

Abstract
The progeroid phenotype of mitochondrial DNA (mtDNA) mutator mice has been nebulously attributed to general mitochondrial “dysfunction”, though few studies have rigorously defined the bioenergetic consequences of accumulating mtDNA mutations. Comprehensive mitochondrial diagnostics was employed to interrogate the bioenergetic properties of isolated cardiac mitochondria from mtDNA mutator mice and wild type littermates. Assessment of respiratory flux in conjunction with parallel measurements of mitochondrial free energy all point to the cause of respiratory flux limitations observed in mtDNA mutator mouse mitochondria being due to impairments within the energy transduction step catalyzed by the electron transport system in which NAD(P)H/NAD(P)+ free energy is transduced to the proton motive force (∆P). The primary bioenergetic consequence of this limitation appears to be hyper-reduction of NAD(P)H/NAD(P)+ redox poise across multiple substrate conditions, particularly evident at moderate to high respiration rates. This hyper-reduced phenotype appears to result from specific reductions in both complex I and complex IV expression, presumably due to compromised mtDNA integrity. Translation of these findings to the working heart would suggest that the primary biological consequence of accumulated mtDNA damage is accelerated electron leak driven by an increase in electron redox pressure for a given rate of oxygen consumption.
Keywords: Bioenergetics, mitochondrial DNA, DNA polymerase gamma, heart, reactive oxygen species
INTRODUCTION1
Although the vast majority of proteins involved in mitochondrial function are encoded within the nuclear genome of the mammalian cell, the mitochondria maintain a separate genome, consisting of thousands of copies of circular mitochondrial DNA (mtDNA). In mice and humans, each double-stranded circle of mtDNA encodes 13 protein subunits of the electron transport system (ETS), including key subunits of both complex I (CI) and complex IV (CIV). This genome is replicated continuously through mechanisms separate from the cell cycle, using a single, unique, nuclear-encoded DNA polymerase (DNA polymerase gamma; Polg) [1]. The catalytic subunit of Polg (Polga) has DNA polymerase, 3’−5’ exonuclease, and 5’ deoxyribose phosphate lyase activities, making it the sole subunit responsible for synthesizing and proofreading new mtDNA.
The importance of preserving the integrity of the mitochondrial genome was first recognized through the identification of human diseases such as Leber’s hereditary optic neuropathy, the etiology of which was traced to a specific mtDNA substitution mutation [2]. Later, two separate labs bred transgenic mice overexpressing an exonuclease-deficient version of Polg (Polgm/m) in which the catalytic residue of the Polga exonuclease domain was mutated from aspartic acid to alanine (D257A) [3,4]. Loss of proofreading capabilities lead to the accumulation of single base substitution and deletion mutations randomly within the mitochondrial genome of the Polgm/m mice, inspiring the name “mtDNA mutator” mice. The resultant phenotype was akin to accelerated aging, including premature hair loss and wrinkled skin, osteoporosis, sarcopenia, anemia, cardiomyopathy, presbycusis, and a reduced lifespan [3,4].
In the literature, the Polgm/m phenotype has been nebulously attributed to general mitochondrial “dysfunction”. Evidence of this “dysf unction” includes reported abnormalities in the size and shape of the mitochondria [3], reduced maximal oxidative ATP production [3], elevated apoptotic activity [4], and increased production of reactive oxygen species (ROS) [5,6]. In particular, increased ROS generation appears to contribute significantly to the progeroidphenotype of the Polgm/m mice, as increasing the redox scavenging capacity of the mitochondria has been shown to lengthen their lifespan, slow the accumulation of mtDNA deletions, as well as rescue some aspects of cardiac function [6,7]. However, no study to date has directly identified the source(s) of ROS in Polgm/m mice, nor sufficiently characterized how an increased frequency of mtDNA mutations may alter the underlying bioenergetic efficiency of Polgm/m mitochondria in favor of accelerated ROS production (i.e., electron leak).
