

Abstract
Background:
Matched related donor hematopoietic stem cell transplant (HSCT) is a successful treatment for chronic granulomatous disease (CGD), but the safety and efficacy of HSCT from unrelated donors is less certain.
Objective:
We evaluated the outcomes and overall survival in patients with CGD after HSCT.
Methods:
We report the outcome for eleven children undergoing HSCT from matched related donor (MRD) (n=4) or an HLA matched unrelated donor (MUD) (n=7); nine were males and the median age was 3.8 years (range: 1–13). We treated both X-linked (n=9) and autosomal recessive (n=2) disease. Nine children had serious clinical infections before transplant. The conditioning regimens contained busulfan, cyclophosphamide, cytarabine, or fludarabine according to the donor used. All patients received alemtuzumab (anti-CD52 antibody). Additional graft vs host disease (GvHD) prophylaxis included cyclosporine and methotrexate (MTX) for MUD recipients and cyclosporine and prednisone for MRD recipients.
Results:
Neutrophil recovery took a median of 16 days (range, 12–40 days) and 18 days (range, 13–24 days) for MRD and MUD recipients respectively. Full donor neutrophil engraftment occurred in 9 patients, while 2 developed stable mixed chimerism; all patients had sustained correction of neutrophil oxidative burst (NOB) defect. Four patients developed grade I skin acute GVHD responding to topical treatment. No patient developed grade II-IV acute GvHD or chronic GvHD. All patients are alive between 1 to 8 years post HSCT.
Conclusion:
We conclude that for CGD, equivalent outcomes can be obtained using MRD or MUD stem cells and that HSCT should be considered an early treatment option.
Keywords: chronic granulomatous disease, primary immunodeficiencies, bone marrow transplant, graft vs. host disease
Chronic granulomatous disease (CGD) is an inherited immunodeficiency estimated to occur in one in 250,000 individuals.1 The disease is caused by mutations in any of the genes that encode the proteins of the phagocytic NADPH oxidase enzyme complex (gp91phox, p47phox, p67 phox p22, phox, and p40 phox).2 The disease is X-linked in 65% of affected individuals (gp91phox ) and autosomal recessive in others. Defects in this enzyme complex render neutrophils incapable of phagocytic microbial killing, leading to severe and recurrent infections. Patients with CGD have an impaired quality of life with frequent hospitalization, recurrent diarrhea, infections and inflammatory organ damage.3 Furthermore, established infections (fungal and bacterial organisms including Staphylococcal aureus, Burkholderia cepacia and Aspergillus fumigatus ) are difficult to eradicate and remain a significant cause of mortality. In a large European study of over400 CGD patients followed over 50 years, the mean age at death for X-linked CGD patients was 38 years.1 Other reports suggest a life expectancy of 25–30 years for X-linked CGD patients.4 The annual rate of death due to CGD in the US is 2–5% and only 50% of the patients will survive to 30 years of age.5–6 The standard of care for CGD includes infection prophylaxis with antibiotics, antifungal agents and γ-interferon(IFN).7–9 Gallin et al. have shown that Itraconazole prophylaxis therapy has been widely used and proved to be safe and effective in children and adults with CGD to prevent fungal infections.10 Despite these measures, morbidity remains significant in CGD patients Patients may develop drug associated toxicity and suboptimal compliance, especially Patients may develop drug associated toxicity and suboptimal compliance, especially among adolescents and young adults, compromising the efficacy of prophylaxis measures For these reasons there is need for better and definitive therapies
The optimal treatment for most patients with severe primary immunodeficiencies (PIDs) is hematopoietic stem cell transplantation (HSCT) from an HLA-matched related donor (MRD).11 Unfortunately, such donors are available for only a minority of patients. Matched unrelated donor (MUD) HSCT has been successfully used for other PIDs and phagocytic disorders (including leukocyte adhesion defect), with overall survival of about 80%. 12–14 Unfortunately, these studies have also shown a high incidence of graft versus host disease (GvHD). 12–14
Since most children with CGD lack a related donor, Soncini et al.15 described the European experience in a 10 patient CGD cohort, who received stem cells from an HLA-matched unrelated donor. They reported an overall survival of 90% with a 30% incidence of acute GvHD grade II, and 1 patient developing chronic GvHD. In this cohort, one patient developed graft failure and required a second transplant.15 Recent data from the European consortium (SCETIDE) described a total of 41 CGD patients transplanted with an overall survival of 81% at 5 years with the deaths occurring early in the first 6 month post transplant, verbal communication kindly given by Paul Landais and Nizar Mahlaoui (September 8, 2011). An unpublished survey of North American centers treating patients with CGD found that 59 patients hadundergone allogeneic transplantation with 71% survival outcome2. We now report our single US center experience of treating 11 CGD patients with HLA-matched related and unrelated donors
METHODS
Patients
Eleven patients with CGD and history of significant morbidity with HLA-matched stem cell donors were eligible for HSCT according to a study approved by our Institutional Review Board (Table 1). CGD was confirmed by the absence of oxidase activity in neutrophils by dihydrorhodamine (DHR) oxidation analysis in all patients. Nine of these patients had X-linked CGD (by identification of a carrier mother and/or by gp91phox mutation analysis); one girl had autosomal recessive CGD (p67phox) and a mutation could not be identified for one girl (Table I). Likewise mutations could not be identified for three boys but are likely CYBB mutations as maternal oxidative burst studies suggested a carrier state for this mutation. Irrespective of the genetic mutations, all patients had very low stimulation indices at diagnosis, suggestive of high risk disease.2
Table 1.
