14).
Figure 1.

Lineage progression of the supporting and interstitial cells in the mouse testis. Somatic progenitor cells in the XY gonad (green) give rise to supporting/Sertoli cells (blue) or an interstitial progenitor population (pale orange). The supporting/Sertoli cell lineage is defined by the upregulation of Sry, followed by Sox9, that transforms them into a polarized epithelial cell type in the testis. As Sertoli cells mature postnatally (dark blue), they downregulate Amh expression and acquire androgen responsiveness. The interstitial progenitor cell population is defined by expression of Hes1 and other genes (Arx, CouptfII/Nr2f2, Mafb). The interstitial progenitor cells begin to express Gli1 in response to hedgehog signaling from Sertoli cells and differentiate into fetal Leydig cells (orange). A population of the interstitial progenitor cells remains as a nonsteroidogenic progenitor population that gives rise to adult Leydig cells (dark orange) postnatally. A second population of interstitial progenitor cells is specified in the mesonephros and enters the gonad during fetal development. These cells are poorly characterized, but they potentially express similar markers as the gonadal interstitial progenitors. The mesonephric interstitial progenitors differentiate into a population of fetal Leydig cells that express Nr5a1 and Cyp17a1, but do not express Gli1, and thus develop into Leydig cells independently of hedgehog signaling. The mesonephric interstitial progenitor cells do not contribute to the adult Leydig cell population.
Figure 2.
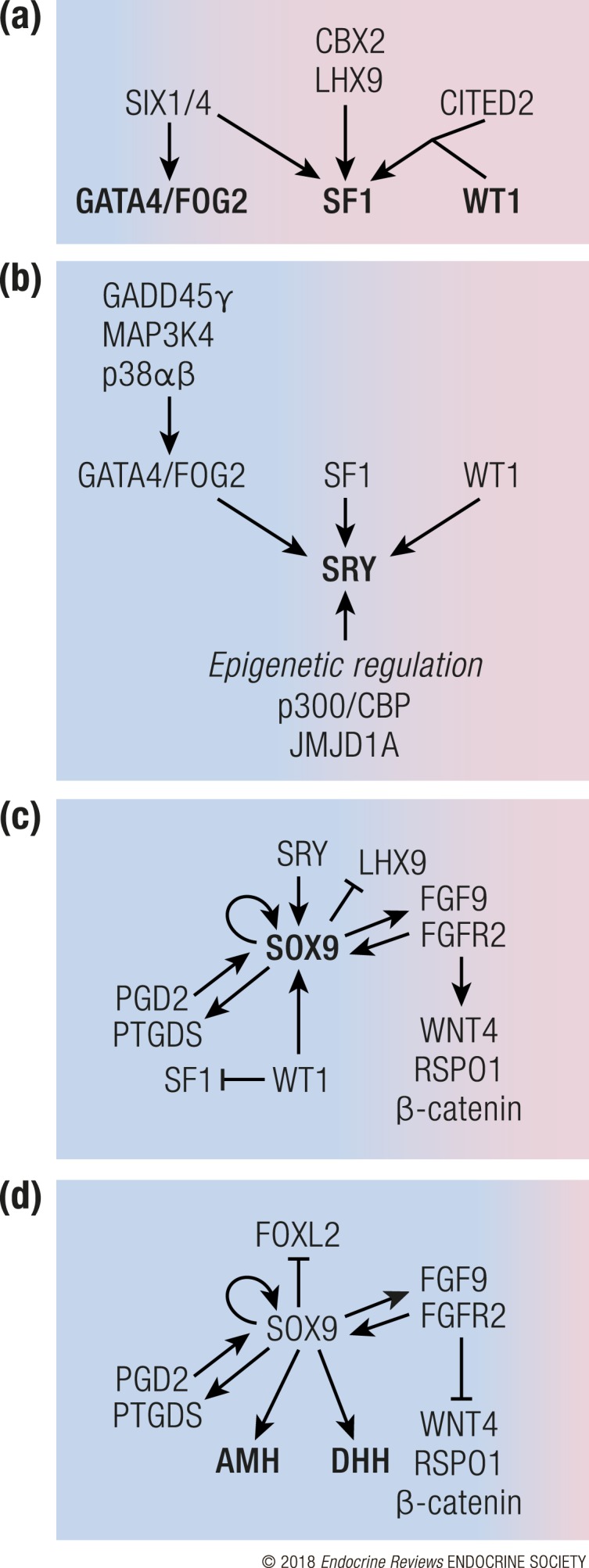
Pathways that govern Sertoli cell differentiation in the mouse. Sertoli cell differentiation steers the bipotential gonad from a female-biased fate (pink) to male fate (blue). (a) As the gonad begins to form, the MAPK pathway, SIX1/4, CBX2, LHX9, and CITED2 upregulate expression of GATA4/FOG, SF1, and WT1, which are necessary for the survival and expansion of the gonadal primordium. (b) At the beginning of testis determination, SF1, GATA4, and WT1 trigger Sry expression. Sry expression is also under epigenetic regulation by p300/CBP and JMJD1A. (c) SRY upregulates Sox9, which leads to upregulation of additional pro-testis pathways (FGF9 and PGD2) while simultaneously inhibiting the pro-ovarian WNT4/RSPO1/β-catenin pathway. WT1 enforces supporting cell fate by suppressing SF1 and promoting SOX9. (d) Under the control of SOX9, differentiating Sertoli cells begin to secrete DHH and AMH that control the appearance of Leydig cell lineage and dimorphic differentiation of the reproductive tract. SOX9 further promotes male fate by repressing the pro-ovarian forkhead box L2 (FOXL2).
Following the initial thickening of the coelomic epithelium, the progenitor cell population depends on sufficient expression of Wt1 and Nr5a1 for their continued survival (15, 18). WT1 promotes expression of Nr5a1 together with the transcriptional coregulator CITED2 (19, 20). If the progenitor cells fail to induce sufficient Nr5a1 due to loss of Emx2, Cbx2, Lhx9, or Six1/Six4, the gonadal primordium does not expand and becomes severely hypoplastic (17, 20–23), suggesting that gonadal primordium relies on Nr5a1 for survival. WT1 and NR5A1 control early gonadal development also in humans, evident by the observation that some WT1 mutations in human patients are associated with 46,XY gonadal dysgenesis with streak gonads (severely dysgenetic fibrotic gonads) (24). Moreover, mutations in NR5A1 lead to highly variable phenotypes in humans, some of which are associated with a complete gonadal and adrenal agenesis, consistent with its role as a survival factor in progenitor population (25, 26) [for a review on NR5A1 mutations in humans, see (27)]. Also, sufficient proliferation of the adrenogonadal primordium is necessary for gonad and adrenal development. This proliferation is promoted by insulin signaling, and loss of insulin/IGF1 signaling leads to agenesis of adrenal glands as well as testis-to-ovary sex reversal (28, 29).
While the coelomic epithelium proliferates and expands, the basement membrane underlying the epithelium disintegrates, thereby permitting ingression of the coelomic epithelial cells to form the gonad. Coelomic epithelial cells undergo asymmetric cell division where one daughter cell remains in the epithelium and the other ingresses into the gonad. The ingression of these coelomic epithelial cells requires proper polarization of the cells, which is primarily controlled by Notch signaling (30). The proliferating progenitor cells therefore have a high Notch activity, whereas the daughter cells that ingress into the gonad downregulate Notch activity through expression of the Notch antagonist NUMB to become intermediate progenitor cells that are capable of differentiating into the somatic cell lineages (30). If Numb is lost, the coelomic epithelial cells continue to proliferate and ingress into the gonad but cannot properly differentiate. As a result, large patches of undifferentiated progenitor cells aggregate in the gonads (30). In addition to the progenitors originating from the coelomic epithelium, gonadal somatic cells also arise from cells that migrate into the gonad from the adjacent mesonephros (this is discussed in detail in the context of the development of each of the somatic cell lineages).
Whereas gonadal somatic cells arise at the site of gonadal primordium, primordial germ cells (PGCs) are specified far from the gonad in the epiblast prior to gonad formation [reviewed in (31, 32)]. As the embryo develops, PGCs migrate through the allantois, hindgut, and mesentery toward the genital ridges. Migration of PGCs is guided by active movement toward chemotactic signals along the migratory path and passively by expansion of the embryo (33). In the human, the PGCs migrate along nerve fibers to the gonad (34). Once the PGCs arrive in the gonad, they become enclosed by the supporting cell progenitors and undergo several successive rounds of rapid proliferation to establish the germ cell pool (35–37). Despite their importance as a vector for transmitting genetic information to the next generation, germ cells are not essential for the establishment of various somatic cell lineages in the gonads, based on the fact that loss of germ cells does not affect somatic cell lineage specification (38–40).
Lineage Separation of the Somatic Cell Populations in the Gonads
The daughter cells from the coelomic epithelium give rise to two main somatic cell populations in the gonads: the supporting cells and the steroidogenic cells (Fig. 1; see Fig. 4). The supporting cells, which are the first to differentiate, orchestrate sex determination of the gonads and support gametogenesis. Additionally, they are responsible for the differentiation of the steroidogenic cell lineage. The supporting cells become Sertoli cells in the testis and granulosa cells in the ovary. In the testis, the first wave of cells ingressing into the gonad gives rise to Sertoli cells, whereas the progenitor cells that ingress later are destined to become interstitial cells, which include Leydig cells (41). Cell ingression into the testis ends soon after sex determination occurs in the mouse, when tunica albuginea begins to form on the border between the gonad and coelomic epithelium, blocking ingression of the coelomic epithelium (41). In the ovary, the coelomic epithelium continues to proliferate past E12.5, and these progenitor cells give rise to both theca and granulosa cells (41–43).
Figure 4.

Lineage progression of the supporting and stromal cells in the mouse ovary. Somatic progenitor cells in the XX gonad (green) commit to either supporting/granulosa cells (pink/lilac) or a stromal progenitor population (pale orange). Granulosa cells (GC) differentiate in two asynchronous waves. The first population of the granulosa cells (pink) is specified by upregulation of the cell cycle inhibitor Cdkn1b/p27 and subsequent induction of Foxl2, and it eventually gives rise to granulosa cells that surround the follicle in the medullary region of the ovary (dark pink). The second population of granulosa cells (lilac) expresses the RSPO1 receptor Lgr5 and actively proliferates to expand the cell pool prior to cell cycle arrest and differentiation through induction of Cdkn1b/p27 and Foxl2. The second wave of granulosa cell differentiation gives rise to granulosa cells of both medullary (dark pink) and cortical follicles (purple). The stromal progenitor cells in the XX gonads (light orange) remain as undifferentiated Lhx9-positive cells in the fetal ovary, but they potentially express markers for interstitial cell fate (Arx, CouptfII/Nr2f2, and Mafb). Near birth the hedgehog signaling from granulosa cells transforms the progenitor cells into the steroidogenic Gli1-positive theca cells (dark orange). Stromal progenitor cells from the mesonephros also contribute to the theca cell population. These cells potentially express genes similar to the gonadal stromal progenitors. Some of the stromal cell progenitors might remain in the postnatal ovary as a nonsteroidogenic stromal cell population, which expresses CouptfII and Arx, similarly to the nonsteroidogenic interstitial cells in the testis.
The coelomic epithelial progenitor pool is positive for both WT1 and SF1 prior to the divergence of supporting and steroidogenic cell lineages (15, 16, 42, 44–46). In the testis, the supporting cell lineage retains WT1 expression and downregulates SF1 expression as they gain Sertoli cell identity. In contrast, the steroidogenic cell lineage loses WT1 expression as a high level of SF1 expression is maintained upon acquisition and differentiation of steroidogenic potential, whereas the nonsteroidogenic interstitial progenitor cells express a low level of SF1 (47–49). This lineage-specific pattern of WT1 and SF1 expression is essential for the maintenance of their respective cell identities (50). When Wt1 is ablated from the mouse gonadal primordium prior to sex determination, the progenitor cells fail to turn on the program for supporting cell development in both XX and XY gonads. Instead, they become steroidogenic cells with a high expression of SF1 and steroidogenic enzymes (50). The presence of WT1 thus tilts the somatic cell progenitor fate toward supporting cell lineage by decreasing the transcription of Nr5a1, which is known to stimulate steroidogenic enzyme expression (50). Furthermore, in the fetal mouse ovary, where steroidogenesis is limited, Nr5a1 is downregulated after sex determination, implicating the importance of maintaining low levels of SF1 in a steroidogenically quiescent state (51).
After establishment of the gonadal primordium and the first lineage decision, the gonad is poised to embark on the journey toward either a testis or an ovary. In the XY embryo, the presence of testis-determining gene SRY steers the supporting cells toward the Sertoli cell program, which consequently leads to organization of distinct testis structures (testis cords and interstitium), appearance of a steroidogenic cell population, and production of male hormones [testosterone and anti-Müllerian hormone (AMH)]. In the XX embryos where SRY is absent, supporting cells differentiate into granulosa cells, which facilitate germ cell meiosis and folliculogenesis.
Roadmap to Maleness—Differentiation of Somatic Cell Populations in the Testis
Molecular pathways that govern Sertoli cell development
Differentiation of the somatic cell progenitors into Sertoli cells is initiated by the SRY gene on the Y chromosome. SRY is transiently upregulated in the supporting cells at the beginning of sex determination around GW6 in humans and on E10.5 in mice (Fig. 2) (52–54). Sry expression in the mouse is silenced after sex determination, whereas in humans a low level of SRY expression persists even in the mature testis (52, 55, 56). In the mouse, progenitors of Sertoli cells (pre-Sertoli cells) in the center of the gonad are the first to express Sry, followed by extension of its expression toward cells in the anterior and posterior poles (56).
Induction of Sry is controlled by a number of factors, including WT1, SF1, GATA4, and the MAPK pathway (Fig. 2B) [for detailed information, please see (57–62)]. Additionally, Sry expression is a target of epigenetic control by DNA methylation, histone modifications, and chromatin repression (63). In the mouse, the Sry promoter region is hypermethylated prior to sex determination and becomes hypomethylated in the beginning of sex determination (64). Sry expression is also sensitive to histone modifications as the repressive histone 3 on lysine 9 (H3K9me2) mark is associated with silencing of Sry expression. Removal of this repressive histone mark by the demethylase JMJD1A is crucial for Sry induction based on the observation that loss of Jmjd1a results in decreased expression of Sry in the somatic cells of the XY gonad and a partial male-to-female sex reversal (65). Conversely, the active histone mark (H3K27Ac), which is controlled by the histone/lysine acetyltransferases CREB-binding protein (CBP, CREBBP, or KAT3A) and its closely related paralog p300 (EP300 or KAT3B), is essential for Sry induction. Loss of Cbp or p300 causes a partial male-to-female sex reversal, whereas loss of both factors results in a complete sex reversal (66). In humans, CBP/p300 is also implicated in control of SRY expression, as variants in these genes have been identified by next-generation sequencing studies in patients with DSDs (11, 66, 67). Altered SRY expression is a relatively frequent phenomenon in human patients with DSDs, and up to 20% of cases presenting 46,XY gonadal dysgenesis can be attributed to mutations in the coding region of SRY (68). In ~90% of patients with 46,XX testicular DSDs, the cause is translocation of a Y chromosome fragment containing the SRY gene (69).
SRY induction marks the beginning of the sexually dimorphic development of the gonads. This initiation of testicular development can be detected on the transcriptomic level first in the supporting cell lineage at E11.5 in the mouse (45). SRY, a DNA-binding protein, stimulates genes critical for testis development and leads to repression of genes responsible for ovarian differentiation. The major target of SRY is the SRY-related high-mobility group (HMG) box 9 (Sox9) (56, 70, 71). SRY and SOX9 expression overlap transiently in mouse Sertoli cells. Induction of SRY in the pre-Sertoli cell lineage releases a cascade of transcriptional events, leading to upregulation of ~1500 genes and repression of ~1300 genes (45). As SRY expression dwindles, SOX9 persists in Sertoli cells, taking over the role of SRY in controlling a similar set of genes (71, 72). On a genome-wide level, promoter regions of genes associated with both male and female supporting cells have open chromatin in Sertoli cells; however, active enhancer marks (H3K26ac) are associated with genes that are only expressed in Sertoli cells. Thus, the open chromatin likely facilitates recruitment of repressive complexes to promoters of granulosa cell–associated genes, whereas Sertoli cell–associated promoters are active (73). A large proportion of this sexual dimorphism stems directly from the induction of SOX9, where 38% of upregulated and 44% of downregulated genes are potential direct targets of SOX9 (74).
SOX9 is necessary and sufficient for specification of the Sertoli cell lineage (75–77). Once SOX9 is induced in the progenitor cells, the expression of LHX9, a marker associated with an undifferentiated state of gonadal somatic cell progenitors, is reciprocally downregulated, demonstrating a potential mutual antagonism between expression of SOX9 and undifferentiated somatic cell fate (78). Expression of Sox9 is initiated by binding of SRY and SF1 to the testis-specific enhancer core element (TESCO) in the promoter of Sox9 (79). SOX9 also binds to the TESCO region as a self-maintaining mechanism when Sry expression ceases. The TESCO enhancer is sufficient to confer Sertoli cell–specific expression when introduced together with a reporter gene in a transgenic mouse model (79). However, mutation of TESCO in the mouse only partially reduces Sox9 expression, and no testis-to-ovary sex reversal occurs, implying the involvement of other regulatory elements (80). In humans, a second distal enhancer of SOX9 RevSex, rather than TESCO, is associated with cases of 46,XY DSDs (81–83). Testis-specific expression of Sox9 is thus under several layers of control by enhancer elements, which cooperatively ensure its timely and sufficient expression during testis determination.