The objective of the present study was to evaluate the bioenergetic properties of isolated cardiac mitochondria from Polgm/m mice using a mitochondrial diagnostics assay platform [8]. The beauty of this platform lies in its ability to provide unbiased diagnostic coverage of a given mitochondrial network at a level approaching that of traditional molecular omics. In brief, multiple distinct bioenergetic readouts are produced from a single mitochondrial preparation using a modified version of the creatine kinase energetic clamp. These include mitochondrial respiratory control, membrane potential, NAD(P)H/NAD(P)+, and dehydrogenase flux/enzyme activity. With the entire collection of assays, a given change in respiratory control across disparate mitochondrial populations can be reasonably assigned to one of three main bioenergetic control nodes: 1. Matrix Dehydrogenases, 2. Electron transport system (ETS), 3. ATP synthesis. Additional interrogation within each control node is made possible by comparing results from the disparate substrate combinations (each substrate mix activates a unique set of dehydrogenases and ETS complexes), as well as the additional measurements of dehydrogenase flux and enzyme activity.
MATERIALS AND METHODS
All animal studies were approved by the East Carolina University Institutional Animal Care and Use Committee. Polgm/m (PolgaD257A/D257A) and wildtype (WT) littermates were aged to ~13 months prior to experimentation. All mice were housed in a temperature (22°C) and light controlled (12h light/12h dark) room and given free access to food and water. At the time of tissue harvest, mice were anesthetized with isofluorane, and hearts (complete left and right ventricles) were removed and subjected to mitochondrial isolation.
Chemical & Reagents:
Unless otherwise stated, all chemicals were purchased from Sigma-Aldrich. Tris salts of phosphocreatine (Cat #P1937) and ATP (Cat #A9062) were purchased from Sigma-Aldrich. Potassium pyruvate was purchased from Combi-Blocks (Cat #QA-1116). Potassium NADP+ was purchased from Ark-Pharm (Cat #AK671068). Amplex UltraRed (Cat #A36006), and Tetramethylrhodamine methyl ester (Cat #T6428; TMRM) were purchased from Thermo Fisher Scientific.
Mitochondrial Isolation:
Differential centrifugation was employed to prepare isolated mitochondria from heart tissue. The following buffers were utilized for all isolations: Buffer A - MOPS (50mM; pH=7.1), KCl (100mM), EGTA (1mM), MgSO4 (5mM); Buffer B - Buffer A, supplemented with bovine serum albumin (BSA; 2g/L). Hearts were excised and immediately placed in ice-cold Buffer B. All tissues were minced and then homogenized via a Teflon pestle and borosilicate glass vessel. Tissue homogenates were centrifuged at 600 × g for 10 minutes at 4°C. Supernatant from each tissue was then filtered through thin layers of gauze and subjected to an additional centrifugation at 10,000 × g for 10 minutes at 4°C. Mitochondrial pellets were washed in 1.4ml of Buffer A, transferred to microcentrifuge tubes and centrifuged at 10,000 × g for 10 minutes at 4°C. Buffer A was aspirated from each tube and final mitochondrial pellets were suspended in 100–200μL of Buffer A. Protein content was determined via the Pierce BCA protein assay. Functional assays involving isolated mitochondria were carried out in the following buffers: Buffer C - Potassium-MES (105mM; pH=7.2), KCl (30mM), KH2PO4 (10mM), MgCl2 (5mM), EGTA (1mM), BSA (2.5g/L); Buffer D - HEPES (20mM; pH=8.0), KCl (100mM), KH2PO4 (2.5mM), MgCl2 (2.5mM), Glycerol (1%).
Preparation of mouse mitochondrial pellets for western blotting:
A portion of each mitochondrial preparation was aliquoted and subsequently lysed in CelLytic M (Sigma-Aldrich; C2978) supplemented with protease inhibitor cocktail. The lysate was then separated into two aliquots (one for enzyme activity assays and one for Western blotting). Following centrifugation at 10,000 × g for 10min at 4°C, the supernatant from the Western blot lysate was subjected to sonication. Protein concentration was determined via the BCA method and the samples were diluted in Laemmli’s loading buffer, supplemented with dithiothreitol (DDT; 5mM). Thirty micrograms of protein were resolved by SDS-PAGE, transferred to nitrocellulose, blocked for ~1h in 5% milk prepared with TBS followed by Western blotting with specific antibodies. Antibodies employed herein were: OXPHOS cocktail (abcam: #ab110413). Band intensity was normalized to total protein via the use of TGX Stain-Free precast gels (Bio-Rad).
