

Apical infection of endocervical cells by Neisseria gonorrhoeae drives basolateral-to-apical neutrophil migration. Neutrophil transepithelial migration requires bacterial–endocervical cell contact. Two eicosanoid-producing pathways coordinate neutrophil migration: epithelial 12R-lipoxygenase (LOX) and eLOX3 hepoxilin synthase, and neutrophil 5-LOX–dependent production of leukotriene B4.
Keywords: Neisseria gonorrhoeae, neutrophil, inflammation, eicosanoid, migration
Abstract
Background
Infection with Neisseria gonorrhoeae (GC) is characterized by robust neutrophil influx that is insufficient to clear the bacteria. Sustained neutrophilic inflammation contributes to serious clinical sequelae that particularly affect women, including pelvic inflammatory disease and infertility.
Methods
We established a 3-component system using GC, End1 polarized human endocervical cells, and primary human neutrophils to investigate neutrophil transepithelial migration following infection.
Results
Neutrophil migration across endocervical monolayers increased with the infectious dose and required GC-epithelial cell contact. Epithelial protein kinase C, cytosolic phospholipase A2, 12R-lipoxygenase (LOX), and eLOX3 hepoxilin synthase were required for neutrophil transmigration to GC, and migration was abrogated by blocking the MRP2 efflux pump and by adding recombinant soluble epoxide hydrolase. These results are all consistent with epithelial cell production of the neutrophil chemoattractant hepoxilin A3 (HXA3). Neutrophil transmigration was also accompanied by increasing apical concentrations of leukotriene B4 (LTB4). Neutrophil 5-lipoxygenase and active BLT1 receptor were required for apical LTB4 and neutrophil migration.
Conclusions
Our data support a model in which GC-endocervical cell contact infection stimulates HXA3 production, driving neutrophil migration that is amplified by neutrophil-derived LTB4. Therapeutic targeting of these pathways could limit inflammation and deleterious clinical sequelae in women with gonorrhea.
Gonorrhea is caused by the human-specific pathogen Neisseria gonorrhoeae (GC) and is an important sexually transmitted infection worldwide, with 78 million cases estimated per year [1]. Lack of a protective immune response, emergence of drug-resistant strains, and lack of a vaccine have prompted the classification of GC as an urgent threat [1]. The hallmark of GC infection is influx of polymorphonuclear leukocytes (neutrophils, or PMNs) [2]. GC evades a protective adaptive immune response and instead skews the immune response toward neutrophilic inflammation [3]. While male urethritis commonly presents with symptoms, up to 80% of female cervical infections are asymptomatic [4]. As a result, clinical complications are most common and serious in women. Cervicitis still occurs in women with asymptomatic infection and, despite neutrophil recruitment, GC can evade neutrophil killing and is not cleared [2, 5]. Sustained neutrophil influx that does not clear infection contributes to severe clinical sequelae including pelvic inflammatory disease, tubal scarring, ectopic pregnancy, and infertility. Although gonococcal infection and resulting neutrophilic inflammation cause significant morbidity in women, to date the mechanisms underlying GC-cervical cell interactions that stimulate neutrophil influx are poorly understood.
During GC infection, mucosal epithelial and resident immune cells secrete cytokines including interleukin 8 (CXCL8; IL-8) that recruit neutrophils from the bloodstream into infected tissue [5]. These cytokines may not be sufficient for the terminal step of neutrophil migration across the epithelium into the apical/luminal compartment, termed neutrophil transmigration [6]. Instead, mucosal epithelial cells release additional chemotactic factors that drive transmigration [7, 8]. Recent work has identified an important role for eicosanoids during neutrophil migration to mucosal bacterial pathogens [6, 8–21]. Eicosanoids are arachidonic acid (AA)–derived bioactive lipids [22]. The rate-limiting step in eicosanoid generation is liberation of AA by phospholipase A2 (PLA2) [22]. AA can then be oxidized by lipoxygenases (LOXs) or cyclooxygenases (COXs) to generate eicosanoids, including hepoxilins, leukotrienes, and prostaglandins [22]. Of these, hepoxilin A3 (HXA3) and leukotriene B4 (LTB4) are particularly potent neutrophil chemoattractants that coordinate neutrophil migration to sites of infection and inflammation [19, 23, 24]. HXA3 is produced by different 12-lipoxygenase enzymes that are cell-type specific [25]. Neutrophil chemotactic activity of HXA3 requires its epoxide ring, which is rapidly degraded by an acidic environment and/or soluble epoxide hydrolase (sEH) [26, 27]. LTB4 is produced by sequential activity of 5-lipoxygenase (5-LOX) and LTA4-hydrolase [28]. LTB4 is primarily produced by myeloid cells and amplifies neutrophil influx in a number of inflammatory disease processes [23, 24]. Recent work revealed an interplay between bacterial promotion of eicosanoid production and eicosanoid regulation of innate immune responses in the context of host–pathogen interactions [21, 23].
Neutrophilic inflammation is associated with mucosal infection by pathogenic bacteria including Salmonella, Shigella flexneri, enteroaggregative Escherichia coli, Streptococcus pneumoniae, and Pseudomonas aeruginosa, which use HXA3 and/or LTB4 to coordinate neutrophil transepithelial migration [8–10, 14–18, 20, 21]. Each of these pathogens stimulates eicosanoid production in a distinct way, suggesting that individual bacteria have evolved unique mechanisms to co-opt eicosanoid-driven neutrophilic inflammation. In contrast, the signals driving neutrophil influx to gonococcal infection are poorly understood. This is particularly problematic in the context of female infection, where unresolved infection and sustained neutrophilic inflammation contribute to severe clinical consequences. In this work we established a 3-component GC-endocervical cell–neutrophil model, and found evidence to support the hypothesis that eicosanoids are required for the coordination of neutrophil transepithelial migration in response to infection.
MATERIALS AND METHODS
Bacterial Strains and Growth Conditions
A piliated, OpaD-expressing derivative of GC strain FA1090 [29] was used for these studies unless otherwise indicated. Growth conditions are described in the Supplementary Methods.
Human Neutrophil Isolation
Venous blood was collected from healthy human donors who provided informed consent. All human subject research was conducted in accordance with a protocol approved by the University of Virginia Institutional Review Board for Health Sciences Research. The neutrophil preparation protocol is detailed in the Supplementary Methods. Replicate experiments used neutrophils from different donors on separate days.
Cell Culture
Human End1/E6E7 (End1) cells (ATCC CRL-2615) were maintained and polarized End1 monolayers established on inverted Corning Transwell inserts as described previously [30] and in the Supplementary Methods.
Microscopy
End1 monolayers were fixed and stained as described in the Supplementary Methods.
Neutrophil Transmigration Assays
End1 monolayers were washed and inverted into a humidified chamber. Monolayers were apically infected with 2.3 × 106 GC colony-forming unit (CFU) equivalents (multiplicity of infection [MOI] = 10) for 1 hour (unless otherwise indicated) at 37°C, 5% carbon dioxide (CO2). To assay neutrophil migration across an uninfected End1 monolayer with GC in the apical reservoir, GC was added to the bottom of a tissue culture well for 1 hour, followed by addition of an End1 monolayer on a Transwell insert immediately prior to addition of neutrophils. Neutrophil migration was assayed as previously described [31], and detailed in the Supplementary Methods. For all transmigration assays, migration was compared to buffer alone as a negative control or an imposed apical gradient of N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLP) (1 μM) over 2 hours as positive control for neutrophil migratory capacity. In all experiments, >1 × 105 neutrophils underwent transmigration to fMLP.