The ability of SOX9 to program gonadal progenitor cells into Sertoli cells requires the synergistic action of several additional transcription factors, including GATA4, WT1, doublesex and mab-3–related transcription factor 1 (DMRT1), and SF1. Combined expression of Wt1, Gata4, Sf1, Dmrt1, and Sox9 transforms human fibroblasts into embryonic Sertoli cell–like cells, showing that they are sufficient for the establishment of the Sertoli cell lineage (84). SOX9 likely cooperates with GATA4 and DMRT1 to induce Sertoli cell differentiation in vivo (74), based on the observation that the regulatory elements of SOX9 target genes contain not only the consensus SOX9 binding motif, but also a combination of binding motifs for GATA4 and DMRT1 in both mouse and bovine Sertoli cells (74).
Male vs female fate in the supporting cell lineage is determined through a balancing act between SOX9 and the pro-ovarian [R-spondin 1 (RSPO1)/β-catenin] signaling. In the absence of the main testis- and ovary-promoting signaling factors, SOX9 and RSPO1/β-catenin, respectively, gonad differentiation is delayed in both XY and XX gonads (85, 86). As a sign of this delay, the mutant XY gonads retain Sry expression longer than in wild-type counterparts (85, 86). This is followed by initiation of pro-testis signaling by other SOX factors and DMRT1 that are activated in both XX and XY supporting cells to push the supporting cells toward a male fate in both sexes. However, the XY gonad becomes more masculinized than the XX gonad due to additional SRY-mediated activation of pro-testis signaling (85, 86).
Sry and Sox9 belong to the same gene family and interact with DNA via the HMG box; therefore, it is not surprising that they control similar sets of genes during sex determination (72). Other SOX family members, specifically Sox3 and Sox10, are able to induce Sertoli cell differentiation and testis development when they are ectopically induced in the XX gonads (87, 88). Sox9 is a member of the SoxE group that includes the closely related Sox8 and Sox10 genes. Owing to their conserved structures, it is predicted that there is functional redundancy between the SoxE factors (89). Although SOX9 is essential for initial fate specification of Sertoli cells, it is dispensable for fate maintenance after sex determination (E14.5) (90). However, when Sox8 is ablated along with Sox9, Sertoli cells fail to maintain their male fate, resulting in disruption of testis morphology and infertility (91, 92). This indicates that SOX8 has an overlapping function with SOX9 during fetal testis development following sex determination. The other SoxE family member SOX10 is also able to induce ovary-to-testis sex reversal in XX mice following transgenic expression (88). Sox10 is expressed at low levels in both XX and XY somatic cells prior to sex determination, but it becomes upregulated in the XY gonad after E11.5 (88). Furthermore, SOX10 interacts directly with the Amh promoter and TESCO to promote their activity in the XY cells (88). In accordance, the human SOX10 gene is mapped to chromosome 22q13, and duplications of this region are associated with 46,XX gonadal dysgenesis (93). Additional members of the SOX gene family, such as Sox4, a member of the SoxC family, are also involved in gonad development (94). Ablation of Sox4 in the mouse leads to the development of an abnormally oblong form of both testes and ovaries. In contrast to Sox9, loss of Sox4 does not affect the supporting cell fate in either the XX or XY gonad; instead, it results in an upregulation of both Sry and Sox9 in the XY gonads, suggesting a role as a negative regulator for these two testis-determining factors (94).
Despite the exhaustive evidence for an essential role of Sry in triggering testis determination, Sry and the rest of the Y chromosome are not prerequisite for Sertoli cell development. In XX/XY chimeric embryos, where gonadal somatic cells were composed of both XX and XY cells (95), ~10% of the Sertoli cells were XX and hence lacked Sry, implying that Sertoli cell differentiation can be triggered by paracrine signaling from the adjacent Sry-bearing XY Sertoli cells. The threshold for the chimeric gonad to develop as a testis was ~30% of XY cells; with fewer XY cells, the gonad developed as an ovary (95). Paracrine and autocrine amplification of Sertoli cell population is achieved through fibroblast growth factor (FGF) 9 and prostaglandin D2 (PGD2) (Fig. 2C). SOX9 induces Fgf9 expression and together they form a positive regulatory loop that promotes expression of each other. FGF9, whose mRNA expression follows the center to pole pattern of Sox9, acts as a diffusible inducer of Sertoli cell fate across gonadal field (96). Furthermore, FGF9 suppresses the ovarian WNT4/RSPO1/β-catenin pathway (discussed later in the review) and thus promotes testis fate. In the absence of Fgf9, XY mouse gonads become sex reversed with incomplete or absent testis structures and decreased expression of Sox9 (97, 98). FGF9 signals through the c isoform of FGF receptor 2 (FGFR2c) in Sertoli cells (99). In accordance, a heterozygous missense mutation, c.1025G>C (p.Cys342Ser) in the human FGFR2c gene, was associated with a complete 46,XY gonadal dysgenesis. The patient developed as a phenotypic female, supporting that the function of FGFR2 in testicular development is conserved between humans and mice (67).
The second autocrine/paracrine factor amplifying SOX9 signaling in Sertoli cells is PGD2 (100–102). PGD2 is the metabolic product of lipocalin prostaglandin D synthase (100). The l-pgds gene is induced in the fetal testis by SOX9, and, conversely, PGD2 produced by L-PGDS enhances expression of Sox9 independent of FGF9 (100, 102). Furthermore, PGD2 promotes SOX9-mediated gene expression by inducing nuclear localization of SOX9 (103). Before sex determination, low levels of SOX9 are detected in the cytoplasm of both XX and XY gonads, but at the onset of sex determination SOX9 is shuttled into the nucleus in the XY gonad (104). In fact, inducing nuclear localization of SOX9 in XX gonads results in expression of AMH, which is characteristic of Sertoli cell differentiation (104).
In addition to paracrine induction of Sox9 expression by FGF9/PDG2 signaling, the Sertoli cell population expands though rapid proliferation. Proliferation of Sertoli cells in mouse XY gonads accelerates 24 hours after the induction of the testis program and can be detected before any other morphological signs of testicular development, such as testis cord formation and testis-specific vascularization (49). When proliferation is suppressed during the critical window that coincides with induction of Sry expression, XY gonads fail to develop into a testis (105). However, when proliferation is blocked before or after this critical window, the resulting XY gonad is smaller, but it still differentiates into a testis (105). The proliferation occurs primarily in the coelomic epithelium in several bursts. The first burst of proliferation happens at E11.0 to E11.5 and is promoted by Sertoli cell–secreted FGF9. The cell progenies resulting from this burst give rise to Sertoli and interstitial cells (41, 106). The second burst of proliferation occurs after E11.5, and is fueled by platelet-derived growth factor (PDGF). The progenies of the second wave contribute only to the interstitial cell population (41, 107). As a result, the testis rapidly increases in size during E11.5 to E13.5, with the width of the gonad doubling every 24 hours during this window (108). The third burst of Sertoli cell proliferation, which occurs after E15.5, is promoted by activin A from fetal Leydig cells (109). When Inhba, which encodes the subunit that forms activin A as a dimer, is ablated from fetal Leydig cells, Sertoli cell proliferation is significantly decreased after E15.5, leading to testicular dysgenesis (109). This phenotype is a consequence of impaired canonical TGFβ signaling in the Sertoli cells, as deletion of Smad4, a central component of the TGFβ signaling pathway, in fetal Sertoli cells recapitulates the testicular phenotype in activin A knockout mice (109). Sufficient proliferation of Sertoli cells is necessary for not only initiation of the testis determination program, but also the size of both the steroidogenic and germ cell populations (110).
In the human testis, the first phase of rapid expansion that corresponds to the differentiation of pre-Sertoli cells occurs around GW6 to GW7 (111). The human fetal Sertoli cells continue to proliferate until GW14, subsequently resuming proliferation first neonatally and then prepubertally (112). In mouse testes, Sertoli cell proliferation gradually ceases by postnatal day 18 (113, 114).
In addition to FGF9 and PDGFs, Sertoli cells secrete AMH, which plays a critical role in sexual differentiation (Fig. 2D). AMH, whose expression is triggered by SOX9, induces regression of the female reproductive tract progenitor Müllerian ducts (45, 52, 115–118). In the absence of functional AMH or its receptor AMHR2, male embryos retain both female and male reproductive tracts (persistent Müllerian duct syndrome) in mice and humans (117, 119–122).
Sertoli cell fate is maintained through adulthood by Dmrt1 (123). Even though Dmrt1 is dispensable for the establishment of Sertoli cell fate in mice, it is capable of inducing male fate in XX somatic cells (124, 125). In humans, loss of DMRT1 function is most often associated with deletions of chromosome 9p24, which can result in varying degrees of 46,XY gonadal dysgenesis (126), suggesting that DMRT1 also contributes to the establishment of male fate in human supporting cells.
Fate decisions of the interstitial cells in the testis
Sertoli cell differentiation initiates the separation of the testicular somatic cells into the supporting cell and interstitial cell populations (Fig. 1). The first group of interstitial cell progenitors arises at the time of sex determination, when cells from the coelomic epithelium ingress into the gonad and become a part of the interstitium (41). Similar to Sertoli cell lineage, these progenitor cells initially express WT1, but instead of acquiring SOX9 expression, they activate Notch signaling, downregulate WT1 expression, and become interstitial cells (46, 127). These Notch-activated interstitial precursor cells maintain the progenitor cell characteristics (LHX9-positive) until a subpopulation acquires Leydig cell properties on E12.5 in the mouse (78). A separate population of interstitial cell progenitors arises from cells that migrate into the gonad from the neighboring mesonephros (78, 128–131). Most of these migrating cells are endothelial cells of the vasculature, and only a small population of perivascular cells from the gonad/mesonephric border eventually become steroidogenic Leydig cells (78). In contrast to the progenitor cells from the mesonephros, the gonad-derived progenitors contribute to the bulk of the functional fetal Leydig cell population (78, 129, 130).
“ARX mutations are associated with undervirilization phenotypes also in humans.”
Most of the interstitial cells are in fact nonsteroidogenic. They are negative for SF1 and steroidogenic enzymes, but express the hedgehog receptor Patched 1 (Ptch1) and other markers associated with a progenitor-like state, such as aristaless-related homeobox (ARX) and chicken ovalbumin upstream promoter–transcription factor II (COUP-TFII; also known as NR2F2) (48, 132, 133). As the interstitial progenitors commit to a steroidogenic cell fate, they gradually downregulate expression of genes associated with the progenitor-like state and then upregulate SF1 and steroidogenic enzymes (46, 47, 78). In the human fetal testis, the number of COUP-TFII–expressing cells decreases with increasing gestational age, suggesting that cells are recruited from the precursor population to form steroidogenic cells (132). The precise role of COUP-TFII in controlling the balance between steroidogenic and nonsteroidogenic cell populations has not been clearly defined. In the adult testis, Coup-tfII is important for adult Leydig cell differentiation, and its loss in the postnatal mouse testis leads to a spermatogenic failure due to hypoandrogenism (134). A proper lineage separation between supporting and interstitial cells and maintenance of the interstitial progenitor pool requires suppression of SF1 expression by Tcf21 (also known as Pod1) (135). In the absence of Tcf21, the SF1-positive steroidogenic cell population expands in both the XX and XY gonads (135). Furthermore, it is the interstitial progenitor cell pool that determines whether sufficient numbers of fetal Leydig cells can eventually develop to support male sexual development. ARX is important for the survival of the progenitor cells, and loss of Arx results in fewer fetal Leydig cells and impaired testosterone production. In accordance, ARX mutations are associated with undervirilization phenotypes also in humans (47, 136, 137).
The first sign of fetal Leydig cell development is the upregulation of genes encoding steroidogenic enzymes in preparation for subsequent steroidogenesis (52, 118, 138). Fetal Leydig cells appear shortly after the Sertoli cells around GW7 to GW8 in humans and on E12.5 in mice (139). Fetal Leydig cells are the main source of androgens, which are responsible for the establishment of male internal and external genitalia. Androgen production begins around GW8 in human testis and reaches a peak at GW12 to GW14 (140). Additionally, fetal Leydig cells support male reproductive tract development not only by producing androgens, but also by secreting insulin-like 3 that is essential for testicular descent in both mice and humans (141–145).
The balance between undifferentiated progenitors and differentiated fetal Leydig cells is controlled by Notch signaling. Inactivation of the Notch pathway (i.e., loss of Notch receptors Notch2 or Notch3) in the somatic progenitor population increases the number of fetal Leydig cells and decreases the progenitor pool. Alternatively, ectopic activation of the Notch pathway has the opposite effect (46, 127). The Notch pathway seems to also be involved in human Leydig cell development. A mutation in the mastermind-like domain–containing protein 1 (MAMLD1) gene, a transcriptional cofactor responsible for activation of the Notch target genes, was identified in a patient with complete 46,XY gonadal dysgenesis (146). Microdeletions in the MAMLD1 gene in humans are associated with variable undervirilization phenotypes (hypospadias, cryptorchidism, micropenis, and decreased anogenital distance), indicating defective fetal Leydig cell differentiation and subsequent reduced levels of androgens (147). However, in the mouse, loss of Mamld1 only causes a mild decrease in the expression of steroidogenic enzymes in the testis, which does not disrupt development of male characteristics during sexual differentiation (148, 149).
Fetal Leydig cells do not express the testis-determining gene SRY; therefore, their differentiation must rely on paracrine signals from the SRY-positive Sertoli cells, including the PDGF and hedgehog signaling pathways (Fig. 3A). The steroidogenic progenitor population expands in response to PDGFs secreted by Sertoli cells and loss of Pdgfra leads to decreased Leydig cell numbers and defective testis morphogenesis (41, 107). Immediately after their specification, Sertoli cells begin to produce hedgehog ligand desert hedgehog (DHH) as a result of SRY and SOX9 action (44, 72, 150). DHH targets the interstitial progenitor cell population by binding to its receptor PTCH1, which consequently promote gene expression via the downstream effectors glioma-associated oncogene (GLI) transcription factors, GLI transcription factor 1 (GLI1). Subsequently, Gli1 expression is triggered in a subpopulation of the interstitial cells at E12.5 (46). Thus, acquisition of Gli1 expression serves as a marker of hedgehog pathway responsiveness. In the human testis, an increase in GLI1 expression coincides with fetal Leydig cell differentiation (118). DHH induces upregulation of SF1 and P450 side chain cleavage enzyme (SCC, Cyp11a1) in fetal Leydig cells as a sign of steroidogenic cell fate (44, 151). However, DHH is not the only signal capable of inducing fetal Leydig cell differentiation, because some interstitial cells become steroidogenic even in the absence of Dhh (44, 46). Sertoli cells also negatively regulate the size of the Leydig cell population by secreting AMH, and loss of Amh leads to Leydig cell hyperplasia (117).
Figure 3.

Paracrine regulation of steroidogenic cell development. (a) In the fetal mouse testis, Sertoli cells produce paracrine factors PDGFα and DHH that control the appearance and expansion of fetal Leydig cells in the interstitium after E12. Conversely, AMH from Sertoli cells negatively regulates the steroidogenic cell population. Fetal Leydig cells secrete activin A, which promotes proliferation of Sertoli cells via SMAD4 activation. Germ cells do not contribute to paracrine regulation of steroidogenic cells in the testis. (b) Unlike Leydig cells, which differentiate in the testis right after sex determination, the ovarian steroidogenic theca cells differentiate near birth and full steroidogenesis begins after birth. Also, oocytes participate in theca cell recruitment in the ovary through secreting GDF9, which induces expression of the hedgehog ligands DHH and IHH in the granulosa cells. DHH and IHH transform stromal progenitors into the theca cells that surround the follicles around birth in the mouse. Furthermore, granulosa cells promote theca cell recruitment by also producing PDGFα.
In humans, mutations in the components of the hedgehog pathway are implicated in various cases of DSDs. DHH mutations are associated with 46,XY gonadal dysgenesis (152–156). A patient with 46,XY gonadal dysgenesis and chondrodysplasia was shown to have a homozygous missense mutation in the hedgehog acyltransferase (HHAT) gene (157), which is responsible for posttranscriptional palmitoylation of hedgehog ligands. Palmitoylation enhances both short- and long-range action of the hedgehog ligands by multimerizing them. Loss of Hhat in a mouse model recapitulated the human phenotypes with chondrodysplasia, 46,XY testicular dysgenesis, and absence of fetal Leydig cells (157).
Leydig cells have two distinct populations: fetal Leydig cells that form after sex determination, and adult Leydig cells that emerge after birth. Most of the fetal Leydig cells involute after birth, and adult Leydig cells assume responsibility of androgen production. However, a proportion of fetal Leydig cells persist in the adult testis and they represent ~10% of all Leydig cells in the adult mouse (158). Fetal Leydig cells that persist in the postnatal testis gradually shift their gene expression profiles to resemble adult Leydig cells, possibly as a response to changes in the endocrine environment after birth (159). In the mouse, the main androgen produced by fetal Leydig cells is androstenedione. Sertoli cells convert androstenedione to testosterone by hydroxysteroid 17β dehydrogenase 3 (HSD17B3) action (160). The conversion of androstenedione to testosterone is crucial for sufficient androgen action to ensure normal development of the male reproductive tract in humans also: mutations in HSD17B3 result in development of female external genitalia in 46,XY individuals and accumulation of the HSD17B3 substrate androstenedione in the blood (161–163). Autocrine androgen action is dispensable for differentiation of fetal and adult Leydig cells, but it is required for final adult Leydig cell maturation and acquisition of full steroidogenic capacity (164). Adult Leydig cell progenitors remain dormant in the fetal gonads and are recruited to differentiate as steroidogenic cells only after birth. These adult progenitor cells start as a part of the pool of nonsteroidogenic interstitial cells in the fetal testis (44, 46, 133). Interestingly, the adult Leydig cell progenitors respond to hedgehog signaling. However, they simultaneously retain expression of progenitor cell markers and resist differentiation into steroidogenic cells by an unknown mechanism.