Mitochondrial Respiratory Control:
High-resolution O2 consumption measurements were conducted using the Oroboros Oxygraph-2K (Oroboros Instruments). All experiments were carried out at 37°C in a 2mL reaction volume. Steady-state oxygen consumption rates (JO2) were determined within individual experiments using a modified version of the creatine kinase energetic clamp technique [9,10]. In this assay, the free energy of ATP hydrolysis (∆G’ATP, depicted throughout the manuscript as ∆GATP) can be calculated based on known amounts of creatine (Cr), phosphocreatine (PCr) and ATP in combination with excess amounts of creatine kinase (CK) and the equilibrium constant for the CK reaction (i.e., KCK). Calculation of ∆G’ATP was performed according to the following formula:
where ∆G’°ATP is the standard apparent transformed Gibbs energy (under a specified pH, ionic strength, free magnesium and pressure), R is the gas constant (8.3145 J/kmol) and T is temperature in kelvin (310.15). Given that experiments were performed via sequential additions of PCr, both the ∆G’°ATP and K’CK were determined at each titration step based on the changes in buffer ionic strength and free magnesium, as previously described [11,12]. For complete details regarding the calculation of ∆G’ATP at each titration point see [8]. Buffer for all assays was Buffer C, supplemented with ATP (5mM), Cr (5mM), PCr (1mM), and CK (20U/mL). To begin, isolated mitochondria (0.05mg/mL) were added to assay buffer, followed by the addition of respiratory substrates. The following substrate conditions were tested: pyruvate/malate - (Pyr/M; 5/2.5mM), palmitoyl-carnitine/malate - (Pc/M; 0.02/2.5mM), succinate/rotenone - (Succ/R; 10/0.005mM). Following substrate additions, sequential additions of PCr to 6, 9,15 and 21mM were performed to gradually slow JO2 back toward baseline. Plotting the calculated ∆GATP against the corresponding JO2 reveals a linear force-flow relationship, the slope of which represents the conductance/sensitivity of the entire respiratory system under specified substrate constraints as previously described [8].
Mitochondrial membrane potential (∆Ψ) and NAD(P)H/NAD(P)+ Redox:
Fluorescent determination of ∆Ψ and NAD(P)H/NAD(P)+ were carried out simultaneously via a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific). Determination of ∆Ψ via TMRM was done by recording the fluorescence ratio of the following excitation/emission parameters [Ex/Em, (572/590)/(551/590)] [13]. The 572/551 ratio was then converted to millivolts via a KCl standard curve performed in the presence of valinomycin as described previously [8,14]. In this protocol, isolated mitochondria energized with Succ/R were incubated in a potassium-free buffer in the presence of valinomycin, a potassium-specific ionophore. Assuming a matrix potassium concentration of 120mM, ΔΨ can be reasonably estimated by applying the Nernst equation and buffer ion concentrations resulting from sequential additions of KCl [14]. NAD(P)H excitation/emission parameters were 340/450. All experiments were carried out at 37°C in a 0.2mL reaction volume with continuous stirring. Buffer for all assays was Buffer C, supplemented with Cr (5mM), PCr (1mM), ATP (5mM), CK (20U/mL) and TMRM (0.2μM). To begin, isolated mitochondria (0.1mg/mL) were added to the assay buffer, followed by the addition of respiratory substrates (Pyr/M, Pc/M and Succ/R) and then sequential PCr additions to a final of 6, 9,15 and 21mM. Following the final PCr addition, cyanide (10mM) was added to induce a state of 100% reduction within the NAD(P)H/NAD(P)+ couple. The fluorescence (Ex/Em, 340/450) signal recorded in the presence of mitochondria alone without respiratory substrates was used as the 0% reduction state for the NAD(P)H/NAD(P)+ couple. NAD(P)H/NAD(P)+ during the entire experiment was expressed as a percentage of reduction according to the following formula: % Reduction = (F - F0%)/(F100% - F0%).