Enzyme-Linked Immunosorbent Assay
Supernatants from GC-infected End1 monolayers ± neutrophils were collected, passed through a 0.2-μm filter, and stored at −80°C until processed. Enzyme-linked immunosorbent assays (ELISAs) for IL-8 (R&D Systems) and LTB4 (Cayman Chemical) were performed per the manufacturers’ protocols.
Inhibitor and Drug Treatments
End1 monolayers were pretreated with inhibitors or vehicle controls diluted in Hanks’ Balanced Salt Solution with calcium and magnesium, 10mM HEPES, and 5mM NaHCO3 (HBSS+) and incubated at 37°C, 5% CO2 for the indicated times. Monolayers were then thoroughly washed in HBSS+ before infection. Inhibitor concentrations are provided in the Supplementary Methods and figure legends. The inhibitors used did not affect GC viability or adherence to End1 monolayers, as assayed by CFU enumeration after saponin lysis, serial dilution, and plating on gonococcal base agar (Supplementary Table 1). End1 and neutrophil viability, as monitored by Trypan blue (HyClone) exclusion, was >95% after treatment with inhibitor or vehicle. In all cases, inhibitors or vehicle treatment did not affect neutrophils’ ability to migrate toward an imposed apical gradient of fMLP.
Short Hairpin RNA Knockdown
A lentiviral delivery system was used for short hairpin RNA (shRNA) knockdown of 12R-LOX, eLOX-3, or 5-LOX in End1 cells using MISSION (Sigma-Aldrich) shRNA clones for GenBank accession numbers NM_001139.2-2340s21c1 (12R-LOX), NM_021628.1-1945s1c1 (eLOX-3), and NM_000698.1-405s1c1 (5-LOX) as detailed in the Supplementary Methods.
Quantitative Reverse-Transcription Polymerase Chain Reaction
RNA was extracted from 3 biological replicate End1 monolayers per condition and analyzed by quantitative reverse-transcription polymerase chain reaction (qRT-PCR), as detailed in the Supplementary Methods.
Lipid Extractions
Lipids were isolated from End1 supernatants using solid phase extraction as described [17, 20] and detailed in the Supplementary Methods.
Statistical Analysis
For neutrophil transmigration assays, results are expressed as the mean ± standard error of the mean for ≥3 independent experiments, with 3 transwells per condition. Statistics were calculated using a 2-tailed Student t test, either unpaired for neutrophil migration and qRT-PCR or paired for ELISA and transmigration to lipid-extracted infection supernatants (Excel). For analysis of MOI-dependent neutrophil transmigration, statistics were calculated using a 1-way analysis of variance with Tukey post hoc test (GraphPad Prism). In all cases, a P value <.05 was considered statistically significant compared either to the negative control (buffer alone) or between groups as indicated.
RESULTS
Contact-Dependent GC Infection of Polarized Endocervical Cell Monolayers Stimulates Neutrophil Migration
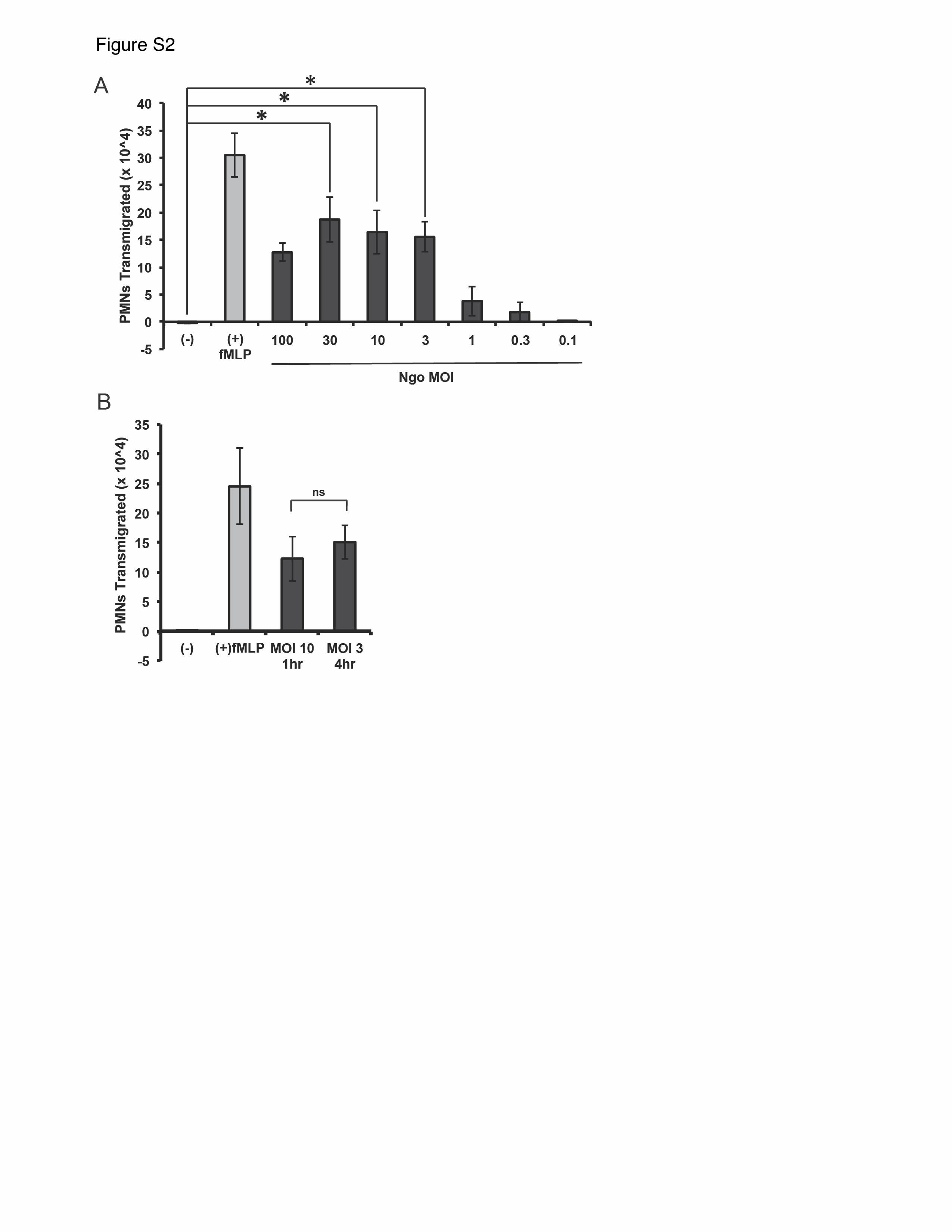
To define factors governing neutrophil migration in response to GC infection, we established an in vitro 3-component system to model infection and neutrophil migration at the endocervix (Figure 1A). End1 cells maintain stable expression of endocervical epithelial differentiation markers and upregulate inflammatory biomarkers when exposed to GC [32, 33], providing a physiologically relevant cell line to model human endocervix. End1 monolayers exhibited apicobasal polarity and junctional integrity, as reflected by increased transepithelial electrical resistance (Supplementary Figure 1A), inhibition of paracellular flux of 10-kDa fluorescein isothiocyanate–dextran in a divalent cation-dependent manner (Supplementary Figure 1B), and lateral localization of the tight junctional protein ZO-1 (Supplementary Figure 1C). These findings agree with previous reports using polarized End1 cells [30, 34]. A piliated, OpaD-expressing derivative of FA1090 GC avidly colonized the apical surface of inverted End1 monolayers 1 hour after inoculation (Supplementary Figure 1D). Primary human neutrophils were then added to the basal aspect of End1 monolayers, and transepithelial migration into the apical reservoir was measured as cell equivalents of neutrophil myeloperoxidase. GC infection elicited neutrophil transepithelial migration over 2 hours that increased with increasing bacterial inoculum (Figure 1B). In all experiments, 2 hours of exposure to fMLP was included as a positive control and buffer alone (no apical stimulus) as a negative control (Figure 1B). MOI-dependent neutrophil migration to GC was also observed after 4 hours of apical infection at lower inocula (Supplementary Figure 2A), and migration was similar between infection with an MOI of 10 after 1 hour and an MOI of 3 after 4 hours (Supplementary Figure 2B). After 1 hour of infection at an MOI of 10, migration that was significantly greater than the negative control was first observed 75 minutes after adding neutrophils and increased at times thereafter (Figure 1C), with migration to fMLP measured after 2 hours (Figure 1C). The following experiments were performed with 1 hour GC infection at MOI of 10 and 2 hours’ neutrophil migration, unless otherwise noted. Neutrophils that had migrated across GC-infected End1 monolayers appeared highly polarized and protrusive by scanning electron microscopy (Supplementary Figure 1E).
