

Abstract
Importance:
Synovial cell sarcoma of the head and neck (SCSHN) is a rare tumor associated with significant morbidity and mortality. The literature regarding these tumors is limited to case series and case reports. We utilized data from the population-based United States SEER cancer registry to determine factors affecting both overall survival and disease specific survival of patients with SCSHN.
Objective:
To determine the clinicopathologic and therapeutic factors determining survival in patients with SCSHN.
Design, Setting, and Participants:
The Surveillance, Epidemiology, and End Results registry was reviewed for patients with primary SCSHN from 1973–2011.
Interventions: None
Main Outcomes and Measures:
Overall survival (OS) and disease-specific survival (DSS).
Results:
A total of 167 cases of SCSHN were identified. The median age at diagnosis was 37 and 59.9% of patients were males. The mono-phasic spindle cell and biphasic variants were the most common histological subtypes. Surgical resection and radiation therapy were performed in 89.8% and 64.7% of cases, respectively. The median overall survival (OS) at 2, 5, and 10 years was 77%, 66%, and 53%, respectively. Univariate Kaplan-Meier survival analysis revealed that age, race, stage, and size were associated with improved survival. Histological subtype was not associated with significant differences in survival. Radiotherapy was associated with improved disease-specific survival (p=0.003), while surgical management was not associated with improved survival. Multivariate Cox regression analysis revealed that size >5 cm (p=0.007) and stage at presentation (p=0.003) were independent determinants of OS. When separately analyzing cohorts with tumors ≤5 cm and >5 cm, stage at presentation was found to be a significant predictor of survival in both (p=0.003, p=0.010); surgical resection and radiotherapy were not associated with differential survival outcomes using this model.
Conclusion and Relevance:
SCSHN is an extremely rare malignancy. Independent significant determinants of survival include size >5 cm and stage at presentation. Histological subtype of the tumor is not a significant predictor of survival. Surgical resection and radiation therapy were not found to be independent determinants of survival.
INTRODUCTION
Synovial cell sarcomas are rare malignant tumors of mesenchymal stem cell origin that are now recognized to be unrelated to synovial tissue.1,2 These tumors have propensity for the lower limbs and most commonly afflict males in the third to fifth decade of life.3–5 Synovial cell tumors are spindle cell tumors with variable epithelial differentiation. These tumors are further classified based on the degree of epithelial differentiation into either mono-phasic or biphasic subtypes. The mono-phasic subtype is the most common subtype and is characterized by either predominantly spindle or less commonly epithelial cell morphology. The biphasic subtype is composed of both distinct epithelial and spindle cell components. In >95% of cases, synovial cell sarcoma is associated with the specific t(x; 18) (p11.2; q11.2) translocation, which results in the SYT-SSX fusion protein that has been used to aid in the diagnosis of this histopathologically complex malignancy.6–9
Synovial cell sarcoma of the head and neck (SCSHN) represent less than 0.1% of all head and neck malignancies, and only 3–10% of synovial sarcomas present in the head and neck region.10 These tumors are among the rarest in the head and neck region and our knowledge of them clinical behavior as it pertains to this region is limited to case reports and small case series. Management and treatment algorithms for the treatment of SCSHN have been highly dependent on the behavior of these tumors in other anatomic regions, most notably the limbs.11–15 Though SCSHN is morphologically similar to synovial cell sarcoma at other subsites, there is increasing evidence that SCSHN is a unique pathological entity with potentially distinct mechanisms of tumorigenesis.16 The most common site of presentation reported in the literature is the parapharyngeal space, followed by the hypopharynx.17,18 The overall 5-year survival has been reported to range from 40–70%.11,19–21 Lymph node involvement is noted on presentation in 15–20% of patients, but does not appear to affect overall survival.19 Likewise, histological subtype does not appear to affect survival.19,22 Clinicopathological factors linked to poor survival in previous reports include a high mitotic index, high grade, metastatic or recurrent disease, and the size of the tumor at presentation.22–24 While tumor size has been clearly associated with decreased survival, there is conflicting information regarding the pivot point for precipitous decrease in survival, and whether this occurs at 4 or 5 cm remains debated in the literature.17,19,20,22,24
Management of SCSHN is controversial, with no controlled studies that define optimal treatment protocols. At present, the mainstay of treatment includes complete resection with wide margins to limit local recurrence.5,11–15,17,19,23–28 The extent of the resection must factor in the propensity of these tumors to spread beyond the visible or palpable limits of tumor. Radiotherapy is typically employed to improve local control if resection is inadequate. The role of chemotherapy is also controversial with questionable survival benefits when combined with adjuvant radiation therapy.26,29 Regardless of treatment modality these tumors continue to portend a more serious prognosis, with a reported 5-year overall survival ranging from 40–70%, and a recurrence rate of 40%.19–21,27,30
While there are a myriad of small case series and reports, fewer than 100 cases have been reported in the international literature and population based data is scarce.17 The purpose of this study was to determine the incidence and survival determinants of patients with SCSHN. We utilized data from the population-based United States SEER cancer registry to analyze a number of patient and disease related characteristics to determine factors affecting both overall and disease-specific survival.
