

Abstract
Background:
In humans, deficiency of coagulation factor XI (FXI) may be associated with a bleeding disorder, but until recently FXI-deficient mice did not appear to have a hemostatic defect. A recent study, however, indicated that FXI-deficient mice display a moderate hemostatic defect in a saphenous vein bleeding (SVB) model.
Objectives:
To study the effect of FXI on bleeding in mice with normal levels of the FXI-substrate factor IX (FIX) and in mice lacking FIX (a murine model of hemophilia B).
Methods:
Wild type mice and mice lacking either FIX (F9-) or FXI (F11−/−) were tested in the SVB model. The plasma levels of FXI in F11−/− mice were manipulated by infusion of FXI or its active form FXIa, or by overexpressing FXI using hydrodynamic tail vein injection.
Results:
F9- mice demonstrated a significant defect in the SVB model, while F11−/− mice and wild type mice were indistinguishable. Intravenous infusion of FXI or FXIa into, or overexpression of FXI in, F9- mice improved hemostasis in the SVB model. Overexpression of a FXI variant lacking a FIX binding site also improved hemostasis in F9- mice.
Conclusions:
While we were unable to demonstrate a hemostatic defect in F11−/− mice in the SVB model, our results support the premise that supraphysiologic levels of FXI improve hemostasis in F9- mice through FIX-independent pathways.
Keywords: Bleeding, Factor IX, Factor XI, Hemostasis, Saphenous vein
INTRODUCTION
Factor XI (FXI) is the precursor of a plasma protease (FXIa) that contributes to thrombin generation primarily through activation of factor IX (FIX) [1]. FXI-deficiency was first described in humans in 1953 as plasma thromboplastin antecedent deficiency, a condition associated with a significant defect in surface-induced plasma coagulation, but a relatively mild propensity for abnormal bleeding [2–4]. FXI deficient patients seldom experience unprovoked hemorrhage, and excessive bleeding typically follows trauma to tissues with high intrinsic fibrinolytic activity such as the mouth, nose or urinary tract. Experience with FXI deficiency in humans indicates that the protein is required for hemostasis in some individuals for certain types of hemostatic challenges. But, it is clear that some people with severe FXI deficiency do not experience abnormal hemostasis, suggesting that variables unrelated to the plasma FXI level contribute to bleeding in FXI-deficient individuals.
FXI-deficiency has been linked to abnormal hemostasis in domesticated animals (cattle, dogs, and cats) [5–7], but not in several species commonly used in preclinical research. Rodents, rabbits and primates treated with a variety of agents that inhibit FXI/FXIa activity [8–10] or block FXI synthesis [11,12] do not exhibit an unusual bleeding propensity. We, and others, did not observe abnormal hemostasis in FXI-deficient (F11−/−) mice after tail transection [13,14]. These observations raise the possibility that FXI does not function as a hemostatic protein in all species. Given this, we were interested in the recent report by Ay and coworkers describing abnormal hemostasis in F11−/− mice after saphenous vein injury [15]. Here we describe our experience with a saphenous vein bleeding (SVB) model, and report that FXI can affect bleeding in mice with hemophilia B (FIX deficiency).
METHODS
Mice.
Studies with mice were approved by the Vanderbilt University Medical Center IACUC. C57Bl/6 male mice lacking FIX (F9-) or FXI (F11−/−) have been described [13,16]. The F9- and F11−/− genotypes were back-crossed to wild type C57Bl/6 mice (Jackson Laboratory) through ten generations. Males age 12 to 16 weeks were used. Wild type mice used in these studies were not littermates of F9- or F11−/− mice, and the investigators were not blinded to genotype.
Saphenous vein bleeding model (SVB) [17,18].
Mice were anesthetized with pentobarbital, and the ventral surface of the right hind limb was shaved. An incision was made along the length of the limb, and connective tissue was removed from around the saphenous vein. An entry hole in the vein midway between the femoral vein and distal saphenous branch was made with a 23-G needle. Blood was absorbed with gauze. Immediately after initial bleeding stopped (1–2 min), one blade of a Student Vannas spring scissors was introduced through the original entry hole, creating an incision in the vessel, and bleeding was observed for 30 min. Each time bleeding stopped, the formed clot was disrupted by wiping with gauze in the direction of blood flow to restart bleeding. Numbers of clots formed in 30 min and duration of bleeding episodes were recorded. No animal was allowed to bleed more than 30 minutes. Mice were sacrificed 30 min after incision by intracardiac injection of pentobarbital.