Decreased androgen action in the XY male or ectopic action in the XX female commonly leads to development of atypical external genitalia, which often serve as the first sign of DSDs. Mutations in the androgen receptor can lead to either partial or complete androgen insensitivity (CAIS). Cases of partial androgen insensitivity and CAIS exhibit a range of phenotypes, with CAIS as the most severe, causing 46,XY individuals to develop female external genitalia without Müllerian structures, despite functioning testes that secrete testosterone and AMH (165).
Androgen production is stimulated by LH in the postnatal testis, but LH signaling is not required for fetal Leydig cell differentiation and androgen production in mice (166–168). However, mutations in LHCGR in humans lead to 46,XY DSD due to lack of androgens (169). Human fetal Leydig cells are LH responsive, and their androgen production depends on placental chorionic gonadotropin early in gestation and LH secreted by the fetal pituitary toward the end of gestation (52, 170, 171).
Becoming Female—Somatic Cell Fate Decisions in the Fetal Ovary
Establishing granulosa cell fate
Both granulosa and Sertoli cells arise from common somatic progenitor cells in the gonadal primordium (Fig. 4) (70, 172). Similar to the XY gonads, the supporting cell lineage in the XX gonad requires a core of regulatory components, including GATA4/FOG2, WT1, SF1, and insulin, to maintain the survival and expansion of the progenitor pool (29, 50, 173, 174). In XX somatic progenitors, which lack Sry, these core regulators also facilitate the progression of the pro-ovarian WNT4/RSPO1/β-catenin signaling pathway (Fig. 5). WNT4 and RSPO1, which are produced by XX somatic progenitors as autocrine/paracrine regulators, bind to membrane-bound WNT receptor Frizzled and G protein–coupled receptors LGR4 and LGR5, respectively. WNT4 and RSPO1 trigger a cascade of intracellular signaling pathways that leads to stabilization and nuclear translocation of cytoplasmic catenin β1 (β-catenin) in the granulosa cell progenitors (175). Before the onset of sex determination, expression of Wnt4 and Rspo1 is not sexually dimorphic, and the β-catenin pathway is active in both XX and XY gonads (176). On a global transcriptomic level, somatic cells in the bipotential gonad exhibit a female bias prior to sex determination (45, 177, 178). Around the window of sex determination, the ovary diverges from the bipotential state to ovarian differentiation though induction/maintenance of pro-ovarian genes and repression of pro-testis genes (45, 177). However, the fate acquisition process continues to be malleable during the sex determination window (E11 to E11.25 in mice). Ectopic expression of Sry between E11 and E11.25 in the mouse ovary leads to a stable induction of SOX9 expression, but when ectopic Sry expression is induced after this window (E12.0 to E12.5), only a subpopulation of XX somatic cells continues to express SOX9 (179). This suggests that after the window of sex determination, most ovarian somatic cells have already acquired a robust pro-ovarian program, which resists Sry-mediated SOX9 activation (52, 177, 178, 179).
Figure 5.

Pathways that govern granulosa cell differentiation in the mouse. (a) As the gonad begins to form, SIX1/4, CBX2, LHX9, and CITED2 upregulate expression of GATA4/FOG2, SF1, and WT1, which are necessary for the survival and expansion of the adrenogonadal primordium. (b) In the XX somatic progenitor cells, where Sry is absent, WT1 and GATA4 promote maintenance of WNT4/RSPO1/β-catenin signaling. β-Catenin forms a feedforward loop by stimulating Wnt4 expression. (c) WNT4/RSPO1/β-catenin signaling inhibits the protestis signaling by SOX9 and FGF9 and induces female fate by promoting secretion of FST. (d) As pregranulosa cells begin to differentiate, they upregulate p27 and exit the cell cycle. In granulosa cells FOXL2 expression is initiated, which further strengthens female fate by antagonizing SOX9 and promoting FST expression.
As sex determination is initiated, the WNT4/RSPO1/β-catenin pathway is silenced in the XY somatic cells as a result of combined action of WT1, FGF9, and SOX9 (90, 98, 180). Conversely, activation of the WNT4/RSPO1/β-catenin pathway in the ovary suppresses Sox9 and Fgf9 expression, leading to stabilization of ovarian fate in the somatic progenitor cells (98, 180). When the WNT4/RSPO1/β-catenin pathway is compromised, such as in Wnt4, Rspo1, or Ctnnb1 knockout mouse models, the XX gonad undergoes partial female-to-male sex reversal characterized by upregulation of Sox9 and appearance of testicular vasculature (128, 175, 181–185). Alternatively, when the WNT4/RSPO1/β-catenin pathway is ectopically activated in the XY gonad through β-catenin stabilization, the testicular program is partially overridden (180, 186). However, when only Wnt4 is overexpressed in the XY gonad, Sertoli cell fate is not altered, suggesting that WNT4 and β-catenin may have differential roles in promoting female fate (184, 187). Furthermore, loss of Sox9 in XY somatic cells results in upregulation of the WNT4/RSPO1/β-catenin pathway and testis-to-ovary sex reversal, but complete sex reversal is prevented by additional loss of Rspo1 or Ctnnb1, thereby demonstrating that in the absence of SOX9, WNT4/RSPO1/β-catenin signaling is able to drive granulosa cell differentiation (85, 86).
In human fetal gonads, WNT4 expression is identical in male and female gonads with no temporal fluctuation, whereas RSPO1 expression is ovary specific (118, 188). Nevertheless, both WNT4 and RSPO1 are important in inducing ovarian development in humans. 46,XX patients with loss-of-function mutations in WNT4 are virilized and lack Müllerian structures (189, 190), whereas RSPO1 loss-of-function mutations lead to 46,XX DSD with complete sex reversal (185, 191). Conversely, gain of function through duplication of chromosome 1p31–p35, which contains both WNT4 and RSPO1 loci, causes 46,XY male-to-female sex reversal due to induction of granulosa cell fate in the supporting cell lineage (187).
Following initiation of the female fate by the WNT4/RSPO1/β-catenin pathway, granulosa cell fate is enforced by expression of the transcription factor forkhead box L2 (FOXL2) (Fig. 5) (192–196) [for a review on FOXL2, see (197)]. It is likely that the WNT4/RSPO1/β-catenin pathway contributes to the upregulation of FOXL2 in the granulosa cells. First, the WNT4 pathway is active in the mouse ovary prior to Foxl2 expression (177). Second, activation of WNT signaling by stabilization of β-catenin leads to expression of FOXL2 in the XY gonad, whereas loss of Ctnnb1 results in a decrease of Foxl2 expression in the XX gonad (172, 180). Loss of FOXL2 in goats leads to polled intersex syndrome where the XX gonad undergoes a complete female-to-male sex reversal (198, 199). In humans, an autosomal-dominant mutation in FOXL2 is associated with premature ovarian failure, but not sex reversal (200, 201). In the mouse, despite the early upregulation of Foxl2 in ovarian supporting cells, FOXL2 is not essential for initiation of granulosa cell development, but it is crucial for maintenance of granulosa cell fate after birth (195, 202). Loss of Foxl2 leads to derepression of Sox9 and granulosa-to-Sertoli cell transdifferentiation (195, 203). However, FOXL2 is capable of inducing ovarian differentiation in the XY gonad and male-to-femal sex reversal, following transgenic upregulation of Foxl2 or upregulation of Foxl2 due to loss of Fgfr2c in the XY gonad (99, 204). These observations together support that FOXL2 and the WNT4/RSPO1/β-catenin pathway play complementary roles in promoting granulosa cell fate in the fetal gonad. Indeed, a combined loss of Foxl2 and Wnt4 or Rspo1 results in a female-to-male sex reversal that occurs earlier and with a more severe phenotype than sex reversal resulting from loss of only Foxl2, Wnt4, or Rspo1 (204, 205).
After the establishment of the first pool of granulosa cell progenitors, their subsequent differentiation occurs asynchronously as a gradient from the medulla to the cortex (Fig. 4). Somatic cell progenitors from the gonadal primordium give rise to the first pool of granulosa cells in the medulla (43, 179, 206, 207). The medullary granulosa cells are mitotically arrested with upregulation of several cyclin-dependent kinase inhibitors such as CDKN1b/p27 and subsequent downregulation of cyclin A1 (Ccna1) (45, 84, 178, 192). Expression of p27 is maintained in granulosa cells by FOXL2, but loss of Foxl2 does not result in cell cycle re-entry, suggesting that other factors besides p27 and FOXL2 contribute to maintenance of granulosa cell quiescence in fetal life (192). The coelomic epithelium, alternatively, continues to proliferate and provides progenitors for the second wave of granulosa cells (43). The coelomic epithelium, which expresses both RSPO1 receptors Lgr5 and Lgr4, retains a more undifferentiated state compared with the medullary granulosa cell population, and it gives rise to new cortical granulosa cells until birth (43, 192, 206, 207, 208). Consistent with the role of WNT4/RSPO1/β-catenin signaling in promoting proliferation of granulosa cell progenitors, ectopic activation of the WNT pathway in the ovarian somatic cells through stabilization of β-catenin results in an expansion of the granulosa cell population and increased size of the ovary (180).
In the XY gonad, the coelomic epithelium loses Lgr5 expression after sex determination, coinciding with the cessation of cell recruitment from the coelomic epithelium to the testis (41, 185). The continued presence of LGR in the coelomic epithelial domain in the XX gonad maintains its ability to respond to RSPO1 and fuels the continuous recruitment of granulosa cells throughout fetal life. Furthermore, RSPO1 responsiveness is retained through LGR expression in differentiated medullary granulosa cells after they become mitotically arrested (192, 208). Medullary granulosa cells downregulate Lgr5 expression as they further differentiate (192). Moreover, when granulosa cell recruitment from the cortex ceases in the postnatal ovary, Lgr5 is restricted to the ovarian epithelial stem cells, which participate in repair of ovulation wounds in the mature ovary (209). Genetic inactivation of Lgr5 in mouse fetal ovaries does not alter granulosa cell fate, whereas loss of Lgr4 results in a partial female-to-male sex reversal due to decreased WNT/β-catenin signaling (206, 208), suggesting partially compensatory and redundant roles of these two receptors.
Similar to their counterpart Sertoli cells, granulosa cells were originally thought to commit to a permanent fate once they are formed, but an additional layer of pro-ovarian signaling in the postnatal granulosa cell is achieved through estrogen signaling. In the mouse, estrogens are crucial for the maintenance of granulosa cell fate in the adult, since loss of estrogen receptors (ERα and ERβ) causes transdifferentiation of granulosa cells to Sertoli cell–like cells (210). Furthermore, the estrogen receptor cooperates with FOXL2 in adult granulosa cells to repress expression of Sox9 (195, 203). These models indicate that, similar to Sertoli cell fate, granulosa cell fate is malleable after their initial differentiation and needs to be actively maintained throughout life by pro-ovarian signaling.
Granulosa cells orchestrate ovarian development through secretion of paracrine factors. One of the early paracrine factors secreted by XX somatic cells is follistatin (FST), which is a downstream target of WNT4/β-catenin and FOXL2 (211, 212). FST inhibits migration of endothelial cells into the fetal ovary by neutralizing the binding ability of activins to their receptors (213, 214). In the testis, activin B promotes formation of testis-specific vasculature, whereas in the ovary, activin activity is suppressed by FST, rendering a lack of such vasculature (213). Furthermore, granulosa cells recruit stromal progenitor cells to differentiate into theca cells around birth through secretion of hedgehog ligands and PDGFα (42, 215). Granulosa cells are thus the central organizers that lead ovarian morphogenesis, support germ cell development, and promote differentiation of the steroidogenic cell lineage.
Establishment of the theca cell lineage
Theca cells, the ovarian counterparts of testicular Leydig cells, become morphologically distinguishable after birth as the follicles assemble. Similar to Leydig cells, theca cells are located in the interstitium, separated from the supporting granulosa cells by a basal membrane. The major function of theca cells is to produce androgens, particularly androstenedione, which is aromatized to estrogens by CYP19 expressed in granulosa cells (216). Theca cells originate from at least two progenitor populations: most theca cells arise from WT1-positive somatic progenitors in the gonadal primordium, and a smaller population of theca progenitors migrates to the ovary from the mesonephros (Fig. 4) (42). In the mouse, stromal progenitor cells for the theca cell lineage can be isolated from the postnatal ovary and subsequently induced to proliferate and differentiate into theca-like cells in vitro by exposure to serum, LH, IGF1, and stem cell factor (217). When these theca-like cells are injected into an adult ovary, they integrate into the proper location of the theca layer surrounding the follicles.
Based on the similar functions and characteristics between theca cells and Leydig cells, candidates for identifying the stromal progenitor cells have been established, including MAFB, ARX, and COUP-TFII, all of which have been shown to mark Leydig cell progenitors (47, 78, 132). In the fetal mouse ovary, ARX is expressed in the coelomic epithelial cells and other unknown cell types in the gonadal parenchyma and gonad/mesonephric border at the time of sex determination (47). Around birth when assembly of the medullary follicles occurs, ARX-expressing cells are recruited to the medullary region of the ovary (47). However, it is not known whether the ARX-positive cells give rise to theca cells, granulosa cells, or both. Interestingly, COUP-TFII–positive cells form a distinct population that is negative for granulosa cell markers FOXL2 and LGR5 (206). The lack of granulosa cell marker expression in these cells suggests either an undifferentiated state or an acquisition of stromal cell fate. It is not known whether ARX and COUP-TFII mark the same cell population in the ovary or whether they contribute to the heterogeneity of the stromal cell population. COUP-TFII–expressing cells likely contribute to the steroidogenic capacity of the postnatal ovary, as haploinsufficiency for CouptfII results in delayed puberty and impaired progesterone production in mice (218).
An additional indication of the parallel developmental path of theca cells and Leydig cells is that fetal Leydig cells and theca cell progenitors use the same enhancer region to control expression of Nr5a1, the master regulator of steroidogenesis (219, 220). When this enhancer sequence is used to drive expression of reporter gene Gfp in female mice, some theca cells and steroidogenic stromal cells become GFP-positive postnatally, suggesting that a similar set of transcription factors controls Nr5a1 expression in fetal Leydig cells and theca cells (220). A subpopulation of theca cells is negative for Nr5a1 enhancer-driven GFP, implying that these cells could represent a population similar to the adult Leydig cells, which is recruited to acquire a steroidogenic cell fate later in development (220).
Theca and granulosa cells have a shared origin in the coelomic epithelial progenitor population, but mechanisms responsible for the divergence of granulosa and theca cell lineages are not known. In the fetal testis, the separation between supporting Sertoli cells and interstitial cells is well defined, as expression of Sry/Sox9 specifies the supporting cell lineage. In the fetal ovary, the presence of FOXL2 defines the granulosa cell lineage, which is negative for the potential interstitial cell lineage markers Mafb and CouptfII (78, 206). The FOXL2-negative interstitial cells, which are positive for Mafb, express very few genes that are unique to this population when compared with pregranulosa cells, suggesting that they are closely related to the supporting cell lineage in the ovary (45).
Theca cell recruitment and differentiation is under paracrine control by the granulosa cells, through mechanisms that parallel fetal Leydig cell differentiation in the testis (Fig. 3B). In the mouse, granulosa cells in the neonatal ovary produce two hedgehog ligands, DHH and indian hedgehog (IHH) (42, 221). In mice, the theca cell progenitors begin to express the hedgehog receptor PTCH1 and GLI1 around the time-of-birth pathway in response to granulosa cell–derived DHH and IHH (42, 221). At the same time, another population of Gli1-positive theca cell progenitors migrates from the neighboring mesonephros into the ovary (42). These mesonephric theca cell progenitors respond to the hedgehog ligand sonic hedgehog in the mesonephros first, and then migrate into the ovary around birth (42, 44, 150). Transformation of both types of progenitor cells into theca cells requires Dhh and Ihh, as inactivation of both hedgehog ligands results in absence of the theca layer, impaired steroidogenesis, and arrested follicle growth (42). Production of Dhh and Ihh in granulosa cells is stimulated by oocyte-derived growth differentiation factor 9 (GDF9) (42, 222). In the absence of Gdf9, theca cells are not recruited to surround the follicles so that granulosa cells activate steroidogenesis in an attempt to compensate for the theca cell loss (222). An additional level of paracrine control of theca cell differentiation is achieved through PDGFRα signaling, and ablation of Pdgfra in the somatic progenitor cells results in impaired theca cell development (215). Theca cells arise though paracrine recruitment of several populations of progenitor cells, and the mesonephros-derived theca cells show signs of stronger steroidogenic activity than do the gonad-derived theca cells (42). However, whether this heterogeneity contributes to significant functional differences between mature theca cells remains to be determined.