JNADH and JNADPH Production:
Mitochondrial lysates prepared in CelLytic M and supplemented with protease inhibitor cocktail were utilized for all assays. Rates of NADH and NADPH production were determined as described previously [8,15]. Buffer for the assays was Buffer C, supplemented with rotenone (0.005mM) and NAD+ (2mM) or NADP+ (2mM). For experiments designed to assess JNADH from the pyruvate dehydrogenase complex (PDH), the alpha-ketoglutarate dehydrogenase complex (AKGDH), and the branched-chain keto-acid dehydrogenase complex (BCKDH), cofactors coenzyme A (0.1mM) and thiamine pyrophosphate (0.3mM) were included. Assay buffer (200μL) was loaded into individual wells of96-well plate, followed by mitochondrial lysate (2–60μg/well). The assay was initiated with the addition of enzymatic substrates. In the assay, NADH and NADPH were determined via autofluorescence (Ex:Em 340/450). Fluorescence values were converted to pmol of NADH or NADPH via an NADH/NADPH standard curve. The following substrates were tested in parallel for each assay: pyruvate (5mM), alpha-ketoglutarate (10mM), α-keto-β-methylvalerate (5mM), glutamate (10mM), malate (5mM), isocitrate (5mM).
Complex V Activity Assay:
Complex V (CV) activity was assessed as described previously [8,16]. Buffer for the assay was Buffer D, supplemented with lactate dehydrogenase/pyruvate kinase (10U/mL), phosphoenoyl-pyruvate (5mM), rotenone (0.005mM) and NADH (0.2mM). Assay buffer (200μL) was loaded into individual wells of a 96-well plate, followed by mitochondrial lysate (2μg/well). Assays were done in the absence and presence of oligomycin (0.005mM) to calculate the oligomycin-sensitive rates of ATP hydrolysis. The assay was initiated with the addition of ATP (5mM). In the assay, NADH oxidation and ATP hydrolysis occur at a 1:1 stoichiometry and thus CV activity (pmol of ATP/sec/mg) was determined via tracking the degradation in the NADH autofluorescence (Ex:Em 376/450) signal upon ATP addition. Fluorescence values were converted to pmol of NADH via an NADH standard curve.
Hydroxyacyl-CoA Dehydrogenase Activity:
Buffer for the assay was Buffer D, supplemented with rotenone (0.005mM) and NADH (0.2mM). Assay buffer (200μL) was loaded into individual wells of a 96-well plate, followed by mitochondrial lysate (5μg/well). The assay was initiated with the addition of acetoacetyl-CoA (0.2mM). The activity of hydroxyacyl-CoA dehydrogenase was determined via tracking the degradation in the NADH autofluorescence (Ex:Em 340/450) signal upon acetoacetyl-CoA addition. Fluorescence values were converted to pmol of NADH via an NADH standard curve.
Quantification and Statistical Analysis:
Data are presented as mean ± SEM. Statistical analysis was performed using t-tests. Figures were generated using GraphPad Prism (Version 7.0). The level of significance was set at P < 0.05.
RESULTS & DISCUSSION
Loss of mitochondrial DNA integrity limits respiratory flux by shifting ETS stoichiometry.
To determine the bioenergetic impact of compromised mtDNA integrity, cardiac mitochondria were isolated from Polgm/m and wild-type (WT) littermates and subsequently interrogated using a mitochondrial diagnostics workflow [8]. In this system, mitochondrial oxygen consumption is assessed under multiple substrate combinations across a physiological span of clamped ATP/ADP (i.e., free energy of ATP hydrolysis; ∆GATP), effectively spanning the full range of respiratory flux (i.e., rest to exercise). The resulting linear relationship between ∆GATP (i.e., force) and JO2 (i.e., flux) represents respiratory “conductance” (i.e., reciprocal of resistance), wherein a steeper slope indicates greater respiratory sensitivity or responsiveness. Independent of substrate combination (Pyr/M, Pc/M and Succ/R), absolute JO2 across all values of ∆GATP was lower in Polgm/m mitochondria (Fig. 1A). Interestingly, although respiratory sensitivity quantified on an absolute scale was lower in mitochondria derived from Polgm/m mice (Fig 1B), normalization to the starting JO2 revealed no differences across genotypes in relative sensitivity (Fig 1C). These data imply that the lower absolute JO2 in Polgm/m mitochondria most likely arises from decreases in the number of active ETS complexes or ATP synthetic machinery, rather than inherent limitations of individual components per se. Consistent with this, immunoblot analysis of the respiratory complexes revealed specific reductions in complex I (CI) and complex IV (CIV) within Polgm/m mitochondria (Fig 1D & 1E). The expression (Fig 1E) and activity (Fig 1F) of complex V (CV) were unaffected by genotype. Based on these data, it appears that compromised mtDNA integrity primarily impacts respiratory complex stoichiometry, represented by CI and CIV depletion. Specificity with respect to CI and CIV likely stems from the fact that of the 13 protein-coding genes in the mitochondrial genome, 10 correspond to subunits of CI (7 genes) or CIV (3 genes).