Patient characteristics of 11 children with CGD
| Age at CGD diagnosis (dx) |
Age at HSCT |
Gender | Ethnicity 1 |
Genetics | Stimulation Index (SI) at dx |
Pre-HSCT Infections (isolation;age at diagnosis) |
Comorbidities | HSCT Type/ Risk |
Outcome Post-HSCT |
|
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 weeks | 30 mos | M | C |
X-linked gp91phox |
2 |
S. aureus Otitis (Culture; 2 yrs ) |
Molluscuma, URTI associated wheezinga |
MRD |
-Mixed chimerism |
| 2 | 14 months | 45 mos | M | C |
X-linked gp91phox |
2 |
Burkholderia cepacia Pneumonia (Lung bx; 11 mos ) |
Asthmaa,b, Pulmonary nodulesa,b |
MUD |
-Skin grade 1 aGvHD |
| 3 | 8 months | 41 mos | F | H |
AR gp67phox |
1 |
Serratia Marcescens Osteomyelitis (Bone aspirate; 8 mos) |
Iron deficiency anemiaa, Transaminitisa, Chronic lung cystsa,b |
MRD * |
-Adenovirus in plasma (resolved w/o intervention) |
| 4 | 36 months | 8.3 years | M | H |
X-linked gp91phox |
1 |
Aspergillus species Pneumonia (Lung biopsy; 5 years) |
G tube for poor feedinga, Chronic pulmonary nodulesa,b, Asthmaa,b |
MUD * |
-CMV reactivation (Rx meds) -Aspergillus Pneumonia |
| 5 | 6 months | 11 mos | M | H |
X-linked gp91phox |
1 |
S. aureus Abcess (Surgical wound I&D; 6 mos) |
Perirectal abcessesa, Chronic diarrheaa |
MUD |
-Skin grade 1 aGvHD |
| 6 | 6 weeks | 6.4 years | F | A |
AR (no molecular testing)2 |
3 |
B. cepacia Pneumonia (Lung biopsy; 5 yrs) Aspergillus niger Pneumonia (Lung biopsy; 5.5 yrs) |
Neonatal HIV exposurea, Chronic cystic lung diseasea,b |
MUD * |
-EBV reactivation (resolved w/o intervention) |
| 7 | 4.5 years | 5.9 years | M | C |
X-linked gp91phox |
1 |
B. cepacia Osteomyelitis, Bacteremia (Bone I&D; 4.5 yrs) |
Weaknessa, Voriconazole sensitivitya, Chronic lung diseasea,b |
MUD * |
-AIHA tx with steroids po (14 months post HSCT) |
| 8 | 2.5 months | 18 mos | M | H |
X-linked (no molecular testing)2 |
2 |
Candida albicans Abcess & Bacteremia (Surgical I&D; 1 yr) |
Lymphadenitisa, Perirectal abcessa, Transaminitisa |
MRD * |
-CMV reactivation (Meds RX) -Mixed Chimerism |
| 9 | 9 months | 7.4 years | M | C |
X-linked (no molecular testing)2 |
1 |
B. gladioli Osteomyeletis (Bone biopsy; 5.8yrs) |
Hearing lossa,b, Eosinophilic cystitisa, Drug induced lupusa |
MUD |
-Skin grade 1 aGvHD |
| 10 | 3 weeks | 50 mos | M | C |
X-linked gp91phox |
1 |
Serratia marcescens Liver Abcess (Liver biopsy; 2 weeks) Aspergillus fumigatus Pneumonia (Lung biopsy;1 mo) |
CGD colitisa, Transaminitisa, Asthmaa,b |
MRD * |
-Skin grade 1 aGvHD -Busulfan related seizures -Adenovirus of stool ( resolved w/o intervention) |
| 11 | 2.5 months | 13 years | M | C |
X-linked ( negative molecular testing, negative sequencing)2 |
1 |
Presumed Aspergillus Pneumonia (Lung biopsy with hyphae; 8 yrs ); S.Aureus Perirectal Abcess (Surgical I&D; 10 yrs) |
Perirectal abcessa, Recurrent lymph- adenitisa, Chronic pulmonary nodulesa,b |
MUD * |
-Hashimoto’s Thyroiditis (17 months post HSCT) |
1: C =Caucasian, H= Hispanic, A=African American
2: Pt #6 had no molecular testing and no family history of CGD. Pts # 8, 9, 11 all had mothers with NBT findings consistent with X-linked carrier status with 2 populations of granulocytes (normal and poor oxidative burst). Pt#9 with younger brother with demise at 1 year of age with B. cepacia and granulomas in liver/lungs. Pt#11 with negative CYBB/CYBA mutations and full sequencing did not identify a known CGD associated mutation.
Comorbidities pre HSCT
Comorbidities post HSCT
High risk patients (ongoing treatement/prophylaxis for known infections and/or significant pulmonary inflammation by imaging: ongoing granulomas)
URTI (Upper Respiratory Tract Infection)
AIHA (Autoimmune Hemolytic Anemia)
All patients had at least one invasive infection of lung, liver, lymph node, blood, gastrointestinal tract, or bone, requiring prolonged intravenous antimicrobial therapy (Table 1). Moreover, by the parameter of intractable infections or steroid dependent chronic granulomatous disease 16, 70% of our patients had high risk disease at the time of transplantation. Three of 11 patients had required mechanical ventilation for respiratory failure.The mean age at transplantation was 3.8 years with a range of 11 months to 13 years.
