

Abstract
Voltage-dependent anion-selective channels (VDACs) are primarily located in the mitochondrial outer membrane (MOM). They are essential for the regulation of ion and metabolite exchanges. In particular, their role in energy-related nucleotide exchange has many implications in apoptosis, cancer, and neurodegenerative diseases. It has been proposed that VDACs’ functions are regulated by mobility of the N-terminal helical domain, which is bound to the inner wall of the main β-barrel domain but exists in equilibrium between the bound-folded and the unbound-unfolded state. When the N-terminal domain detaches from the channel’s wall and eventually leaves the lumen, it can either stay exposed to the cytosolic environment or interact with the outer leaflet of the MOM; then, it may also interact with other protein partners. In humans, three different VDAC isoforms are expressed at different tissue-specific levels with evidence of distinct roles. Although the N-terminal domains share high sequence similarity, important differences do exist, with the functionality of the entire protein mostly attributed to them. In this work, the three-dimensional structure and membrane affinity of the three isolated hVDAC N-terminal peptides have been compared through Fourier-transform infrared and NMR spectroscopy in combination with molecular dynamics simulations, and measurement of the surface pressure of lipid monolayers. Although peptides were studied as isolated from the β-barrel domain, the observed differences are relevant for those whole protein’s functions in which a protein–protein interaction is mediated by the N-terminal domain.
1. Introduction
The voltage-dependent anion-selective channel (VDAC) is a 30 kDa pore-forming protein located in the mitochondrial outer membrane (MOM). VDACs are mainly responsible for ion and metabolite exchange between the cytosol and the intermembrane space of mitochondria, being particularly important for the regulation of adenosine nucleotide exchange, such as ATP/ADP and NADH/NAD+.1−5 In addition to the pore activity, VDACs interact with many cytosolic proteins, such as hexokinases6 and Bcl-2 family members,7 playing a crucial role in the regulation of apoptosis8,9 with many implications in cancer10,11 and in neurodegenerative diseases.12,13 On the intermembrane space side, VDAC interacts with both creatine kinase and adenine nucleotide translocator, playing a fundamental role in the generation of membrane potentials in mitochondria.14−16
Higher eukaryotes express three different isoforms of VDAC, encoded by three distinct genes.4,5 VDAC1 is the most abundant and ubiquitously expressed of the three isoforms, with VDAC2 and VDAC3 10 and 100 times less expressed, respectively.4,5,17 The electrophysiological features of the three VDAC isoforms were studied in detail in artificial membranes. Mammalian VDAC1 and VDAC2 are easily incorporated into phospholipid bilayers and are sensitive to the applied voltage. On the other hand, VDAC3 hardly forms pores in artificial membranes and shows a reduced sensitivity to voltage.18,19 In particular, at low voltages, VDAC1 and VDAC2 show high conductance and selectivity for anions over cations, whereas they switch to the so-called “closed state” at higher voltages, which is characterized by low conductance and cation selectivity.
The three-dimensional (3D) structures of both human and mouse VDAC1 were solved by X-ray crystallography, solution-state NMR, and a hybrid approach.20−23 A few years later, the zebrafish VDAC2 X-ray structure was also released.24 More recently, two new crystal structures of VDAC1 have been released.23 Overall, the results indicate a common structure for the three VDAC proteins. VDAC is a large transmembrane channel formed by 19 amphipathic antiparallel β-strands, with the only exception of the parallel pairing between strands 1 and 19. The outer diameter of this β-barrel is 4.5 nm, whereas the inner diameter is about 3.0 nm, but it is reduced down to 2.0 nm by the invagination of the N-terminal domain into the channel’s lumen.20−22,24,25 All of the reported 3D structures show VDAC in the open state, representing the basis to characterize its permeability and selectivity at the atomic level through molecular dynamics (MD) simulations.26−28 However, simulations were not able to show the closed state.29,30 The N-terminal domain of VDAC is proposed to modulate the two states but, unless specific conditions are applied, the “closed” states are transiently and rather infrequently visited, with their structural nature still unclear.30
In both mouse and human VDAC1 structures, the N-terminal domain consists of 25 amino acid residues. The central portion is folded as a relatively short helix (ca. 15 residues).21−23 A similar structure was found in zfVDAC2, in spite of the additional 11 N-terminal residues.24 In the five structures, the N-terminal helix is located half-way along the channel, such that it restricts the lumen and provides steric filtering to metabolite passage. In addition, its net positive charge characterizes the electrostatics of the whole channel, both in terms of permeability and selectivity.31 However, the exact position inside the lumen and the folding of the N-terminal domain were questioned. On the one hand, the X-ray structures showed that the helix is attached to the channel’s wall,21,22,24 and, on the other hand, the NMR structure of human VDAC1 at room temperature revealed a mostly unfolded N-terminal domain unbound from the channel’s wall and partially blocking the lumen.20 This is particularly important since the stability of its folding and adhesion to the internal wall of the channel were proposed to be fundamental for barrel stability and, in turn, channel permeability and gating.26,27,29,32−36 In addition, the N-terminal domain was also shown to be primarily responsible for VDAC interactions with apoptosis-related proteins,4,9,10,37 for which it is assumed to detach from the β-barrel wall and move out of the channel. A straightforward experiment showed that unbinding of the N-terminal domain from the channel wall was promoted by destabilizing its helical structure and that, in turn, its exposure to the cytosol outside the lumen was favored.38
The three human VDAC isoforms share high sequence similarity, with the N-terminal domain having slight but significant differences. All of the three N-terminal fragments show an amphipathic helical profile,35 but whereas hVDAC1 has no cysteine residues, two of them are present in the sequence of both hVDAC2 and hVDAC3. Furthermore, VDAC2 has 11 additional residues on the N-term side. In light of these differences, as well as of the different expression level, the hypothesis of complementary roles was put forward.5,10,17 This is well supported by the work of Reina and colleagues,39 which showed that the swapping of the N-terminal domain of VDAC3 with the corresponding sequence of VDAC1 completely changed the channel activity, indicating that the N-terminal domain is critical for channel functionality.
Two main factors contribute to stabilize the folded state of the N-terminal fragment when it is bound to the channel wall: its amphipathicity31 and several specific hydrophobic contacts and hydrogen bonds.20−22,31,36 Most of the models proposed in the literature have the N-terminal fragment unbinding from the internal wall as the very first step. Then, the N-terminal domain can move out of the channel and either stay unfolded and solvent-exposed or possibly bind to the external leaflet of the MOM.9,10,29,32 In this scenario, the amphipathic profile, which is strictly interconnected to both folded structure and binding stability, is of primary importance. However, when widely used hydropathicity scales were applied,31,35 no significant differences were found among the N-terminal fragments of the three hVDAC isoforms because of their high sequence homology (Figure 1). Nevertheless, a different folding tendency has recently been reported, even in isotropic buffered solutions, with helical propensity following the order hVDAC2(12–36)a > hVDAC3(1–25) > hVDAC1(1–25).35 Therefore, it is fundamental to characterize possible differences at a higher level of details to shed some light on the different functionalities of the three isoforms.
Figure 1.

Sequence alignment of the three N-terminal hVDAC peptides. Differences are highlighted in bold.