Supplementing F9- mice with FXI or FXIa.
F9- mice received infusions of human plasma-derived FXI or FXIa (Haematologic Technologies) in 50µL phosphate buffered saline (PBS) through the jugular vein to give an estimated final plasma concentration of 60 nM. Mice were tested in the SVB model within 5 min of infusion.
Expressing human FXI in mice by hydrodynamic tail vein injection (HTI).
Expression of human FXI was induced in F9- mice by HTI. cDNAs for wild type FXI (FXI-WT) [19] or FXI with alanine replacements for amino acids 183 to 185 (FXI-A183–185) [20] were introduced into an EEV600A expression vector (System Biosciences). FXI/EEV600A or empty vector (control) constructs (3.5µg) were diluted in 2 mL Lactated Ringer Solution, and infused into tail veins of F9- mice over 30 seconds. Mice were tested in the SVB model 72 hrs post-HTI. Blood was obtained at the completion of experiments to assess FXI expression by western blot using a monoclonal IgG (14E11) that recognizes mouse and human FXI [21].
Carotid artery thrombosis model [21].
FXI-WT, FXI containing the prekallikrein A3 domain (FXI/PKA3) or FXI-A183–185 was expressed in F11−/− or F9−/− mice using HTI. Twenty-four hrs post-HTI, the right common carotid artery of an anesthetized mouse was exposed and fitted with a Doppler probe (Model 0.5 VB, Transonic System). Two 1 × 1.5 mm filter papers (GB003, Schleicher & Schuell) saturated with 3.5% FeCl3 were applied to opposite sides of the artery for 3 min, followed by rinsing with PBS. Flow was monitored for 30 min. Blood was obtained at the completion of the experiment for assessing FXI expression.
Clotting assay.
FIX-deficient human plasma (35µL, George King Biomedical) was mixed with 35µL Tris-Buffered Saline containing 1% albumin with or without human TF (1pM, Innovin, Dade) and varying concentrations of FXIa (0 to 120 nM). After 3 min incubation, 35µL of 25mM CaCl2 was added and time to clot formation was measured on a Start4 coagulation analyzer (Diagnostica Stago).
Thrombin generation assay.
Thrombin generation was measured in plasma as described [22,23]. Briefly, FIX-deficient plasma (80μl) containing 415 μM Z-Gly-Gly-Arg-AMC (Bachem, Torrance, CA) was supplemented with TF (1pM) with or without FXIa (15nM). Additives were in 20μl volumes, and concentrations given are final concentrations. Thrombin generation was initiated by adding 10μl of 20 mM HEPES pH 7.4, 100 mM CaCl2, 6% BSA and fluorescence was monitored over 60 min on a Fluoroskan Ascent® fluorometer (Thermolab Systems OY, Helsinki). Thrombin generation was calculated using Calibrated Automated Thrombogram software Thrombinoscope ®.
RESULTS AND DISCUSSION
Bleeding after saphenous vein injury was significantly more severe in F9- mice than in wild type mice (p<0.05,) as reflected by fewer bleeding episodes (Fig.1A, white) and longer duration of each bleed (Fig.1B). The results are consistent with published data [15,17]. In contrast to results reported by Ay et al. [15], we did not observe significant differences between C57Bl/6 wild type and F11−/− mice in the number of clots (Fig.1A), average bleeding time (Fig.1B), time to first clot formation (Fig.1C), or average times of subsequent bleeding events ( Fig.1D). The differences between our results and those of Ay et al. could easily reflect subtle differences in technique, including the degree of injury and the manner in which clots were disturbed. Also, while F11−/− mice used in the two studies are of the same lineage [13], the colonies at the two institutions involved have been separate for at least four years. This may have allowed genetic drift to introduce factors that render members of one colony more susceptible to bleeding than the other.
Figure 1. Effects of FXI/FXIa on hemostasis in F9- mice and coagulation in FIX-deficient human plasma. Saphenous vein bleeding model (SVB).