Transplantation
Four of 11 patients received MRD SCT from 6/6 HLA-identical siblings. Seven patients received a 10/10 HLA-genoidentical graft from an unrelated donor without clinical evidence of CGD. All related donors had normal oxidative burst activity and no evidence of the carrier state. γ-IFN was discontinued in all patients 7 to 10 days prior to HSCT
All patients received a busulfan-based myeloblative conditioning regimen combined with cyclophosphamide and cytarabine for MRD transplant recipients or fludarabine for MUD transplant recipients. Busulfan (Bu) was administered on days −9 to −6 before transplantation at a starting dose of 0.8–1mg/kg, and cyclophosphamide was administered at a dose of 45 mg/kg at days −3 and −2 for MRD and at a total dose of 50 mg/kg at days −5 to −2 for MUD recipients. Dosing of Bu was based upon actual weight unless actual weight exceeded ideal weight by 30%. For these patients, we calculated adjusted weight(ideal body weight plus 25%). Bu was administered intravenously every 6 hours for 16 doses. Blood samples were obtained with the first and ninth dose to modify the dose of Bu to an AUC of 900 – 1200 µmol/min/L. All patients received anticonvulsant therapy while receiving Bu. Cytarabine was administered at 2 g/m² for four doses on days −6 to −4 for MRD. Fludarabine was administered at 30 mg/m² at days −5 to −2 for MUD. All patients received alemtuzumab (anti-CD52) at 3mg (if <15kg), 5mg (if >15 kg but<30Kg) or 10mg (if >30kg) at days −5 to −2 to improve engraftment and decrease GvHD Additional GvHD prophylaxis consisted of cyclosporine A and prednisone in patients receiving a MRD graft and cyclosporine A and methotrexate in patients receiving a MUD graft. Bone marrow grafts had a median total nucleated cell dose of 6 × 108/kg, with a range of 5.0 × 107/kg – 1.5 × 1010/kg.
Chimerism was established either by fluorescent in situ hibridization for sex chromosome or by short tandem repeats for allele DNA sequence. The presence of oxidase-positive neutrophils was detected by flow cytometry with the use of DHR oxidation assay and reported as geometric mean fluorescence or stimulation index (SI). Following HSCT, recovery of B and T cells was measured by flow cytometric analyses as described by Fleisher and Oliveira.17 Lymphoproliferative responses were measured using isolated mononuclear cells. These cells were cultured in micro well plates loaded with diluted mitogen or specific antigens. The phytohemaglutinin (PHA) and Concanavalin A (ConA) response were measured by ³H-thymidine incorporation. A PHA or ConA response was considered normal if ≥75,000 cpm ³H-thymidine incorporation. Following transplantation, we evaluated specific antigen responses to tetanus and candida in all patients. Specific antigen results were considered normal if the stimulation index was 2 or greater
Statistical analysis
The times to neutrophil engraftment and platelet engraftment were defined as the time from the transplantation to the time when the neutophil count reached 500cells/µl for three consecutive days and the time when the unsupported platelet count reached 20,000 cell/mm³, respectively. The cumulative probability for the times to neutrophil engraftment and platelet engraftment were estimated and plotted by the Kaplan-Meier method. The median times to engraftment were compared between MRD and MUD by the Wilcoxon method. The immune reconstitution data (CD3, CD4 T cell or PHA responses) were repeated measurements and analyzed by nonlinear mixed-effects models with autoregressive correlation of log 1. The patterns of immune reconstitution data over times suggest a nonlinear logistic growth model.18 The estimated curves fit the data reasonably well as shown in Figure 4. The times for the curves to hit a fixed boundary were estimated by the delta method and the difference between MRD and MUD were compared by the Wald asymptotic test.
Figure 4. Immuno reconstituion.
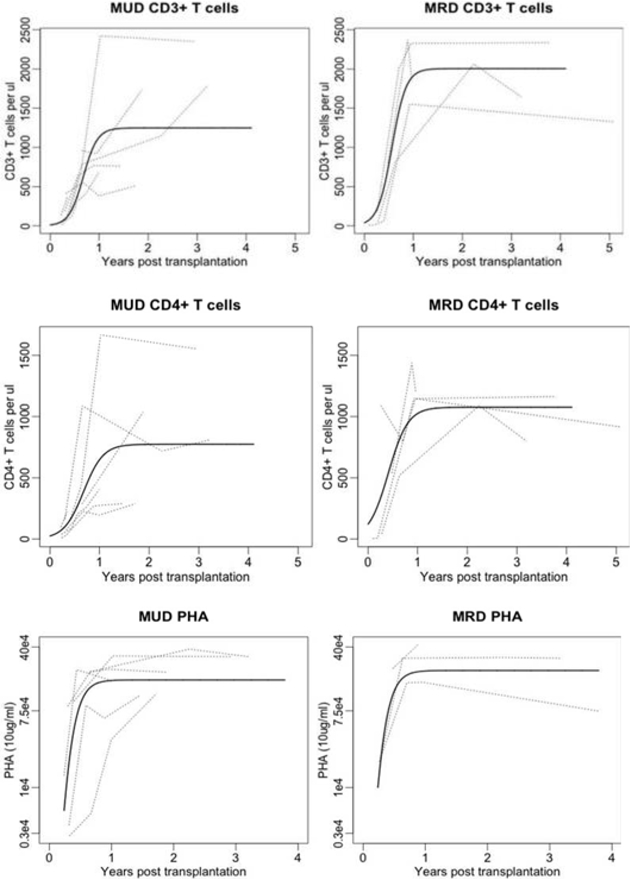
a. CD3 T cell and b. CD4 T cell absolute number recovery, and c. function measured by proliferative responses to mitogen(PHA 10 µg/mL) after HSCT.