METHODS
A population-based search for patients diagnosed with SCSHN was performed using the case-listing session protocol of the National Cancer Institute’s Surveillance Epidemiology and End Results (SEER) 18 database (www.seer.cancer.gov); the database is a widely-used cancer registry that covers an estimated 28% of the US population, including 23% of African Americans and 40% of Hispanics. Geographic regions covered include San Francisco-Oakland, Connecticut, metropolitan Detroit, Hawaii, Iowa, New Mexico, Seattle (Puget Sound), Utah, metropolitan Atlanta, San Jose-Monterey, Los Angeles, Alaska, rural Georgia, California, Kentucky, Louisiana, New Jersey, and greater Georgia. No internal review board approval was required in this study because the database uses publicly available information with no personal identifiers.
Patients diagnosed with synovial sarcoma from 1973–2011, the widest date ranges available in the latest version of the SEER software at the time of publication. Histologic ICD-0–3 codes were used to include: synovial sarcoma, NOS (9040/3); synovial sarcoma, spindle cell (9041/3); synovial sarcoma, epithelioid cell (9042/3); and synovial sarcoma, biphasic (9043/3). Site specific codes were used to confirm that the tumor originated in the head and neck. The following primary data were extracted from the database for analysis: age at diagnosis, sex, race, histologic subtype (ICD), tumor extent and tumor size from both extent of disease (EOD) and collaborative stage (CS) coding methods, tumor grade, tumor stage, treatment with surgery and/or radiation therapy, cause of death, and survival months. Well- and moderately-differentiated histologies were grouped as low grade (grades I/II), while poorly differentiated and undifferentiated histologies were grouped as high grade (grades III/IV). Where available, TNM staging was recorded as explicitly listed in the SEER registries for all patients diagnosed from 2003–2011. For cases diagnosed prior to 2003, TNM stage was retroactively determined, where possible without ambiguity, using CS and EOD staging codes for tumor size, extent, lymph node involvement, and evidence of distant metastasis using classification criteria determined by the American Joint Committee on Cancer (AJCC). TNM staging and grade classification were then used to determine stage at presentation (I-IV).
Primary outcome was defined as time in months from diagnosis to death from any cause for OS, and time from diagnosis to death specific to the cancer-related diagnosis for DSS. Descriptive statistics were calculated for all variables. OS and DSS curves were calculated using the Kaplan-Meier method. Univariate analysis to determine differences were formally tested for using the log-rank test. Covariates were assessed for independent predictive performance with multivariate Cox proportional hazards regression models, using hazard ratios with corresponding 95% confidence intervals, with regard to OS and DSS. Comparisons between groups were deemed statistically significant at the p<0.05 threshold. Covariates were chosen for multivariate analysis based on factors identified as significant on univariate analysis with histological subtype, surgical resection, and radiation therapy included by default. This method was chosen to minimize the total number of covariates thus improving the generalizability of the findings and minimizing instability in the model, with a target of at least 10 events per covariate where possible. Statistical analyses were performed using SPSS 21 software (IBM Corp., Armonk, NY).
RESULTS
The SEER database search revealed 167 patients with primary SCSHN from 1973 to 2011. Of these, 59.9% were males and 81.4% were white (Table 1). The mean age of diagnosis was 37.9 years with ages ranging from 5 to 75. The most common primary sites of disease were the oral cavity (5.4%), and parotid gland (4.8%), although the tumor site was not specified in the majority (64.1%) of cases. The most common histologic subtypes were synovial sarcoma, spindle cell (31.1%), and biphasic (22.8%); no cases of the purely epithelioid cell variant were identified and 46.1% of cases were listed as synovial sarcoma, NOS. Tumor grade was available for 45.5% of cases; 29.9% of all tumors were histologically confirmed to be poorly differentiated or undifferentiated tumors of high grade. Definitive staging was possible in 43.1% of cases with the majority of cases presenting as Stage I (12.6%) or Stage II (18.0%) tumors. The mean and median tumor size at the time of diagnosis was 5.0 and 4.3 cm, respectively, with sizes ranging from 0.3 to 23 cm. 60.5% of patients received bimodal therapy, 26.9% underwent surgical resection only, and 3.6% underwent radiation therapy only, while 4.2% of patients received no therapy. The treatment regimen was unknown in 4.8% of patients.