Figure 1.

Contact-dependent Neisseria gonorrhoeae (GC) infection of polarized endocervical cell monolayers stimulates neutrophil transepithelial migration. Human End1/E6E7 (End1) monolayers were grown on the underside of collagen-coated Transwell inserts (6.5 mm diameter, 3.0-μm pores). Neutrophil transepithelial migration was quantified by myeloperoxidase (MPO) assay. A, Schematic of in vitro 3-component system depicting apical infection and neutrophil transepithelial migration. Polarized End1 monolayers were inverted in a humidified chamber and apically infected with GC. Following infection, infected monolayers were thoroughly washed in Hanks’ balanced salt solution with calcium and magnesium, 10mM HEPES, and 5mM NaHCO3 (HBSS+) to remove nonadherent bacteria and reverted into a 24-well tissue culture well containing 1 mL HBSS+. Primary human neutrophils (1 × 106) were added to the basolateral reservoir and transmigration was allowed to proceed for 2 hours unless otherwise noted. Monolayers were then removed and the number of neutrophils in the apical reservoir quantified by MPO assay. B, Polarized End1 monolayers were infected at various multiplicities of infection (MOI) for 1 hour and then assayed for neutrophil transepithelial migration. C, Polarized End1 monolayers were apically infected with GC at an MOI = 10 for 1 hour, and then neutrophil transmigration was assayed after 60, 75, 90, and 120 minutes. D, End1 monolayers were apically infected with GC at MOI = 10, an equivalent number of GC was added to the apical reservoir without interacting with End1 cells, or an equivalent number of GC was added to the apical reservoir an acellular Transwell insert and incubated for 1 hour before assaying for neutrophil transmigration. B–D, White bars represent neutrophil transepithelial migration to buffer alone with no apical stimulus (–), gray bars represent neutrophil transepithelial migration to an imposed apical gradient of N-formyl-l-methionyl-l-leucyl-l-phenylalanine, and black bars represent neutrophil migration to GC. Results are expressed as a mean ± standard error of the mean for at least 3 independent experiments per condition. Statistics for (B) were calculated using a 1-way analysis of variance with Tukey post hoc test, and statistics for (C) and (D) were calculated using a 2-tailed, unpaired Student t test. *P < .05. Abbreviations: Ap., apical; Ba., basolateral; End1, End1 E6/E7 cells; fMLP, fMLP, N-formyl-l-methionyl-l-leucyl-l-phenylalanine; GC, Neisseria gonorrhoeae; MOI, multiplicity of infection; ns, not significant; PMN, polymorphonuclear cell/neutrophil.
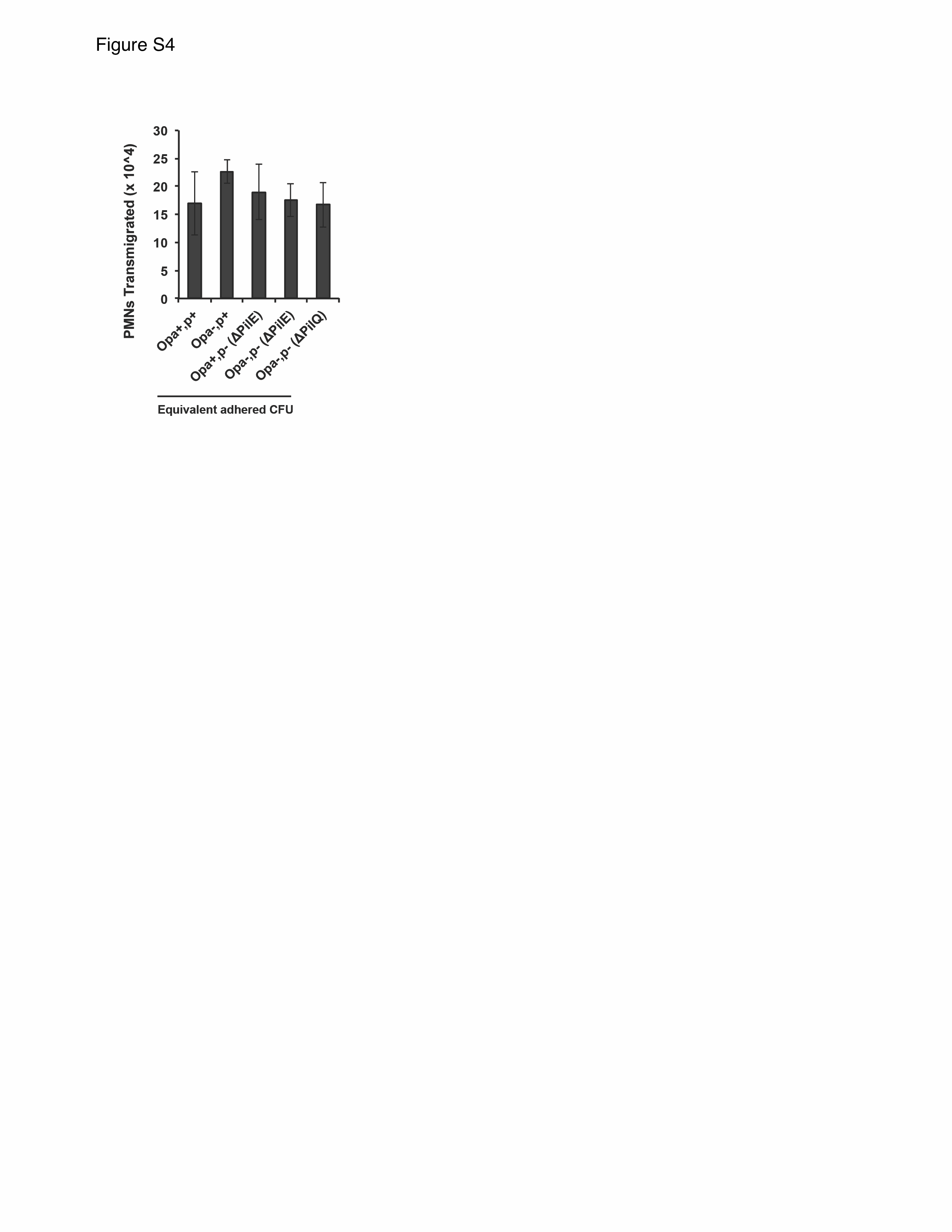
We hypothesized that GC-endocervical cell contact is required to stimulate neutrophil migration. In support of this hypothesis, there was no neutrophil transepithelial migration when GC was in the apical reservoir but not in contact with the End1 monolayers (Figure 1D). Under these conditions, GC did not associate with the End1 apical surface, as assessed by CFU enumeration (Supplementary Table 1) and confocal microscopy (Supplementary Figure 3). Also in support of this hypothesis, neutrophils did not migrate across acellular inserts toward equivalent CFU of GC (Figure 1D). In both conditions, neutrophils still migrated toward fMLP; migration across the End1 monolayer was greater than that observed across an acellular insert, likely because the presence of the monolayer helped maintain a concentration gradient of fMLP. Type IV pili and opacity-associated (Opa) proteins, major GC adhesins [29], were dispensable for neutrophil transepithelial migration in this system, when normalized to equivalent adhered CFU (Supplementary Figure 4; Supplementary Table 1). These results show that polarized endocervical cells are necessary and sufficient to coordinate the migration of neutrophils after apical infection with GC, without requiring pili and Opa proteins.