RESULTS
Engraftment
A neutrophil count greater than 500cells/µl (Figure 1) was reached at a median of 18 days for the cohort with a median of 17 days (range, 13–24 days) and 18 days (range, 16–21 days) for MRD and MUD recipients, respectively (MRD vs. MUD, p=0.65). A platelet count greater than 20,000cells/mm³ (Figure 2) was reached at a median of 16 days for the cohort and at a median of 16 days (range, 14–22 days) and 21 days (range, 12–40 days) for MRD and MUD recipients, respectively (MRD vs. MUD, p=0.52). All patients achieved greater than 95% donor chimerism prior to day 100. Beyond day +100, donor chimerism for two MRD recipients declined, but stabilized at a mean of 70% at 22 and 59 months post HSCT. Donor-derived chimerism has remained stable in all patients with no further stem cell infusions required to improve engraftment, with a median follow up time of 4 years (range, 1 – 8 years).
Figure 1. Neutrophil engraftment.
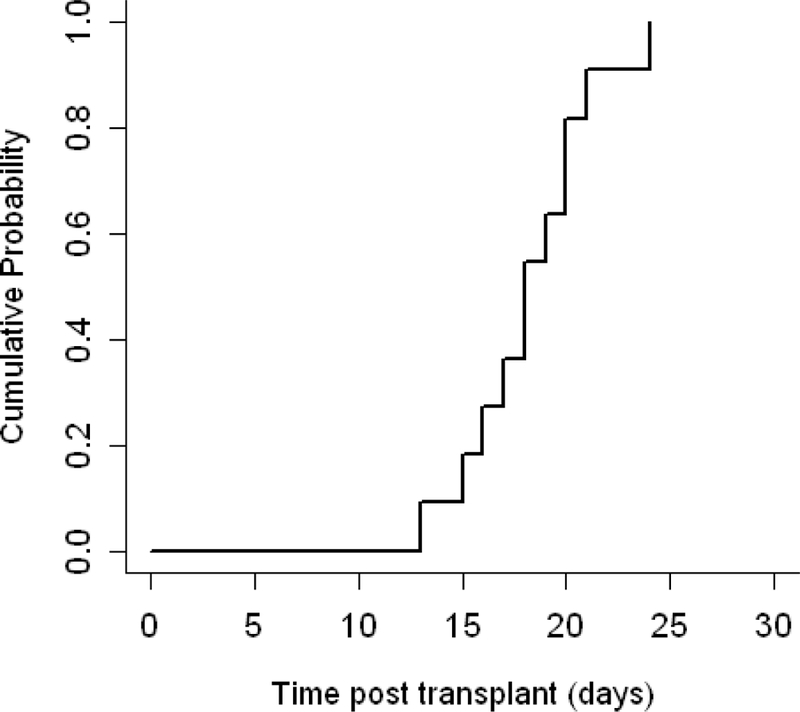
Cumulative incidence of neutrophil engraftment(defined as a neutrophil count greater than 500/ul) occurred at a median time of 18 days (range, 13–24).
Figure 2. Platelet engraftment.
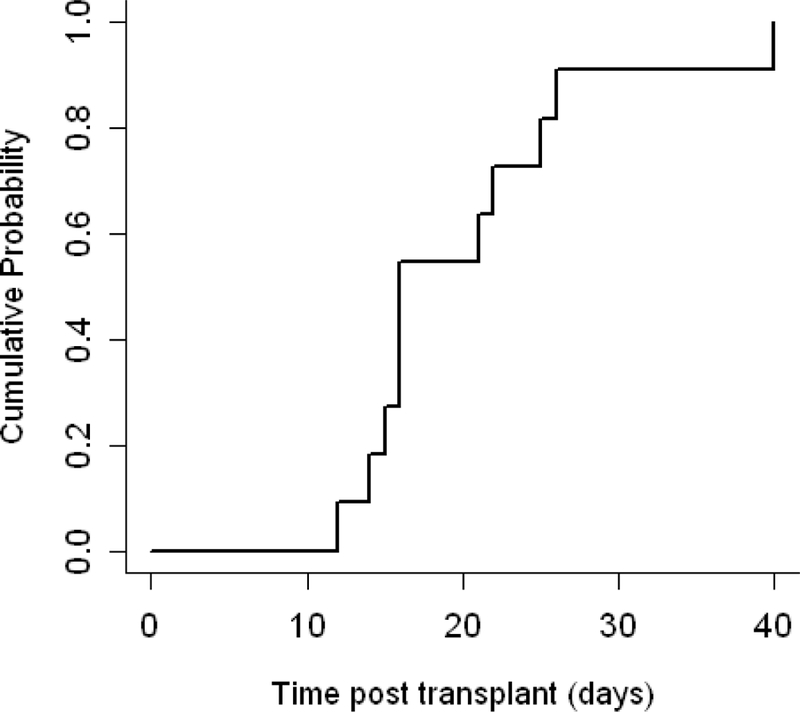
Cumulative incidence of platelet engraftment(defined as a platelet count greater than 20,000 per cubic millimeter) occurred at a median time of 16 days (range, 12–40).
Neutrophil oxidative burst activity post HSCT was assayed by DHR and was normal by day 100 for all patients. Figure 3 shows pre and post HSCT mean DHR stimulation indices (SI) for our cohort. All patients including those with mixed chimerism status post transplant had sustained normal DHR activity.