In this work, the same three peptides have been fully characterized in terms of the 3D structure adopted upon binding to a membrane-mimicking model using both experimental and computational techniques. Structure differences were also correlated with the evidenced difference in membrane affinity. The observed differences appeared to be due to subtle variation in the amino acid sequence of the three peptides. By studying the isolated peptides, we focused on the unspecific interaction they have with the lipid bilayer by virtue of the intrinsic possibility to form an amphipathic 3D folded structure. Direct information about the relative stability of the channel-bound state could not be obtained (in the absence of the β-barrel domain). However, after the N-terminal domain detaches from the pore’s lumen, its availability to interact with the various proteins related to VDAC functions will ultimately depend on its membrane affinity, such that the reported differences between the three hVDAC N-terminal peptides potentially underlie the different functionalities of the whole proteins.
2. Results and Discussion
2.1. Peptide Conformation on Lipid Vesicles
Attenuated total reflection (ATR)-FTIR was employed for the conformational analysis of the three hVDAC N-terminal peptides in the presence of large unilamellar vesicles (LUVs). The composition of the latter was 20%mol 1-palmitoyl-2-oleyl-sn-glycero-3-phosphoglycerol (POPG) (negatively charged) and 80%mol 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) (zwitterionic), which was selected to mimic the overall content of negative lipids in the MOM.40Figure 2 shows the amide I band region of the three peptides together with the model α-helical peptide model amphipathic peptide (MAP), whose secondary structure was previously characterized through oriented circular dichroism.41
Figure 2.

(A) Stacking plot of the normalized amide I band from samples of hVDAC1(1–25) in red, hVDAC2(12–36) in green, hVDAC3(1–25) in blue, and MAP in black. Spectra were acquired in the presence of LUVs with 20%mol POPG and 80%mol POPC, at a lipid/peptide molar ratio of 10 and a total peptide concentration of 2 mM. The table reports the position of the maximum and a dashed line centered to that of MAP is shown as a reference. (B) The same spectral region for the four samples in A are superimposed.
The three hVDAC peptides showed a comparable profile for the amide I band, with the most intense component at ∼1650 cm–1, which is indicative of α-helical folding.42−44 The shoulder at ∼1580 cm–1, which was almost absent in the spectrum of MAP, mostly originated from the stretching modes of tyrosine and aspartate side chains.42,43 Although, at the first sight, the three hVDAC peptides appeared similar, they showed significant differences. In particular, the position of the maximum of hVDAC2(12–36) was identical to that of the model peptide MAP, indicating a regular α-helical folding. Differently, the position of the maximum observed for the other peptides was lower (Figure 2A) and comparable to each other, suggesting a less regular backbone conformation.42,43 Then, all three hVDAC peptides showed a band broader than the model peptide MAP (Figure 2B), indicating superior conformational flexibility and, possibly, plasticity.42,43 This was not surprising since the MAP peptide was specifically designed to fold as a highly amphipathic α-helix in the presence of lipid layers. Interestingly, hVDAC2(12–36) showed the smallest full-width at half-maximum (FWHM) among the three hVDAC peptides, with a value of 48 cm–1, whereas the amide I bands of hVDAC3(1–25) and hVDAC1(1–25) were slightly larger, with FWHM of 50 and 53 cm–1, respectively. For the sake of comparison, MAP showed a FWHM of 31 cm–1.
However, the intrinsic low resolution of infrared spectroscopy did not allow us to obtain more details about structural differences among the three hVDAC peptides. Although the difference in the amide I band’s position was indicative, the FWHM was quite large, and we did not want to attempt a quantitative deconvolution. We preferred to move to higher resolution techniques, instead, as reported below.
2.2. Three-Dimensional Structure
Liquid-state NMR was coupled to computational techniques to solve the 3D structure of the three hVDAC peptides in the presence of DPC micelles. A series of two-dimensional (2D) experiments, namely DQF-COSY, TOCSY, NOESY, and 1H13C HSQC were used to assign 1H and 13C resonances45,46 for hVDAC1(1–25), hVDAC2(12–36), and hVDAC3(1–25) (Tables S1–S3). The software TALOS+47 was applied to analyze the experimental chemical shift of peptides’ backbone to estimate values for the Φ and Ψ angles. Figure 3 shows the results together with the corresponding uncertainties (see Section 4 for definition of “good”, “warn”, and “dynamic”).
Figure 3.

Backbone Φ and Ψ angles predicted through the software TALOS + on the basis of the experimental chemical shift values of both 1H and 13C for (A) hVDAC1(1–25), (B) hVDAC2(12–36), and (C) hVDAC3(1–25). Prediction ranked as good and “warning” (see Section 4) are reported in green and orange, respectively. The few dynamic predictions are not shown for the sake of clarity. Selected amino acid residues are explicitly labeled in the picture to help the discussion provided in the text.
In each peptide, the last five residues were classified as dynamic, and for this reason they were excluded from Figure 3. This was an expected result, due to the presence of three glycine residues located in the C-terminal portion of each peptide. Concerning the other residues, most of them resulted in a good ranking. Predictions of the few residues marked as warn were excluded from both structure calculations and MD simulations.
In the case of hVDAC1(1–25), residues from P5 to K20 populated the α-helix region of the Ramachandran plot with the only exception of residue A8 (Figure 3A). Residue T6 resulted in a warn attribution due to a single outlier (see Section 4) but, regardless, it has to be noted that up to four consecutive residues with proper Φ/Ψ values are needed to define at least one round of a α-helix. Thus, by taking the reported results into account, hVDAC1(1–25) was predicted to fold as α-helix only from residue D9 to K20 on the basis of the experimental chemical shifts.
Also for hVDAC2(12–36), the α-helix region of the Ramachandran plot was populated by residues from P5 to K20 with the only exception of residue G11 in this case. Up to four residues in this range were designated as warn, namely, P5, S6, 7Y, and N19, i.e., at the beginning and end of the range. Thus, peptide hVDAC2(12–36) was predicted to fold as a longer α-helix, seemingly starting from the fifth residue, with a kink at the level of G11.
Finally, in the case of hVDAC3(1–25), only N3 was ranked as warn. A lower number of residues populated the α-helix region of the Ramachandran plot. These corresponded to the positions from L10 to K20 without exceptions. Thus, the prediction for this peptide’s fold was not dissimilar from hVDAC1(1–25).
Only good-ranked Φ/Ψ estimated values were included as restraints in both structure calculations and MD simulations, together with NOE-derived interproton distances. Only unambiguous assignments were taken into account. Figure 4 shows the typical representation of sequential interproton distances.
Figure 4.

Sequential interproton through-space dipolar coupling (NOEs) for (A) hVDAC1(1–25), (B) hVDAC2(12–36), and (C) hVDAC3(1–25). Short (i,i + 1) and medium-range (i,i + 2; i,i + 3; i,i + 4) NOEs are shown as lines connecting the two residues involved. Three different line thicknesses are used to indicate the relative intensity of the corresponding NOESY cross-peaks, which were categorized as weak, medium, and strong.
The results found for hVDAC1(1–25) and hVDAC3(1–25) were similar to each other, characterized by short- and medium-range NOEs spreading all along the peptide sequence but with the highest density approximately from the 10th to the 20th position. The number of medium-range i,i + 3 and i,i + 4 interactions was higher for hVDAC3(1–25) than that for hVDAC1(1–25), although the NOE intensity was lower on average. Peptide hVDAC2(12–36) showed the lowest number of sequential NOEs among the three peptides under investigation. However, these were concentrated in the region between the 6th and the 20th position of the sequence, and they were of medium intensity for the most part.