TABLE 1:
Demographics of patients with synovial cell sarcoma of the head and neck
| Age | Years |
|---|---|
| Mean | 37.9 ± 17.3 |
| Median | 37 |
| Min | 5 |
| Max | 75 |
| Characteristic | Percentage (n) |
| Sex | |
| Female | 40.1% (67) |
| Male | 59.9% (100) |
| Race | |
| White | 81.4% (136) |
| Black | 11.4% (19) |
| Asian | 4.8% (8) |
| Pacific Islander | 1.8% (3) |
| Native American/Alaskan Native | 0.6% (1) |
| Histological subtype | |
| Synovial sarcoma, NOS | 46.1% (77) |
| Synovial sarcoma, spindle cell | 31.1% (52) |
| Synovial sarcoma, epithelioid cell | 0.0% (0) |
| Synovial sarcoma, biphasic | 22.8% (38) |
| Tumor grade | |
| Low | 15.6% (26) |
| High | 29.9% (50) |
| Unknown | 54.5% (91) |
| Stage at presentation | |
| Stage I | 12.6% (21) |
| Stage II | 18.0% (30) |
| Stage III | 6.6% (11) |
| Stage IV | 6.0% (10) |
| Unknown | 56.9% (72) |
| Surgery performed | |
| Yes | 89.8% (150) |
| No | 8.4% (14) |
| Unknown | 1.8% (3) |
| Radiation therapy | |
| Yes | 64.7% (108) |
| No | 31.7% (53) |
| Unknown | 3.6% (6) |
| Treatment modality | |
| Surgery + Radiation | 60.5% (101) |
| Surgery only | 26.9% (45) |
| Radiation only | 3.6% (6) |
| No therapy | 4.2% (7) |
| Unknown | 4.8% (8) |
| Size (cm) | |
| Mean | 5.0 ± 3.3 |
| Median | 4.3 |
Survival analysis from Kaplan-Meier curves (Figure 1A–B) revealed that the 5-year OS and DSS for all SCSHN was 66% and 71%, respectively (Table 2); the median OS was 14.9 years. Both OS and DSS showed no statistically significant difference in survival based on histological subtype (Table 3, 1C–D). Similarly, size >5 cm was associated with worse OS and DSS (p=0.005, p=0.009, Table 3, Figure 1E–F). Tumors >5 cm were associated with a poor prognosis (OS 4.4 years, DSS 5.5 years), whereas less than 50% of patients with tumors ≤5 cm died during the study period. This difference in prognosis was not evident for either OS or DSS when stratifying cases by tumor sizes ≤4 cm and >4 cm (OS p=0.244, DSS p=0.420). In addition, the Kaplan-Meier univariate survival analysis revealed that increased age, race, and stage at presentation were also associated with significantly better OS. Age, race, and radiation therapy were also associated with significantly improved DSS. Sex, primary site, tumor grade, and surgical resection were not associated with significant differences in survival (Table 3).
FIGURE 1: Survival analysis of patients with synovial cell sarcoma of the head and neck using Kaplan-Meier analysis.
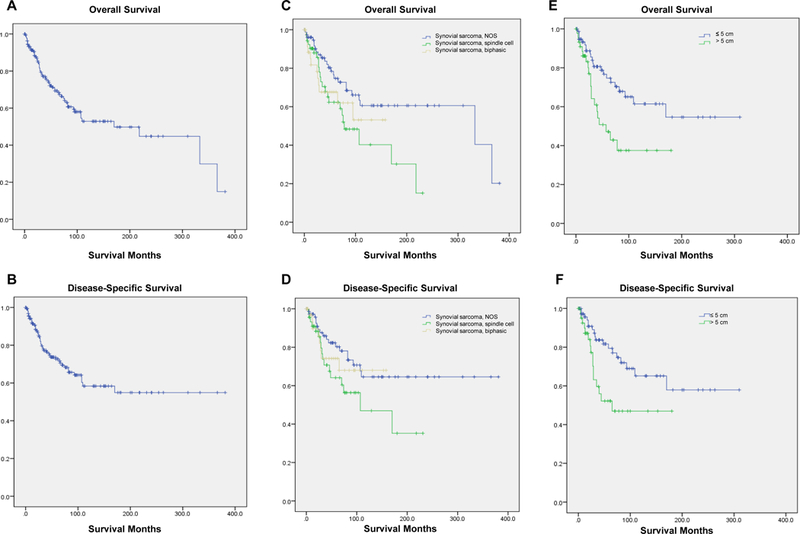
(A) Kaplan-Meier estimates of overall survival (OS) and (B) disease-specific survival (DSS) are shown for all cases. Kaplan-Meier analysis of (C) OS and (D) DSS when stratified by histological subtype. Kaplan-Meier analysis of (E) OS and (F) DSS when stratified by tumor size above and below a critical size cutoff of 5 cm.