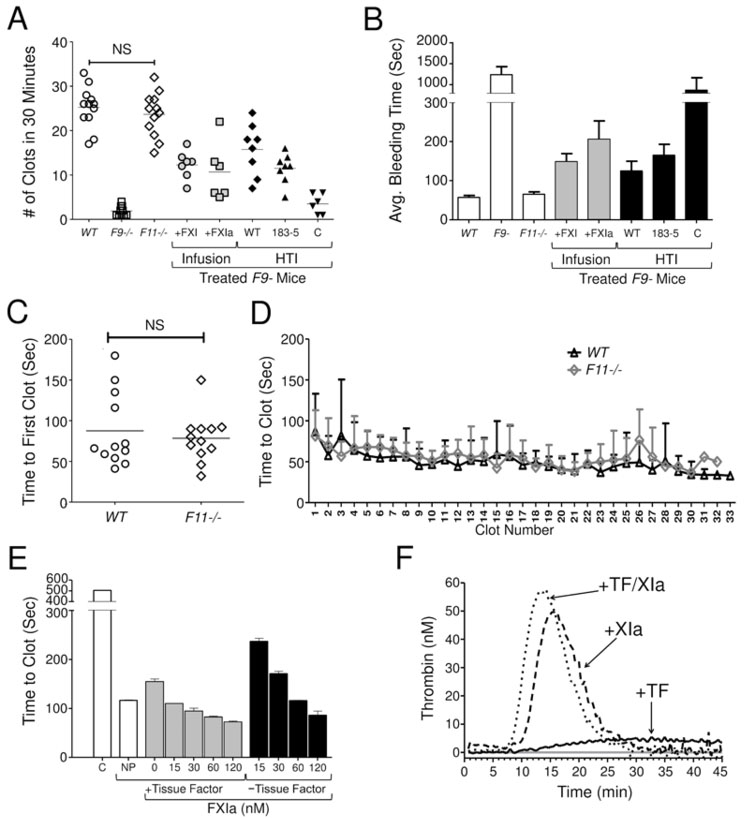
Each symbol indicates (A) the number of clotting events in one mouse over 30 min following vessel injury or (B) the average duration of bleeding events. For panels A and B, White Symbols/Bars indicate values for wild type (WT), FIX-deficient (F9-) and FXI-deficient (F11−/−) C57Bl/6 mice. The results for WT and F11−/− mice were not statistically different (p=0.4, NS). Gray Symbols/Bars represent F9- mice infused with human FXI or FXIa to an estimated plasma concentration of 60 nM tested in the SBV model starting five minutes after infuson. Black Symbols/Bars represent F9- mice tested 72 hours after HTI with expression constructs for FXI-WT (WT), FXI-Ala183–185 (183–5) or empty EEV600A vector (C - control). In panel A, each symbol indicates results for one mouse, and horizontal bars indicate means for each group. For panel B bars indicate average bleeding times ± one standard deviation. (D) Comparison of the time to first clot between WT and F11−/− mice. Results for the groups were not significantly different (p=0.7, NS) between the groups. (E) Comparison of the average durations of each specific bleeding event for WT and F11−/− mice. Human Plasma Clotting Assay. (E) Shown are average times (± one standard deviation) to clot formation in human FIX-deficient plasma after recalcification in the absence tissue factor or FXIa (white bar, C - control); or in the presence (gray) or absence (black) of 1 pM human tissue factor and varying concentrations of human FXIa. NP indicates results with normal human plasma supplemented with 1 pM tissue factor. Thrombin Generation Assay. (F) Shown are curves representing averages of two separate experiments for thrombin generation in recalcified human FIX-deficient plasma either in the absence of supplements (gray line) or in the presence of tissue factor (1 pM), FXIa (15 nM), or both tissue factor and FXIa. For statistical analysis in panels A and D, groups were compared by unpaired t-test, with the level of signficance set at 0.05.
Previously, Whelihan et al. and our group observed that FXIa activates plasma coagulation factors other than FIX (factors V, VIII and X) [24,25]. This may explain the observation that mice lacking both FIX and FXI are more resistant to arterial thrombosis than mice lacking only FIX [25], and supports the notion that FXIa can promote thrombin generation independently of FIX. We did not find evidence for a role for FXI in hemostasis in C57Bl/6 mice using the SVB model. However, based on our experience with thrombosis models, we reasoned that we might be able to detect a pro-hemostatic effect in mice in the absence of FIX. The data presented here support the notion that FXIa can influence the coagulation mechanism “downstream” from FIX.
Infusing FXIa increased the number of clotting episodes and reduced bleeding duration in F9- mice (Fig.1A, gray). A similar effect can be produced in plasma clotting (Fig.1E) and thrombin generation (Fig.1F) assays using small amounts of tissue factor to start thrombin generation. In F9- mice given FXIa, the duration of bleeding was shortest for early bleeds, and became longer with time (data not shown), consistent with protease neutralizing FXIa. In contrast, raising the plasma FXI level to >200% had a more sustained pro-hemostatic effect, suggesting that infusion of FXI enhanced FXIa generation over time.