Three-dimensional structure calculation was performed without any water or detergent molecule, at first, through a simulated annealing (SA) scheme. Thousand-peptide conformers were independently generated from the same starting random configuration. The 100 conformers with the lowest potential energy were selected and their secondary structure analyzed (Figure 5A). To perform a structure refinement, the conformer with the minimum average root mean square deviation (RMSD) from among the others was selected as the starting configuration for MD simulations with explicit solvent and detergent micelles. The last 50 ns of the trajectory were analyzed in terms of the secondary structure (Figure 5B).
Figure 5.

Secondary structure analysis from computer simulations with NMR-derived geometrical restraints. Results are shown for (A) the 100 conformers with the lowest potential energy from SA, and (B) the last 50 ns of MD trajectories. Selected positions along the sequences are marked with dashed lines and corresponding Arabic number. Whenever occurrence was lower than 100% for a given portion, this is explicitly indicated.
Despite the high sequence similarity, significant folding differences emerged. The hVDAC1(1–25) showed the shortest and less regular folded fraction. A helical structure comprising residues from 9 to 19 resulted from the SA. However, a first 310-helix round was present only in 50% of the conformers. After a long MD equilibration in the presence of explicit solvent and the detergent micelle, a unique α-helix was found for the same portion, with an additional round of 310-helix between the 5th and the 7th position only in 50% of the conformers.
The α-helical folding was more regular and stable for hVDAC3(1–25). A unique α-helix comprising residues from 10 to 21 resulted from SA, then MD revealed an additional helical round up to the 24th position in 80% of the frames. Peptide hVDAC2(12–36) was definitely the one with the highest occurrence of α-helical conformation for most of the amino acid residues. A rather long α-helix was observed already from the SA, with a kink at the level of G11. The N-terminal portion of the helix started from P5 but had an occurrence of 50%. This was almost completely stabilized during the MD.
However, it is important to note that the overall helicity of hVDAC2(12–36) and hVDAC3(1–25) was remarkably comparable, with the most important difference being the localization of the folded portion along the amino acid sequence. In particular, the N-terminal half was almost completely unfolded in hVDAC3(1–25), whereas this showed some folding ability at the C-terminal end. In addition, a slight but significant difference was also found in the probability distribution of the helix length (Figure 6).
Figure 6.
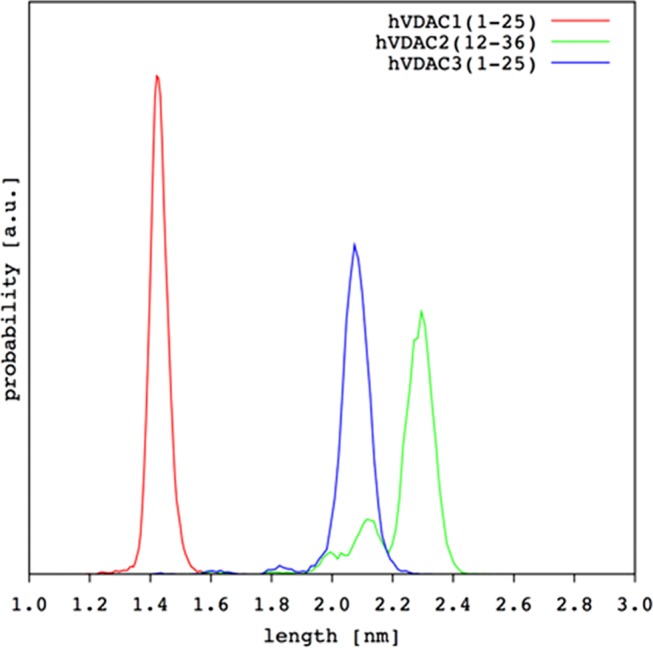
Helix length distribution from MD simulations for the three hVDAC N-terminal peptides.
Although, again, these results demonstrate the marked difference of hVDAC1(1–25), some interesting differences also emerged between the other two isoforms. To finally compare helix flexibility, we selected the portion always folded as α-helix in all three hVDAC peptides already in the SA conformers. This was the region comprising residues from 11 to 19, for which we calculated the backbone RMSD using the frame corresponding to 50 ns of our MD production runs as the reference structure. Results were in the order hVDAC1(1–25) > hVDAC3(1–25) > hVDAC2(12–36), i.e., 0.3(2) ± 0.1(1), 0.26 ± 0.09, and 0.13 ± 0.06 Å, respectively. Even if the standard deviation showed overlapping results, it was really interesting to find a perfect correlation with the order found based on the FWHM of the amide I band in infrared spectra (Section 2.1). Finally, Figure 7 shows the 3D structure obtained for the three N-terminal hVDAC peptides.
Figure 7.

Three-dimensional structure of (A) hVDAC1(1–25), (B) hVDAC2(12–36), and (C) hVDAC3(1–25) are shown. The final frame from the MD simulation is reported on the left hand, where only the backbone trace is shown for clarity. On the right hand, the superposition of the last 50 ns of the MD is shown (10k frames in step of 100) with both backbone and side chains represented. Structures were rotated to have the N-terminus on the left.
2.3. Affinity to Lipid Monolayers
Surface pressure (π) measurements are able to provide information about peptide tendency to insert through lipid molecules in a monolayer, indicating thus, their ability to perturb the lipid package. By measuring the difference of surface pressure (Δπ) of a given lipid monolayer at different initial values (πi) after injection of the peptide in the aqueous subphase, important information can be derived. A straight line for Δπ vs πi is typically obtained, from which the maximum surface pressure variation (Δπmax) and the exclusion pressure (πex) can be extrapolated as the intercept with the y- and x-axis, respectively. The composition of the lipid monolayer was identical to that of the liposomes used for FTIR measurements (Section 2.1), i.e., 20%mol POPG (negatively charged) and 80%mol POPC (zwitterionic), to mimic the overall content of negative lipids in the MOM.40Figure 8 shows the results obtained for the three hVDAC peptides.
Figure 8.
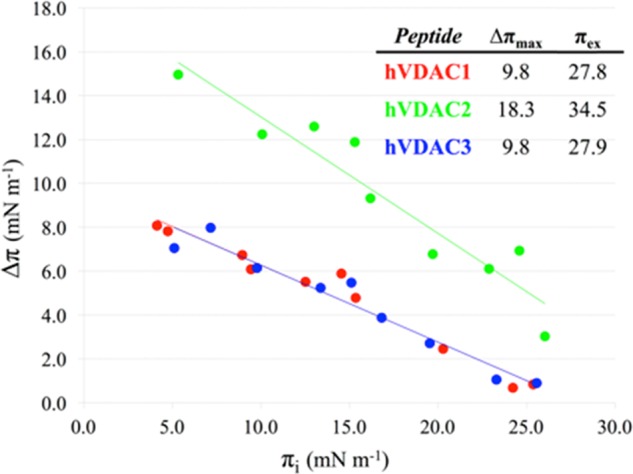
Surface pressure difference (Δπ) as a function of initial surface pressures (πi) for hVDAC1(1–25) in red, hVDAC2(12–36) in green, and hVDAC3(1–25) in blue is shown. The corresponding linear curve fitting obtained by least-squares minimization is reported. In the table, the values of both Δπmax and πex are given. Monolayers were prepared with 20%mol POPG and 80%mol POPC. Peptide concentration in the subphase was 1.5 μM.