TABLE 2:
Survival data of patients with synovial cell sarcoma of the head and neck
| Median survival (years) | Overall (OS) | Disease-specific (DSS) |
|---|---|---|
| Overall | 14.9 | UNDEF |
| Histological subtype | ||
| Synovial sarcoma, NOS | 27.5 | UNDEF |
| Synovial sarcoma, spindle cell | 6.8 | 8.7 |
| Synovial sarcoma, biphasic | UNDEF | UNDEF |
| Tumor size | ||
| ≤ 5cm | UNDEF | UNDEF |
| > 5cm | 4.4 | 5.5 |
| Percent survival (%) | ||
| at 2 years | 77% | 79% |
| at 5 years | 66% | 71% |
| at 10 years | 53% | 60% |
TABLE 3:
Univariate analysis of variables of overall and disease-specific survival of patients with synovial cell sarcoma of the head and neck
| Overall survival (OS) | Disease-specific survival (DSS) | |||
|---|---|---|---|---|
| Characteristic | HR (95% CI) | log rank p-value | HR (95% CI) | log rank p-value |
| Age | 1.02 (1.01–1.04) | <0.001 | 1.11 (1.08–1.15) | <0.001 |
| Sex | 1.01 (0.60–1.72) | 0.963 | 1.02 (0.55–1.88) | 0.948 |
| Race | ||||
| White vs. Black | 0.88 (0.53–1.23) | 0.398 | 0.74 (0.34–1.68) | 0.150 |
| White vs. Asian | 0.99 (0.91–1.06) | 0.218 | 1.03 (0.27–2.52) | 0.351 |
| White vs. Pacific Islander | 0.53 (0.31–0.75) | 0.004 | 0.22 (0.11–0.63) | <0.001 |
| White vs. Native American | 0.84 (0.49–2.01) | 0.517 | 0.77 (0.26–1.43) | 0.567 |
| Black vs. Asian | 1.77 (0.33–2.45) | 0.154 | 2.19 (0.07–8.43) | 0.156 |
| Black vs. Pacific Islander | 1.34 (1.17–1.51) | 0.049 | 1.48 (0.35–9.11) | 0.079 |
| Black vs. Native American | 0.73 (0.22–1.34) | 0.512 | 0.84 (0.08–2.33) | 0.477 |
| Asian vs. Pacific Islander | 0.58 (0.32–0.74) | 0.004 | 0.48 (0.31–0.79) | 0.004 |
| Asian vs. Native American | 0.26 (0.11–0.42) | <0.001 | 0.30 (0.07–0.89) | <0.001 |
| Pacific Islander vs. Native American | 1.11 (0.22–2.03) | 0.257 | 1.23 (0.16–3.55) | 0.257 |
| Histological type | 1.18 (0.95–1.47) | 0.051 | 1.10 (0.85–1.42) | 0.133 |
| Primary site | 1.03 (0.96–1.10) | 0.845 | 1.02 (0.94–1.11) | 0.927 |
| Grade | 1.62 (0.68–3.84) | 0.267 | 1.79 (0.66–4.85) | 0.248 |
| Stage | 2.10 (1.38–3.20) | 0.001 | 1.76 (1.03–3.03) | 0.187 |
| Surgery performed | 0.54 (0.21–1.35) | 0.177 | 0.52 (0.19–1.46) | 0.205 |
| Radiation therapy | 1.49 (0.83–2.66) | 0.176 | 0.29 (0.12–0.68) | 0.003 |
| Treatment modality | ||||
| Surgery + Radiation vs. Surgery only | 0.76 (0.42–1.38) | 0.359 | 0.36 (0.15–0.86) | 0.016 |
| Surgery + Radiation vs. Radiation only | 2.44 (0.75–7.96) | 0.116 | 2.70 (0.82–8.87) | 0.094 |
| Surgery + Radiation vs. No therapy | 0.58 (0.08–4.20) | 0.582 | UNDEFINED | 0.207 |
| Surgery only vs. Radiation only | 0.33 (0.29–2.01) | 0.063 | UNDEFINED | 0.112 |
| Surgery only vs. No therapy | 0.76 (0.10–5.89) | 0.760 | UNDEFINED | 0.446 |
| Radiation only vs. No therapy | 0.24 (0.05–2.28) | 0.273 | UNDEFINED | 0.072 |
| Size > 5 cm | 2.35 (1.26–4.37) | 0.005 | 2.45 (1.22–4.90) | 0.009 |
In our multivariate analysis model, stage at presentation (aHR 3.86, CI 2.01–7.44), and size >5 cm (aHR 3.60, CI 1.43–9.08) were found to be independent predictors of OS (Table 4). No variables were significant predictors of DSS. The multivariate analysis model was also used to ascertain the independent effects of case variables on patients with patients with tumors ≤5 cm and >5 cm as separate cohorts. For tumors ≤5 cm (n=77), stage at presentation (aHR 3.10, CI 1.46–6.60) alone was an independent predictor of OS. No variables were significant predictors of DSS in this cohort. Similarly for tumors >5 cm (n=45), stage at presentation (aHR 5.32, CI 1.49–18.98) was the sole independent predictor of OS. Again, no variables were significant predictors of DSS in this cohort. Because surgical resection was performed in a significant majority of the patients, the majority of multivariate analysis models have an undefined hazard ratio and confidence interval because of constant or linearly dependent covariates reducing the degrees of freedom for surgical resection as an independent determinant.