In the mouse fetal ovary, granulosa cells do not produce hedgehog ligands. Consequently, the hedgehog pathway is silenced and no theca cell differentiation is detected in the fetal ovary. This unique developmental time frame is crucial, as the interstitial progenitor cells remain plastic prior to birth. When hedgehog signaling is prematurely activated in Sf1-expressing somatic cell progenitors, which give rise to both supporting and steroidogenic lineages, some of these cells differentiate into androgen-producing fetal Leydig-like cells in the ovary (223). Interestingly, if the hedgehog pathway is ectopically induced later in development, exclusively in the supporting cell lineage, it is insufficient to transform supporting cells to steroidogenic cells (224). The effect of hedgehog signaling on ovarian somatic cell differentiation thus depends on the timing and the cell type where the signaling is activated. Mutations in the hedgehog signaling pathway have not been reported to be associated with defects in ovarian development or female fertility in humans, but activation of hedgehog signaling is associated with ovarian carcinoma, suggesting a significance for properly tuned hedgehog signaling in ovarian somatic cell populations (225).
Despite the lack of theca cells, fetal mouse ovaries show a low level of steroidogenesis. Both mouse and human fetal ovaries express key steroidogenic enzymes (3βHSD and CYP19) in granulosa cells and oocytes and consequently produce low levels of progesterone and estradiol (226–228). The significance of this low level of steroidogenesis for ovarian development is not known. It has been hypothesized that fetal ovarian steroidogenesis inhibits follicle formation prenatally in the mouse, and withdrawal of both the maternal source of estradiol and fetal ovarian estradiol synthesis at birth initiates follicle formation (227, 229). However, loss of endogenous estradiol (through loss of aromatase function) or estrogen responsiveness have not been reported to alter follicle formation, suggesting that steroidogenesis in the fetal ovary has little functional significance for prenatal ovarian development (210, 230–235). Ovaries reach full steroidogenic capacity only after theca cell recruitment, formation of the first antral follicles, and acquisition of gonadotropin-responsiveness on postnatal day 5 in the mouse and near birth in humans (236–238).
“In humans with congenital adrenal insufficiency, steroidogenic cells form adrenal rest tumors in gonads and reproductive tract.”
Swapping Fate Between Adrenal and Gonad—Shared Origin of Adrenal and Gonadal Steroidogenic Cells
Gonads and adrenal glands, specifically the adrenal cortex, derive from a common origin, the adrenogonadal primordium (Fig. 6) (239). Around E10.5 in the mouse, the SF1-positive adrenogonadal primordium splits into two organs (240): a gonadal primordium that retains WT1 and GATA4 expression, and an adrenal primordium that downregulates WT1 and GATA4 expression (19, 240). Downregulation of WT1 is a prerequisite for the adrenal primordium to differentiate into steroidogenic cells in the adrenal cortex (240). However, a few WT1-positive undifferentiated progenitor cells remain in the adrenal even in the adult (240), and upon gonadectomy or chronic gonadotropin exposure, these WT1-positive progenitor cells differentiate into gonad-like steroidogenic cells (241). A similar phenomenon is observed in mice that are genetically susceptible to gonadotropin-induced adrenal tumorigenesis such as DBA/2J inbred mice, inhibin knockout mouse (Inha−/−), and mice with transgenic expression of GATA4 in the adrenal (242–246). These mice develop adrenal tumors after gonadectomy, which are characterized by expression of granulosa and theca cell markers, and rely on gonadotropin stimulation for expansion and estrogen production (241–243, 247). Gonadotropin-dependent adrenal hyperplasia and adenomas have been also reported in women during pregnancy or menopause when chorionic gonadotropin or LH levels are elevated (248, 249). A reverse scenario is found in the gonads where adrenal-like progenitors are present (250). These adrenal-like cells are adrenal progenitors that are left behind in the gonads when the two primordia separate. The adrenal-like cells reside near the gonad/mesonephric border and migrate into the gonad during fetal development (250). Similar to adrenal progenitor cells, the residual adrenal cells in the gonads are responsive to both adrenocorticotropic hormone and LH (250). The appearance of adrenal-like cells in the gonad are negatively regulated by WNT4 in both sexes (251). WNT4 may elicit this effect through inhibiting migration of the adrenal-like cells into the gonad or by promoting separation between adrenal and gonadal primordium (128, 252). The switch in fate from adrenal progenitor to gonadal-like steroidogenic cells is associated with a change in expression of the adrenal-specific Gata6 factor to the gonad-specific Gata4 factor in the subcapsular adrenal progenitor cell population (241). Gata6 and Gata4 are both expressed in adrenocortical cells and gonadal cells during fetal development. However, Gata4 is downregulated at birth, whereas Gata6 persists in the adrenocortical cells, and this change maintains adrenocortical fate and represses gonadal-like differentiation (253, 254). Adrenocortical progenitors require both Gata4 and Gata6 for survival, and deletion of both factors results in adrenal aplasia (255). Interestingly, gonads still form even in the absence of Gata4 and Gata6. However, without Gata4 and Gata6 the steroidogenic population in the testis consists of adrenal-like cells instead of fetal Leydig cells. These observations suggest that GATA4 and GATA6 are required for the adrenal vs Leydig cell fate decision (256). In addition to the GATA factors, sumoylation of SF1 is involved in controlling the adrenal vs Leydig cell fate decision. Small ubiquitin-like modifier conjugation is a posttranslational modification that modulates the action of proteins such as histones, transcription factors, and chromatin modifiers. When SF1 fails to be sumoylated, steroidogenic cell fate is reciprocally disrupted: gonadal-like cells are detected in the adrenals, and adrenal-like cells become absent in the gonads (257). Interestingly, in humans with congenital adrenal insufficiency, steroidogenic cells form adrenal rest tumors in gonads and reproductive tract, suggesting that a similar adrenal-like cell population might exist in human gonads (258–260).
Figure 6.

Lineage separation of the adrenogonadal primordium to adrenal and gonadal steroidogenic cells. Cells in the adrenogonadal primordium (green) express SF1, WT1. and GATA4. As separation of the adrenal and gonadal populations begins in the mouse, the gonadal progenitors (purple) retain expression of these factors, whereas adrenal progenitors (blue) downregulate WT1 and GATA4 and upregulate GATA6 expression. Few undifferentiated gonadal progenitor-like cells (green) remain in the adrenal gland. When LH levels are increased in the mouse by gonadectomy or administration of ectopic LH/human chorionic gonadotropin, these progenitor cells differentiate into gonad-like steroidogenic cells in the adrenal (purple). Some adrenal-like cells (light blue) reside around the gonad mesonephric border. Upon loss of WNT4 signaling, this population expands in the testes and ovaries.
Conclusions and Future Perspectives
Gonadal somatic cells undergo several lineage decisions on the path to form a functional gonad. The coelomic epithelial cells surrounding the gonadal primordium become specified as the somatic cell progenitors proliferate and ingress into the gonad. Ingression of cells into the gonadal parenchyma is coupled with a fate decision process that separates the supporting cell progenitors from the interstitial/stromal cell progenitors. Sex-specific identities of supporting cells (Sertoli vs granulosa cells) are established first through a balancing and antagonizing act of transcriptional regulation and paracrine/autocrine signaling. The supporting cells then facilitate the differentiation of steroidogenic cell progenitors into Leydig cells in the testis and theca cells in the ovary, which subsequently shape the sex-specific endocrine environment.
Although a rough roadmap for the lineage derivation of major somatic cell types has come to light, several avenues remain to be explored. It is likely that in addition to the cell types that have been identified so far, intermediate cell populations and novel progenitor subpopulations are present in the developing gonads. Such cell populations could be identified and studied using single-cell RNA-sequencing technologies. So far this approach has identified three differentiation states of human fetal granulosa cells and shed light on the signaling pathways that operate between somatic cells and germ cells to control gametogenesis in human fetal gonads (261). Additionally, single-cell RNA sequencing of Sf1-positive cells isolated from E10.5 to E16.5 mouse testis confirmed the presence of a common Sertoli and Leydig cell progenitor population and elucidated dynamic gene expression changes that occur during lineage specification of the supporting and steroidogenic cells in the testis (262). However, in both of these studies only previously known somatic cell populations were captured due to targeted analysis of a single isolated lineage and/or limited numbers of cells sequenced for each cell type and age (261, 262). With the improvement of single-cell sequencing techniques and sequencing capacity, it is expected that the lineage progression of fetal somatic cells will be described with even more granularity in the near future, especially with respect to the constitution of the interstitial/stromal cell populations. The interstitial/stromal populations express a set of common lineage markers such as Ptch1, Gli1, Arx, and Coup-tfII; however, only a subset of the interstitial/stromal cells becomes steroidogenic cells and peritubular myoid cells, which surround the seminiferous tubules. How is the steroidogenic fate defined? Does a distinct Leydig cell progenitor population exist already before sex determination? How does the testis-specific peritubular myoid cell population arise from the interstitial/stromal progenitors, and why do they not differentiate in the ovary? Single-cell analyses will enable us to gain unprecedented insight into the contribution of different populations into the undifferentiated cell pool and to decipher signaling pathways that could be responsible for the lineage separation.
Matching transcriptome data to cell-specific epigenetic status of the chromatin is a powerful tool to study gene networks that govern cell fate determination. DNase sequencing on isolated fetal Sertoli cells and SOX9 chromatin immunoprecipitation sequencing on whole testis have provided tremendous molecular insights in the elucidation of the fate decision process of Sertoli cells (72–74). Similar approaches on various cell types in the fetal ovary will help the field answer several key questions, particularly on how the cortical and medullary granulosa cells emerge from a common source, and how they contribute to meiosis and the formation of follicles.
Knowledge of the normal development of fetal gonadal somatic cells provides invaluable information for generating in vitro models, such as organoids and in vitro differentiation models, to study gonad development. In vitro differentiation of human gonadal somatic cells from stem cells will enable understanding not only the basic biology of these cells, but also how patient-specific mutations and chemical exposures influence their differentiation (263). Patients with endocrine disorders as a result of DSDs, hypogonadism, and/or adrenal insufficiency often need to rely on life-long hormone replacement therapy. Gene therapy and in vitro differentiation of steroidogenic cells could possibly offer a new therapy to these patients through regenerative medicine and stem cell biology. The gonad is a complex tissue with multiple cell types and sex-specific three-dimensional structures. These complex structures can be modeled using organoid cultures and microphysiologic systems that contain not only the gonad but also the reproductive tract and metabolic organs (264–266). These systems will provide powerful tools to tease out how interactions between different cell types and organ systems are altered in humans as a result of DSDs or environmental exposures.
Acknowledgments
We thank Dr. Barbara Nicol, Dr. Karina Rodriguez, and Paula R. Brown for carefully reviewing the manuscript.
Financial Support: This work was supported in part by National Institute of Environmental Health Sciences/National Institutes of Health Intramural Research Fund Grant ZIAES102965 (to H.H.-C.Y.).
Disclosure Summary: The authors have nothing to disclose.
Glossary
Abbreviations
- AMH
anti-Müllerian hormone
- ARX
aristaless-related homeobox
- CAIS
complete androgen insensitivity
- CBP
CREB-binding protein
- COUP-TFII
chicken ovalbumin upstream promoter–transcription factor II
- DHH
desert hedgehog
- DMRT1
doublesex and mab-3–related transcription factor 1
- DSD
disorder of sexual development
- E
embryonic day
- FGF
fibroblast growth factor
- FOXL2
forkhead box L2
- FST
follistatin
- GATA4
GATA binding protein 4
- GDF9
growth differentiation factor 9
- GLI
glioma-associated oncogene
- GLI1
glioma-associated oncogene transcription factor 1
- GW
gestational week
- HHAT
hedgehog acyltransferase
- HSD17B3
hydroxysteroid 17β dehydrogenase 3
- IHH
indian hedgehog
- MAMLD1
mastermind-like domain–containing protein 1
- PGC
primordial germ cell
- PDGF
platelet-derived growth factor
- PGD2
prostaglandin D2
- PTCH1
Patched1
- RSPO1
R-spondin 1
- SF1
steroidogenic factor 1
- SIX
sine oculis homeobox
- SOX9
SRY-related high-mobility group box 9
- TESCO
testis-specific enhancer core element
- WT1
Wilms tumor 1
References
- 1. Bale TL, Epperson CN. Sex differences and stress across the lifespan. Nat Neurosci. 2015;18(10):1413–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ober C, Loisel DA, Gilad Y. Sex-specific genetic architecture of human disease. Nat Rev Genet. 2008;9(12):911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Arnold AP. Sex chromosomes and brain gender. Nat Rev Neurosci. 2004;5(9):701–708. [DOI] [PubMed] [Google Scholar]
- 4. Morselli E, Santos RS, Criollo A, Nelson MD, Palmer BF, Clegg DJ. The effects of oestrogens and their receptors on cardiometabolic health. Nat Rev Endocrinol. 2017;13(6):352–364. [DOI] [PubMed] [Google Scholar]
- 5. Wells JC. Sexual dimorphism of body composition. Best Pract Res Clin Endocrinol Metab. 2007;21(3):415–430. [DOI] [PubMed] [Google Scholar]
- 6. McCarthy MM, Arnold AP. Reframing sexual differentiation of the brain. Nat Neurosci. 2011;14(6):677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, Baratz Dalke K, Liao LM, Lin-Su K, Looijenga LH III, Mazur T, Meyer-Bahlburg HF, Mouriquand P, Quigley CA, Sandberg DE, Vilain E, Witchel S; Global DSD Update Consortium . Global disorders of sex development update since 2006: perceptions, approach and care [published correction appears in Horm Res Paediatr. 2016;85(3):180]. Horm Res Paediatr. 2016;85(3):158–180. [DOI] [PubMed] [Google Scholar]
- 8. Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology . Consensus statement on management of intersex disorders. Pediatrics. 2006;118(2):e488–e500. [DOI] [PubMed] [Google Scholar]