It is immediately evident that hVDAC2(12–36) had a remarkably different affinity for the monolayer, when compared to either hVDAC1(1–25) or hVDAC3(1–25). In particular, πex, i.e., the monolayer surface pressure at which the peptide is no more able to penetrate, was 124% of that extrapolated for the other two hVDAC peptides. This result reflects the inherent stronger ability of hVDAC2(12–36) to intercalate between the lipids. On the other hand, hVDAC1(1–25) and hVDAC3(1–25) showed an almost identical behavior, with lower Δπ values in comparison to hVDAC2(12–36) at any πi.
When these results are taken together with the folding structure and conformational flexibility, the positive correlation between membrane affinity, folding ability, and structure rigidity emerges clearly. Despite the high sequence identity shared by the three hVDAC N-terminal peptides, isoform 2 has a distinct behavior. When bioinformatic tools were applied to analyze peptide sequence’s amphipathicity, no significant differences were found among the three peptides.31,35 Results previously obtained in buffered solutions without any membrane model or artificial folding support35 were absolutely in agreement with the evidence reported here, bolstering the distinct properties of the N-terminal domain of isoform 2, even without the first additional 11 residues.a The fundamental reasons for this remain to be deciphered, but it appeared that the subtle combination of a few single-point mutations (Figure 1) made the difference in the behavior of the hVDAC2 peptide. The available scoring functions for hydropathic profiling were unable to predict the reported differences. Further studies are needed to shed light on this point.
3. Concluding Remarks
Whether the three human isoforms of VDAC have distinct and complementary roles is still unclear.5,17,39 Most of the available 3D structures of VDAC show that the N-terminal domains are folded in a helical conformation and bound to the β-barrel’s internal wall, half-way through the hydrophobic portion of the membrane.21−24 As such, the N-terminal domain plays a major role in the channel functionality as a pore by (i) forming a restricted zone that acts as the main steric filter for molecules, and (ii) by determining the electrostatic potential inside the channel and, thus, its ion selectivity.26,27,31,48 The N-terminal domain is stabilized in the folded-bound state by two main factors: first, the helix amphipathicity31 and, second, several specific hydrophobic contacts and hydrogen bonds.20−22,31 Detachment of this domain from the channel wall is widely recognized to be the very first step of the channel’s gating mechanism, but this is also related to other important functions of VDACs, i.e., interaction with anti- and pro-apoptotic proteins like hexokinase and Bcl-2 family members.10,37 In addition, evidence was provided about N-terminal domain’s role in the oligomerization of VDAC1, which, upon apoptotic stimuli, is believed to be responsible for the formation of a larger conduit, allowing cytochrome c release to the cytosol.49
After unbinding the channel’s wall, unfolding and movement of the N-terminal domain will be promoted. Leaving the lumen will turn into the eventual exposure to the cytosol, from where the N-terminal domain might bind to the external leaflet of the MOM9,10,26,29,31,32,50 by assuming again its specific amphipathic conformation. The relative probability of staying mostly unstructured and solvent-exposed, or adhering and folding onto the MOM, ultimately depends on the affinity for the latter. Thus, the affinity of the N-terminal peptide for the MOM can have a profound impact on those whole VDAC’s roles in which a protein–protein interaction is mediated by this domain.
In this work, remarkable differences were shown for the shared portion of the N-terminal domain of the three human VDAC isoforms. In particular, the interaction with surfaces that favored amphipathic conformations revealed their dissimilarities. The hVDAC1(1–25) peptide was the one with the lowest helical propensity and folding stability. The other two peptides, hVDAC3(1–25) and hVDAC2(12–36), shared a higher helicity, but the structured portion was remarkably different. The latter was definitely the one with the highest membrane affinity. Although simplified membrane models were used in our experiments, the results clearly highlighted interesting differences between the three N-terminal hVDAC domains. The present investigation was performed on isolated peptides but, nevertheless, important information can be inferred. Of course, the absence of the β-barrel domain in our experiments prevented the possibility of assessing possible specific differences as far as the channel’s wall-bound state is concerned, while we focused on the different behavior after detachment.
The expression level of the three hVDAC isoforms was different and appeared also to be tissue-specific, with evidence that bolstered possible different roles.5,17 The most expressed (and studied) VDAC1 is reported to be probably the isoform mainly responsible for both ion and metabolite permeation through the MOM. This “classic” channel function is related to its gating, which requires an N-terminal domain in equilibrium between the bound-folded and unbound-unfolded state,29 in agreement with the highest conformational plasticity found for hVDAC1(1–25) in this report. In addition, the relatively low affinity for the membrane model we used suggests a high probability for the N-terminal domain to stay mostly unstructured and solvent-exposed when leaving the lumen. In this configuration, this domain can be expected to interact with the cytosolic protein hexokinase more easily than if it was bound onto the MOM’s external leaflet. Comparative studies with the other isoforms are very few but the information collected so far indicates that VDAC2 might have a predominant role in apoptosis as the Bak receptor, as well as in the modulation of the interactions among antagonist proteins of the Bcl-2 family. These pro- and anti-apoptotic proteins are located in the MOM. Interestingly, the higher membrane affinity we found for hVDAC2(12–36) in this work supports the putative primary role of the corresponding isoform. Finally, VDAC3 appeared to have an intermediate behavior within the present work, with the N-terminal peptide being more rigid than VDAC1 but sharing the same low affinity for the membrane model. This is the least expressed and least characterized isoform and further investigations are needed to understand the differences from the other two isoforms.
4. Materials and Methods
4.1. Materials
The three peptides, namely, hVDAC1(1–25) [MAVPPTYADLGKSARDVFTKGYGFG], hVDAC2(1–36) [MCIPPSYADLGKAARDIFNKGFGFG], and hVDAC3(1–25) [MCNTPTYCDLGKAAKDVFNKGYGFG], were purchased from Peptide Protein Research Ltd. (Fareham, U.K.) as HCl salt, with amidated C-terminus and a purity of 98%. Peptide model amphipathic peptide (MAP) [KLALKLALKALKAALKLA], HCl salt, 95% purity with amidated C-terminus was kindly provided by Prof. Anne S. Ulrich and Dr. Parvesh Wadhwani from the Karlsruhe Institute of Technology, Germany. Glycerophospholipids, 1-palmitoyl-2-oleyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleyl-sn-glycero-3-phosphoglycerol (POPG), were purchased from Avanti Polar Lipids (Alabaster, AL) with a purity grade of >99%. The reducing agent 1,4-dithio-d-threitol (DTT) was used at a 1:3 peptide/DTT molar ratio in all experiments with both hVDAC2(12–36) and hVDAC3(1–25) to prevent oxidation of cysteine residues and the formation of either intra- or intermolecular disulfide bridges. Perdeuterated dodecylphosphocholine (DPC-d38) and 3-(trimethylsilyl)-2,2′,3,3′-tetradeutero-propionic acid (TSP-d4) were purchased from Cambridge Isotope Laboratories with a purity of 98%. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless differently indicated. A complete list follows (with used name/abbreviation): chloroform, ≥99.8% (CHCl3); methanol, ≥99.8% (CH3OH); perdeuterated water, 99% D (D2O); phosphate buffer saline (PBS) was prepared at 10 mM total phosphate concentration (KH2PO4 + K2HPO4) and 150 mM NaCl, pH 7.4.