Neutrophil Transepithelial Migration to GC Requires Neutrophil-Derived Leukotriene B4
Given that human gonococcal infection is characterized by neutrophil recruitment, we examined the possibility that the eicosanoid neutrophil chemoattractant LTB4 is involved. LTB4 accumulated over time in the apical medium of GC-infected End1 monolayers following neutrophil transepithelial migration (Figure 2A). LTB4 concentrations were significantly higher in apical compared to basal medium after 2 hours, suggesting establishment of an apical-to-basolateral gradient of LTB4 following infection (Figure 2A). To evaluate if LTB4 contributed to neutrophil transepithelial migration, LY223982, an antagonist to the high-affinity LTB4 receptor BLT1, was added to the apical medium [20]. LY223982 significantly inhibited neutrophil transepithelial migration following GC infection in a concentration-dependent manner (Figure 2B). LTB4–BLT1 interaction increases the liberation of AA to consequently stimulate more LTB4 production [23, 24].These results show that LTB4 is released during neutrophil migration across GC-infected End1 monolayers, and implicate LTB4 signaling through BLT1 as a contributor to potent neutrophil recruitment during GC infection.
Figure 2.

Neutrophil migration to Neisseria gonorrhoeae (GC) requires leukotriene B4 (LTB4) but not epithelial 5-lipoxygenase activity. A, Supernatants from apical and basolateral compartments following GC infection and/or neutrophil transepithelial migration were collected at the indicated time points and conditions and passed through a 0.2-μm filter, and LTB4 was quantified by enzyme-linked immunosorbent assay. B, Polarized human End1/E6E7 (End1) monolayers were infected with GC at a multiplicity of infection (MOI) = 10 for 1 hour. During neutrophil transepithelial migration, LY223982, an antagonist to the high-affinity LTB4 receptor BLT1, was added to the apical reservoir immediately prior to addition of neutrophils at the indicated concentrations (μg/mL). Gray bars represent neutrophil transepithelial migration to an imposed apical gradient of LTB4 and black bars represent neutrophil migration to GC. C, Polarized End1 monolayers were pretreated with 5-lipoxygenase inhibitor Zileuton (25 μM), caffeic acid (50 μM), or an equivalent concentration of the vehicle control (dimethyl sulfoxide) diluted in Hanks’ Balanced Salt Solution with calcium and magnesium, 10mM HEPES, and 5mM NaHCO3 for 1 hour prior to GC infection, thoroughly washed, and infected at an MOI = 10 and neutrophil transepithelial migration. There was no significant difference in neutrophil migration. D, Neutrophil transepithelial migration was assayed across End1 monolayers infected with GC at an MOI = 10 for 1 hour, with End1 cells either stably transformed with vector short hairpin RNA (shRNA) or shRNA against alox5. For all panels, results are expressed as a mean ± standard error of the mean for at least 3 independent experiments per condition. Statistics were calculated using a 2-tailed, paired (A) or unpaired (B and C) Student t test. *P < .05. Abbreviations: Ap., apical; Ba., basolateral; DMSO, dimethyl sulfoxide; LTB4, leukotriene B4; LY, LY223982; PMN, polymorphonuclear cell/neutrophil; shRNA, short hairpin RNA.
We next sought to determine the source of LTB4 released during GC infection. While neutrophils are major producers of LTB4, epithelial cells can potentially make this eicosanoid [35, 36]. However, expression of alox5, which encodes 5-LOX, was poorly detected in uninfected and infected End1 cells (Supplementary Table 3). Pretreatment of End1 cells with the 5-LOX inhibitors Zileuton or caffeic acid did not affect neutrophil transepithelial migration following GC infection (Figure 2C). Furthermore, stable lentiviral-mediated delivery of shRNA targeting alox5 to End1 cells did not affect neutrophil migration (Figure 2D). Additionally, no LTB4 was detected in the apical compartment of infected End1 monolayers in the absence of neutrophils (Figure 2A). Together these results indicate that End1 cells do not produce LTB4 during GC infection. Expression of BLT1 by End1 cells was also negligible compared to the positive control of differentiated HL60 human promyelocytes, in keeping with reports that BLT1 is primarily expressed on myeloid cells (Supplementary Table 2) [23, 24].
We then tested the hypothesis that neutrophils were the main source of LTB4 in this system. In support of this hypothesis, GC-induced transepithelial migration was abrogated in neutrophils that were pretreated with Zileuton (Figure 3A). This was not due to general effects on neutrophil health or migratory ability, since Zileuton treatment did not inhibit neutrophil migration toward fMLP. Since we had observed neutrophil migration to GC as early as 75 minutes (Figure 1C), we investigated whether we could inhibit LTB4-mediated amplification of GC-induced neutrophil transepithelial migration over time by targeting BLT1. When LY223982 was added to the apical medium at 0, 60, or 75 minutes following basolateral addition of neutrophils, the number of apically migrated neutrophils was significantly reduced compared to the untreated condition (Figure 3B). This correlated with significantly less LTB4 in the apical medium when LY223982 was added at 0 or 75 minutes, compared with no treatment (Figure 3C). We conclude that transepithelially migrated neutrophils produce LTB4 over time to establish a chemotactic gradient for neutrophils.