Figure 3. Neutrophil oxidative burst by DHR.
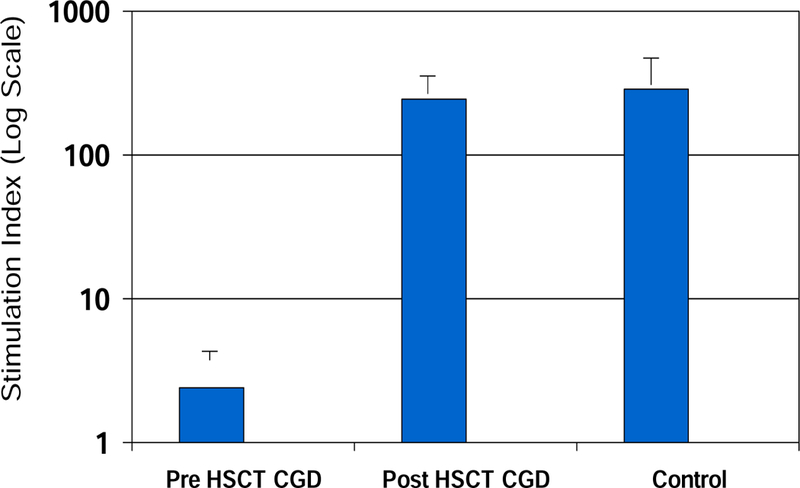
Pre-HSCT mean stimulation indices (SI) averaged less than 2 prior to HSCT and corrected to and were sustained normal following HSCT for all patients.
Acute GvHD grade I of the skin developed in 4 of 11 patients. Three of these patients received an unrelated product but all of them responded to topical steroids. No patient developed grade II or greater acute GvHD or chronic GvHD.
Clinical outcomes and adverse events
The conditioning regimen was well tolerated apart from one patient, who developed seizures during busulfan administration. The median busulfan AUC after the first dose for the group was 934 µmol/min/L. Four patients needed dose adjustments (2 in the sibling donor and 2 in the unrelated donor group), in 3 of whom the dose was increased by 30%. A requirement for dose adjustment had no discernible effects on engraftment kinetics, GvHD or complications after transplant
One patient had a relapse of Aspergillus pneumonia prior to engraftment (patient #4, Table 1), developing bilateral pulmonary infiltrates, high fevers and impaired respiratory function. Treatment with amphotericin, echinocandin, and imidazole was combined with granulocyte infusions and additional donor CD34-selected cells, leading to complete and sustained pulmonary recovery. No other patient had serious infection or other grade 4 toxicities.
Immune reconstitution
The estimated time for CD3+ T cells reaching 300/μl was 114 days (95% CI, [44.6, 183.4]) for MRD, and 185.9 days (95%CI, [123.4, 248.4]) for MUD (p = [NS] 0.12). The estimated time for CD3+ T cells reaching 500/μl was 147.5 days (95%CI, [89.0, 206.0]) for MRD, and 225.3 days (95%CI, [168.9, 281.8]) for MUD (p = [NS] 0.13). The estimated time for CD4+ T cells reaching 300/μl was 81.2 days (95% CI, [−17.1, 179.4]) for MRD, and 215.1 days (95%CI, [123.4, 248.4]) for MUD (p= [NS] 0.09). The estimated time for CD4+ T cells reaching 500/μl is 139.5 days (95%CI, [50.9, 228.1]) for MRD, and 291.3 days (95%, [194.8, 387.9]) for MUD (p = [NS] 0.12). The data and the estimated curves are shown in Figure 4 a–b. The function of these T cells was measured using the in vitro proliferation responses to mitogens (PHA) and specific antigens(tetanus). Responses to log counts per minute (cpm) of PHA at a concentration of 10ug/ml are shown in Figure 4c. The estimated time to normalization was 148.2 days (95%CI, [85.8, 210.5]) for MRD, and 169.7 days (95%CI, [108.9, 230.5]) for MUD (p =[NS] 0.96). Confidence intervals to indicate the variability of the T cell recovery and function are wide likely secondary to small sample size. Hence we cannot correlate cell numbers with strength of immune responses. Specific antigen responses following exposure to tetanus and candida and mitogen responses to ConA in addition to other immune parameters are shown in Table II and show normalization for the majority of patients at last follow up.
Table. 2. Immune Parameters at last follow up.