TABLE 4:
Cox proportional hazards model for overall and disease-specific survival of patients with synovial cell sarcoma of the head and neck
| Overall survival (OS) | Disease-specific survival (DSS) | |||
|---|---|---|---|---|
| Characteristic | aHR (95% CI) | p-value | aHR (95% CI) | p-value |
| Overall (n=167) | ||||
| Age | 1.03 (1.00–1.07) | 0.080 | 0.99 (0.95–1.03) | 0.540 |
| Race | 1.09 (0.50–2.38) | 0.832 | 0.88 (0.40–1.96) | 0.756 |
| Histological subtype | 0.84 (0.56–1.24) | 0.371 | 0.85 (0.54–1.33) | 0.466 |
| Surgery performed | UNDEFINED | 0.984 | UNDEFINED | 0.986 |
| Radiation therapy | 0.70 (0.24–2.07) | 0.519 | 1.20 (0.32–4.52) | 0.792 |
| Stage | 3.86 (2.01–7.44) | <0.001 | 2.14 (1.00–4.58) | 0.051 |
| Size >5 cm | 3.60 (1.43–9.08) | 0.007 | 2.87 (0.98–8.35) | 0.054 |
| Size ≤5 cm (n=77) | ||||
| Age | 1.03 (0.98–1.08) | 0.198 | 1.01 (0.95–1.07) | 0.749 |
| Race | 1.29 (0.44–3.79) | 0.648 | 1.13 (0.39–3.26) | 0.818 |
| Histological subtype | 0.90 (0.48–1.67) | 0.732 | 0.84 (0.42–1.67) | 0.615 |
| Surgery performed | UNDEFINED | UNDEF | UNDEFINED | UNDEF |
| Radiation therapy | 0.86 (0.13–5.50) | 0.873 | 1.44 (0.15–13.96) | 0.754 |
| Stage | 3.10 (1.46–6.60) | 0.003 | 2.23 (0.89–5.61) | 0.089 |
| Size >5 cm (n=45) | ||||
| Age | 0.98 (0.92–1.05) | 0.565 | 0.95 (0.89–1.02) | 0.169 |
| Race | 0.40 (0.09–1.78) | 0.229 | 0.46 (0.10–2.18) | 0.325 |
| Histological subtype | 0.43 (0.18–1.00) | 0.060 | 0.51 (0.19–1.36) | 0.179 |
| Surgery performed | UNDEFINED | 0.977 | UNDEFINED | 0.984 |
| Radiation therapy | 0.56 (0.11–2.72) | 0.470 | 0.94 (0.16–5.54) | 0.941 |
| Stage | 5.32 (1.49–18.98) | 0.010 | 2.79 (0.60–13.08) | 0.193 |
DISCUSSION
SCSHN is an extremely rare oncological entity that arises from primitive pluripotent mesenchymal stem cells unrelated to synovial tissue and accounts for less than 10% of all soft tissue sarcomas of the head and neck. It typically afflicts young adults with slight predilection for males and carries a poor prognosis. Optimal treatment of these tumors is controversial and is likely to vary according to the size of the tumor on presentation. Currently, our knowledge of the behavior of these tumors is based on case reports and single-institution studies as well as treatment of these tumors in the extremities where they more commonly present.11,16,19,21,23,25 Use of the SEER database allows for the analysis of treatment and outcomes of rare malignancies such as SCSHN with a greater statistical power than would be achievable through conventional chart review. The SEER database has been used to find determinants of survival in many types of head and neck cancer and has been validated in previous studies.31–35
There are inherent limitations in studies based on analysis of the SEER database. The database lacks information on patient comorbidities, margin status, and administration of chemotherapy, and there are also concerns regarding misclassification, as there is no centralized review by a head and neck pathologist. Furthermore, there is limited data on tumor histopathology – particularly mitotic indices, nuclear atypia, and gene and tumor marker expression – which is known to have important prognostic implications with such a sarcoma of primitive cell origin.36–39 This report represents, to our knowledge, the first effort analyzing SCSHN from an epidemiological perspective at the population level.
Our study cohort, which found a median age of 37 years at diagnosis, reinforces that SCSHN typically presents in the third to fifth decade of life.5 Our data also closely mirrors previous reports of a male:female predominance of 3:2.3,4 The median OS was 14.9 years and the median overall 2-, 5-, and 10-year survival was 77%, 66%, and 53%, respectively. This data is in concordance with previous studies performed that have demonstrated a 5-year survival of 40–70%.19–21,27,30 While age was found to significantly affect survival in univariate Kaplan-Meier analysis, it was not found to be an independent predictor of survival using a more robust Cox multivariate regression model. Of note, age has never been explicitly identified as a prognostic indicator for synovial sarcoma, though such a correlation has been reported with other head and neck malignancies.31 The most common subsites of SCSHN represented in the SEER data were the oral cavity and parotid gland; previous reports have indicated that the most common site in the head and neck is the parapharyngeal space.18 However, it should be noted that the primary location of the majority of the tumors (64.1%) were coded non-specifically as head and neck soft tissue.