- 9. Hughes IA, Houk C, Ahmed SF, Lee PA LWPES Consensus GroupESPE Consensus Group . Consensus statement on management of intersex disorders. Arch Dis Child. 2006;91(7):554–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brauner R, Picard-Dieval F, Lottmann H, Rouget S, Bignon-Topalovic J, Bashamboo A, McElreavey K. Familial forms of disorders of sex development may be common if infertility is considered a comorbidity. BMC Pediatr. 2016;16(1):195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eggers S, Sadedin S, van den Bergen JA, Robevska G, Ohnesorg T, Hewitt J, Lambeth L, Bouty A, Knarston IM, Tan TY, Cameron F, Werther G, Hutson J, O’Connell M, Grover SR, Heloury Y, Zacharin M, Bergman P, Kimber C, Brown J, Webb N, Hunter MF, Srinivasan S, Titmuss A, Verge CF, Mowat D, Smith G, Smith J, Ewans L, Shalhoub C, Crock P, Cowell C, Leong GM, Ono M, Lafferty AR, Huynh T, Visser U, Choong CS, McKenzie F, Pachter N, Thompson EM, Couper J, Baxendale A, Gecz J, Wheeler BJ, Jefferies C, MacKenzie K, Hofman P, Carter P, King RI, Krausz C, van Ravenswaaij-Arts CM, Looijenga L, Drop S, Riedl S, Cools M, Dawson A, Juniarto AZ, Khadilkar V, Khadilkar A, Bhatia V, Dũng VC, Atta I, Raza J, Thi Diem Chi N, Hao TK, Harley V, Koopman P, Warne G, Faradz S, Oshlack A, Ayers KL, Sinclair AH. Disorders of sex development: insights from targeted gene sequencing of a large international patient cohort. Genome Biol. 2016;17(1):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Arboleda VA, Sandberg DE, Vilain E. DSDs: genetics, underlying pathologies and psychosexual differentiation. Nat Rev Endocrinol. 2014;10(10):603–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wilhelm D, Yang JX, Thomas P. Mammalian sex determination and gonad development. Curr Top Dev Biol. 2013;106:89–121. [DOI] [PubMed] [Google Scholar]
- 14. Hu YC, Okumura LM, Page DC. Gata4 is required for formation of the genital ridge in mice. PLoS Genet. 2013;9(7):e1003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell. 1993;74(4):679–691. [DOI] [PubMed] [Google Scholar]
- 16. Ikeda Y, Shen WH, Ingraham HA, Parker KL. Developmental expression of mouse steroidogenic factor-1, an essential regulator of the steroid hydroxylases. Mol Endocrinol. 1994;8(5):654–662. [DOI] [PubMed] [Google Scholar]
- 17. Fujimoto Y, Tanaka SS, Yamaguchi YL, Kobayashi H, Kuroki S, Tachibana M, Shinomura M, Kanai Y, Morohashi K, Kawakami K, Nishinakamura R. Homeoproteins Six1 and Six4 regulate male sex determination and mouse gonadal development. Dev Cell. 2013;26(4):416–430. [DOI] [PubMed] [Google Scholar]
- 18. Luo X, Ikeda Y, Parker KL. A cell-specific nuclear receptor is essential for adrenal and gonadal development and sexual differentiation. Cell. 1994;77(4):481–490. [DOI] [PubMed] [Google Scholar]
- 19. Buaas FW, Val P, Swain A. The transcription co-factor CITED2 functions during sex determination and early gonad development. Hum Mol Genet. 2009;18(16):2989–3001. [DOI] [PubMed] [Google Scholar]
- 20. Wilhelm D, Englert C. The Wilms tumor suppressor WT1 regulates early gonad development by activation of Sf1. Genes Dev. 2002;16(14):1839–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kusaka M, Katoh-Fukui Y, Ogawa H, Miyabayashi K, Baba T, Shima Y, Sugiyama N, Sugimoto Y, Okuno Y, Kodama R, Iizuka-Kogo A, Senda T, Sasaoka T, Kitamura K, Aizawa S, Morohashi K. Abnormal epithelial cell polarity and ectopic epidermal growth factor receptor (EGFR) expression induced in Emx2 KO embryonic gonads. Endocrinology. 2010;151(12):5893–5904. [DOI] [PubMed] [Google Scholar]
- 22. Katoh-Fukui Y, Miyabayashi K, Komatsu T, Owaki A, Baba T, Shima Y, Kidokoro T, Kanai Y, Schedl A, Wilhelm D, Koopman P, Okuno Y, Morohashi K. Cbx2, a polycomb group gene, is required for Sry gene expression in mice. Endocrinology. 2012;153(2):913–924. [DOI] [PubMed] [Google Scholar]
- 23. Birk OS, Casiano DE, Wassif CA, Cogliati T, Zhao L, Zhao Y, Grinberg A, Huang S, Kreidberg JA, Parker KL, Porter FD, Westphal H. The LIM homeobox gene Lhx9 is essential for mouse gonad formation. Nature. 2000;403(6772):909–913. [DOI] [PubMed] [Google Scholar]
- 24. Moorthy AV, Chesney RW, Lubinsky M. Chronic renal failure and XY gonadal dysgenesis: “Frasier” syndrome—a commentary on reported cases. Am J Med Genet Suppl. 1987;3:297–302. [DOI] [PubMed] [Google Scholar]
- 25. Achermann JC, Ito M, Ito M, Hindmarsh PC, Jameson JL. A mutation in the gene encoding steroidogenic factor-1 causes XY sex reversal and adrenal failure in humans. Nat Genet. 1999;22(2):125–126. [DOI] [PubMed] [Google Scholar]
- 26. Lin L, Gu WX, Ozisik G, To WS, Owen CJ, Jameson JL, Achermann JC. Analysis of DAX1 (NR0B1) and steroidogenic factor-1 (NR5A1) in children and adults with primary adrenal failure: ten years’ experience. J Clin Endocrinol Metab. 2006;91(8):3048–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Domenice S, Machado AZ, Ferreira FM, Ferraz-de-Souza B, Lerario AM, Lin L, Nishi MY, Gomes NL, da Silva TE, Silva RB, Correa RV, Montenegro LR, Narciso A, Costa EM, Achermann JC, Mendonca BB. Wide spectrum of NR5A1-related phenotypes in 46,XY and 46,XX individuals. Birth Defects Res C Embryo Today. 2016;108(4):309–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nef S, Verma-Kurvari S, Merenmies J, Vassalli JD, Efstratiadis A, Accili D, Parada LF. Testis determination requires insulin receptor family function in mice. Nature. 2003;426(6964):291–295. [DOI] [PubMed] [Google Scholar]
- 29. Pitetti JL, Calvel P, Romero Y, Conne B, Truong V, Papaioannou MD, Schaad O, Docquier M, Herrera PL, Wilhelm D, Nef S. Insulin and IGF1 receptors are essential for XX and XY gonadal differentiation and adrenal development in mice. PLoS Genet. 2013;9(1):e1003160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lin YT, Barske L, DeFalco T, Capel B. Numb regulates somatic cell lineage commitment during early gonadogenesis in mice. Development. 2017;144(9):1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Richardson BE, Lehmann R. Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat Rev Mol Cell Biol. 2010;11(1):37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tang WW, Kobayashi T, Irie N, Dietmann S, Surani MA. Specification and epigenetic programming of the human germ line. Nat Rev Genet. 2016;17(10):585–600. [DOI] [PubMed] [Google Scholar]
- 33. Harikae K, Miura K, Kanai Y. Early gonadogenesis in mammals: significance of long and narrow gonadal structure. Dev Dyn. 2013;242(4):330–338. [DOI] [PubMed] [Google Scholar]
- 34. Mamsen LS, Brøchner CB, Byskov AG, Møllgard K. The migration and loss of human primordial germ stem cells from the hind gut epithelium towards the gonadal ridge. Int J Dev Biol. 2012;56(10–12):771–778. [DOI] [PubMed] [Google Scholar]
- 35. Lei L, Spradling AC. Mouse primordial germ cells produce cysts that partially fragment prior to meiosis. Development. 2013;140(10):2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tam PPL, Snow MHL. Proliferation and migration of primordial germ cells during compensatory growth in mouse embryos. J Embryol Exp Morphol. 1981;64:133–147. [PubMed] [Google Scholar]
- 37. Motta PM, Nottola SA, Makabe S. Natural history of the female germ cell from its origin to full maturation through prenatal ovarian development. Eur J Obstet Gynecol Reprod Biol. 1997;75(1):5–10. [DOI] [PubMed] [Google Scholar]
- 38. Handel MA, Eppig JJ. Sertoli cell differentiation in the testes of mice genetically deficient in germ cells. Biol Reprod. 1979;20(5):1031–1038. [DOI] [PubMed] [Google Scholar]
- 39. McCoshen JA. In vivo sex differentiation of congeneic germinal cell aplastic gonads. Am J Obstet Gynecol. 1982;142(1):83–88. [DOI] [PubMed] [Google Scholar]
- 40. Maatouk DM, Mork L, Hinson A, Kobayashi A, McMahon AP, Capel B. Germ cells are not required to establish the female pathway in mouse fetal gonads. PLoS One. 2012;7(10):e47238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Karl J, Capel B. Sertoli cells of the mouse testis originate from the coelomic epithelium. Dev Biol. 1998;203(2):323–333. [DOI] [PubMed] [Google Scholar]
- 42. Liu C, Peng J, Matzuk MM, Yao HH. Lineage specification of ovarian theca cells requires multicellular interactions via oocyte and granulosa cells. Nat Commun. 2015;6(1):6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mork L, Maatouk DM, McMahon JA, Guo JJ, Zhang P, McMahon AP, Capel B. Temporal differences in granulosa cell specification in the ovary reflect distinct follicle fates in mice. Biol Reprod. 2012;86(2):37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yao HH, Whoriskey W, Capel B. Desert Hedgehog/Patched 1 signaling specifies fetal Leydig cell fate in testis organogenesis. Genes Dev. 2002;16(11):1433–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jameson SA, Natarajan A, Cool J, DeFalco T, Maatouk DM, Mork L, Munger SC, Capel B. Temporal transcriptional profiling of somatic and germ cells reveals biased lineage priming of sexual fate in the fetal mouse gonad. PLoS Genet. 2012;8(3):e1002575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Liu C, Rodriguez K, Yao HH. Mapping lineage progression of somatic progenitor cells in the mouse fetal testis. Development. 2016;143(20):3700–3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Miyabayashi K, Katoh-Fukui Y, Ogawa H, Baba T, Shima Y, Sugiyama N, Kitamura K, Morohashi K. Aristaless related homeobox gene, Arx, is implicated in mouse fetal Leydig cell differentiation possibly through expressing in the progenitor cells. PLoS One. 2013;8(6):e68050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McClelland KS, Bell K, Larney C, Harley VR, Sinclair AH, Oshlack A, Koopman P, Bowles J. Purification and transcriptomic analysis of mouse fetal Leydig cells reveals candidate genes for specification of gonadal steroidogenic cells. Biol Reprod. 2015;92(6):145. [DOI] [PubMed] [Google Scholar]
- 49. Schmahl J, Eicher EM, Washburn LL, Capel B. Sry induces cell proliferation in the mouse gonad. Development. 2000;127(1):65–73. [DOI] [PubMed] [Google Scholar]
- 50. Chen M, Zhang L, Cui X, Lin X, Li Y, Wang Y, Wang Y, Qin Y, Chen D, Han C, Zhou B, Huff V, Gao F. Wt1 directs the lineage specification of Sertoli and granulosa cells by repressing Sf1 expression. Development. 2017;144(1):44–53. [DOI] [PubMed] [Google Scholar]
- 51. Ikeda Y. SF-1: a key regulator of development and function in the mammalian reproductive system. Acta Paediatr Jpn. 1996;38(4):412–419. [DOI] [PubMed] [Google Scholar]
- 52. del Valle I, Buonocore F, Duncan AJ, Lin L, Barenco M, Parnaik R, Shah S, Hubank M, Gerrelli D, Achermann JC. A genomic atlas of human adrenal and gonad development. Wellcome Open Res. 2017;2:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Koopman P, Münsterberg A, Capel B, Vivian N, Lovell-Badge R. Expression of a candidate sex-determining gene during mouse testis differentiation. Nature. 1990;348(6300):450–452. [DOI] [PubMed] [Google Scholar]
- 54. Gubbay J, Collignon J, Koopman P, Capel B, Economou A, Münsterberg A, Vivian N, Goodfellow P, Lovell-Badge R. A gene mapping to the sex-determining region of the mouse Y chromosome is a member of a novel family of embryonically expressed genes. Nature. 1990;346(6281):245–250. [DOI] [PubMed] [Google Scholar]
- 55. Hanley NA, Hagan DM, Clement-Jones M, Ball SG, Strachan T, Salas-Cortés L, McElreavey K, Lindsay S, Robson S, Bullen P, Ostrer H, Wilson DI. SRY, SOX9, and DAX1 expression patterns during human sex determination and gonadal development. Mech Dev. 2000;91(1–2):403–407. [DOI] [PubMed] [Google Scholar]
- 56. Bullejos M, Koopman P. Spatially dynamic expression of Sry in mouse genital ridges. Dev Dyn. 2001;221(2):201–205. [DOI] [PubMed] [Google Scholar]
- 57. de Santa Barbara P, Méjean C, Moniot B, Malclès MH, Berta P, Boizet-Bonhoure B. Steroidogenic factor-1 contributes to the cyclic-adenosine monophosphate down-regulation of human SRY gene expression. Biol Reprod. 2001;64(3):775–783. [DOI] [PubMed] [Google Scholar]
- 58. Hossain A, Saunders GF. The human sex-determining gene SRY is a direct target of WT1. J Biol Chem. 2001;276(20):16817–16823. [DOI] [PubMed] [Google Scholar]
- 59. Miyamoto Y, Taniguchi H, Hamel F, Silversides DW, Viger RSA. A GATA4/WT1 cooperation regulates transcription of genes required for mammalian sex determination and differentiation. BMC Mol Biol. 2008;9(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tevosian SG, Albrecht KH, Crispino JD, Fujiwara Y, Eicher EM, Orkin SH. Gonadal differentiation, sex determination and normal Sry expression in mice require direct interaction between transcription partners GATA4 and FOG2. Development. 2002;129(19):4627–4634. [DOI] [PubMed] [Google Scholar]
- 61. Warr N, Carre G-A, Siggers P, Faleato JV, Brixey R, Pope M, Bogani D, Childers M, Wells S, Scudamore CL, Tedesco M, del Barco Barrantes I, Nebreda AR, Trainor PA, Greenfield A. Gadd45γ and Map3k4 interactions regulate mouse testis determination via p38 MAPK-mediated control of Sry expression. Dev Cell. 2012;23(5):1020–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gierl MS, Gruhn WH, von Seggern A, Maltry N, Niehrs C. GADD45G functions in male sex determination by promoting p38 signaling and Sry expression. Dev Cell. 2012;23(5):1032–1042. [DOI] [PubMed] [Google Scholar]
- 63. Tachibana M. Epigenetic regulation of mammalian sex determination. J Med Invest. 2015;62(1–2):19–23. [DOI] [PubMed] [Google Scholar]
- 64. Nishino K, Hattori N, Tanaka S, Shiota K. DNA methylation-mediated control of Sry gene expression in mouse gonadal development. J Biol Chem. 2004;279(21):22306–22313. [DOI] [PubMed] [Google Scholar]
- 65. Kuroki S, Matoba S, Akiyoshi M, Matsumura Y, Miyachi H, Mise N, Abe K, Ogura A, Wilhelm D, Koopman P, Nozaki M, Kanai Y, Shinkai Y, Tachibana M. Epigenetic regulation of mouse sex determination by the histone demethylase Jmjd1a. Science. 2013;341(6150):1106–1109. [DOI] [PubMed] [Google Scholar]
- 66. Carré GA, Siggers P, Xipolita M, Brindle P, Lutz B, Wells S, Greenfield A. Loss of p300 and CBP disrupts histone acetylation at the mouse Sry promoter and causes XY gonadal sex reversal. Hum Mol Genet. 2018;27(1):190–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bagheri-Fam S, Ono M, Li L, Zhao L, Ryan J, Lai R, Katsura Y, Rossello FJ, Koopman P, Scherer G, Bartsch O, Eswarakumar JV, Harley VR. FGFR2 mutation in 46,XY sex reversal with craniosynostosis. Hum Mol Genet. 2015;24(23):6699–6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Koopman P. The curious world of gonadal development in mammals. Curr Top Dev Biol. 2016;116:537–545. [DOI] [PubMed] [Google Scholar]
- 69. Knarston I, Ayers K, Sinclair A. Molecular mechanisms associated with 46,XX disorders of sex development. Clin Sci (Lond). 2016;130(6):421–432. [DOI] [PubMed] [Google Scholar]
- 70. Albrecht KH, Eicher EM. Evidence that Sry is expressed in pre-Sertoli cells and Sertoli and granulosa cells have a common precursor. Dev Biol. 2001;240(1):92–107. [DOI] [PubMed] [Google Scholar]
- 71. Sekido R, Bar I, Narváez V, Penny G, Lovell-Badge R. SOX9 is up-regulated by the transient expression of SRY specifically in Sertoli cell precursors. Dev Biol. 2004;274(2):271–279. [DOI] [PubMed] [Google Scholar]
- 72. Li Y, Zheng M, Lau YF. The sex-determining factors SRY and SOX9 regulate similar target genes and promote testis cord formation during testicular differentiation. Cell Reports. 2014;8(3):723–733. [DOI] [PubMed] [Google Scholar]
- 73. Maatouk DM, Natarajan A, Shibata Y, Song L, Crawford GE, Ohler U, Capel B. Genome-wide identification of regulatory elements in Sertoli cells. Development. 2017;144(4):720–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Rahmoun M, Lavery R, Laurent-Chaballier S, Bellora N, Philip GK, Rossitto M, Symon A, Pailhoux E, Cammas F, Chung J, Bagheri-Fam S, Murphy M, Bardwell V, Zarkower D, Boizet-Bonhoure B, Clair P, Harley VR, Poulat F. In mammalian foetal testes, SOX9 regulates expression of its target genes by binding to genomic regions with conserved signatures. Nucleic Acids Res. 2017;45(12):7191–7211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Bishop CE, Whitworth DJ, Qin Y, Agoulnik AI, Agoulnik IU, Harrison WR, Behringer RR, Overbeek PA. A transgenic insertion upstream of Sox9 is associated with dominant XX sex reversal in the mouse. Nat Genet. 2000;26(4):490–494. [DOI] [PubMed] [Google Scholar]