| Pt | Age at HSCT |
Age at F/U |
ANC | CD13 | CD4 (abs#) |
CD19 (abs#) |
PHA + 10ug/ml |
ConA + 10ug/ml |
Candida (SI) |
Tetanus (SI) |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 30 mo | 5 yrs | 2262 | 1.647 (1.4–3.7) |
709 (0.7–2.2) |
538 (0.4–1.4) |
297328 | 267247 | 4516 | 30944 |
| 2 | 45 mo | 8 yrs | 1367 | 1.789 (1.2–2.6) |
810 (0.65–1.5) |
0.635 (0.2–.86) |
304466 | 313285 | 2586 | 43710 |
| 3 | 41 mo | 6 yrs | 2690 | 1.874 (1.4–3.7) |
1.192 (0.7–2.2) |
2.121 (0.4–1.4) |
426433 | 324072 | N/A | 763 |
| 4 | 8.3 yrs | 10 yrs | 2650 | 1.034 (1.2–2.6) |
0.423 (0.65–1.5) |
0.103 (0.4–1.4) |
152145 | N/A | N/A | N/A |
| 5 | 11 mo | 3 yrs | 4080 | 2.531 (1.4–3.7) |
1.554 (0.7–2.2) |
0.446 (0.4–1.4) |
309721 | 113562 | 5931 | 749 |
| 6 | 6.4 yrs | 9 yrs | 10301 | 1.801 (1.4–3.7) |
0.959 (0.65–1.5) |
0.773 (0.4–1.4) |
166508 | 141519 | 11093 | 61579 |
| 7 | 5.9 yrs | 6 yrs | 1820 | 0.367** (1.4–3.7) |
0.222** (0.7–2.2) |
0.403** (0.4–1.4) |
306364 | 242380 | 770 | 118 |
| 8 | 18 mo | 5 yrs | 2690 | 2.336 (1.4–3.7) |
1.164 (0.7–2.2) |
1.267 (0.4–1.4) |
74320 | 80528 | 281 | 304 |
| 9 | 7.4 yrs | 10 yrs | 3040 | 0.507 (1.2–2.6) |
0.288 (0.65–1.5) |
0.879 (0.4–1.4) |
118145 | 50525 | 247 | 14350 |
| 10 | 50 mo | 9 yrs | 2770 | 1.326 (1.2–2.6) |
0.916 (0.65–1.5) |
0.781 (0.4–1.4) |
253455 | 83238 | 2342 | 24138 |
| 11 | 13 yrs | 15 yrs | 2429 | 1054 (1–2.2) |
0.645 (0.5–1.3) |
0.679 (0.1–.6) |
277627 | 178714 | N/A | N/A |
( ) Normal values are presented as 10th and 90th percentiles. Subset counts (number of cell per micro liter x 10–3). 19
studies performed while on steroids for AIHA (Autoimmune hemolytic anemia).
Proliferation responses expressed as log of counts per minute (cpm)
Survival, activity level and educational status
All patients are well with a mean follow up of 4 years (range, 1−8 years). Quality of life has improved for all patients, reaching normal activity without special care (Lansky score of 100%). All but one patient currently attends school (9 at elementary school, 1 at high school). One patient is receiving home schooling secondary to family social needs
DISCUSSION
The long-term survival of patients with CGD remains poor in spite of improvements in conventional therapies. Although gene therapy holds promise as a curative option, success has been limited with CGD patients losing gene-corrected cells within 6 months of treatment or developing myelodysplastic syndrome and acute myelogenous leukemia.20,22 Hence, HSCT currently remains the only curative treatment. To date HSCT has largely been recommended only to CGD patients with an HLA matched related donor who also had more than one life-threatening infection in the past or intractable infections, severe granulomatous disease with organ dysfunction or steroid-dependence, non-availability of specialist care, or non-compliance with antibiotic prophylaxis.16 We now report 100% survival for 11 patients undergoing HSCT, for whom 7 received grafts from matched unrelated donors. Stable engraftment with full donor chimerism was observed in nine of eleven patients with a median follow up time of 2.5 years (range; 1–9). There was no acute GvHD beyond grade I or chronic GvHD or graft failure and in all but one patient with recurrent aspergillosis, the HSCT was uneventful.
Seger et al. previously reported 85% overall survival in 27 CGD patients receiving a HSCT mainly consisting of genotypically identical related grafts (n=25)22, while Soncini et al. reported survival in 9 of 10 European patients with CGD following MUD HSCT with myeloablative conditioning and standard aGvHD prophylaxis mainly consiting of cyclosporine and methotrexate with an incidence of grade II aGvHD of approximated 30%.15
Our MUD conditioning regimen of busulfan, cyclophosphamide, fludarabine and alemtuzumab is a regimen that has been associated with high engraftment rates and a low risk of significant GvHD when no matched sibling is available and to be well tolerated when used as conditioning for patients with primary immunodeficiencies. 23 Incorporation of cytarabine as part of triple chemotherapy conditioning for primary immunodeficiencies in MRD has been used by our group for more than a decade. When combined with lower doses of cyclophosphamide and alemtuzumab (a humanized monoclonal antibody that eliminates cells expressing CD52, including T and B lymphocytes, eosinophils, monocytes, natural killer cells, and some dendritic cells), these agents have been well tolerated and produced a high level of engraftment and low GvHD in patients transplanted for non-malignant diseases. Because alemtuzumab given prior to transplantation remains at lytic levels in peripheral blood for over 21 days after administration, it produces depletion of both recipient and donor immune system cells, favoring engraftment and a low rate of GvHD, respectively.24,25 Such immune depletion can be associated with a high level of post transplant infection.25–27 In this series, we monitored viral reactivation routinely, and observed the expected rate. Reactivation was controlled with medical treatment where feasible (Table I) and no viral disease occurred
In this patient population our main goal is engrafment so alemtuzumab was not adjusted according to graft type to reduce the incidence of graft failure. To date we have not had any graft failure or graft rejection in our CGD population. We adjust alemtuzumab dosage according to graft type when transplant is used to treat patients with leukemias, since graft failure is less common in these heavily pre-treated patients We observed a faster immune recovery for CD4+ T cells compared to earlier reports of recovery after using alemtuzumab conditioning,24,25 perhaps because of lower dose used in our trial and younger age of our patient population. Although there was a trend for a more prompt CD3+ and CD4+ T cell recovery after transplant in the MRD group compared to the MUD group, this trend did not reach statistical significance. There was similar T cell function in both transplant groups measured by responses to PHA. Consistent with these observations, we observed no discernible increase in the incidence of infections in the MUD versus the MRD recipients. Immunoglobulin supplementation was suspended by 12 months post HSCT, and specific antibody response to vaccine challenge was documented for most patients. Although virus-specific cytotoxic T lymphocytes derived from the stem cell donor or a third party28, 29 may be of benefit for post transplant infections in intensely lymphodepleted patients, these cells were not required or used in this patient cohort
There is no reason to believe that the excellent outcome we observed was attributalble to inadvertent selection bias in the patients in terms of clinical severity or mutational status. The majority of patients were X-linked gp91phox with baseline DHR of less than 2 with high incidence of severe intractable infections and granulomas.