Figure 3.

Neutrophils produce leukotriene B4 (LTB4) to amplify neutrophil transepithelial migration to Neisseria gonorrhoeae (GC). A, Primary human neutrophils were pretreated with the 5-lipoxygenase inhibitor Zileuton (25 μM) or vehicle (dimethyl sulfoxide) prior to assaying neutrophil transepithelial migration across polarized human End1/E6E7 (End1) monolayers infected with GC at a multiplicity of infection = 10 for 1 hour. B, LY223982 (2.5 μg/mL), an antagonist to the high-affinity LTB4 receptor BLT1, was added to the apical reservoir of infected End1 monolayers at the indicated time points during neutrophil transmigration (0, 60, 75, and 90 minutes). Neutrophil transmigration after addition of LY223982 is compared to the maximum migration seen to apical GC infection after 120 minutes with no treatment. C, Supernatants from apical and basolateral compartments following GC infection and/or neutrophil transepithelial migration were collected at the indicated time points and conditions and passed through a 0.2-μm filter, and LTB4 was quantified by enzyme-linked immunosorbent assay. Results are expressed as a mean ± standard error of the mean for at least 3 independent experiments per condition. Statistics for were calculated using a 2-tailed, unpaired (A and B) or paired (C) Student t test. *P < .05. Abbreviations: Ap., apical; DMSO, dimethyl sulfoxide; GC, Neisseria gonorrhoeae; LTB4, leukotriene B4; NT, no treatment; PMN, polymorphonuclear cell/neutrophil.
Gc Infection Stimulates Endocervical Cell Signaling Pathways Leading To 12-Lox Activation And Consequent Neutrophil Transepithelial Migration
The observations above led us to hypothesize that infected endocervical cells produce a non-LTB4 chemoattractant that coordinates neutrophil transmigration, which is then amplified by neutrophil-derived LTB4. In support of this hypothesis, we identified a signaling pathway involving epithelial enzymes protein kinase C (PKC) and PLA2 during neutrophil migration to GC. Pretreatment of End1 monolayers with the pan-PKC inhibitor chelerythrine chloride or the pan-PLA2 inhibitor ONO-RS-081 completely inhibited neutrophil migration to GC (Figure 4A and 4B). Neutrophil migration to GC was sensitive to inhibition of cytosolic PLA2α (cPLA2α), but not soluble PLA2 (Figure 4B). Inhibition of PLA2 in End1 cells reduced apical LTB4 to uninfected, negative control concentrations (Supplementary Figure 5), further supporting our hypothesis that transmigrated neutrophils are the major source for apical LTB4.
Figure 4.

Neutrophil transepithelial migration to Neisseria gonorrhoeae (GC) requires an epithelial pathway involving protein kinase C and phospholipase A2 (PLA2), but not apically directed interleukin 8 (IL-8). For treatment with inhibitors of epithelial cell targets, epithelial cells were pretreated with the inhibitor and then washed thoroughly before infection with GC at a multiplicity of infection (MOI) = 10 and neutrophil transepithelial migration. Polarized human End1/E6E7 (End1) monolayers were pretreated with the pan–protein kinase C inhibitor chelerythrine chloride (CCL) (5 μM) (A), or the pan-PLA2 inhibitor ONO-RS-082 (5 μM), cytosolic PLA2α inhibitor (cPLA2 inhib) (6 μM), or soluble PLA2 inhibitor 2,4ʹ-dibromoacetophenone (sPLA2 inhib) (7 μM) (B). C, Supernatants from apical and basolateral compartments following a 3-hour GC infection or mock infection were collected and passed through a 0.2-μm filter, and IL-8 was quantified by enzyme-linked immunosorbent assay. D, Polarized End1 monolayers were infected with GC at an MOI = 10 for 1 hour. During neutrophil transepithelial migration, an IL-8 blocking antibody (20 μg/mL), was added to the apical and basolateral reservoirs immediately prior to addition of neutrophils. Gray bars represent neutrophil transepithelial migration to an imposed apical gradient of IL-8, and black bars represent neutrophil migration to GC. Results are expressed as mean ± standard error of the mean for at least 3 independent experiments per condition. Statistics were calculated using a 2-tailed, unpaired (A, B, and D) or paired (C) Student t test. *P < .05. Abbreviations: Ap., apical; Ba., basolateral; CCL, chelerythrine chloride; cPLA2, cytosolic phospholipase A2; DMSO, dimethyl sulfoxide; GC, Neisseria gonorrhoeae; IL-8, interleukin 8; inhib, inhibitor; ns, not significant; NT, no treatment; PMN, polymorphonuclear cell/neutrophil; sPLA2, soluble phospholipase A2.
IL-8 is elevated in acute gonorrhea and is produced by End1 cells infected with GC [32, 37]. In agreement, we found that apically infected End1 monolayers released IL-8, with concentrations significantly higher in the basolateral than apical medium (Figure 4C). Addition of a neutralizing antibody against IL-8 to both the apical and basolateral medium had no effect on neutrophil migration to GC, but inhibited migration across uninfected End1 cells to exogenously added apical IL-8 (Figure 4D). Therefore IL-8, while produced by GC-infected epithelial cells, is not required for basolateral-to-apical neutrophil migration in this system.
The requirement for cPLA2 implied the involvement of epithelial-derived AA and its metabolite(s) in neutrophil migration to GC. In particular, AA-derived eicosanoids made by 12-LOX are neutrophil chemoattractants important for neutrophil influx to multiple mucosal bacterial pathogens [19, 20, 26]. Pretreatment of End1 monolayers with the 12-LOX inhibitor cinnamyl-3,4-dihydroxy-α-cyanocinnamate completely inhibited GC-dependent neutrophil transpeithelial migration (Figure 5A). AA can also be converted to prostaglandins via COX-2; however, pretreatment of End1 monolayers with the COX-2 inhibitor NS398 had no effect on neutrophil transepithelial migration to GC (Supplementary Figure 6). We therefore focused on 12-LOX downstream of PKC and PLA2 in epithelial cells.
Figure 5.