4.2. Vesicle Preparation
Large unilamellar vesicles (LUVs) were used as the membrane model in the infrared spectroscopy experiments. LUVs were prepared by the extrusion method with the mini-extruder from Avanti Polar Lipids. Appropriate amounts of POPC and POPG were weighed in a dark glass vial and dissolved in CHCl3/CH3OH 1/1 v/v. The organic solvent was removed under a gentle stream of nitrogen and the obtained lipid film was placed under vacuum pumping overnight. PBS was used to rehydrate the film, and after five vortex cycles of 1 min separated by 1 min rest, a suspension of large multilamellar vesicles was obtained. Then, five freeze–thaw cycles were performed by placing the vial in liquid N2 and in a 323 K water bath. Finally, extrusion was carried out by passing the vesicle suspension (1 mL) 11 times through a polycarbonate filter (Whatman). In particular, the first extrusion was performed with a 400 nm pore size filter, then with a second 100 nm filter. To mimic the net charge of MOM,40 a composition of 20%mol negatively charged POPG and 80%mol zwitterionic POPC was chosen.
4.3. Infrared Spectroscopy
Fourier-transform infrared (FTIR) spectroscopy in the attenuated total reflection mode (ATR-FTIR) was chosen to investigate the peptide conformation and plasticity. Spectra were acquired at room temperature with a Bruker Vector-22 equipped with a diamond single-reflection platinum ATR module and a liquid nitrogen-cooled MCT detector. The software OPUS (Bruker, V6.5) was used both for spectra acquisition and data analysis. All three peptides were investigated in PBS solution prepared with D2O as solvent, at a final concentration of 2 mM in the presence of LUVs at a lipid/peptide molar ratio of 10. For each peptide, 10 μL of solution were placed on the ATR crystal and dried with a gentle stream of nitrogen, to reduce the disturbance by the intense solvent absorption bands and to attain a significant improvement of the signal/noise ratio (S/N). The height of the peptide amide I band (∼1650 cm–1) was about 10 times that due to the D2O stretching mode (∼2500 cm–1). After the spectrum was acquired on the dried film, several droplets of D2O were dispensed all around the sample area and then covered with a Petri dish. Adsorption of D2O vapor and rehydration of the sample were monitored for about 60 s. Spectrum acquisition was started when the height of the D2O stretching band was 3 times that of the peptide amide I band. All spectra were recorded with a spectral window of 3500–600 cm–1, resolution of 4 cm–1, automatic atmosphere compensation, and 32 scans. Before each measurement, the clean ATR crystal was acquired as background and automatically subtracted by the software.
4.4. Nuclear Magnetic Resonance
Peptides were dissolved in 700 μL of a dispersion of DPC-d38 micelles in PBS at a final concentration of 2 mM and at a peptide/detergent molar ratio of 1:50. The methyl resonance of TSP-d4 (∼2 mM) was used as the internal reference for both 1H and 13C chemical shift scales. NMR spectra were acquired at 300 K with a Unity Inova 500 NM high-resolution spectrometer (Agilent Technologies, CA), operated at a 1H frequency of 500 MHz and equipped with an indirect detection probe. 1H spectra were acquired using a 6.7 μs pulse (90°), 1 s delay time, 1 s acquisition time, and a spectral width of 6 kHz. The WET sequence51,52 (100 Hz-wide uburp shape centered at water resonance) was applied to suppress the water signal. Two-dimensional (2D) experiments, 1H–1H double-quantum filtered correlation (DQF-COSY), 1H–1H total correlation (TOCSY), and 1H–1H nuclear overhauser effect (NOESY) spectra were recorded over the same spectral window, by each of the 512 increments with 2048 complex points and 48 scans. Mixing time was 80 and 200 ms for the TOCSY and NOESY, respectively. The 1H–13C heteronuclear single-quantum correlation (HSQC) spectra were recorded using a spectral window of 6 kHz for 1H and 21 kHz for 13C.
4.5. NMR-Based Structure Calculation
The peptide 3D structure was obtained through the simulated annealing (SA) protocol implemented in the software Dynamo.53 In addition to Dynamo force-field potentials, experimental restraints were applied to interproton distances and backbone torsion angles as explained herein. NOESY cross-peaks (NOEs) were classified as strong, medium, and weak on the basis of their relative intensity, and upper limits of 0.27, 0.33, and 0.50 nm were applied, respectively, to restrain the corresponding interproton distance. The potential energy contribution was zero below the upper limit, whereas a harmonic potential was applied above. Only unambiguously assigned NOEs were considered. Either Φ or Ψ backbone angle restraint was obtained from the statistical analysis performed on the 1Hα, 1Hβ, 13Cα, and 13Cβ chemical shift values implemented in the software TALOS+.47 Briefly, TALOS + analyzes the input peptide sequence in terms of successive [i – 1, i, i + 1] residue triplets. Both experimental chemical shift values, as well as the residue types, are compared to the 10 best-matched triplets in a high-resolution structure database and to the prediction output of the implemented artificial neural network.47 Final prediction ranking is stringent. The estimated Φ/Ψ couple is ranked as consistent or good only if the central residues of all of the 10 best-matched triplets cluster in the same region of the Ramachandran plot, otherwise it is designated as ambiguous or warn. Residues with a calculated random coil index-derived order parameter54,55S2 < 0.5 are ranked as dynamic anyway. Only the predictions ranked as good were used as torsional angle restraints. One thousand structures were calculated with 5k steps at 4000 K and 20k steps of cooling down to zero. The 100 conformers with the lowest potential energy were selected for the analysis. Neither solvent nor detergent molecules were included in the calculations.
4.6. Molecular Dynamics Simulations
Molecular dynamics (MD) simulations were performed through the GROMACS package version 4.6.3,56,57 in the presence of one DPC micelle and explicit solvent. The GROMOS-53A6 force-field58 was used for the peptide and the SPC model59 for water. Force-field parameters and the relaxed configuration of a DPC micelle with 54 molecules60 were downloaded from http://moose.bio.ucalgary.ca/. The starting peptide configuration was obtained through the NMR-based SA reported above (Section 4.5). The 100 conformers with the lowest potential energy were aligned on the basis of their backbone; the one with the minimum average root mean square deviation (RMSD) from among the others was selected and placed in a random orientation at 2 nm from the micelle surface. Water and the minimum number of counterions needed to neutralize the total charge filled the 8 × 8 × 8 nm3 cubic simulation box. Thousand steps of energy minimization were performed using the steepest descent algorithm. Positional restraints were then applied on the heavy atoms for 100 ps. Finally, experimentally derived NMR restraints (interproton distances and backbone torsion angles; see Section 4.5) were introduced in the system topology. As Hα and Hβ are not explicitly represented in the united-atom force-field of choice, corresponding interproton restraints were elongated by 0.1 nm. An equilibration run of 10 ns was performed, followed by 100 ns trajectory production. The time step was 2 fs, and system coordinates were recorded every 5 ps. All simulations were carried out in the NPT ensemble at 300 K and 1 bar. The velocity-rescale algorithm61 was used for temperature coupling with τT = 1.0 ps. The Berendsen62 and Parrinello–Rahman63,64 algorithms were used for pressure coupling with τP = 1.0 ps during the equilibration and production run, respectively. A twin-range cut-off (1.0 and 1.4 nm) was applied to describe Lennard-Jones nonbonded interactions. The particle mesh Ewald summation was used for the electrostatics with a cut-off of 1.0 nm.