Our series reports the use of HLA-matched unrelated donors as an alternative stem cells ource, but umbilical cord blood or haploidentical donor sources may also be suited for individuals lacking a fully HLA-matched donor, and addition of alemtuzumab conditioning regimen may be beneficial in this setting as well. 24 There has been a debate among clinical immunologists about whether to treat CGD patients with antimicrobial agents and preserve their lives or whether to pursue a more definitive mode of therapy with HCST. Previous concerns over the failure of HSCT for CGD other than from HLA-identical siblings have placed definitive therapy on hold, which encourage physicians to continue to treat the numerous and serious infection with appropriate antibiotics antifungal agents, and antiviral drugs. But these prolonged treatments are frequently insufficient to prevent infection of lymph nodes, lungs, and liver and often demand surgical removal of diseased tissue. In addition to the physical problems of such patients, CGD adolescents and young adults may decline strict adherence to drug therapies and often express giving up on life because of their inability to lead normal lives free of often express giving up on life because of their inability to lead normal lives free of frequent infections, clinic visits, and hospital admissions. The excellent outcome with low complication rates observed in our patient cohort supports the argument for early SCT in young CGD patients. While our report is of a single center retrospective study with a small number of patients and will clearly require confirmation in multicenter prospective studies, it is now our practice to consider HSCT (both MRD and MUD) after the first life threatening infection and prior to onset of end organ damage, allowing permanent cures of CGD to become more commonplace
Clinical Implications:
Matched unrelated donors have been proven to be as good as matched related donors in HSCT, broadening the choice of definitive therapy for all patients with CGD.
Capsule summary:
Matched unrelated donor and matched related donor HSCT have comparable outcomes in patients with CGD and should be considered prior to the onset of end organ damage.
Acknowledgements
This research was supported by grants from the National Institutes of Health: Primary Immune Deficiency Treatment Consortium (AI082979)
We thank the supporting faculty of the Allergy and Immunology Section and the Hematology and Oncology Section, Department of Pediatrics, Baylor College of Medicine. Also, we thank the supporting personnel of the Stem Cell Transplant Unit at Texas Children’s Hospital. Finally, we thank the courageous families who entrusted the care of their children to us in a search for a better life for their children
We also thank: Bobby Gaspar, Luigi Notarangelo, Paul Landais, Nizar Mahlaoui, Alain Fischer and Reinhard Seger for their verbal communications regarding the most current European data of outcomes of CGD patients after stem cell transplantation
Abbreviations:
- HSCT
Hematopoietic Stem Cell Transplant
- MRD
Matched related donor
- MUD
Matched unrelated donor
- GvHD
Acute graft vs host disease
- CGD
Chronic granulomatous disease
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors declare no conflict of interest. No honorarium, grant or other form of payment was given to authors to produce the manuscript.
References:
- 1.Merlijn van den Berg J, van Doppen E, Ahlin A, Belohradsky B, Bernatowska E, Corbeel L, et al. Chronic Granulomatous Disease: The European Experience PloS ONE 2009; 4:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kang EM, Marciano BE, DeRavin S, Zarember K, Holland S, Malech HL. Chronic granulomatous disease: Overview and hematopoietic stem cell transplantation. J Allergy Clin Immunol 2011;127:1319–26392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liese J, Kloos S, Jendrossek V, Petropoulou T, Wintergerst U, Notheis G, et al. Long-term follow-up and outcome of 39 patients with chronic granulomatous disease. J Pediatr 2000;137:687–93. [DOI] [PubMed] [Google Scholar]
- 4.Martire B, Rondelli R, Soresina A, Pignata C, Broccoletti T et al. Clinical features, long-term follow up and outcome of a large cohort of patients with Chronic Granulomatous Disease: An Italian multicenter study. Clin Immunol 2008;126:155–164. [DOI] [PubMed] [Google Scholar]
- 5.Jones R, McGrogan P, Flood T, Gennery AR, Morton L, Thrasher A, et al. 400 Special article: Chronic granulomatous disease in the United Kingdom and . Ireland: a comprehensive national patient-based registry. Clin Exp Immunol 2008; 152: 211–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winkesltein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, Gallin J, et al. Chronic granulomatous disease: report on a national registry of 368 patients Medicine 2000; 79:155–69. [DOI] [PubMed] [Google Scholar]
- 7.The International Chronic Granulomatous Disaease Cooperative Study Group. A controlled trial of interferon gamma to prevent infection in chronic granulomatous disease. N Engl J Med 1991; 324:509–16. [DOI] [PubMed] [Google Scholar]