Figure 1. Comprehensive assessment of mitochondrial respiratory control.
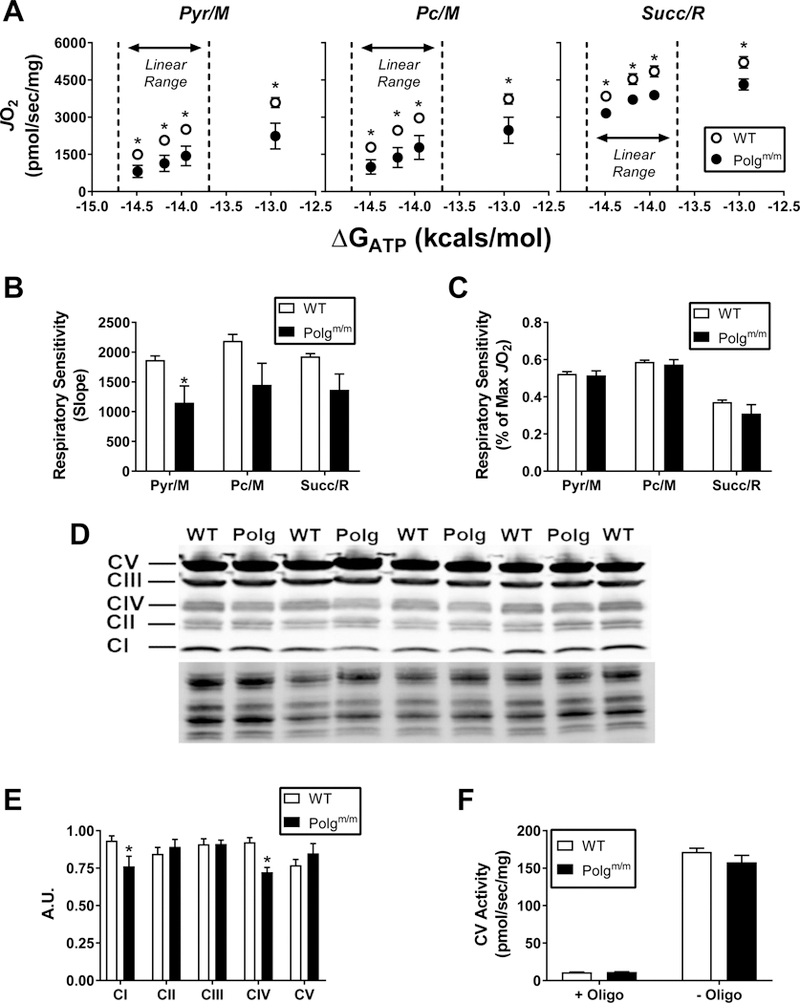
Cardiac isolated mitochondria were utilized for all experiments. (A) Relationship between mitochondrial JO2 versus ATP free energy (∆GATP) in mitochondria energized with Pyr/M, Pc/M andSucc/R. (B) Calculated slopes from the linear portions of the data depicted in panel A. Linear portions are located to the left of the dotted line on each graph. (C) Relative respiratory sensitivity whereby slopes were calculated after normalizing data to the starting JO2. (D) Western blotdepicting abundance of all ETS complexes. (E) Quantification of individual complex-specific bands normalized to total protein. (F) Complex V (CV) activity in the presence and absence of oligomycin, (Oligo). Data are mean ± SEM, N=5 (WT), N=4 (Polgm/m), *P<0.05.
Loss of CI/CIV abundance shifts the relationship between respiratory flux and mitochondrial free energy in favor of increased electron leak.