Results similar to FXI infusion were achieved by over-expressing human FXI in F9- mice using HTI (Fig.1A, black) [26]. The CAG promoter in the EEV600A vector supports sustained FXI expression for at least 4 weeks (Fig.2A), and results in plasma concentrations of human FXI that are 5- to 7-fold higher than the endogenous FXI level of mice (Fig.2B & 2C) [27]. The FXI variant FXI-A183–185 has alanine replacements for three residues that are required to form a FIX-binding exosite [20]. FXIa-A183–185 has a severe defect in FIX activation [20], but activates factors X [25] and V (data not shown) similar to FXIa-WT. The defect in FXI-A183–185 is obvious in a model in which carotid artery occlusion is induced by FeCl3 exposure [14,21]. Thrombosis in this model depends on FIX and FXI. Consistent with this, the resistance of F11−/− mice to thrombus formation is reversed by FXI-WT but not by FXI-A183–185 (Fig.2D). FXI with its A3 domain (which contains residues 183–185) replaced with the A3 domain from the homologous protein prekallikrein has a similar defect (Fig.2D). Furthermore, the resistance of F9- mice to FeCl3-induced thrombosis cannot be reversed by over-expressing FXI-WT. These findings emphasize the importance of FXIa activation of FIX in the thrombosis model. In contrast, in the SVB model, FXI-WT and FXI-A183–185 improved hemostasis in F9- mice similarly, consistent with FXIa exerting a pro-hemostatic effect independent of FIX (Fig.1A&B).
Figure 2. Expression of human FXI in F9- and F11−/− mice by hydrodynamic tail vein injection.
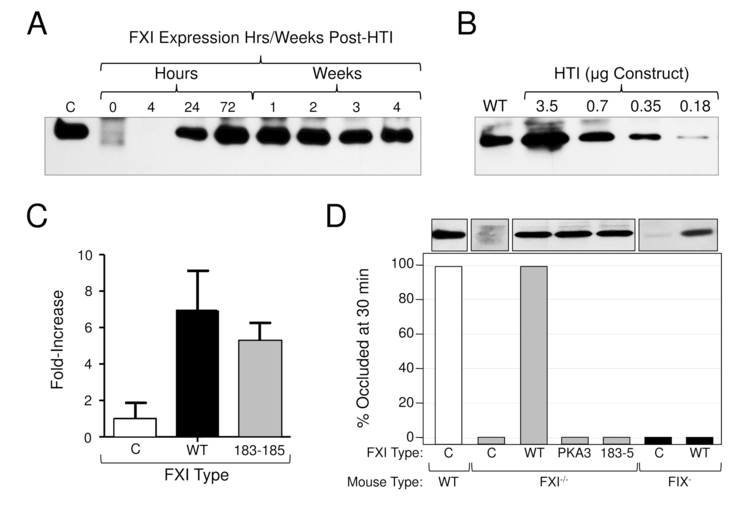
(A) HTI in F11−/− mice - duration of expression. Shown is a western blot of 1 μl samples of mouse plasma at various times after HTI with a Human FXI/ EEV600A construct (3.5 μg). C (control) is a 100 ng sample of purified human FXI. (B) HTI in F11−/− mice - dose effect. Shown is a western blot of 1 μl samples of mouse plasma 24 hrs after HTI with varying concentrations of human FXI/ EEV600A construct. WT indicates a sample of plasma from a normal mouse. (C) Expression of human FXI in F9−/− mice undergoing testing in the saphenous vein bleeding model. Fold-increase in plasma FXI level as determined by densitometry evaluation of western blots. Results for FXI-WT and FXI-Ala183–185 are shown relative to the average value for mice treated with empty vector, which was assigned a value of 1. The value for empty vector control reflects endogenous FXI in F9- mice. Results are +/− 1 SD. (D) Effect of FXI-WT and FXI-Ala183–185 in a carotid artery thrombosis model in F11−/− and F9- mice. Shown are the percent of F11−/− and F9- mice tested in a FeCl3-carotid artery thrombosis model with occluded carotid arteries 30 minutes after vessel injury. Twenty-four hours prior to testing, F11−/− mice (gray bars) underwent HTI with empty vector (C) or with vectors containing cDNAs for wild type human FXI (WT) or FXI-Ala183–185 (183–5). Also shown are results for mice expressing FXI containing the A3 domain of prekallikrein (PKA3), like FXI-Ala183–185, removes the FIX-binding site on FXIa. F9- mice (black bars) underwent HTI with empty vector (C) or vector containing cDNA for wild type human FXI (WT). Results for wild type mice (white bar) are shown for comparison. For each bar n = 6 animals. The western blots at the top of the figure were prepared with 1 μl samples of plasma from representative mice. For all panels, blots were developed with the monoclonal IgG 14E11 [21], which was raised against mouse FXI in a F11−/− mouse, and which recognizes mouse and human FXI.