4.7. Lipid Monolayer Preparation
Appropriate amounts of POPC and POPG were weighed and dissolved in CHCl3/CH3OH 1/1 v/v to obtain a homogeneous mixture with 20%mol POPG and 80%mol POPC. This specific composition was the same as that used for vesicle preparation (Section 4.2), and it was chosen to mimic the net charge of MOM.40 The total lipid concentration of the stock solution was about 2 × 10–5 M. In a circular well of 19 mm diameter, a 500 μL drop of PBS was deposited and aliquots of the lipid stock solution were spread at the air/buffer interface using a Hamilton microsyringe. A lipid monolayer was easily formed as organic solvents spontaneously evaporated, due to self-association of lipid molecules at the air/buffer interface.
4.8. Surface Pressure Measurements
The surface pressure of the lipid monolayer was measured at 300 ± 1 K with a tensiometer Kibron “DeltaPi”, which exploits the Wilhelmy method, described in detail elsewhere.65 The instrument makes use of a high-resolution electrobalance (resolution of 0.01 mN m–1) attached to a platinum wire with 0.5 mm diameter. All measurements were performed at constant area. Filtered deionized water was used during the calibration stage by measuring the voltage difference (probe at the interface and at air) corresponding to a surface tension of 500 μL (71.7 mN m–1) in a circular glass well (diameter of 19 mm). Under the same experimental conditions, the surface pressure of 500 μL of PBS was measured and set as the reference zero. By spreading different amounts of the lipid stock solution (in small aliquots) as described herein before (Section 4.7), monolayers with a different initial surface pressure (πi) were prepared in the 5–25 mN m–1 range. After the lipid monolayer reached a stable πi, a small volume of the peptide stock solution (50 μM in PBS) was injected into the subphase at a peptide final concentration of 1.5 μM. The surface pressure was monitored over time up to a stable final surface pressure (πf). The surface pressure variation induced by the peptide intercalating the lipids was obtained as Δπ = πf – πi.
Acknowledgments
This work was supported by the Italian Ministero dell’Istruzione, dell’Università e della Ricerca, MIUR, PRIN Project 2015795S5W_005. The Sardinia Regional Government (http://www.regione.sardegna.it/) is greatly acknowledged for the fellowship provided to G.M. and I.S. (L.R. 7/2007) and for the project RAS/FdS (CUP F72F16003070002). A.M. and V.D.P. acknowledge the grant FIR-UNICT project 2017. Prof. Anne S. Ulrich and Dr. Parvesh Wadhwani of the Karlsruhe Institute of Technology, Germany, are greatly acknowledged for having provided the model peptide MAP.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01536.
1H and 13C resonance assignments (Tables S1–S3) (PDF)
Author Present Address
Institute of Pharmaceutical Science, King’s College London, 150 Stamford Street, SE1 9NH London, United Kingdom.
Author Contributions
⊥ G.M. and I.S. contributed equally to this work
The authors declare no competing financial interest.
Footnotes
hVDAC2 has 11 additional residues [MATHGQTCARP] on the N-terminal end of the sequence. Sequence alignment showed that the hVDAC2(12–36) sequence is similar to both hVDAC1(1–25) and hVDAC3(1–25).
Supplementary Material
References
- Benz R. Permeation of Hydrophilic Solutes through Mitochondrial Outer Membranes: Review on Mitochondrial Porins. Biochim. Biophys. Acta, Rev. Biomembr. 1994, 1197, 167–196. 10.1016/0304-4157(94)90004-3. [DOI] [PubMed] [Google Scholar]
- Rostovtseva T.; Colombini M. VDAC Channels Mediate and Gate the Flow of ATP: Implications for the Regulation of Mitochondrial Function. Biophys. J. 1997, 72, 1954–1962. 10.1016/S0006-3495(97)78841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombini M. VDAC: The Channel at the Interface between Mitochondria and the Cytosol. Mol. Cell. Biochem. 2004, 256–257, 107–115. 10.1023/B:MCBI.0000009862.17396.8d. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V.; De Pinto V.; Zweckstetter M.; Raviv Z.; Keinan N.; Arbel N. VDAC, a Multi-Functional Mitochondrial Protein Regulating Cell Life and Death. Mol. Aspects Med. 2010, 31, 227–285. 10.1016/j.mam.2010.03.002. [DOI] [PubMed] [Google Scholar]
- Messina A.; Reina S.; Guarino F.; De Pinto V. VDAC Isoforms in Mammals. Biochim. Biophys. Acta, Biomembr. 2012, 1818, 1466–1476. 10.1016/j.bbamem.2011.10.005. [DOI] [PubMed] [Google Scholar]
- Beutner G.; Ruck A.; Riede B.; Brdiczka D. Complexes between Hexokinase, Mitochondrial Porin and Adenylate Translocator in Brain: Regulation of Hexokinase, Oxidative Phosphorylation and Permeability Transition Pore. Biochem. Soc. Trans. 1997, 25, 151–157. 10.1042/bst0250151. [DOI] [PubMed] [Google Scholar]
- Cheng E. H. Y.; Sheiko T. V.; Fisher J. K.; Craigen W. J.; Korsmeyer S. J. VDAC2 Inhibits BAK Activation and Mitochondrial Apoptosis. Science 2003, 301, 513–517. 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- Tomasello F.; Messina A.; Lartigue L.; Schembri L.; Medina C.; Reina S.; Thoraval D.; Crouzet M.; Ichas F.; De Pinto V.; et al. Outer Membrane VDAC1 Controls Permeability Transition of the Inner Mitochondrial Membrane in Cellulo during Stress-Induced Apoptosis. Cell Res. 2009, 19, 1363–1376. 10.1038/cr.2009.98. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V.; Keinan N.; Abu-Hamad S.; Tyomkin D.; Aram L. Apoptosis Is Regulated by the VDAC1 N-Terminal Region and by VDAC Oligomerization: Release of Cytochrome c, AIF and Smac/Diablo. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 1281–1291. 10.1016/j.bbabio.2010.03.003. [DOI] [PubMed] [Google Scholar]
- Shoshan-Barmatz V.; Ben-Hail D. VDAC, a Multi-Functional Mitochondrial Protein as a Pharmacological Target. Mitochondrion 2012, 12, 24–34. 10.1016/j.mito.2011.04.001. [DOI] [PubMed] [Google Scholar]
- Maldonado E. N.; Lemaster J. J. Warburg Revisited: Regulation of Mitochondrial Metabolism by Voltage-Dependent Anion Channels in Cancer Cells. J. Pharmacol. Exp. Ther. 2012, 342, 637–641. 10.1124/jpet.112.192153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magrì A.; Belfiore R.; Reina S.; Tomasello M. F.; Di Rosa M. C.; Guarino F.; Leggio L.; De Pinto V.; Messina A. Hexokinase I N-Terminal Based Peptide Prevents the VDAC1-SOD1 G93A Interaction and Re-Establishes ALS Cell Viability. Sci. Rep. 2016, 6, 34802 10.1038/srep34802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magri A.; Messina A. Interactions of VDAC with Proteins Involved in Neurodegenerative Aggregation: An Opportunity for Advancement on Therapeutic Molecules. Curr. Med. Chem. 2017, 24, 4470–4487. 10.2174/0929867324666170601073920. [DOI] [PubMed] [Google Scholar]