- 76. Morais da Silva S, Hacker A, Harley V, Goodfellow P, Swain A, Lovell-Badge R. Sox9 expression during gonadal development implies a conserved role for the gene in testis differentiation in mammals and birds. Nat Genet. 1996;14(1):62–68. [DOI] [PubMed] [Google Scholar]
- 77. Vidal VPI, Chaboissier M-C, de Rooij DG, Schedl A. Sox9 induces testis development in XX transgenic mice. Nat Genet. 2001;28(3):216–217. [DOI] [PubMed] [Google Scholar]
- 78. DeFalco T, Takahashi S, Capel B. Two distinct origins for Leydig cell progenitors in the fetal testis. Dev Biol. 2011;352(1):14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sekido R, Lovell-Badge R. Sex determination involves synergistic action of SRY and SF1 on a specific Sox9 enhancer. Nature. 2008;453(7197):930–934. [DOI] [PubMed] [Google Scholar]
- 80. Gonen N, Quinn A, O’Neill HC, Koopman P, Lovell-Badge R. Normal levels of Sox9 expression in the developing mouse testis depend on the TES/TESCO enhancer, but this does not act alone [Published correction appears in PLoS Genet. 2017;13(2):e1006584]. PLoS Genet. 2017;13(1):e1006520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Benko S, Gordon CT, Mallet D, Sreenivasan R, Thauvin-Robinet C, Brendehaug A, Thomas S, Bruland O, David M, Nicolino M, Labalme A, Sanlaville D, Callier P, Malan V, Huet F, Molven A, Dijoud F, Munnich A, Faivre L, Amiel J, Harley V, Houge G, Morel Y, Lyonnet S. Disruption of a long distance regulatory region upstream of SOX9 in isolated disorders of sex development. J Med Genet. 2011;48(12):825–830. [DOI] [PubMed] [Google Scholar]
- 82. Smyk M, Szafranski P, Startek M, Gambin A, Stankiewicz P. Chromosome conformation capture-on-chip analysis of long-range cis-interactions of the SOX9 promoter. Chromosome Res. 2013;21(8):781–788. [DOI] [PubMed] [Google Scholar]
- 83. Georg I, Bagheri-Fam S, Knower KC, Wieacker P, Scherer G, Harley VR. Mutations of the SRY-responsive enhancer of SOX9 are uncommon in XY gonadal dysgenesis. Sex Dev. 2010;4(6):321–325. [DOI] [PubMed] [Google Scholar]
- 84. Buganim Y, Itskovich E, Hu YC, Cheng AW, Ganz K, Sarkar S, Fu D, Welstead GG, Page DC, Jaenisch R. Direct reprogramming of fibroblasts into embryonic Sertoli-like cells by defined factors. Cell Stem Cell. 2012;11(3):373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Lavery R, Chassot AA, Pauper E, Gregoire EP, Klopfenstein M, de Rooij DG, Mark M, Schedl A, Ghyselinck NB, Chaboissier MC. Testicular differentiation occurs in absence of R-spondin1 and Sox9 in mouse sex reversals. PLoS Genet. 2012;8(12):e1003170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Nicol B, Yao HH. Gonadal identity in the absence of pro-testis factor SOX9 and pro-ovary factor beta-catenin in mice. Biol Reprod. 2015;93(2):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Bergstrom DE, Young M, Albrecht KH, Eicher EM. Related function of mouse SOX3, SOX9, and SRY HMG domains assayed by male sex determination. Genesis. 2000;28(3–4):111–124. [PMC free article] [PubMed] [Google Scholar]
- 88. Polanco JC, Wilhelm D, Davidson TL, Knight D, Koopman P. Sox10 gain-of-function causes XX sex reversal in mice: implications for human 22q-linked disorders of sex development. Hum Mol Genet. 2010;19(3):506–516. [DOI] [PubMed] [Google Scholar]
- 89. She ZY, Yang WX. Sry and SoxE genes: how they participate in mammalian sex determination and gonadal development? Semin Cell Dev Biol. 2017;63:13–22. [DOI] [PubMed] [Google Scholar]
- 90. Chang H, Gao F, Guillou F, Taketo MM, Huff V, Behringer RR. Wt1 negatively regulates β-catenin signaling during testis development. Development. 2008;135(10):1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Barrionuevo FJ, Hurtado A, Kim GJ, Real FM, Bakkali M, Kopp JL, Sander M, Scherer G, Burgos M, Jiménez R. Sox9 and Sox8 protect the adult testis from male-to-female genetic reprogramming and complete degeneration. eLife. 2016;5:e15635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Barrionuevo F, Georg I, Scherthan H, Lécureuil C, Guillou F, Wegner M, Scherer G. Testis cord differentiation after the sex determination stage is independent of Sox9 but fails in the combined absence of Sox9 and Sox8. Dev Biol. 2009;327(2):301–312. [DOI] [PubMed] [Google Scholar]
- 93. Falah N, Posey JE, Thorson W, Benke P, Tekin M, Tarshish B, Lupski JR, Harel T. 22q11.2q13 duplication including SOX10 causes sex-reversal and peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, Waardenburg syndrome, and Hirschsprung disease. Am J Med Genet A. 2017;173(4):1066–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Zhao L, Arsenault M, Ng ET, Longmuss E, Chau TC, Hartwig S, Koopman P. SOX4 regulates gonad morphogenesis and promotes male germ cell differentiation in mice. Dev Biol. 2017;423(1):46–56. [DOI] [PubMed] [Google Scholar]
- 95. Palmer SJ, Burgoyne PS. In situ analysis of fetal, prepuberal and adult XX–XY chimaeric mouse testes: Sertoli cells are predominantly, but not exclusively, XY. Development. 1991;112(1):265–268. [DOI] [PubMed] [Google Scholar]
- 96. Hiramatsu R, Harikae K, Tsunekawa N, Kurohmaru M, Matsuo I, Kanai Y. FGF signaling directs a center-to-pole expansion of tubulogenesis in mouse testis differentiation. Development. 2010;137(2):303–312. [DOI] [PubMed] [Google Scholar]
- 97. Colvin JS, Green RP, Schmahl J, Capel B, Ornitz DM. Male-to-female sex reversal in mice lacking fibroblast growth factor 9. Cell. 2001;104(6):875–889. [DOI] [PubMed] [Google Scholar]
- 98. Kim Y, Kobayashi A, Sekido R, DiNapoli L, Brennan J, Chaboissier MC, Poulat F, Behringer RR, Lovell-Badge R, Capel B. Fgf9 and Wnt4 act as antagonistic signals to regulate mammalian sex determination. PLoS Biol. 2006;4(6):e187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bagheri-Fam S, Bird AD, Zhao L, Ryan JM, Yong M, Wilhelm D, Koopman P, Eswarakumar VP, Harley VR. Testis determination requires a specific FGFR2 isoform to repress FOXL2. Endocrinology. 2017;158(11):3832–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Moniot B, Declosmenil F, Barrionuevo F, Scherer G, Aritake K, Malki S, Marzi L, Cohen-Solal A, Georg I, Klattig J, Englert C, Kim Y, Capel B, Eguchi N, Urade Y, Boizet-Bonhoure B, Poulat F. The PGD2 pathway, independently of FGF9, amplifies SOX9 activity in Sertoli cells during male sexual differentiation. Development. 2009;136(11):1813–1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Adams IR, McLaren A. Sexually dimorphic development of mouse primordial germ cells: switching from oogenesis to spermatogenesis. Development. 2002;129(5):1155–1164. [DOI] [PubMed] [Google Scholar]
- 102. Wilhelm D, Hiramatsu R, Mizusaki H, Widjaja L, Combes AN, Kanai Y, Koopman P. SOX9 regulates prostaglandin D synthase gene transcription in vivo to ensure testis development. J Biol Chem. 2007;282(14):10553–10560. [DOI] [PubMed] [Google Scholar]
- 103. Malki S, Nef S, Notarnicola C, Thevenet L, Gasca S, Méjean C, Berta P, Poulat F, Boizet-Bonhoure B. Prostaglandin D2 induces nuclear import of the sex-determining factor SOX9 via its cAMP-PKA phosphorylation. EMBO J. 2005;24(10):1798–1809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Gasca S, Canizares J, De Santa Barbara P, Mejean C, Poulat F, Berta P, Boizet-Bonhoure B. A nuclear export signal within the high mobility group domain regulates the nucleocytoplasmic translocation of SOX9 during sexual determination. Proc Natl Acad Sci USA. 2002;99(17):11199–11204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Schmahl J, Capel B. Cell proliferation is necessary for the determination of male fate in the gonad. Dev Biol. 2003;258(2):264–276. [DOI] [PubMed] [Google Scholar]
- 106. Schmahl J, Kim Y, Colvin JS, Ornitz DM, Capel B. Fgf9 induces proliferation and nuclear localization of FGFR2 in Sertoli precursors during male sex determination. Development. 2004;131(15):3627–3636. [DOI] [PubMed] [Google Scholar]
- 107. Brennan J, Tilmann C, Capel B. Pdgfr-α mediates testis cord organization and fetal Leydig cell development in the XY gonad. Genes Dev. 2003;17(6):800–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nel-Themaat L, Vadakkan TJ, Wang Y, Dickinson ME, Akiyama H, Behringer RR. Morphometric analysis of testis cord formation in Sox9-EGFP mice. Dev Dyn. 2009;238(5):1100–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Archambeault DR, Yao HH. Activin A, a product of fetal Leydig cells, is a unique paracrine regulator of Sertoli cell proliferation and fetal testis cord expansion. Proc Natl Acad Sci USA. 2010;107(23):10526–10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Rebourcet D, Darbey A, Monteiro A, Soffientini U, Tsai YT, Handel I, Pitetti JL, Nef S, Smith LB, O’Shaughnessy PJ. Sertoli cell number defines and predicts germ and Leydig cell population sizes in the adult mouse testis. Endocrinology. 2017;158(9):2955–2969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ostrer H, Huang HY, Masch RJ, Shapiro E. A cellular study of human testis development. Sex Dev. 2007;1(5):286–292. [DOI] [PubMed] [Google Scholar]
- 112. Jørgensen A, Rajpert-De Meyts E. Regulation of meiotic entry and gonadal sex differentiation in the human: normal and disrupted signaling. Biomol Concepts. 2014;5(4):331–341. [DOI] [PubMed] [Google Scholar]
- 113. Sharpe RM, McKinnell C, Kivlin C, Fisher JS. Proliferation and functional maturation of Sertoli cells, and their relevance to disorders of testis function in adulthood. Reproduction. 2003;125(6):769–784. [DOI] [PubMed] [Google Scholar]
- 114. Vergouwen RP, Jacobs SG, Huiskamp R, Davids JA, de Rooij DG. Proliferative activity of gonocytes, Sertoli cells and interstitial cells during testicular development in mice. J Reprod Fertil. 1991;93(1):233–243. [DOI] [PubMed] [Google Scholar]
- 115. Arango NA, Lovell-Badge R, Behringer RR. Targeted mutagenesis of the endogenous mouse Mis gene promoter: in vivo definition of genetic pathways of vertebrate sexual development. Cell. 1999;99(4):409–419. [DOI] [PubMed] [Google Scholar]
- 116. Behringer RR, Cate RL, Froelick GJ, Palmiter RD, Brinster RL. Abnormal sexual development in transgenic mice chronically expressing Müllerian inhibiting substance. Nature. 1990;345(6271):167–170. [DOI] [PubMed] [Google Scholar]
- 117. Behringer RR, Finegold MJ, Cate RL. Müllerian-inhibiting substance function during mammalian sexual development. Cell. 1994;79(3):415–425. [DOI] [PubMed] [Google Scholar]
- 118. Mamsen LS, Ernst EH, Borup R, Larsen A, Olesen RH, Ernst E, Anderson RA, Kristensen SG, Andersen CY. Temporal expression pattern of genes during the period of sex differentiation in human embryonic gonads. Sci Rep. 2017;7(1):15961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Mishina Y, Rey R, Finegold MJ, Matzuk MM, Josso N, Cate RL, Behringer RR. Genetic analysis of the Müllerian-inhibiting substance signal transduction pathway in mammalian sexual differentiation. Genes Dev. 1996;10(20):2577–2587. [DOI] [PubMed] [Google Scholar]
- 120. Imbeaud S, Faure E, Lamarre I, Mattéi MG, di Clemente N, Tizard R, Carré-Eusèbe D, Belville C, Tragethon L, Tonkin C, Nelson J, McAuliffe M, Bidart JM, Lababidi A, Josso N, Cate RL, Picard JY. Insensitivity to anti-Müllerian hormone due to a mutation in the human anti-Müllerian hormone receptor. Nat Genet. 1995;11(4):382–388. [DOI] [PubMed] [Google Scholar]
- 121. Imbeaud S, Carré-Eusèbe D, Rey R, Belville C, Josso N, Picard JY. Molecular genetics of the persistent Müllerian duct syndrome: a study of 19 families. Hum Mol Genet. 1994;3(1):125–131. [DOI] [PubMed] [Google Scholar]
- 122. Knebelmann B, Boussin L, Guerrier D, Legeai L, Kahn A, Josso N, Picard JY. Anti-Müllerian hormone Bruxelles: a nonsense mutation associated with the persistent Müllerian duct syndrome. Proc Natl Acad Sci USA. 1991;88(9):3767–3771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Matson CK, Murphy MW, Sarver AL, Griswold MD, Bardwell VJ, Zarkower D. DMRT1 prevents female reprogramming in the postnatal mammalian testis [Published correction appears in Nature. 2011;477(7363):238]. Nature. 2011;476(7358):101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Zhao L, Svingen T, Ng ET, Koopman P. Female-to-male sex reversal in mice caused by transgenic overexpression of Dmrt1. Development. 2015;142(6):1083–1088. [DOI] [PubMed] [Google Scholar]
- 125. Lindeman RE, Gearhart MD, Minkina A, Krentz AD, Bardwell VJ, Zarkower D. Sexual cell-fate reprogramming in the ovary by DMRT1. Curr Biol. 2015;25(6):764–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Eggers S, Sinclair A. Mammalian sex determination—insights from humans and mice. Chromosome Res. 2012;20(1):215–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Tang H, Brennan J, Karl J, Hamada Y, Raetzman L, Capel B. Notch signaling maintains Leydig progenitor cells in the mouse testis. Development. 2008;135(22):3745–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jeays-Ward K, Hoyle C, Brennan J, Dandonneau M, Alldus G, Capel B, Swain A. Endothelial and steroidogenic cell migration are regulated by WNT4 in the developing mammalian gonad. Development. 2003;130(16):3663–3670. [DOI] [PubMed] [Google Scholar]
- 129. Cool J, Carmona FD, Szucsik JC, Capel B. Peritubular myoid cells are not the migrating population required for testis cord formation in the XY gonad. Sex Dev. 2008;2(3):128–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Merchant-Larios H, Moreno-Mendoza N, Buehr M. The role of the mesonephros in cell differentiation and morphogenesis of the mouse fetal testis. Int J Dev Biol. 1993;37(3):407–415. [PubMed] [Google Scholar]
- 131. Martineau J, Nordqvist K, Tilmann C, Lovell-Badge R, Capel B. Male-specific cell migration into the developing gonad. Curr Biol. 1997;7(12):958–968. [DOI] [PubMed] [Google Scholar]
- 132. van den Driesche S, Walker M, McKinnell C, Scott HM, Eddie SL, Mitchell RT, Seckl JR, Drake AJ, Smith LB, Anderson RA, Sharpe RM. Proposed role for COUP-TFII in regulating fetal Leydig cell steroidogenesis, perturbation of which leads to masculinization disorders in rodents. PLoS One. 2012;7(5):e37064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Kilcoyne KR, Smith LB, Atanassova N, Macpherson S, McKinnell C, van den Driesche S, Jobling MS, Chambers TJ, De Gendt K, Verhoeven G, O’Hara L, Platts S, Renato de Franca L, Lara NL, Anderson RA, Sharpe RM. Fetal programming of adult Leydig cell function by androgenic effects on stem/progenitor cells. Proc Natl Acad Sci USA. 2014;111(18):E1924–E1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Qin J, Tsai MJ, Tsai SY. Essential roles of COUP-TFII in Leydig cell differentiation and male fertility. PLoS One. 2008;3(9):e3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Cui S, Ross A, Stallings N, Parker KL, Capel B, Quaggin SE. Disrupted gonadogenesis and male-to-female sex reversal in Pod1 knockout mice. Development. 2004;131(16):4095–4105. [DOI] [PubMed] [Google Scholar]
- 136. Sirisena ND, McElreavey K, Bashamboo A, de Silva KS, Jayasekara RW, Dissanayake VH. A child with a novel de novo mutation in the aristaless domain of the aristaless-related homeobox (ARX) gene presenting with ambiguous genitalia and psychomotor delay. Sex Dev. 2014;8(4):156–159. [DOI] [PubMed] [Google Scholar]
- 137. Kitamura K, Yanazawa M, Sugiyama N, Miura H, Iizuka-Kogo A, Kusaka M, Omichi K, Suzuki R, Kato-Fukui Y, Kamiirisa K, Matsuo M, Kamijo S, Kasahara M, Yoshioka H, Ogata T, Fukuda T, Kondo I, Kato M, Dobyns WB, Yokoyama M, Morohashi K. Mutation of ARX causes abnormal development of forebrain and testes in mice and X-linked lissencephaly with abnormal genitalia in humans. Nat Genet. 2002;32(3):359–369. [DOI] [PubMed] [Google Scholar]
- 138. Jameson SA, Lin YT, Capel B. Testis development requires the repression of Wnt4 by Fgf signaling. Dev Biol. 2012;370(1):24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Svechnikov K, Landreh L, Weisser J, Izzo G, Colón E, Svechnikova I, Söder O. Origin, development and regulation of human Leydig cells. Horm Res Paediatr. 2010;73(2):93–101. [DOI] [PubMed] [Google Scholar]