To determine the impact of partial depletion of CI/CIV on mitochondrial free energy maintenance, parallel measurements of mitochondrial membrane potential (∆Ψ) and NAD(P)H/NAD(P)+ redox poise were performed under experimental conditions identical to those used for respirometry assays. Measurements of ∆Ψ revealed no differences across genotypes for all substrates (Fig 2A). That said, normalization of ∆Ψ to respiratory flux revealeda leftward shift in Polgm/m mitochondria, particularly evident at the top end of flux (Fig 2B). Relative depolarization of ∆Ψ for a given rate of oxygen consumption (i.e., leftward shift in JO2 vs ∆Ψ) implies that the source of lower respiratory flux observed in Polgm/m mitochondria originates within the electron transport system (ETS). In line with this, the reduction state ofNAD(P)H/NAD(P)+ was nearly 2-fold higher in Polgm/m mitochondria (Fig 2C - representative trace with Pyr/M; Fig 2D; Pyr/M, Pc/M & Succ/R - quantified data). Unlike that observed for ∆Ψ, plotting NAD(P)H/NAD(P)+ redox against respiratory flux in the presence of Pyr/M and Pc/M revealed a dramatic rightward shift in Polgm/m mitochondria, once again most pronounced at the top end of respiratory flux (Fig 2E). It should be noted that the fluorescent signal corresponding to NAD(P)H gradually decreased over time in Polgm/m mitochondria energized with Pyr/M and Pc/M (Fig 2D). This effect mimics that seen in both genotypes in the presence of Succ/R (Fig 2D; Succ/R). Given that NADH and NADPH fluorescence are indistinguishable, decline in the total fluorescence signal (Ex:Em / 340:450) may reflect NADPH-specific oxidation. The activity of various NAD- (Fig 2F) and NADP-linked (Fig 2G) dehydrogenase enzymes were completely unaffected by genotype.
Figure 2. Parallel determinations of mitochondrial free energy maintenance and dehydrogenase flux.
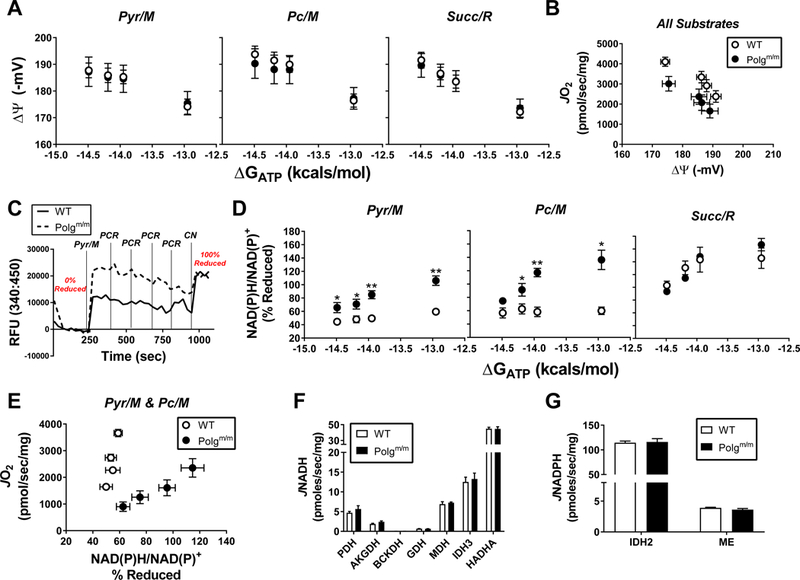
Relationship between (A) mitochondrial ΔΨ and (D) NAD(P)H/NAD(P)+ redox state versus ATP free energy (∆GATP) in mitochondria energized with Pyr/M, Pc/M and Succ/R. (B) Mitochondrial JO2 plotted against ΔΨ. Data from all substrates were pooled to illustrate the underlying relationship between JO2 and ΔΨ. (C) Representative trace from an NAD(P)H/NAD(P)+ phosphocreatine (PCR) titration experiment illustrating the normalization in redox state across groups following the addition of cyanide (CN). Experimental additions are indicated on the trace. (E) Mitochondrial JO2 plotted against NAD(P)H/NAD(P)+. Data from both Pyr/M and Pc/M conditions were pooled to illustrate the underlying relationship between JO2 and NAD(P)H/NAD(P)+. Rates of (F) JNADH and (G) JNADPH production from all dehydrogenases studied. Dehydrogenases included pyruvate (PDH), alpha-ketoglutarate (AKGDH), branched-chain ketoacid (BCKDH), glutamate (GDH), malate (MDH), isocitrate (IDH3; NAD-linked), Hydroxyacyl-CoA (HADHA), isocitrate (IDH2; NADP-linked) and malic enzyme (ME). (H) GOT2 activity. Fluorescence data were converted to NADH via standard curve. Data are mean ± SEM, N=5 (WT), N=4 (Polgm/m), *P<0.05, **P<0.001.