Literature indicates that the mono-phasic spindle cell variant is the predominant histological subtype. This was confirmed by our study, which found that histologically confirmed mono-phasic spindle cell and biphasic variants represented 31.1% and 22.8% of cases, respectively. In concordance with previous reports, we found no cases of the mono-phasic, epithelioid cell variant which is noted to be the rarest subtype. Of note, the histologic subtype was not specified as a significant proportion of cases were coded as synovial sarcoma, NOS (46.1%). Although death occurred in fewer than 50% of patients with the biphasic variant and mono-phasic spindle cell variant was associated with worse survival compared to the overall cohort average (OS 6.8 years, DSS 8.7 years), histological subtype was not associated with statistically significant differences in survival in either Kaplan-Meier or multivariate Cox regression analysis (Table 3, Table 4). This supports the consensus of multiple previous reports that showed histologic subtype does not affect survival.19,22
Tumor size at presentation has been identified as a prognostic indicator. The mean size at presentation in our cohort was 5.0 cm, which is comparable to those reported in the literature.18,20,22,24,36,38 While the effect of tumor size on survival likely occurs on a continuous spectrum, there has been some controversy surrounding a critical size cutoff beyond which there is a precipitous change in prognosis. In a meta-analysis of 93 patients with histologically confirmed SCSHN, Wushou and Miao found that tumors >5 cm in diameter had a significantly higher risk of local tumor recurrence, distant metastasis, and overall mortality compared with counterparts ≤5 cm.20 Similar to our study, tumor size >5 cm was the only independent adverse prognostic factor for determining OS in their cohort. Harb et al. also found size >5 cm to be a pivotal prognostic indicator, with systemic neoadjuvant chemotherapy being recommended for tumors exceeding this critical size.22 However, some studies have posited that 4 cm may be a more appropriate critical size cutoff.17,19 Crowson et al.’s analysis of 28 cases at the Mayo Clinic demonstrated that tumor size >4 cm negatively impacted survival, while an analytic pivot point of 5 cm produced equivocal survival distribution results. In our present analysis, size >5 cm was associated with significant negative impact on OS and DSS using univariate Kaplan-Meier analysis (OS p=0.005, DSS p=0.009, Figure 1), and on OS in multivariate Cox regression analysis (HR 3.60, CI 1.43–9.09, p=0.007), while an analytic size cutoff of 4 cm found no significant survival difference. Our multivariate analysis separating tumors at a critical size cutoff of 5 cm did not reveal any differentially significant prognostic indicators; in both cohorts only stage at presentation was an independent predictor of survival (Tumors ≤5 cm p=0.003, tumors >5 cm p=0.010). Age, race, surgical resection, radiation therapy, and histological subtype did not reach statistical significance as independent prognostic indicators in the multivariate analysis.
Surgical therapy was the mainstay of treatment for SCSHN for patients in the SEER database, and was performed in 89.8% of patients. Radiation therapy was performed in 64.7% of patients. This reflects the use of these two modalities as the mainstay of treatment of SCSHN with chemotherapy more frequently used for large tumors, extensive or recurrent disease, and high-risk sites of presentation such as the skull base or paraspinal neck.27,29,30,40 Interestingly, neither surgical resection nor radiotherapy was found to have a statistically significant survival advantage using multivariate analysis on either the overall or cohorts separated by size at the 5 cm critical size cutoff. This may reflect the near universal treatment of these tumors with surgical resection and the high propensity for adjuvant radiotherapy for disease control. Indeed, such a small percentage of patients did not undergo surgical resection that hazard ratios and associated p-values were not defined because of constant or linearly dependent covariates in the model (Table 4). Another potential reason that surgical and radiation therapy did not demonstrate significant survival advantages may be due to recurrent disease, which is known to occur in up to 40% of cases and more frequently with positive margins, acting as a treatment confounder.11,19,21,22 As previously noted, the SEER database does not provide information on the technical approach or extent of surgical resection, margin status, or recurrent disease. Given the controversy surrounding the extent of surgical resection, further studies investigating the optimal surgical approach will be important to optimize treatment. Interestingly, radiation therapy was associated with an improved DSS in our univariate Kaplan-Meier analysis (p=0.003), reflecting the importance of radiotherapy as an adjuvant treatment for SCSHN. Despite limited primary evidence in the literature with such a rare and poorly studied disease, there is near unanimous support for treatment with wide surgical resection and postoperative adjuvant therapy for locoregional control of SCSHN, with the benefit of chemotherapy remaining controversial.5,11–15,17,19,23–28
CONCLUSIONS
In summary, here we present the first population based study of SCSHN. Our study demonstrates a median overall survival of 14.9 years. Significant independent determinants of survival include size of tumor and stage at presentation. Given the mean size at presentation of 5.0 cm and the significantly reduced survival of patient with tumors >5 cm in diameter, it is logical to adopt 5 cm as an analytical pivot point associated with poor prognosis. Radiation therapy was associated with increased DSS on univariate Kaplan-Meier analysis, while surgical resection was not associated with statistically significantly improved survival outcomes. Neither treatment modality was associated with improved survival on multivariate analysis. These results belie the formidable challenge that SCSHN represents as the optimal primary and adjuvant treatment need to be more clearly elucidated.
ACKNOWLEDGMENTS:
This work was funded by the NIH/ National Center for Advancing Translational Science (NCATS) UCLA CTSI grant number ULITR000124. We thank the UCLA Statistical Consulting Group through the Institute for Digital Research and Education (IDRE) for assistance with statistical analysis.