- 140. Tapanainen J, Kellokumpu-Lehtinen P, Pelliniemi L, Huhtaniemi I. Age-related changes in endogenous steroids of human fetal testis during early and midpregnancy. J Clin Endocrinol Metab. 1981;52(1):98–102. [DOI] [PubMed] [Google Scholar]
- 141. Canto P, Escudero I, Söderlund D, Nishimura E, Carranza-Lira S, Gutierrez J, Nava A, Mendez JP. A novel mutation of the insulin-like 3 gene in patients with cryptorchidism. J Hum Genet. 2003;48(2):86–90. [DOI] [PubMed] [Google Scholar]
- 142. Marin P, Ferlin A, Moro E, Rossi A, Bartoloni L, Rossato M, Foresta C. Novel insulin-like 3 (INSL3) gene mutation associated with human cryptorchidism. Am J Med Genet. 2001;103(4):348–349. [PubMed] [Google Scholar]
- 143. Nef S, Parada LF. Cryptorchidism in mice mutant for Insl3. Nat Genet. 1999;22(3):295–299. [DOI] [PubMed] [Google Scholar]
- 144. Tomboc M, Lee PA, Mitwally MF, Schneck FX, Bellinger M, Witchel SF. Insulin-like 3/relaxin-like factor gene mutations are associated with cryptorchidism. J Clin Endocrinol Metab. 2000;85(11):4013–4018. [DOI] [PubMed] [Google Scholar]
- 145. Zimmermann S, Steding G, Emmen JM, Brinkmann AO, Nayernia K, Holstein AF, Engel W, Adham IM. Targeted disruption of the Insl3 gene causes bilateral cryptorchidism. Mol Endocrinol. 1999;13(5):681–691. [DOI] [PubMed] [Google Scholar]
- 146. Ruiz-Arana IL, Hübner A, Cetingdag C, Krude H, Grüters A, Fukami M, Biebermann H, Köhler B. A novel hemizygous mutation of MAMLD1 in a patient with 46,XY complete gonadal dysgenesis. Sex Dev. 2015;9(2):80–85. [DOI] [PubMed] [Google Scholar]
- 147. Fukami M, Wada Y, Miyabayashi K, Nishino I, Hasegawa T, Nordenskjöld A, Camerino G, Kretz C, Buj-Bello A, Laporte J, Yamada G, Morohashi K, Ogata T. CXorf6 is a causative gene for hypospadias [Published correction appears in Nat Genet. 2007;39(1):131]. Nat Genet. 2006;38(12):1369–1371. [DOI] [PubMed] [Google Scholar]
- 148. Miyado M, Yoshida K, Miyado K, Katsumi M, Saito K, Nakamura S, Ogata T, Fukami M. Knockout of murine Mamld1 impairs testicular growth and daily sperm production but permits normal postnatal androgen production and fertility. Int J Mol Sci. 2017;18(6):E1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Miyado M, Nakamura M, Miyado K, Morohashi K, Sano S, Nagata E, Fukami M, Ogata T. Mamld1 deficiency significantly reduces mRNA expression levels of multiple genes expressed in mouse fetal Leydig cells but permits normal genital and reproductive development. Endocrinology. 2012;153(12):6033–6040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Bitgood MJ, Shen L, McMahon AP. Sertoli cell signaling by Desert hedgehog regulates the male germline. Curr Biol. 1996;6(3):298–304. [DOI] [PubMed] [Google Scholar]
- 151. Park SY, Tong M, Jameson JL. Distinct roles for steroidogenic factor 1 and desert hedgehog pathways in fetal and adult Leydig cell development. Endocrinology. 2007;148(8):3704–3710. [DOI] [PubMed] [Google Scholar]
- 152. Canto P, Söderlund D, Reyes E, Méndez JP. Mutations in the desert hedgehog (DHH) gene in patients with 46,XY complete pure gonadal dysgenesis. J Clin Endocrinol Metab. 2004;89(9):4480–4483. [DOI] [PubMed] [Google Scholar]
- 153. Canto P, Vilchis F, Söderlund D, Reyes E, Méndez JP. A heterozygous mutation in the desert hedgehog gene in patients with mixed gonadal dysgenesis. Mol Hum Reprod. 2005;11(11):833–836. [DOI] [PubMed] [Google Scholar]
- 154. Umehara F, Tate G, Itoh K, Yamaguchi N, Douchi T, Mitsuya T, Osame M. A novel mutation of desert hedgehog in a patient with 46,XY partial gonadal dysgenesis accompanied by minifascicular neuropathy. Am J Hum Genet. 2000;67(5):1302–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Werner R, Merz H, Birnbaum W, Marshall L, Schröder T, Reiz B, Kavran JM, Bäumer T, Capetian P, Hiort O. 46,XY gonadal dysgenesis due to a homozygous mutation in desert hedgehog (DHH) identified by exome sequencing. J Clin Endocrinol Metab. 2015;100(7):E1022–E1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Das DK, Sanghavi D, Gawde H, Idicula-Thomas S, Vasudevan L. Novel homozygous mutations in Desert hedgehog gene in patients with 46,XY complete gonadal dysgenesis and prediction of its structural and functional implications by computational methods. Eur J Med Genet. 2011;54(6):e529–e534. [DOI] [PubMed] [Google Scholar]
- 157. Callier P, Calvel P, Matevossian A, Makrythanasis P, Bernard P, Kurosaka H, Vannier A, Thauvin-Robinet C, Borel C, Mazaud-Guittot S, Rolland A, Desdoits-Lethimonier C, Guipponi M, Zimmermann C, Stévant I, Kuhne F, Conne B, Santoni F, Lambert S, Huet F, Mugneret F, Jaruzelska J, Faivre L, Wilhelm D, Jégou B, Trainor PA, Resh MD, Antonarakis SE, Nef S. Loss of function mutation in the palmitoyl-transferase HHAT leads to syndromic 46,XY disorder of sex development by impeding Hedgehog protein palmitoylation and signaling. PLoS Genet. 2014;10(5):e1004340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Shima Y, Matsuzaki S, Miyabayashi K, Otake H, Baba T, Kato S, Huhtaniemi I, Morohashi K. Fetal Leydig cells persist as an androgen-independent subpopulation in the postnatal testis. Mol Endocrinol. 2015;29(11):1581–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Miyabayashi K, Shima Y, Inoue M, Sato T, Baba T, Ohkawa Y, Suyama M, Morohashi KI. Alterations in fetal Leydig cell gene expression during fetal and adult development. Sex Dev. 2017;11(2):53–63. [DOI] [PubMed] [Google Scholar]
- 160. Shima Y, Miyabayashi K, Haraguchi S, Arakawa T, Otake H, Baba T, Matsuzaki S, Shishido Y, Akiyama H, Tachibana T, Tsutsui K, Morohashi K. Contribution of Leydig and Sertoli cells to testosterone production in mouse fetal testes. Mol Endocrinol. 2013;27(1):63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonca BB, Elliston KO, Wilson JD, Russell DW, Andersson S. Male pseudohermaphroditism caused by mutations of testicular 17β-hydroxysteroid dehydrogenase 3. Nat Genet. 1994;7(1):34–39. [DOI] [PubMed] [Google Scholar]
- 162. Rösler A, Silverstein S, Abeliovich D. A (R80Q) mutation in 17 beta-hydroxysteroid dehydrogenase type 3 gene among Arabs of Israel is associated with pseudohermaphroditism in males and normal asymptomatic females. J Clin Endocrinol Metab. 1996;81(5):1827–1831. [DOI] [PubMed] [Google Scholar]
- 163. Lindqvist A, Hughes IA, Andersson S. Substitution mutation C268Y causes 17β-hydroxysteroid dehydrogenase 3 deficiency. J Clin Endocrinol Metab. 2001;86(2):921–923. [DOI] [PubMed] [Google Scholar]
- 164. O’Hara L, McInnes K, Simitsidellis I, Morgan S, Atanassova N, Slowikowska-Hilczer J, Kula K, Szarras-Czapnik M, Milne L, Mitchell RT, Smith LB. Autocrine androgen action is essential for Leydig cell maturation and function, and protects against late-onset Leydig cell apoptosis in both mice and men. FASEB J. 2015;29(3):894–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Mongan NP, Tadokoro-Cuccaro R, Bunch T, Hughes IA. Androgen insensitivity syndrome. Best Pract Res Clin Endocrinol Metab. 2015;29(4):569–580. [DOI] [PubMed] [Google Scholar]
- 166. Cattanach BM, Iddon CA, Charlton HM, Chiappa SA, Fink G. Gonadotrophin-releasing hormone deficiency in a mutant mouse with hypogonadism. Nature. 1977;269(5626):338–340. [DOI] [PubMed] [Google Scholar]
- 167. Ma X, Dong Y, Matzuk MM, Kumar TR. Targeted disruption of luteinizing hormone β-subunit leads to hypogonadism, defects in gonadal steroidogenesis, and infertility. Proc Natl Acad Sci USA. 2004;101(49):17294–17299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Zhang FP, Poutanen M, Wilbertz J, Huhtaniemi I. Normal prenatal but arrested postnatal sexual development of luteinizing hormone receptor knockout (LuRKO) mice. Mol Endocrinol. 2001;15(1):172–183. [DOI] [PubMed] [Google Scholar]
- 169. Themmen AP, Huhtaniemi IT. Mutations of gonadotropins and gonadotropin receptors: elucidating the physiology and pathophysiology of pituitary-gonadal function. Endocr Rev. 2000;21(5):551–583. [DOI] [PubMed] [Google Scholar]
- 170. Lambrot R, Muczynski V, Lécureuil C, Angenard G, Coffigny H, Pairault C, Moison D, Frydman R, Habert R, Rouiller-Fabre V. Phthalates impair germ cell development in the human fetal testis in vitro without change in testosterone production. Environ Health Perspect. 2009;117(1):32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. O’Shaughnessy PJ, Baker PJ, Monteiro A, Cassie S, Bhattacharya S, Fowler PA. Developmental changes in human fetal testicular cell numbers and messenger ribonucleic acid levels during the second trimester. J Clin Endocrinol Metab. 2007;92(12):4792–4801. [DOI] [PubMed] [Google Scholar]
- 172. McLaren A. Development of the mammalian gonad: the fate of the supporting cell lineage. BioEssays. 1991;13(4):151–156. [DOI] [PubMed] [Google Scholar]
- 173. Manuylov NL, Smagulova FO, Leach L, Tevosian SG. Ovarian development in mice requires the GATA4-FOG2 transcription complex. Development. 2008;135(22):3731–3743. [DOI] [PubMed] [Google Scholar]
- 174. Combes AN, Spiller CM, Harley VR, Sinclair AH, Dunwoodie SL, Wilhelm D, Koopman P. Gonadal defects in Cited2-mutant mice indicate a role for SF1 in both testis and ovary differentiation. Int J Dev Biol. 2010;54(4):683–689. [DOI] [PubMed] [Google Scholar]
- 175. Chassot AA, Ranc F, Gregoire EP, Roepers-Gajadien HL, Taketo MM, Camerino G, de Rooij DG, Schedl A, Chaboissier MC. Activation of β-catenin signaling by Rspo1 controls differentiation of the mammalian ovary. Hum Mol Genet. 2008;17(9):1264–1277. [DOI] [PubMed] [Google Scholar]
- 176. Chassot A-A, Bradford ST, Auguste A, Gregoire EP, Pailhoux E, de Rooij DG, Schedl A, Chaboissier MC. WNT4 and RSPO1 together are required for cell proliferation in the early mouse gonad. Development. 2012;139(23):4461–4472. [DOI] [PubMed] [Google Scholar]
- 177. Munger SC, Natarajan A, Looger LL, Ohler U, Capel B. Fine time course expression analysis identifies cascades of activation and repression and maps a putative regulator of mammalian sex determination. PLoS Genet. 2013;9(7):e1003630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Nef S, Schaad O, Stallings NR, Cederroth CR, Pitetti JL, Schaer G, Malki S, Dubois-Dauphin M, Boizet-Bonhoure B, Descombes P, Parker KL, Vassalli JD. Gene expression during sex determination reveals a robust female genetic program at the onset of ovarian development. Dev Biol. 2005;287(2):361–377. [DOI] [PubMed] [Google Scholar]
- 179. Harikae K, Miura K, Shinomura M, Matoba S, Hiramatsu R, Tsunekawa N, Kanai-Azuma M, Kurohmaru M, Morohashi K, Kanai Y. Heterogeneity in sexual bipotentiality and plasticity of granulosa cells in developing mouse ovaries. J Cell Sci. 2013;126(13):2834–2844. [DOI] [PubMed] [Google Scholar]
- 180. Maatouk DM, DiNapoli L, Alvers A, Parker KL, Taketo MM, Capel B. Stabilization of β-catenin in XY gonads causes male-to-female sex-reversal. Hum Mol Genet. 2008;17(19):2949–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Vainio S, Heikkilä M, Kispert A, Chin N, McMahon AP. Female development in mammals is regulated by Wnt-4 signalling. Nature. 1999;397(6718):405–409. [DOI] [PubMed] [Google Scholar]
- 182. Maatouk DM, Mork L, Chassot AA, Chaboissier MC, Capel B. Disruption of mitotic arrest precedes precocious differentiation and transdifferentiation of pregranulosa cells in the perinatal Wnt4 mutant ovary. Dev Biol. 2013;383(2):295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Liu CF, Bingham N, Parker K, Yao HH. Sex-specific roles of β-catenin in mouse gonadal development. Hum Mol Genet. 2009;18(3):405–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Jeays-Ward K, Dandonneau M, Swain A. Wnt4 is required for proper male as well as female sexual development. Dev Biol. 2004;276(2):431–440. [DOI] [PubMed] [Google Scholar]
- 185. Parma P, Radi O, Vidal V, Chaboissier MC, Dellambra E, Valentini S, Guerra L, Schedl A, Camerino G. R-spondin1 is essential in sex determination, skin differentiation and malignancy. Nat Genet. 2006;38(11):1304–1309. [DOI] [PubMed] [Google Scholar]
- 186. Li Y, Zhang L, Hu Y, Chen M, Han F, Qin Y, Chen M, Cui X, Duo S, Tang F, Gao F. β-Catenin directs the transformation of testis Sertoli cells to ovarian granulosa-like cells by inducing Foxl2 expression. J Biol Chem. 2017;292(43):17577–17586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Jordan BK, Mohammed M, Ching ST, Délot E, Chen XN, Dewing P, Swain A, Rao PN, Elejalde BR, Vilain E. Up-regulation of WNT-4 signaling and dosage-sensitive sex reversal in humans. Am J Hum Genet. 2001;68(5):1102–1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Tomaselli S, Megiorni F, Lin L, Mazzilli MC, Gerrelli D, Majore S, Grammatico P, Achermann JC. Human RSPO1/R-spondin1 is expressed during early ovary development and augments β-catenin signaling. PLoS One. 2011;6(1):e16366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Biason-Lauber A, Konrad D, Navratil F, Schoenle EJA. A WNT4 mutation associated with Müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med. 2004;351(8):792–798. [DOI] [PubMed] [Google Scholar]
- 190. Philibert P, Biason-Lauber A, Gueorguieva I, Stuckens C, Pienkowski C, Lebon-Labich B, Paris F, Sultan C. Molecular analysis of WNT4 gene in four adolescent girls with mullerian duct abnormality and hyperandrogenism (atypical Mayer-Rokitansky-Küster-Hauser syndrome). Fertil Steril. 2011;95(8):2683–2686. [DOI] [PubMed] [Google Scholar]
- 191. Tomaselli S, Megiorni F, De Bernardo C, Felici A, Marrocco G, Maggiulli G, Grammatico B, Remotti D, Saccucci P, Valentini F, Mazzilli MC, Majore S, Grammatico P. Syndromic true hermaphroditism due to an R-spondin1 (RSPO1) homozygous mutation. Hum Mutat. 2008;29(2):220–226. [DOI] [PubMed] [Google Scholar]
- 192. Gustin SE, Hogg K, Stringer JM, Rastetter RH, Pelosi E, Miles DC, Sinclair AH, Wilhelm D, Western PS. WNT/β-catenin and p27/FOXL2 differentially regulate supporting cell proliferation in the developing ovary. Dev Biol. 2016;412(2):250–260. [DOI] [PubMed] [Google Scholar]
- 193. Ottolenghi C, Omari S, Garcia-Ortiz JE, Uda M, Crisponi L, Forabosco A, Pilia G, Schlessinger D. Foxl2 is required for commitment to ovary differentiation. Hum Mol Genet. 2005;14(14):2053–2062. [DOI] [PubMed] [Google Scholar]
- 194. Uda M, Ottolenghi C, Crisponi L, Garcia JE, Deiana M, Kimber W, Forabosco A, Cao A, Schlessinger D, Pilia G. Foxl2 disruption causes mouse ovarian failure by pervasive blockage of follicle development. Hum Mol Genet. 2004;13(11):1171–1181. [DOI] [PubMed] [Google Scholar]
- 195. Uhlenhaut NH, Jakob S, Anlag K, Eisenberger T, Sekido R, Kress J, Treier AC, Klugmann C, Klasen C, Holter NI, Riethmacher D, Schütz G, Cooney AJ, Lovell-Badge R, Treier M. Somatic sex reprogramming of adult ovaries to testes by FOXL2 ablation. Cell. 2009;139(6):1130–1142. [DOI] [PubMed] [Google Scholar]
- 196. Schmidt D, Ovitt CE, Anlag K, Fehsenfeld S, Gredsted L, Treier AC, Treier M. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131(4):933–942. [DOI] [PubMed] [Google Scholar]
- 197. Pannetier M, Chassot AA, Chaboissier MC, Pailhoux E. Involvement of FOXL2 and RSPO1 in ovarian determination, development, and maintenance in mammals. Sex Dev. 2016;10(4):167–184. [DOI] [PubMed] [Google Scholar]
- 198. Boulanger L, Pannetier M, Gall L, Allais-Bonnet A, Elzaiat M, Le Bourhis D, Daniel N, Richard C, Cotinot C, Ghyselinck NB, Pailhoux E. FOXL2 is a female sex-determining gene in the goat. Curr Biol. 2014;24(4):404–408. [DOI] [PubMed] [Google Scholar]
- 199. Pailhoux E, Vigier B, Vaiman D, Servel N, Chaffaux S, Cribiu EP, Cotinot C. Ontogenesis of female-to-male sex-reversal in XX polled goats. Dev Dyn. 2002;224(1):39–50. [DOI] [PubMed] [Google Scholar]
- 200. Crisponi L, Deiana M, Loi A, Chiappe F, Uda M, Amati P, Bisceglia L, Zelante L, Nagaraja R, Porcu S, Ristaldi MS, Marzella R, Rocchi M, Nicolino M, Lienhardt-Roussie A, Nivelon A, Verloes A, Schlessinger D, Gasparini P, Bonneau D, Cao A, Pilia G. The putative forkhead transcription factor FOXL2 is mutated in blepharophimosis/ptosis/epicanthus inversus syndrome. Nat Genet. 2001;27(2):159–166. [DOI] [PubMed] [Google Scholar]