While the plasma FXI/XIa levels used in our studies were supraphysiologic, the results suggest that differences in FXI plasma levels, or differences in ability to generate FXIa, could contribute to variability in bleeding tendency in patients with FIX deficiency (hemophilia B). They also raise the possibility that supraphysiologic FXIa or FXI could be used in patients with FIX or FVIII inhibitors to promote hemostasis in a manner similar to recombinant factor VIIa or activated prothrombin complex preparations. Indeed, the long plasma half-life of FXI (~50 hours) would be an advantage over currently used preparations. However, we doubt that infusions of wild type FXI or FXIa would be practical. The use of FXI concentrates in FXI deficient patients has been associated with thrombotic events, particularly in older individuals with cardiovascular risk factors [28,29]. With these concentrates, contaminating FXIa is suspected to be the prothrombotic culprit, suggesting FXIa infusions would not be safe. Perhaps a modified FXIa, such as FXIa-A183–185, that does not activate FIX might be safer.
While our results differ from those reported by Ay et al. regarding the behavior of F11−/− mice in the SVB model, the data generated with F9- animals do support their conclusion that FXI can contribute to hemostasis with some types of injury in mice [15]. However, we suspect that differences in sensitivity to FXI of the SVB assays run in different laboratories, and the relatively small differences in bleeding tendency observed, will make this method difficult to use for studying the role of FXI in hemostasis. It is notable that Ay et al. did not detect differences between F11−/− and wild type mice in a tail-bleeding assay or with abdominal surgical wounds [15]. In the past, we did not observe differences in tail bleeding between these mouse lines [14], nor in a liver laceration model (data not shown). This is consistent with the observation that hemorrhage in FXI-deficient humans, if it occurs, tends to involve certain types of tissues [2–4].
ESSENTIALS.
Mice lacking factor IX (FIX) or factor XI (FXI) were tested in a saphenous vein bleeding model.
FIX-deficient mice displayed a hemostatic defect. FXI-deficient mice were similar to wild type.
Infusion of FXI or over-expression of FXI in FIX-deficient mice improved hemostasis.
FXI may affect the phenotype of FIX-deficiency (hemophilia B).
ACKNOWLEDGMENTS
The authors wish to acknowledge support from awards HL81326, HL58837 and HL140025 from the National Institutes of Health, and grant PPOP-14–01 from the Netherlands Ministry of Health.
Footnotes
ADDENDUM
B. M. Mohammed performed the clotting assay, saphenous vein bleed experiments, FXI quantification and wrote the manuscript. Q. Cheng performed the hydrodynamic tail injection, and quantified FXI levels. A. Matafonov performed the thrombin generation assays. D. M. Monroe and J.C.M. Meijers contributed to study design and writing of the manuscript. D. Gailani was responsible for oversight of the project and preparation of the manuscript
DISCLOSURE OF CONFLICT OF INTEREST
J.C.M. Meijers is a consultant for Bayer Pharma for which his employer receives consultant fees. D. Gailani is a consultant for several pharmaceutical companies (Bayer, Bristol-Myers Squibb, Dyax, Instrument Laboratory, Ionis, and Ono) and receives consultant fees or research support. The other authors state that they have no conflict of interest.