- Lemeshko V. V. VDAC Electronics: 1. VDAC-Hexo(Gluco)Kinase Generator of the Mitochondrial Outer Membrane Potential. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1362–1371. 10.1016/j.bbamem.2014.01.001. [DOI] [PubMed] [Google Scholar]
- Lemeshko V. V. VDAC Electronics: 2. A New, Anaerobic Mechanism of Generation of the Membrane Potentials in Mitochondria. Biochim. Biophys. Acta, Biomembr. 2014, 1838, 1801–1808. 10.1016/j.bbamem.2014.02.007. [DOI] [PubMed] [Google Scholar]
- Lemeshko V. V. VDAC Electronics: 3. VDAC-Creatine Kinase-Dependent Generation of the Outer Membrane Potential in Respiring Mitochondria. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1411–1418. 10.1016/j.bbamem.2016.04.005. [DOI] [PubMed] [Google Scholar]
- De Pinto V.; Guarino F.; Guarnera A.; Messina A.; Reina S.; Tomasello F. M.; Palermo V.; Mazzoni C. Characterization of Human VDAC Isoforms: A Peculiar Function for VDAC3?. Biochim. Biophys. Acta, Bioenerg. 2010, 1797, 1268–1275. 10.1016/j.bbabio.2010.01.031. [DOI] [PubMed] [Google Scholar]
- Checchetto V.; Reina S.; Magrì A.; Szabo I.; De Pinto V. Recombinant Human Voltage Dependent Anion Selective Channel Isoform 3 (HVDAC3) Forms Pores with a Very Small Conductance. Cell. Physiol. Biochem. 2014, 34, 842–853. 10.1159/000363047. [DOI] [PubMed] [Google Scholar]
- Reina S.; Checchetto V.; Saletti R.; Gupta A.; Chaturvedi D.; Guardiani C.; Guarino F.; Scorciapino M. A.; Magrì A.; Foti S.; et al. VDAC3 as a Sensor of Oxidative State of the Intermembrane Space of Mitochondria: The Putative Role of Cysteine Residue Modifications. Oncotarget 2016, 7, 2249–2268. 10.18632/oncotarget.6850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller S.; Garces R. G.; Malia T. J.; Orekhov V. Y.; Colombini M.; Wagner G. Solution Structure of the Integral Human Membrane Protein VDAC-1 in Detergent Micelles. Science 2008, 321, 1206–1210. 10.1126/science.1161302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayrhuber M.; Meins T.; Habeck M.; Becker S.; Giller K.; Villinger S.; Vonrhein C.; Griesinger C.; Zweckstetter M.; Zeth K. Structure of the Human Voltage-Dependent Anion Channel. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 15370–15375. 10.1073/pnas.0808115105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujwal R.; Cascio D.; Colletier J.-P.; Faham S.; Zhang J.; Toro L.; Ping P.; Abramson J. The Crystal Structure of Mouse VDAC1 at 2.3 A Resolution Reveals Mechanistic Insights into Metabolite Gating. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 17742–17747. 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka T.; Okazaki M.; Kimura-Someya T.; Ishizuka-Katsura Y.; Ito K.; Yokoyama S.; Dodo K.; Sodeoka M.; Shirouzu M. Crystal Structural Characterization Reveals Novel Oligomeric Interactions of Human Voltage-Dependent Anion Channel 1. Protein Sci. 2017, 26, 1749–1758. 10.1002/pro.3211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schredelseker J.; Paz A.; López C. J.; Altenbach C.; Leung C. S.; Drexler M. K.; Chen J.-N.; Hubbell W. L.; Abramson J. High Resolution Structure and Double Electron-Electron Resonance of the Zebrafish Voltage-Dependent Anion Channel 2 Reveal an Oligomeric Population. J. Biol. Chem. 2014, 289, 12566–12577. 10.1074/jbc.M113.497438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomasello M. F.; Guarino F.; Reina S.; Messina A.; De Pinto V. The Voltage-Dependent Anion Selective Channel 1 (VDAC1) Topography in the Mitochondrion Outer Membrane as Detected in Intact Cell. PLoS One 2013, 8, e81522 10.1371/journal.pone.0081522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui H.; Lee K. I.; Pastor R. W.; Im W. Molecular Dynamics Studies of Ion Permeation in VDAC. Biophys. J. 2011, 100, 602–610. 10.1016/j.bpj.2010.12.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary O. P.; Ujwal R.; Kowallis W.; Coalson R.; Abramson J.; Grabe M. The Electrostatics of VDAC: Implications for Selectivity and Gating. J. Mol. Biol. 2010, 396, 580–592. 10.1016/j.jmb.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guardiani C.; Magrì A.; Karachitos A.; Di Rosa M. C.; Reina S.; Bodrenko I.; Messina A.; Kmita H.; Ceccarelli M.; De Pinto V. YVDAC2, the Second Mitochondrial Porin Isoform of Saccharomyces Cerevisiae. Biochim. Biophys. Acta, Bioenerg. 2018, 1859, 270–279. 10.1016/j.bbabio.2018.01.008. [DOI] [PubMed] [Google Scholar]
- Zachariae U.; Schneider R.; Briones R.; Gattin Z.; Demers J.; Giller K.; Maier E.; Zweckstetter M.; Griesinger C.; Becker S.; et al. B-Barrel Mobility Underlies Closure of the Voltage-Dependent Anion Channel. Structure 2012, 20, 1540–1549. 10.1016/j.str.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones R.; Weichbrodt C.; Paltrinieri L.; Mey I.; Villinger S.; Giller K.; Lange A.; Zweckstetter M.; Griesinger C.; Becker S.; et al. Voltage Dependence of Conformational Dynamics and Subconducting States of VDAC-1. Biophys. J. 2016, 111, 1223–1234. 10.1016/j.bpj.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amodeo G. F.; Scorciapino M. A.; Messina A.; De Pinto V.; Ceccarelli M. Charged Residues Distribution Modulates Selectivity of the Open State of Human Isoforms of the Voltage Dependent Anion-Selective Channel. PLoS One 2014, 9, e103879 10.1371/journal.pone.0103879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijido O.; Ujwal R.; Hillerdal C.-O.; Kullman L.; Rostovtseva T. K.; Abramson J. Affixing N-Terminal α-Helix to the Wall of the Voltage-Dependent Anion Channel Does Not Prevent Its Voltage Gating. J. Biol. Chem. 2012, 287, 11437–11445. 10.1074/jbc.M111.314229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Midson C.; Blachly-Dyson E.; Forte M.; Colombini M. The Sensor Regions of VDAC Are Translocated from within the Membrane to the Surface during the Gating Processes. Biophys. J. 1998, 74, 2926–2944. 10.1016/S0006-3495(98)78000-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannella C. A. Conformational Changes in the Mitochondrial Channel Protein, VDAC, and Their Functional Implications. J. Struct. Biol. 1998, 121, 207–218. 10.1006/jsbi.1997.3954. [DOI] [PubMed] [Google Scholar]
- Guardiani C.; Scorciapino M. A.; Amodeo G. F.; Grdadolnik J.; Pappalardo G.; De Pinto V.; Ceccarelli M.; Casu M. The N-Terminal Peptides of the Three Human Isoforms of the Mitochondrial Voltage-Dependent Anion Channel Have Different Helical Propensities. Biochemistry 2015, 54, 5646–5656. 10.1021/acs.biochem.5b00469. [DOI] [PubMed] [Google Scholar]