Neutrophil transepithelial migration requires an epithelial 12-lipoxygenase (12-LOX)–dependent chemoattractant that is sensitive to soluble epoxide hydrolase and is secreted through the MRP2 channel. A, Polarized human End1/E6E7 (End1) monolayers were pretreated with 12-LOX inhibitor cinnamyl-3,4-dihydroxy-α-cyanocinnamate (CDC) (50 μM) or an equivalent concentration of the vehicle control (dimethyl sulfoxide) for 1 hour, thoroughly washed, infected at a multiplicity of infection (MOI) = 10, and assayed for neutrophil transepithelial migration. B, Relative expression of alox12b or aloxe3 in End1 monolayers either mock-infected or infected with Neisseria gonorrhoeae (GC) at an MOI = 10 for 3 hours or in End1 monolayers following lentiviral-mediated short hairpin RNA (shRNA) knockdown of alox12b or aloxe3 or vector control. C, Neutrophil transepithelial migration was assayed across End1 monolayers infected with GC at an MOI = 10 for 1 hour, with End1 cells either stably transformed with vector shRNA or shRNA against alox12b or aloxe3. D, Neutrophil transepithelial migration was assayed across End1 monolayers infected with GC at an MOI = 10 for 1 hour, with either 100 μg/mL or 200 μg/mL human recombinant soluble epoxide hydrolase (sEH) added to the apical reservoir prior to and throughout neutrophil transepithelial migration or pretreatment of polarized End1 monolayers with MRP2 inhibitor MK571 (100 μM). A, C, and D, Gray bars represent neutrophil transepithelial migration to an imposed apical gradient of leukotriene B4 (LTB4), and black bars represent neutrophil migration to GC. Results are expressed as a mean ± standard error of the mean for at least 3 independent experiments per condition. Statistics were calculated using a 2-tailed, unpaired Student t test. *P < .05. Abbreviations: CDC, cinnamyl -3,4-dihydroxy-α-cyanocinnamate; DMSO, dimethyl sulfoxide; GC, Neisseria gonorrhoeae; LTB4, leukotriene B4; NT, no treatment; PMN, polymorphonuclear cell/neutrophil; sEH, soluble epoxide hydrolase; shRNA, short hairpin RNA.
The 12-LOX activity in humans can be catalyzed by 15-LOX, 12-LOX, and 12R-LOX/eLOX-3, encoded by alox15, alox12, alox12b, and aloxe3, respectively [17, 38, 39]. Of these, only alox12b and aloxe3 were expressed in End1 cells, with expression significantly increasing following GC infection (Figure 5B; Supplementary Table 3). Unlike other 12-LOXs, 12R-LOX is reported to work in concert with eLOX-3 to produce HXA3 [25, 40–42]. Stable shRNA knockdown of alox12b or aloxe3 (Figure 5B) significantly decreased neutrophil migration to GC (Figure 5C), without affecting migration to apically added LTB4 (data not shown). These results implicate 12R-LOX and eLOX-3 in coordinating the endocervical response to GC, resulting in neutrophil transepithelial migration.
Three lines of evidence suggested HXA3 was the 12-LOX product required for endocervical cells to support neutrophil migration. First, apical addition of recombinant sEH, which hydrolyzes the epoxide ring of HXA3 required for neutrophil chemoattraction to yield the inert trioxilin A3 [26, 27, 43, 44], significantly inhibited neutrophil migration to GC (Figure 5D). sEH addition did not affect neutrophil migration to an imposed apical gradient of LTB4 (Figure 5D), which does not contain an epoxide ring and is thus insensitive to sEH. Second, HXA3 is secreted apically by MRP2 channels in the context of other mucosal bacterial infections [12]. Inhibition of the MRP2 efflux pump on End1 cells with MK571 significantly inhibited neutrophil migration to GC (Figure 5D). Third, lipid-extracted supernatants from infected End1 monolayers stimulated 2- to 5-fold more neutrophil migration across an acellular insert compared to mock-infected supernatants, a statistically significant increase (Supplementary Figure 7). HXA3 is known to partition into the lipid fraction of infection supernatants [17, 20]. Taken together, we conclude that endocervical cells infected with GC stimulate 12R-LOX and eLOX-3 lipoxygenase activity to produce an epoxide-containing eicosanoid, likely HXA3, which is essential for neutrophil transepithelial migration.
DISCUSSION
Neisseria gonorrhoeae infection leads to sustained neutrophil influx, but the mechanisms underlying recruitment of neutrophils across the epithelium in the context of GC infection were not previously understood. Our in vitro system demonstrates that GC–endocervical cell contact stimulates eicosanoid-driven neutrophil migration (Figure 6). Following GC infection, endocervical cells produced an apically directed 12-LOX–derived neutrophil chemoattractant (HXA3) that required efflux through MRP2 and was sensitive to sEH. This pathway was dependent on epithelial PKC and cPLA2 to produce the 12-LOX substrate AA. Transmigrated neutrophils produced the 5-lipoxygenase product LTB4, which amplified the apical-to-basolateral neutrophil chemotactic gradient. To our knowledge, this is the first time eicosanoids have been implicated in neutrophil influx to GC.
Figure 6.

Model of eicosanoid-coordinated neutrophil migration across Neisseria gonorrhoeae (GC)–infected endocervical cells. GC infection stimulates human End1/E6E7 (End1) protein kinase C, phospholipase A2, and lipoxygenases (LOXs) 12R-LOX and eLOX-3 to produce the eicosanoid hepoxilin A3 that is directed apically through the MRP2 channel. Transmigrating neutrophils activate 5-LOX to produce leukotriene B4 that acts in paracrine and autocrine fashion to amplify neutrophil influx. Abbreviations: 5-LOX, 5-lipoxygenase; 12R-LOX, arachidonate 12-lipoxygenase; eLOX-3, epidermis-type lipoxygenase 3; HXA3, hepoxilin A3; LTB4, leukotriene B4; PKC, protein kinase C; PLA2, phospholipase A2.
The LTB4-BLT1 axis is important for directing neutrophil chemotaxis to sites of inflammation [24]. Our results reveal a role for LTB4 in neutrophil influx to GC. In vivo, multiple additional cell types and factors likely influence the inflammatory milieu during GC infection. For example, GC skews the immune response away from a protective adaptive immune response and toward a Th17-driven neutrophilic response during human infection [3]. Interestingly, LTB4 has recently been shown to induce Th17 cell migration to a greater extent than Th1 or Th2 cell migration [45]. Neutrophil migration and LTB4 signaling activate neutrophils to combat invading pathogens [23, 46]. Neutrophils recruited to GC infection appeared protrusive, though their activation status remains to be determined. However, given that GC can evade neutrophil killing, neutrophil activation may not control infection, and instead contributes to host cell damage.
Our results indicate a role for epithelial 12R-LOX and eLOX-3 during neutrophil migration to GC. This was surprising, as other bacterial pathogens studied to date stimulate 15-LOX to produce HXA3 [11, 15]. 12R-LOX is primarily expressed in skin and is unique in that it requires downstream activity of eLOX-3 to generate HXA3 [25, 40–42]. The best described function of 12R-LOX and eLOX-3 is to prevent cellular water loss, with mutations in both linked to dry skin disorders [25, 41, 42]. Interestingly, the first reports of 12R-LOX in the context of disease describe accumulation of its product 12R-HETE in psoriatic scales [47], suggesting that 12R-LOX plays a role during certain inflammatory states. eLOX3 is described as a hepoxilin synthase, and our combined evidence with sEH, the MRP2 efflux pump that exports HXA3, and neutrophil chemotactic activity in lipid-enriched fractions from infected End1 supernatants all suggest that HXA3 coordinates this event. HXA3 is a highly potent yet labile chemoattractant, which has complicated confirmation of this eicosanoid in this in vitro system.
Many laboratories have demonstrated that pathogens stimulate eicosanoid signaling by affecting AA production [6, 8, 9, 11, 18–20]. In agreement, we found that endocervical PKC and PLA2 are required for neutrophil migration to GC. However, GC does not possess the secreted or surface factors employed by these mucosal pathogens to stimulate eicosanoid production [10, 21, 48]. Two major GC adherence factors, Opa proteins and type IV pili, were not required for neutrophil migration. The GC factors that stimulate eicosanoid production will be an important topic for future investigation.