Footnotes
Conflict of Interest: None
Financial Disclosures: None
REFERENCES
- 1.Carrillo R, Rodriguez-Peralto JL, Batsakis JG. Synovial sarcomas of the head and neck. Ann Otol Rhinol Laryngol 1992;101(4):367–370. [DOI] [PubMed] [Google Scholar]
- 2.O’Sullivan PJ, Harris AC, Munk PL. Radiological features of synovial cell sarcoma. Br J Radiol 2008;81(964):346–356. [DOI] [PubMed] [Google Scholar]
- 3.Pai S, Chinoy RF, Pradhan SA, D’Cruz AK, Kane S V, Yadav JN. Head and neck synovial sarcomas. J Surg Oncol 1993;54(2):82–86. [DOI] [PubMed] [Google Scholar]
- 4.Moore DM, Berke GS. Synovial sarcoma of the head and neck. Arch Otolaryngol Head Neck Surg 1987;113(3):311–313. [DOI] [PubMed] [Google Scholar]
- 5.Amble FR, Olsen KD, Nascimento AG, Foote RL. Head and neck synovial cell sarcoma. Otolaryngol Head Neck Surg 1992;107(5):631–637. [DOI] [PubMed] [Google Scholar]
- 6.Turc-Carel C, Dal Cin P, Limon J, Li F, Sandberg AA. Translocation X;18 in synovial sarcoma. Cancer Genet Cytogenet 1986;23(1):93. [DOI] [PubMed] [Google Scholar]
- 7.Cihak RA, Lydiatt WM, Lydiatt DD, Bridge JA. Synovial sarcoma of the head and neck: chromosomal translation (X;18) as a diagnostic aid. Head Neck 1997;19(6):549–553. [DOI] [PubMed] [Google Scholar]
- 8.Birdsall S, Osin P, Lu YJ, Fisher C, Shipley J. Synovial sarcoma specific translocation associated with both epithelial and spindle cell components. Int J Cancer 1999;82(4):605–608. [DOI] [PubMed] [Google Scholar]
- 9.Dimitriadis E, Rontogianni D, Kyriazoglou A, et al. Novel SYT-SSX fusion transcript variants in synovial sarcoma. Cancer Genet Cytogenet 2009;195(1):54–58. [DOI] [PubMed] [Google Scholar]
- 10.Sturgis EM, Potter BO. Sarcomas of the head and neck region. Curr Opin Oncol 2003;15(3):239–252. [DOI] [PubMed] [Google Scholar]
- 11.Salcedo-Hernández RA, Lino-Silva LS, Luna-Ortiz K. Synovial sarcomas of the head and neck: comparative analysis with synovial sarcoma of the extremities. Auris Nasus Larynx 2013;40(5):476–480. [DOI] [PubMed] [Google Scholar]
- 12.Barkan GA, El-Naggar AK. Primary synovial sarcoma of the parotid gland. Ann Diagn Pathol 2004;8(4):233–236. [DOI] [PubMed] [Google Scholar]
- 13.Mamelle G, Richard J, Luboinski B, Schwaab G, Eschwege F, Micheau C. Synovial sarcoma of the head and neck: an account of four cases and review of the literature. Eur J Surg Oncol 1986;12(4):347–349. [PubMed] [Google Scholar]
- 14.Miloro M, Quinn PD, Stewart JC. Monophasic spindle cell synovial sarcoma of the head and neck: report of two cases an review of the literature. J Oral Maxillofac Surg 1994;52(3):309–313. [DOI] [PubMed] [Google Scholar]
- 15.Wang H, Zhang J, He X, Niu Y. Synovial sarcoma in the oral and maxillofacial region: report of 4 cases and review of the literature. J Oral Maxillofac Surg 2008;66(1):161–167. [DOI] [PubMed] [Google Scholar]
- 16.Olsen RJ, Lydiatt WM, Koepsell SA, et al. C-erb-B2 (HER2/neu) expression in synovial sarcoma of the head and neck. Head Neck 2005;27(10):883–892. [DOI] [PubMed] [Google Scholar]
- 17.Bukachevsky RP, Pincus RL, Shechtman FG, Sarti E, Chodosh P. Synovial sarcoma of the head and neck. Head Neck 14(1):44–48. [DOI] [PubMed] [Google Scholar]
- 18.Al-Daraji W, Lasota J, Foss R, Miettinen M. Synovial sarcoma involving the head: analysis of 36 cases with predilection to the parotid and temporal regions. Am J Surg Pathol 2009;33(10):1494–1503. [DOI] [PubMed] [Google Scholar]
- 19.Crowson MG, Lalich I, Keeney MG, Garcia JJ, Price DL. Clinicopathologic factors and adjuvant treatment effects on survival in adult head and neck synovial cell sarcoma. Head Neck 2014. [DOI] [PubMed]
- 20.Wushou A, Miao X-C. Tumor size predicts prognosis of head and neck synovial cell sarcoma. Oncol Lett 2015;9(1):381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kusuma S, Skarupa DJ, Ely KA, Cmelak AJ, Burkey BB. Synovial sarcoma of the head and neck: a review of its diagnosis and management and a report of a rare case of orbital involvement. Ear Nose Throat J 2010;89(6):280–283. [PubMed] [Google Scholar]