- Shuvo S. R.; Ferens F. G.; Court D. A. The N-Terminus of VDAC: Structure, Mutational Analysis, and a Potential Role in Regulating Barrel Shape. Biochim. Biophys. Acta, Biomembr. 2016, 1858, 1350–1361. 10.1016/j.bbamem.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Abu-Hamad S.; Arbel N.; Calo D.; Arzoine L.; Israelson A.; Keinan N.; Ben-Romano R.; Friedman O.; Shoshan-Barmatz V. The VDAC1 N-Terminus Is Essential Both for Apoptosis and the Protective Effect of Anti-Apoptotic Proteins. J. Cell Sci. 2009, 122, 1906–1916. 10.1242/jcs.040188. [DOI] [PubMed] [Google Scholar]
- Geula S.; Ben-Hail D.; Shoshan-Barmatz V. Structure-Based Analysis of VDAC1: N-Terminus Location, Translocation, Channel Gating and Association with Anti-Apoptotic Proteins. Biochem. J. 2012, 444, 475–485. 10.1042/BJ20112079. [DOI] [PubMed] [Google Scholar]
- Reina S.; Palermo V.; Guarnera A.; Guarino F.; Messina A.; Mazzoni C.; De Pinto V. Swapping of the N-Terminus of VDAC1 with VDAC3 Restores Full Activity of the Channel and Confers Anti-Aging Features to the Cell. FEBS Lett. 2010, 584, 2837–2844. 10.1016/j.febslet.2010.04.066. [DOI] [PubMed] [Google Scholar]
- de Kroon A. I.; Dolis D.; Mayer A.; Lill R.; de Kruijff B. Phospholipid Composition of Highly Purified Mitochondrial Outer Membranes of Rat Liver and Neurospora Crassa. Is Cardiolipin Present in the Mitochondrial Outer Membrane?. Biochim. Biophys. Acta 1997, 1325, 108–116. 10.1016/S0005-2736(96)00240-4. [DOI] [PubMed] [Google Scholar]
- Bürck J.; Roth S.; Wadhwani P.; Afonin S.; Kanithasen N.; Strandberg E.; Ulrich A. S. Conformation and Membrane Orientation of Amphiphilic Helical Peptides by Oriented Circular Dichroism. Biophys. J. 2008, 95, 3872–3881. 10.1529/biophysj.108.136085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth A. Infrared Spectroscopy of Proteins. Biochim. Biophys. Acta 2007, 1767, 1073–1101. 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Tamm L. K.; Tatulian S. A. Infrared Spectroscopy of Proteins and Peptides in Lipid Bilayers. Q. Rev. Biophys. 1997, 30, 365–429. 10.1017/S0033583597003375. [DOI] [PubMed] [Google Scholar]
- Haris P. I.; Chapman D. The Conformational Analysis of Peptides Using Fourier Transform IR Spectroscopy. Biopolymers 1995, 37, 251–263. 10.1002/bip.360370404. [DOI] [PubMed] [Google Scholar]
- Cavanagh J.; Fairbrother W. J.; Palmer A. G. III; Rance M.; Skelton N. J.. Protein NMR Spectroscopy - Principles and Practice, 2nd ed.; Elsevier Academic Press: Oxford, 2007. [Google Scholar]
- Wuthrich K.NMR of Proteins and Nucleic Acids; Wiley & Sons: Chichester, 1986. [Google Scholar]
- Shen Y.; Delaglio F.; Cornilescu G.; Bax A. TALOS +: A Hybrid Method for Predicting Protein Backbone Torsion Angles from NMR Chemical Shifts. J. Biomol. NMR 2009, 44, 213–223. 10.1007/s10858-009-9333-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krammer E.-M.; Homblé F.; Prévost M. Concentration Dependent Ion Selectivity in VDAC: A Molecular Dynamics Simulation Study. PLoS One 2011, 6, e27994 10.1371/journal.pone.0027994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keinan N.; Tyomkin D.; Shoshan-Barmatz V. Oligomerization of the Mitochondrial Protein Voltage-Dependent Anion Channel Is Coupled to the Induction of Apoptosis. Mol. Cell. Biol. 2010, 30, 5698–5709. 10.1128/MCB.00165-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller S.; Abramson J.; Mannella C.; Wagner G.; Zeth K. The 3D Structures of VDAC Represent a Native Conformation. Trends Biochem. Sci. 2010, 35, 514–521. 10.1016/j.tibs.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogg R. J.; Kingsley P.; Taylor J. WET, a T1- and B1-Insensitive Water-Suppression Method for in Vivo Localized 1H NMR Spectroscopy. J. Magn. Reson., Ser. B 1994, 104, 1–10. 10.1006/jmrb.1994.1048. [DOI] [PubMed] [Google Scholar]
- Smallcombe S.; Patt S. L.; Keifer P. WET Solvent Suppression and Its Applications to LC NMR and High-Resolution NMR Spectroscopy. J. Magn. Reson., Ser. A 1995, 117, 295–303. 10.1006/jmra.1995.0759. [DOI] [Google Scholar]
- Dynamo: the NMR Molecular Dynamics and Analysis System, 2012. http://spin.niddk.nih.gov/NMRPipe/dynamo.
- Berjanskii M. V.; Wishart D. S. A Simple Method to Predict Protein Flexibility Using Secondary Chemical Shifts. J. Am. Chem. Soc. 2005, 127, 14970–14971. 10.1021/ja054842f. [DOI] [PubMed] [Google Scholar]
- Berjanskii M. V.; Wishart D. S. Application of the Random Coil Index to Studying Protein Flexibility. J. Biomol. NMR 2008, 40, 31–48. 10.1007/s10858-007-9208-0. [DOI] [PubMed] [Google Scholar]
- Van Der Spoel D.; Lindahl E.; Hess B.; Groenhof G.; Mark A. E.; Berendsen H. J. C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- Hess B.; Kutzner C.; van der Spoel D.; Lindahl E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- Oostenbrink C.; Villa A.; Mark A. E.; van Gunsteren W. F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. 10.1002/jcc.20090. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P. M.; Gunsteren W. F. van; Hermans J.. Interaction Models for Water in Relation to Protein Hydration. In Intermolecular Forces; Pullman B., Ed.; The Jerusalem Symposia on Quantum Chemistry and Biochemistry; Springer: Netherlands, 1981; pp 331–342. [Google Scholar]
- Tieleman D. P.; Van der Spoel D.; Berendsen H. J. C. Molecular Dynamics Simulations of Dodecylphosphocholine Micelles at Three Different Aggregate Sizes: Micellar Structure and Chain Relaxation. J. Phys. Chem. B 2000, 104, 6380–6388. 10.1021/jp001268f. [DOI] [Google Scholar]
- Bussi G.; Donadio D.; Parrinello M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Berendsen H. J. C.; Postma J. P.; van Gunsteren W. F.; DiNola A.; Haak J. R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. 10.1063/1.448118. [DOI] [Google Scholar]
- Nosé S.; Klein M. L. Constant Pressure Molecular Dynamics for Molecular Systems. Mol. Phys. 1983, 50, 1055–1076. 10.1080/00268978300102851. [DOI] [Google Scholar]
- Parrinello M.; Rahman A. Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys. 1981, 52, 7182. 10.1063/1.328693. [DOI] [Google Scholar]
- Maget-Dana R. The Monolayer Technique: A Potent Tool for Studying the Interfacial Properties of Antimicrobial and Membrane-Lytic Peptides and Their Interactions with Lipid Membranes. Biochim. Biophys. Acta, Biomembr. 1999, 1462, 109–140. 10.1016/S0005-2736(99)00203-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.