Broadly, our findings highlight the importance of studying context-specific bacterial–epithelial interactions that engage eicosanoid-producing pathways, to identify novel therapeutic targets for infections in which disease is exacerbated by neutrophilic inflammation. Specifically for gonorrhea, sustained neutrophil influx that does not resolve GC infection contributes to host cell damage. With increased failures of antibiotic treatment, we anticipate increased likelihood for inflammatory damage especially in women, leading to pelvic inflammatory disease, ectopic pregnancy, and infertility. Our work suggests targeting this eicosanoid pathway at the cervix would be an attractive adjunctive strategy to mitigate clinical sequelae in infected women. As precedence, immunosuppressive therapy is added to treatment regimens for bacterial meningitis and pneumocystis pneumonia in patients with human immunodeficiency virus [49, 50]. There is also precedence for targeting lipoxygenase pathways in inflammation, with US Food and Drug Administration–approved therapeutics for asthma including Zileuton and the leukotriene receptor antagonist Montelukast. This study reveals novel lines of inquiry into gonorrheal disease pathogenesis to ameliorate the negative, lifelong consequences of gonorrhea in women.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Presented in part: Cold Spring Harbor Laboratories Meeting, Cold Spring Harbor, New York, September 2017, Abstract 185; Gordon Research Conference, Waterville Valley, New Hampshire, June 2017, Presentation XXX; Mid-Atlantic Microbial Pathogenesis Meeting, Wintergreen, Virginia, February 2017, Abstract 87; and International Pathogenic Neisseria Conference, Manchester, United Kingdom, September 2016, Abstract 72.
Notes
Acknowledgments. We thank Beth McCormick (University of Massachusetts) for her advice on and support of this work. We also thank Liz Nelson and Judith White, Beth McKenney and Melissa Kendall, Nicholas Sherman and the W. M. Keck Biomedical Mass Spectrometry Laboratory, Norbert Leitinger, Ryan D’Souza and Jim Casanova, Annie Carlton, Sarbajeet Nagdas, and Michael Schappe (all at UVA) for advice and reagents for various aspects of this project. All experiments with human neutrophils were conducted in accordance with a protocol approved by the UVA Institutional Review Board (IRB) for Health Sciences Research (IRB HSR number 13909).
Author contributions. The authors each reviewed the manuscript and approved it for publication.
Financial support. This work was supported by the National Institutes of Health (NIH) (grant numbers R01 AI097312 to A. K. C. and T32 GM007267 and T32 AI007046 to J. S. S.); the Pinn Scholars Award as well as startup funds from the University of Virginia to A. K. C.; the Robert R. Wagner Fellowship from the University of Virginia to J. S. S.; and the National Institute of Environmental Health Sciences at the NIH (grant number R01 ES002710 to C. M.). This work used the Zeiss LSM 700 and Zeiss Sigma VP field emission scanning electron microscope in the Advanced Microscopy Facility, which is supported by the University of Virginia School of Medicine. For use of the Zeiss scanning electron microscope, we acknowledge NIH S10 OD011966. We also acknowledge use of the Tissue Culture Core Facility, which is supported by the University of Virginia School of Medicine.
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Wi T, Lahra MM, Ndowa F, et al. Antimicrobial resistance in Neisseria gonorrhoeae: global surveillance and a call for international collaborative action. PLoS Med 2017; 14:e1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Johnson MB, Criss AK. Resistance of Neisseria gonorrhoeae to neutrophils. Front Microbiol 2011; 2:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stevens JS, Criss AK. Pathogenesis of Neisseria gonorrhoeae in the female reproductive tract: neutrophilic host response, sustained infection, and clinical sequelae. Curr Opin Hematol 2018; 25:13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Edwards JL, Apicella MA. The molecular mechanisms used by Neisseria gonorrhoeae to initiate infection differ between men and women. Clin Microbiol Rev 2004; 17:965–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Criss AK, Seifert HS. A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol 2012; 10:178–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McCormick BA, Hofman PM, Kim J, Carnes DK, Miller SI, Madara JL. Surface attachment of Salmonella typhimurium to intestinal epithelia imprints the subepithelial matrix with gradients chemotactic for neutrophils. J Cell Biol 1995; 131:1599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. McCormick BA, Parkos CA, Colgan SP, Carnes DK, Madara JL. Apical secretion of a pathogen-elicited epithelial chemoattractant activity in response to surface colonization of intestinal epithelia by Salmonella typhimurium. J Immunol 1998; 160:455–66. [PubMed] [Google Scholar]
- 8. Mrsny RJ, Gewirtz AT, Siccardi D, et al. Identification of hepoxilin A3 in inflammatory events: a required role in neutrophil migration across intestinal epithelia. Proc Natl Acad Sci U S A 2004; 101:7421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hurley BP, Siccardi D, Mrsny RJ, McCormick BA. Polymorphonuclear cell transmigration induced by Pseudomonas aeruginosa requires the eicosanoid hepoxilin A3. J Immunol 2004; 173:5712–20. [DOI] [PubMed] [Google Scholar]
- 10. Zurawski DV, Mitsuhata C, Mumy KL, McCormick BA, Maurelli AT. OspF and OspC1 are Shigella flexneri type III secretion system effectors that are required for postinvasion aspects of virulence. Infect Immun 2006; 74:5964–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mumy KL, Bien JD, Pazos MA, Gronert K, Hurley BP, McCormick BA. Distinct isoforms of phospholipase A2 mediate the ability of Salmonella enterica serotype Typhimurium and Shigella flexneri to induce the transepithelial migration of neutrophils. Infect Immun 2008; 76:3614–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pazos M, Siccardi D, Mumy KL, et al. Multidrug resistance-associated transporter 2 regulates mucosal inflammation by facilitating the synthesis of hepoxilin A3. J Immunol 2008; 181:8044–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hurley BP, Pirzai W, Mumy KL, Gronert K, McCormick BA. Selective eicosanoid-generating capacity of cytoplasmic phospholipase A2 in Pseudomonas aeruginosa-infected epithelial cells. Am J Physiol Lung Cell Mol Physiol 2011; 300:L286–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boll EJ, McCormick BA. A new understanding of enteroaggregative Escherichia coli as an inflammatory pathogen. Cell Adh Migr 2012; 6:413–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boll EJ, Struve C, Sander A, Demma Z, Krogfelt KA, McCormick BA. Enteroaggregative Escherichia coli promotes transepithelial migration of neutrophils through a conserved 12-lipoxygenase pathway. Cell Microbiol 2012; 14:120–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boll EJ, Struve C, Sander A, et al. The fimbriae of enteroaggregative Escherichia coli induce epithelial inflammation in vitro and in a human intestinal xenograft model. J Infect Dis 2012; 206:714–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tamang DL, Pirzai W, Priebe GP, et al. Hepoxilin A(3) facilitates neutrophilic