- 8.Margolis DM, Melnick DA, Alling DW, Gallin JI. Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous 411 disease. J Infect Dis 1990; 162: 723–6. [DOI] [PubMed] [Google Scholar]
- 9.Weening RS, Kabel P, Pijman P, Roos D. Continuous therapy with sulfamethoxasole-trimethoprim in patients with chronic granulomatous disease. J Pediatr 1982; 103: 127–30. [DOI] [PubMed] [Google Scholar]
- 10.Gallin J, Alling D, Malech H, Wesley R, Koziol D, Marciano B, et al. Itraconazole to Prevent Fungal Infections in Chronic Granulomatous Disease. N Engl J Med 2003; 348: 2416–2422). [DOI] [PubMed] [Google Scholar]
- 11.Fisher A, Landais P, Friedrich W, Goldman MD, Morgan MRP, et al. European experience of bone marrow transplantation for severe combined immunodeficiency. Lancet 1990; 336:850–854. [DOI] [PubMed] [Google Scholar]
- 12.Qasim W, Cavazzana-Calvo M, Davies G, Davis J, Duval M, Eames G, et al. Allogeneic Hematopoietic Stem Cell Transplantation for Leukocyte Adhesion Deficiency. Pediatrics 2008; 123(3):836–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gennery A, Slatter M, Grandin L, Taupin P, Cant A, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: Entering a new century, do we do better? J Allergy Clin Immunol 2010; 126(3):602–610e11. [DOI] [PubMed] [Google Scholar]
- 14.Grunebaum E, Mazzolari E, Porta F, Dallera D, Atkinson A, Reid B, et al. Bone Marrow Transplantation for Severe Combined immune Deficiency. JAMA 2006; 295:508–518. [DOI] [PubMed] [Google Scholar]
- 15.Soncini E, Slatter M, Jones LB, Hughes S, Flood T, Barge D, et al. Unrelated donor and HLA-identical sibling haemotopoietic stem cell transplantation cure chronic granulomatous disease with good long-term outcome and growth. Br J Haematol 2009; 145; 73–83. [DOI] [PubMed] [Google Scholar]
- 16.Seger RA. Modern management of chronic granulomatous disease. Br J Haematol 2008; 140: 225–66. [DOI] [PubMed] [Google Scholar]
- 17.Fleisher TA, Oliveira JB. Functional and molecular evaluation of lymphocytes. J Allergy Clin Immunol 2004; 114: 227–234. [DOI] [PubMed] [Google Scholar]
- 18.Pinheiro José and Bates Douglas. Mixed-Effects Models in S and S-PLUS Springer, 2009. [Google Scholar]
- 19.Shearer WT, Rosenblatt HM, Gelman R, Oyomopito R, Plaeger S, Stiehm R, et al. Lymphocyte subsets in healthy children from birth through 18 years of ageThe Pediatric AIDS Clinical Trial Group P1009 study. J Allergy Clin Immuno 2003: 112: 973–80. [DOI] [PubMed] [Google Scholar]
- 20.Malech HL, Maples PB, Whiting-Theobald N, Linton GF, et al. Prolonged production of NADPH oxidase-corrected granulocytes after gene therapy of chronic granulomatous disease. Proc Natl Acad Sci USA 1997; 94;12133–12138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stein S, Ott M, Schultze-Strasser S, Jauch A, Burwinkle B, Kinner A, et al. Genomic Instability and myelodysplasia with monosomy 7 consequent to EVI 1 activation after gene therapy for chronic granulomatous disease. Nat Med 2010;16;198–204. [DOI] [PubMed] [Google Scholar]
- 22.Seger R, Gungor T, Belohradsky B, Blanche S, Bordigoni P, Blance S, et al. an unmodified hemopoietic allograft: a survey of the European experience, 1985–2000. Blood 2002; 100; 4344–4350. [DOI] [PubMed] [Google Scholar]
- 23.Shuetz C, Hoenig M, Gatz S, Specth F, Benninghoff U, Shulz A et al. Hematopoietic stem cell transplantation from matched unrealted donors in chronic granulomatous disease. Immunol Res 2009; 44; 35–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hale G, Jacobs P, Wood L, Fibbe WE, Barge R, Novitzky N, et al. CD 52 Antibodies for prevention of graft vs. host disease and graft rejection following transplantation of allogeneic peripheral blood stem cells. Bone Marrow Transplant 2000; 26:69–76. [DOI] [PubMed] [Google Scholar]
- 25.Shah AJ, Kapoor N, Crooks G, Weinsberg K, Hisham A, Killen R, et al. The effects of Campath 1H upon Graft-versus-Host disease, infection, relapse, and immune reconstitution in recipients of pediatric unrelated transplants. Biol Blood Marrow Transplant 2007; 13:584–593. [DOI] [PubMed] [Google Scholar]
- 26.Chakrabarti S, Mackinnon S, Chopra R, Kottaridis D, Peggs K, O’Gorman P, et al. High incidence of cytomegalovirus infection after nonmyeloablative stem cell transplantation: potential role of Campath-1H in delaying immune reconstitution. Blood 2002; 99: 4357–4363. [DOI] [PubMed] [Google Scholar]
- 27.Kennedy-Nasser A, Leung K, Martinez C, Brenner M, Heslop H, Krance RA, et al. Haploidentical stem cell transplants in children with acute lymphoblastic leukemia in first and second remission. Biol Blood Marrow Transplant 2011:17; S310–311. [DOI] [PubMed] [Google Scholar]
- 28.Heslop HE, Slobod KS, Pule MA, Hale G, Rousseau A, Smith C, et al. Long-term outcome of EBV-specific T-cell infusions to prevent or treat EBV-related lymphoproliferative disease in transplant recipients. Blood 2010;115:925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leen AM, Myers GD, Sili U, Huls H, Weiss H, Leung KS, et al. Monoculture-derived T lymphocytes specific for multiple viruses expand and produce clinically relevant effects in immunocompromised individuals. Nat Med 2006;12:1160–1166. [DOI] [PubMed] [Google Scholar]