- 201. Harris SE, Chand AL, Winship IM, Gersak K, Aittomäki K, Shelling AN. Identification of novel mutations in FOXL2 associated with premature ovarian failure. Mol Hum Reprod. 2002;8(8):729–733. [DOI] [PubMed] [Google Scholar]
- 202. Ottolenghi C, Moreira-Filho C, Mendonça BB, Barbieri M, Fellous M, Berkovitz GD, McElreavey K. Absence of mutations involving the LIM homeobox domain gene LHX9 in 46,XY gonadal agenesis and dysgenesis. J Clin Endocrinol Metab. 2001;86(6):2465–2469. [DOI] [PubMed] [Google Scholar]
- 203. Georges A, L’Hôte D, Todeschini AL, Auguste A, Legois B, Zider A, Veitia RA. The transcription factor FOXL2 mobilizes estrogen signaling to maintain the identity of ovarian granulosa cells. eLife. 2014;3:e04207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ottolenghi C, Pelosi E, Tran J, Colombino M, Douglass E, Nedorezov T, Cao A, Forabosco A, Schlessinger D. Loss of Wnt4 and Foxl2 leads to female-to-male sex reversal extending to germ cells. Hum Mol Genet. 2007;16(23):2795–2804. [DOI] [PubMed] [Google Scholar]
- 205. Auguste A, Chassot AA, Grégoire EP, Renault L, Pannetier M, Treier M, Pailhoux E, Chaboissier MC. Loss of R-spondin1 and Foxl2 amplifies female-to-male sex reversal in XX mice. Sex Dev. 2011;5(6):304–317. [DOI] [PubMed] [Google Scholar]
- 206. Rastetter RH, Bernard P, Palmer JS, Chassot AA, Chen H, Western PS, Ramsay RG, Chaboissier MC, Wilhelm D. Marker genes identify three somatic cell types in the fetal mouse ovary. Dev Biol. 2014;394(2):242–252. [DOI] [PubMed] [Google Scholar]
- 207. Zheng W, Zhang H, Gorre N, Risal S, Shen Y, Liu K. Two classes of ovarian primordial follicles exhibit distinct developmental dynamics and physiological functions. Hum Mol Genet. 2014;23(4):920–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Koizumi M, Oyama K, Yamakami Y, Kida T, Satoh R, Kato S, Hidema S, Oe T, Goto T, Clevers H, Nawa A, Nishimori K. Lgr4 controls specialization of female gonads in mice. Biol Reprod. 2015;93(4):90. [DOI] [PubMed] [Google Scholar]
- 209. Ng A, Tan S, Singh G, Rizk P, Swathi Y, Tan TZ, Huang RY, Leushacke M, Barker N. Lgr5 marks stem/progenitor cells in ovary and tubal epithelia. Nat Cell Biol. 2014;16(8):745–757. [DOI] [PubMed] [Google Scholar]
- 210. Couse JF, Hewitt SC, Bunch DO, Sar M, Walker VR, Davis BJ, Korach KS. Postnatal sex reversal of the ovaries in mice lacking estrogen receptors α and β. Science. 1999;286(5448):2328–2331. [DOI] [PubMed] [Google Scholar]
- 211. Kashimada K, Pelosi E, Chen H, Schlessinger D, Wilhelm D, Koopman P. FOXL2 and BMP2 act cooperatively to regulate follistatin gene expression during ovarian development. Endocrinology. 2011;152(1):272–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Yao HH, Matzuk MM, Jorgez CJ, Menke DB, Page DC, Swain A, Capel B. Follistatin operates downstream of Wnt4 in mammalian ovary organogenesis. Dev Dyn. 2004;230(2):210–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Yao HH, Aardema J, Holthusen K. Sexually dimorphic regulation of inhibin beta B in establishing gonadal vasculature in mice. Biol Reprod. 2006;74(5):978–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Phillips DJ, de Kretser DM. Follistatin: a multifunctional regulatory protein. Front Neuroendocrinol. 1998;19(4):287–322. [DOI] [PubMed] [Google Scholar]
- 215. Schmahl J, Rizzolo K, Soriano P. The PDGF signaling pathway controls multiple steroid-producing lineages. Genes Dev. 2008;22(23):3255–3267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Richards JS, Pangas SA. The ovary: basic biology and clinical implications. J Clin Invest. 2010;120(4):963–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Honda A, Hirose M, Hara K, Matoba S, Inoue K, Miki H, Hiura H, Kanatsu-Shinohara M, Kanai Y, Kono T, Shinohara T, Ogura A. Isolation, characterization, and in vitro and in vivo differentiation of putative thecal stem cells. Proc Natl Acad Sci USA. 2007;104(30):12389–12394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Takamoto N, Kurihara I, Lee K, Demayo FJ, Tsai MJ, Tsai SY. Haploinsufficiency of chicken ovalbumin upstream promoter transcription factor II in female reproduction. Mol Endocrinol. 2005;19(9):2299–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Shima Y, Miyabayashi K, Baba T, Otake H, Katsura Y, Oka S, Zubair M, Morohashi K. Identification of an enhancer in the Ad4BP/SF-1 gene specific for fetal Leydig cells [Published correction appears in Endocrinology. 2012;153(11):5686]. Endocrinology. 2012;153(1):417–425. [DOI] [PubMed] [Google Scholar]
- 220. Miyabayashi K, Tokunaga K, Otake H, Baba T, Shima Y, Morohashi K. Heterogeneity of ovarian theca and interstitial gland cells in mice. PLoS One. 2015;10(6):e0128352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Wijgerde M, Ooms M, Hoogerbrugge JW, Grootegoed JA. Hedgehog signaling in mouse ovary: Indian hedgehog and desert hedgehog from granulosa cells induce target gene expression in developing theca cells. Endocrinology. 2005;146(8):3558–3566. [DOI] [PubMed] [Google Scholar]
- 222. Dong J, Albertini DF, Nishimori K, Kumar TR, Lu N, Matzuk MM. Growth differentiation factor-9 is required during early ovarian folliculogenesis. Nature. 1996;383(6600):531–535. [DOI] [PubMed] [Google Scholar]
- 223. Barsoum IB, Bingham NC, Parker KL, Jorgensen JS, Yao HH. Activation of the Hedgehog pathway in the mouse fetal ovary leads to ectopic appearance of fetal Leydig cells and female pseudohermaphroditism. Dev Biol. 2009;329(1):96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224. Ren Y, Cowan RG, Migone FF, Quirk SM. Overactivation of hedgehog signaling alters development of the ovarian vasculature in mice. Biol Reprod. 2012;86(6):174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. Li H, Li J, Feng L. Hedgehog signaling pathway as a therapeutic target for ovarian cancer. Cancer Epidemiol. 2016;40(Supplement C):152–157. [DOI] [PubMed] [Google Scholar]
- 226. Wilson EA, Jawad MJ. The effect of trophic agents on fetal ovarian steroidogenesis in organ culture. Fertil Steril. 1979;32(1):73–79. [DOI] [PubMed] [Google Scholar]
- 227. Dutta S, Mark-Kappeler CJ, Hoyer PB, Pepling ME. The steroid hormone environment during primordial follicle formation in perinatal mouse ovaries. Biol Reprod. 2014;91(3):68. [DOI] [PubMed] [Google Scholar]
- 228. Fowler PA, Anderson RA, Saunders PT, Kinnell H, Mason JI, Evans DB, Bhattacharya S, Flannigan S, Franks S, Monteiro A, O’Shaughnessy PJ. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J Clin Endocrinol Metab. 2011;96(6):1754–1762. [DOI] [PubMed] [Google Scholar]
- 229. Jefferson W, Newbold R, Padilla-Banks E, Pepling M. Neonatal genistein treatment alters ovarian differentiation in the mouse: inhibition of oocyte nest breakdown and increased oocyte survival. Biol Reprod. 2006;74(1):161–168. [DOI] [PubMed] [Google Scholar]
- 230. Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, Sar M, Korach KS, Gustafsson JA, Smithies O. Generation and reproductive phenotypes of mice lacking estrogen receptor β. Proc Natl Acad Sci USA. 1998;95(26):15677–15682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Morishima A, Grumbach MM, Simpson ER, Fisher C, Qin K. Aromatase deficiency in male and female siblings caused by a novel mutation and the physiological role of estrogens. J Clin Endocrinol Metab. 1995;80(12):3689–3698. [DOI] [PubMed] [Google Scholar]
- 232. Shozu M, Akasofu K, Harada T, Kubota Y. A new cause of female pseudohermaphroditism: placental aromatase deficiency. J Clin Endocrinol Metab. 1991;72(3):560–566. [DOI] [PubMed] [Google Scholar]
- 233. Conte FA, Grumbach MM, Ito Y, Fisher CR, Simpson ER. A syndrome of female pseudohermaphrodism, hypergonadotropic hypogonadism, and multicystic ovaries associated with missense mutations in the gene encoding aromatase (P450arom). J Clin Endocrinol Metab. 1994;78(6):1287–1292. [DOI] [PubMed] [Google Scholar]
- 234. Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. Proc Natl Acad Sci USA. 1993;90(23):11162–11166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Fisher CR, Graves KH, Parlow AF, Simpson ER. Characterization of mice deficient in aromatase (ArKO) because of targeted disruption of the cyp19 gene. Proc Natl Acad Sci USA. 1998;95(12):6965–6970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Rosenfield RL, Ehrmann DA. The pathogenesis of polycystic ovary syndrome (PCOS): the hypothesis of PCOS as functional ovarian hyperandrogenism revisited. Endocr Rev. 2016;37(5):467–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Mannan MA, O’Shaughnessy PJ. Steroidogenesis during postnatal development in the mouse ovary. J Endocrinol. 1991;130(1):101–106, NP. [DOI] [PubMed] [Google Scholar]
- 238. O’Shaughnessy PJ, Dudley K, Rajapaksha WR. Expression of follicle stimulating hormone-receptor mRNA during gonadal development. Mol Cell Endocrinol. 1996;125(1–2):169–175. [DOI] [PubMed] [Google Scholar]
- 239. Piprek RP, Kloc M, Kubiak JZ. Early development of the gonads: origin and differentiation of the somatic cells of the genital ridges. Results Probl Cell Differ. 2016;58:1–22. [DOI] [PubMed] [Google Scholar]
- 240. Bandiera R, Vidal VP, Motamedi FJ, Clarkson M, Sahut-Barnola I, von Gise A, Pu WT, Hohenstein P, Martinez A, Schedl A. WT1 maintains adrenal-gonadal primordium identity and marks a population of AGP-like progenitors within the adrenal gland. Dev Cell. 2013;27(1):5–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Looyenga BD, Hammer GD. Origin and identity of adrenocortical tumors in inhibin knockout mice: implications for cellular plasticity in the adrenal cortex. Mol Endocrinol. 2006;20(11):2848–2863. [DOI] [PubMed] [Google Scholar]
- 242. Matzuk MM, Finegold MJ, Mather JP, Krummen L, Lu H, Bradley A. Development of cancer cachexia-like syndrome and adrenal tumors in inhibin-deficient mice. Proc Natl Acad Sci USA. 1994;91(19):8817–8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Kumar TR, Wang Y, Matzuk MM. Gonadotropins are essential modifier factors for gonadal tumor development in inhibin-deficient mice. Endocrinology. 1996;137(10):4210–4216. [DOI] [PubMed] [Google Scholar]
- 244. Bielinska M, Parviainen H, Porter-Tinge SB, Kiiveri S, Genova E, Rahman N, Huhtaniemi IT, Muglia LJ, Heikinheimo M, Wilson DB. Mouse strain susceptibility to gonadectomy-induced adrenocortical tumor formation correlates with the expression of GATA-4 and luteinizing hormone receptor. Endocrinology. 2003;144(9):4123–4133. [DOI] [PubMed] [Google Scholar]
- 245. Doroszko M, Chrusciel M, Stelmaszewska J, Slezak T, Rivero-Muller A, Padzik A, Anisimowicz S, Wolczynski S, Huhtaniemi I, Toppari J, Rahman NA. Luteinizing hormone and GATA4 action in the adrenocortical tumorigenesis of gonadectomized female mice. Cell Physiol Biochem. 2017;43(3):1064–1076. [DOI] [PubMed] [Google Scholar]
- 246. Chrusciel M, Vuorenoja S, Mohanty B, Rivero-Müller A, Li X, Toppari J, Huhtaniemi I, Rahman NA. Transgenic GATA-4 expression induces adrenocortical tumorigenesis in C57Bl/6 mice. J Cell Sci. 2013;126(8):1845–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Walczak EM, Hammer GD. Regulation of the adrenocortical stem cell niche: implications for disease. Nat Rev Endocrinol. 2015;11(1):14–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Lacroix A, Hamet P, Boutin J-M. Leuprolide acetate therapy in luteinizing hormone–dependent Cushing’s syndrome. N Engl J Med. 1999;341(21):1577–1581. [DOI] [PubMed] [Google Scholar]
- 249. Plöckinger U, Chrusciel M, Doroszko M, Saeger W, Blankenstein O, Weizsäcker K, Kroiss M, Hauptmann K, Radke C, Pöllinger A, Tiling N, Steinmüller T, Huhtaniemi I, Quinkler M, Bertherat J, Lacroix A, Rahman N. Functional implications of LH/hCG receptors in pregnancy-induced Cushing syndrome. J Endocr Soc. 2017;1(1):57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Val P, Jeays-Ward K, Swain A. Identification of a novel population of adrenal-like cells in the mammalian testis. Dev Biol. 2006;299(1):250–256. [DOI] [PubMed] [Google Scholar]
- 251. Heikkilä M, Peltoketo H, Leppäluoto J, Ilves M, Vuolteenaho O, Vainio S. Wnt-4 deficiency alters mouse adrenal cortex function, reducing aldosterone production. Endocrinology. 2002;143(11):4358–4365. [DOI] [PubMed] [Google Scholar]
- 252. Zubair M, Parker KL, Morohashi K. Developmental links between the fetal and adult zones of the adrenal cortex revealed by lineage tracing. Mol Cell Biol. 2008;28(23):7030–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Viger RS, Guittot SM, Anttonen M, Wilson DB, Heikinheimo M. Role of the GATA family of transcription factors in endocrine development, function, and disease. Mol Endocrinol. 2008;22(4):781–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Pihlajoki M, Gretzinger E, Cochran R, Kyrönlahti A, Schrade A, Hiller T, Sullivan L, Shoykhet M, Schoeller EL, Brooks MD, Heikinheimo M, Wilson DB. Conditional mutagenesis of Gata6 in SF1-positive cells causes gonadal-like differentiation in the adrenal cortex of mice. Endocrinology. 2013;154(5):1754–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Tevosian SG, Jiménez E, Hatch HM, Jiang T, Morse DA, Fox SC, Padua MB. Adrenal development in mice requires GATA4 and GATA6 transcription factors. Endocrinology. 2015;156(7):2503–2517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Padua MB, Jiang T, Morse DA, Fox SC, Hatch HM, Tevosian SG. Combined loss of the GATA4 and GATA6 transcription factors in male mice disrupts testicular development and confers adrenal-like function in the testes. Endocrinology. 2015;156(5):1873–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Lee FY, Faivre EJ, Suzawa M, Lontok E, Ebert D, Cai F, Belsham DD, Ingraham HA. Eliminating SF-1 (NR5A1) sumoylation in vivo results in ectopic hedgehog signaling and disruption of endocrine development. Dev Cell. 2011;21(2):315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Auchus RJ. Management considerations for the adult with congenital adrenal hyperplasia. Mol Cell Endocrinol. 2015;408:190–197. [DOI] [PubMed] [Google Scholar]
- 259. Cabrera MS, Vogiatzi MG, New MI. Long term outcome in adult males with classic congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2001;86(7):3070–3078. [DOI] [PubMed] [Google Scholar]
- 260. Reisch N, Flade L, Scherr M, Rottenkolber M, Pedrosa Gil F, Bidlingmaier M, Wolff H, Schwarz HP, Quinkler M, Beuschlein F, Reincke M. High prevalence of reduced fecundity in men with congenital adrenal hyperplasia. J Clin Endocrinol Metab. 2009;94(5):1665–1670. [DOI] [PubMed] [Google Scholar]
- 261. Li L, Dong J, Yan L, Yong J, Liu X, Hu Y, Fan X, Wu X, Guo H, Wang X, Zhu X, Li R, Yan J, Wei Y, Zhao Y, Wang W, Ren Y, Yuan P, Yan Z, Hu B, Guo F, Wen L, Tang F, Qiao J. Single-cell RNA-seq analysis maps development of human germline cells and gonadal niche interactions. Cell Stem Cell. 2017;20(6):858–873.e4. [DOI] [PubMed] [Google Scholar]
- 262. Stévant I, Neirijnck Y, Borel C, Escoffier J, Smith LB, Antonarakis SE, Dermitzakis ET, Nef S. Deciphering cell lineage specification during male sex determination with single-cell RNA sequencing. Cell Reports. 2018;22(6):1589–1599. [DOI] [PubMed] [Google Scholar]
- 263. Sepponen K, Lundin K, Knuus K, Väyrynen P, Raivio T, Tapanainen JS, Tuuri T. The role of sequential BMP signaling in directing human embryonic stem cells to bipotential gonadal cells. J Clin Endocrinol Metab. 2017;102(11):4303–4314. [DOI] [PubMed] [Google Scholar]
- 264. Alves-Lopes JP, Stukenborg J-B. Testicular organoids: a new model to study the testicular microenvironment in vitro? Hum Reprod Update. 2017;24(2):176–191. [DOI] [PubMed] [Google Scholar]
- 265. Baert Y, De Kock J, Alves-Lopes JP, Söder O, Stukenborg J-B, Goossens E. Primary human testicular cells self-organize into organoids with testicular properties. Stem Cell Reports. 2017;8(1):30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Young AN, Moyle-Heyrman G, Kim JJ, Burdette JE. Microphysiologic systems in female reproductive biology. Exp Biol Med (Maywood). 2017;242(17):1690–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]