Comprehensive assessment of respiratory flux in conjunction with parallel measurements of mitochondrial free energy all point to the cause of respiratory flux limitations observed in Polgm/m mitochondria being due to impairments within the energy transduction step catalyzed by the ETS in which NAD(P)H/NAD(P)+ is transduced to ∆Ψ. The primary bioenergetic consequence of this limitation appears to be hyper-reduction of NAD(P)H/NAD(P)+ redox poise, particularly evident at moderate to high respiration rates. Given that NAD(P)H/NAD(P)+ redox has previously been validated as a marker of endogenous electron leak and thus superoxide formation, these data suggest that ETS limitations in Polgm/m mitochondria alter the relationship between respiratory flux and free energy maintenance in favor of heightened free radical leak (Fig 3). Collectively, the data contained herein lend credence to the multiple reports of heightened ROS production in the Polgm/m line by providing a bioenergetic mechanism for elevated electron leak. Translation of these findings to the working heart would suggest that the primary biological consequence of accumulated mtDNA damage is accelerated electron leak driven by an increase in electron redox pressure for a given rate of oxygen consumption.
Figure 3. Summary Diagnostics of Polgm/m mitochondria.
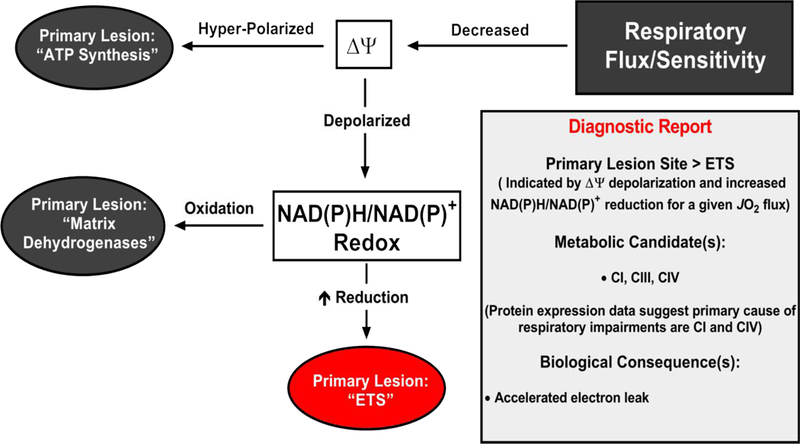
Diagnostic tree diagram depicting the primary bioenergetic lesion(s) induced by loss of mitochondrial DNA repair in the Polgm/m mice. Starting with loss of respiratory flux or sensitivity, potential primary lesion sites are depicted in the grey-filled ovals (“ATP synthesis”, “Matrix Dehydrogenases”) with the Polgm/m specific lesion site indicated in red (“ETS”). The experimental evidence in support of an “ETS” lesion site is provided in the “Diagnostic Report” box.