breach of lipoxygenase-expressing airway epithelial barriers. J Immunol 2012; 189:4960–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bhowmick R, Maung N, Hurley BP, et al. Systemic disease during Streptococcus pneumoniae acute lung infection requires 12-lipoxygenase-dependent inflammation. J Immunol 2013; 191:5115–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Szabady RL, McCormick BA. Control of neutrophil inflammation at mucosal surfaces by secreted epithelial products. Front Immunol 2013; 4:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pazos MA, Pirzai W, Yonker LM, Morisseau C, Gronert K, Hurley BP. Distinct cellular sources of hepoxilin A3 and leukotriene B4 are used to coordinate bacterial-induced neutrophil transepithelial migration. J Immunol 2015; 194:1304–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pazos MA, Lanter BB, Yonker LM, et al. Pseudomonas aeruginosa ExoU augments neutrophil transepithelial migration. PLoS Pathog 2017; 13:e1006548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dennis EA, Norris PC. Eicosanoid storm in infection and inflammation. Nat Rev Immunol 2015; 15:511–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le Bel M, Brunet A, Gosselin J. Leukotriene B4, an endogenous stimulator of the innate immune response against pathogens. J Innate Immun 2014; 6:159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Németh T, Mócsai A. Feedback amplification of neutrophil function. Trends Immunol 2016; 37:412–24. [DOI] [PubMed] [Google Scholar]
- 25. Mashima R, Okuyama T. The role of lipoxygenases in pathophysiology; new insights and future perspectives. Redox Biol 2015; 6:297–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pace-Asciak CR. Pathophysiology of the hepoxilins. Biochim Biophys Acta 2015; 1851:383–96. [DOI] [PubMed] [Google Scholar]
- 27. Morisseau C, Schebb NH, Dong H, Ulu A, Aronov PA, Hammock BD. Role of soluble epoxide hydrolase phosphatase activity in the metabolism of lysophosphatidic acids. Biochem Biophys Res Commun 2012; 419:796–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rouzer CA, Matsumoto T, Samuelsson B. Single protein from human leukocytes possesses 5-lipoxygenase and leukotriene A4 synthase activities. Proc Natl Acad Sci U S A 1986; 83:857–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ball LM, Criss AK. Constitutively Opa-expressing and Opa-deficient Neisseria gonorrhoeae strains differentially stimulate and survive exposure to human neutrophils. J Bacteriol 2013; 195:2982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Fichorova RN, Zhou F, Ratnam V, et al. Anti-human immunodeficiency virus type 1 microbicide cellulose acetate 1,2-benzenedicarboxylate in a human in vitro model of vaginal inflammation. Antimicrob Agents Chemother 2005; 49:323–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parkos CA, Delp C, Arnaout MA, Madara JL. Neutrophil migration across a cultured intestinal epithelium. Dependence on a CD11b/CD18-mediated event and enhanced efficiency in physiological direction. J Clin Invest 1991; 88:1605–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fichorova RN, Desai PJ, Gibson FC III, Genco CA. Distinct proinflammatory host responses to Neisseria gonorrhoeae infection in immortalized human cervical and vaginal epithelial cells. Infect Immun 2001; 69:5840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fichorova RN, Cronin AO, Lien E, Anderson DJ, Ingalls RR. Response to Neisseria gonorrhoeae by cervicovaginal epithelial cells occurs in the absence of Toll-like receptor 4-mediated signaling. J Immunol 2002; 168:2424–32. [DOI] [PubMed] [Google Scholar]
- 34. Sathe A, Reddy KV. TLR9 and RIG-I signaling in human endocervical epithelial cells modulates inflammatory responses of macrophages and dendritic cells in vitro. PLoS One 2014; 9:e83882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Behera AK, Kumar M, Matsuse H, Lockey RF, Mohapatra SS. Respiratory syncytial virus induces the expression of 5-lipoxygenase and endothelin-1 in bronchial epithelial cells. Biochem Biophys Res Commun 1998; 251:704–9. [DOI] [PubMed] [Google Scholar]
- 36. Brock TG. Expression of 5-lipoxygenase in specialized epithelial cells of nasopharyngeal-associated lymphoid tissue. J Mol Histol 2005; 36:475–81. [DOI] [PubMed] [Google Scholar]
- 37. Fichorova RN, Rheinwald JG, Anderson DJ. Generation of papillomavirus-immortalized cell lines from normal human ectocervical, endocervical, and vaginal epithelium that maintain expression of tissue-specific differentiation proteins. Biol Reprod 1997; 57:847–55. [DOI] [PubMed] [Google Scholar]
- 38. Kuhn H, Banthiya S, van Leyen K. Mammalian lipoxygenases and their biological relevance. Biochim Biophys Acta 2015; 1851:308–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuhn H, Walther M, Kuban RJ. Mammalian arachidonate 15-lipoxygenases structure, function, and biological implications. Prostaglandins Other Lipid Mediat 2002; 68–69:263–90. [DOI] [PubMed] [Google Scholar]
- 40. Yu Z, Schneider C, Boeglin WE, Marnett LJ, Brash AR. The lipoxygenase gene ALOXE3 implicated in skin differentiation encodes a hydroperoxide isomerase. Proc Natl Acad Sci U S A 2003; 100:9162–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Epp N, Fürstenberger G, Müller K, et al. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J Cell Biol 2007; 177:173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Krieg P, Rosenberger S, de Juanes S, et al. Aloxe3 knockout mice reveal a function of epidermal lipoxygenase-3 as hepoxilin synthase and its pivotal role in barrier formation. J Invest Dermatol 2013; 133:172–80. [DOI] [PubMed] [Google Scholar]
- 43. Cronin A, Decker M, Arand M. Mammalian soluble epoxide hydrolase is identical to liver hepoxilin hydrolase. J Lipid Res 2011; 52:712–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Morisseau C, Hammock BD. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu Rev Pharmacol Toxicol 2013; 53:37–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lee W, Su Kim H, Lee GR. Leukotrienes induce the migration of Th17 cells. Immunol Cell Biol 2015; 93:472–9. [DOI] [PubMed] [Google Scholar]
- 46. Nadeau WJ, Pistole TG, McCormick BA. Polymorphonuclear leukocyte migration across model intestinal epithelia enhances Salmonella typhimurium killing via the epithelial derived cytokine, IL-6. Microbes Infect 2002; 4:1379–87. [DOI] [PubMed] [Google Scholar]
- 47. Boeglin WE, Kim RB, Brash AR. A 12R-lipoxygenase in human skin: mechanistic evidence, molecular cloning, and expression. Proc Natl Acad Sci U S A 1998; 95:6744–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Silva M, Song C, Nadeau WJ, Matthews JB, McCormick BA. Salmonella typhimurium SipA-induced neutrophil transepithelial migration: involvement of a PKC-alpha-dependent signal transduction pathway. Am J Physiol Gastrointest Liver Physiol 2004; 286:G1024–31. [DOI] [PubMed] [Google Scholar]
- 49. Ewald H, Raatz H, Boscacci R, Furrer H, Bucher HC, Briel M. Adjunctive corticosteroids for Pneumocystis jiroveci pneumonia in patients with HIV infection. Cochrane Database Syst Rev 2015; CD006150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barichello T, Collodel A, Generoso JS, et al. Targets for adjunctive therapy in pneumococcal meningitis. J Neuroimmunol 2015; 278:262–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.