- 22.Harb WJ, Luna MA, Patel SR, Ballo MT, Roberts DB, Sturgis EM. Survival in patients with synovial sarcoma of the head and neck: association with tumor location, size, and extension. Head Neck 2007;29(8):731–740. [DOI] [PubMed] [Google Scholar]
- 23.Simunjak B, Petric V, Bedekovic V, Cupić H, Hat J. Dimensions and outcome of synovial sarcoma of the head and neck: case presentation and review of the literature. J Otolaryngol 2005;34(6):420–423. [DOI] [PubMed] [Google Scholar]
- 24.Stanelle EJ, Christison-Lagay ER, Healey JH, Singer S, Meyers PA, La Quaglia MP. Pediatric and adolescent synovial sarcoma: multivariate analysis of prognostic factors and survival outcomes. Ann Surg Oncol 2013;20(1):73–79. [DOI] [PubMed] [Google Scholar]
- 25.Kartha SS, Bumpous JM. Synovial cell sarcoma: diagnosis, treatment, and outcomes. Laryngoscope 2002;112(11):1979–1982. [DOI] [PubMed] [Google Scholar]
- 26.Kampe CE, Rosen G, Eilber F, et al. Synovial sarcoma. A study of intensive chemotherapy in 14 patients with localized disease. Cancer 1993;72(7):2161–2169. [DOI] [PubMed] [Google Scholar]
- 27.Roth JA, Enzinger FM, Tannenbaum M. Synovial sarcoma of the neck: a followup study of 24 cases. Cancer 1975;35(4):1243–1253. [DOI] [PubMed] [Google Scholar]
- 28.Song S, Park J, Kim HJ, et al. Effects of Adjuvant Radiotherapy in Patients With Synovial Sarcoma. Am J Clin Oncol 2014. [DOI] [PubMed]
- 29.Rosen G, Forscher C, Lowenbraun S, et al. Synovial sarcoma. Uniform response of metastases to high dose ifosfamide. Cancer 1994;73(10):2506–2511. [DOI] [PubMed] [Google Scholar]
- 30.Shmookler BM, Enzinger FM, Brannon RB. Orofacial synovial sarcoma: a clinicopathologic study of 11 new cases and review of the literature. Cancer 1982;50(2):269–276. [DOI] [PubMed] [Google Scholar]
- 31.Mallen-St Clair J, Arshi A, Tajudeen B, Abemayor E, St John M. Epidemiology and treatment of lacrimal gland tumors: a population-based cohort analysis. JAMA Otolaryngol Head Neck Surg 2014;140(12):1110–1116. [DOI] [PubMed] [Google Scholar]
- 32.Tajudeen BA, Arshi A, Suh JD, St John M, Wang MB. Importance of tumor grade in esthesioneuroblastoma survival: a population-based analysis. JAMA Otolaryngol Head Neck Surg 2014;140(12):1124–1129. [DOI] [PubMed] [Google Scholar]
- 33.Peng KA, Grogan T, Wang MB. Head and neck sarcomas: analysis of the SEER database. Otolaryngol Head Neck Surg 2014;151(4):627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mahal BA, Inverso G, Aizer AA, Bruce Donoff R, Chuang S-K. Impact of African-American race on presentation, treatment, and survival of head and neck cancer. Oral Oncol 2014;50(12):1177–1181. [DOI] [PubMed] [Google Scholar]
- 35.Khan MN, Kanumuri V V, Raikundalia MD, et al. Sinonasal melanoma: survival and prognostic implications based on site of involvement. Int Forum Allergy Rhinol 2013. [DOI] [PubMed]
- 36.Meer S, Coleman H, Altini M. Oral synovial sarcoma: a report of 2 cases and a review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 2003;96(3):306–315. [DOI] [PubMed] [Google Scholar]
- 37.Ordóñez NG, Mahfouz SM, Mackay B. Synovial sarcoma: an immunohistochemical and ultrastructural study. Hum Pathol 1990;21(7):733–749. [DOI] [PubMed] [Google Scholar]
- 38.Skytting BT, Bauer HC, Perfekt R, Nilsson G, Larsson O. Ki-67 is strongly prognostic in synovial sarcoma: analysis based on 86 patients from the Scandinavian Sarcoma group register. Br J Cancer 1999;80(11):1809–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coindre J-M, Pelmus M, Hostein I, Lussan C, Bui BN, Guillou L. Should molecular testing be required for diagnosing synovial sarcoma? A prospective study of 204 cases. Cancer 2003;98(12):2700–2707. [DOI] [PubMed] [Google Scholar]
- 40.Lee N, Shin E. Treatment outcomes for patients with synovial sarcoma of the head and neck. Expert Rev Anticancer Ther 2008;8(3):371–373. [DOI] [PubMed] [Google Scholar]