Supplementary Material
AKNOWLEDGMENTS
Funding: This work was supported in part by NIH grant R01HL125695 (JMM)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Abbreviations: AKGDH, alpha-ketoglutarate dehydrogenase complex; BCKDH, branched-chain keto-acid dehydrogenase complex; BSA, bovine serum albumin; CI, complex I; CIV, complex IV; CV, complex V; CK, creatine kinase; Cr, creatine; ∆Ψ, membrane potential; ∆GATP, free energy of ATP hydrolysis; DDT, dithiothreitol; ETS, electron transport system; JNAD(P)H, rate of NAD(P)H production; JO2, oxygen consumption rate; M, malate; mtDNA, mitochondrial DNA; Pc, palmitoyl-carnitine; PCr, phosphocreatine; PDH, pyruvate dehydrogenase complex; Polg, DNA polymerase gamma; Polgm/m, homozygous transgenic mtDNA mutator mouse line; Polga, catalytic subunit of Polg; Pyr, pyruvate; R, rotenone; ROS, reactive oxygen species; Succ, succinate
REFERENCES
- [1].Falkenberg M, Larsson N-G, Gustafsson CM, DNA Replication and Transcription in Mammalian Mitochondria, Annu. Rev. Biochem 76 (2007) 679–699. 10.1146/annurev.biochem.76.060305.152028. [DOI] [PubMed] [Google Scholar]
- [2].Wallace DC, Singh G, Lott MT, Hodge JA, Schurr TG, Lezza AM, Elsas LJ, Nikoskelainen EK, Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy, Science (80-. ) 242 (1988) 1427–1430. 10.1126/science.3201231. [DOI] [PubMed] [Google Scholar]
- [3].Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gldlöf S, Oldfors A, Wibom R, Tör nell J, Jacobs HT, Larsson NG, Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature 429 (2004) 417–423. 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- [4].Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA, Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging, Science (80-. ) 309 (2005) 481–484. 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- [5].Logan A, Shabalina IG, Prime TA, Rogatti S, Kalinovich AV, Hartley RC, Budd RC, Cannon B, Murphy MP, In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice, Aging Cell 13 (2014) 765–768. 10.1111/acel.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS, Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria, Aging Cell 9 (2010) 536–544. 10.1111/j.1474-9726.2010.00581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Shabalina IG, Vyssokikh MY, Gibanova N, Csikasz RI, Edgar D, Hallden-Waldemarson A, Rozhdestvenskaya Z, Bakeeva LE, Vays VB, Pustovidko AV, Skulachev MV, Cannon B, Skulachev VP, Nedergaard J, Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1, Aging (Albany. NY) 9 (2017) 315–339. 10.18632/aging.101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Fisher-Wellman K, Davidson M, Narowski T, Lin C-T, Koves TR, Muoio DM, Mitochondrial diagnostics: A multiplexed assay platform for comprehensive assessment of mitochondrial energy fluxes, Cell Rep In press. [DOI] [PMC free article] [PubMed]
- [9].Glancy B, Willis WT, Chess DJ, Balaban RS, Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria, Biochemistry 52 (2013) 2793– 2809. 10.1021/bi3015983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Messer JI, Jackman MR, Willis WT, Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria., Am. J. Physiol. Cell Physiol 286 (2004) C565–572. 10.1152/ajpcell.00146.2003. [DOI] [PubMed] [Google Scholar]
- [11].Golding EM, Teague WEJ, Dobson GP, Adjustment of K’ to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment, J. Exp. Biol 198 (1995) 1775–1782. [DOI] [PubMed] [Google Scholar]
- [12].Teague WEJ, Golding EM, Dobson GP, Adjustment of K’ for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength., J. Exp. Biol 199 (1996) 509–512. [DOI] [PubMed] [Google Scholar]
- [13].Scaduto RC, Grotyohann LW, Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives., Biophys. J 76 (1999) 469–477. 10.1016/S0006-3495(99)77214-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Krumschnabel G, Eigentler A, Fasching M, Gnaiger E, Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry, Methods Enzymol 542 (2014) 163–181. 10.1016/B978-0-12-416618-9.00009-1. [DOI] [PubMed] [Google Scholar]
- [15].Fisher-Wellman KH, Gilliam LAA, Te Lin C, Cathey BL, Lark DS, Darrell Neufer P, Mitochondrial glutathione depletion reveals a novel role for the pyruvate dehydrogenase complex as a key H2O2-emitting source under conditions of nutrient overload, Free Radic. Biol. Med 65 (2013) 1201–1208. 10.1016/j.freeradbiomed.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barrientos A, Fontanesi F, Díaz F, Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using polarography and spectrophotometric enzyme assays, Curr. Protoc. Hum. Genet 19 (2009) 19.3.1–19.3.14. 10.1002/0471142905.hg1903s63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.