REFERENCES
- 1.Mohammed BM, Matafonov A, Ivanov I, Sun MF, Cheng Q, Dickeson SK, Li C, Sun D, Verhamme IM, Emsley J, Gailani D. An update on factor XI structure and function. Thromb Res 2018. January;161:94–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenthal RL, Dreskin OH, Rosenthal N. New hemophilia-like disease caused by deficiency of a third plasma thromboplastin factor. Pro Soc Exp Biol Med 1953;82:171–4. [DOI] [PubMed] [Google Scholar]
- 3.James P, Salomon O, Mikovic D, Peyvandi F. Rare bleeding disorders - bleeding assessment tools, laboratory aspects and phenotype and therapy of FXI deficiency. Haemophilia 2014;20 Suppl 4:71–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol 2016;9:629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knowler C, Giger U, Dodds WJ, Brooks M. Factor XI deficiency in Kerry Blue Terriers. J Am Vet Med Assoc 1994;205:1557–61. [PubMed] [Google Scholar]
- 6.Troxel MT, Brooks MB, Esterline ML. Congenital factor XI deficiency in a domestic shorthair cat. J Am Anim Hosp Assoc 2002;38:549–53. [DOI] [PubMed] [Google Scholar]
- 7.Marron BM, Robinson JL, Gentry PA, Beever JE. Identification of a mutation associated with factor XI deficiency in Holstein cattle. Anim Genet 2004;35:454–6. [DOI] [PubMed] [Google Scholar]
- 8.Al-Horani RA, Desai UR. Factor XIa Inhibitors: a review of the patent literature. Expert Opin Ther Pat 2016;26:323–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gailani D Future prospects for contact factors as therapeutic targets. Hematology Am Soc Hematol Educ Program 2014 2014;52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tucker EI, Marzec UM, White TC, Hurst S, Rugonyi S, McCarty OJ, Gailani D, Gruber A, Hanson SR. Prevention of vascular graft occlusion and thrombus-associated thrombin generation by inhibition of factor XI. Blood 2009;113:936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yau JW, Liao P, Fredenburgh JC, Stafford AR, Revenko AS, Monia BP, Weitz JI. Selective depletion of factor XI or factor XII with antisense oligonucleotides attenuates catheter thrombosis in rabbits. Blood 2014;123:2102–7. [DOI] [PubMed] [Google Scholar]
- 12.Crosby JR, Marzec U, Revenko AS, Zhao C, Gao D, Matafonov A, Gailani D, MacLeod AR, Tucker EI, Gruber A, Hanson SR, Monia BP. Antithrombotic effect of antisense factor XI oligonucleotide treatment in primates. Arterioscler Thromb Vasc Biol 2013;33:1670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gailani D, Lasky NM, Broze GJ. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis 1997;8:134–44. [DOI] [PubMed] [Google Scholar]
- 14.Wang X, Cheng Q, Xu L, Feuerstein GZ, Hsu MY, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost 2005;3:695–702. [DOI] [PubMed] [Google Scholar]
- 15.Ay C, Hisada Y, Cooley B, Mackman N. Factor XI deficient mice exhibit increased bleeding after injury to the saphenous vein. J Thromb Haemost 2017;15:1829–1833 [DOI] [PubMed] [Google Scholar]
- 16.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood 1997;90:3962–6. [PubMed] [Google Scholar]
- 17.Buyue Y, Whinna HC, Sheehan JP. The heparin-binding exosite of factor IXa is a critical regulator of plasma thrombin generation and venous thrombosis. Blood 2008;112:3234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pastoft AE, Lykkesfeldt J, Ezban M, Tranholm M, Whinna HC, Lauritzen B. A sensitive venous bleeding model in haemophilia A mice: effects of two recombinant FVIII products (N8 and Advate®). Haemophilia 2012;18:782–8. [DOI] [PubMed] [Google Scholar]
- 19.Fujikawa K, Chung D, Hendrickson L, Davie E. Amino acid sequence of human factor XI, a blood coagulation factor with four tandem repeats that are highly homologous with plasma prekallikrein. Biochemistry 1986;25:2417–24. [DOI] [PubMed] [Google Scholar]
- 20.Geng Y, Verhamme IM, Sun MF, Bajaj SP, Emsley J, Gailani D. Analysis of the factor XI variant Arg184Gly suggests a structural basis for factor IX binding to factor XIa. J Thromb Haemost 2013;11:1374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng Q, Tucker EI, Pine MS, Matafonov A, Sun MF, White-Adams TC, Smith SA, Hanson SR, McCarty OJ, Renné T, Gruber A, Gailani D. A role for factor XIIa-mediated factor XI activation in thrombus formation in vivo. Blood 2010;116:3981–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kravtsov DV, Matafonov A, Tucker EI, Sun MF, Walsh PN, Gruber A, Gailani D. Factor XI contributes to thrombin generation in the absence of factor XII. Blood 2009;114:452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Matafonov A, Sarilla S, Sun M, Sheehan JP, Serebrov V, Verhamme IM, Gailani D. Activation of factor XI by products of prothrombin activation. Blood 2011;118:437–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Whelihan MF, Orfeo T, Gissel MT, Mann KG. Coagulation procofactor activation by factor XIa. J Thromb Haemost 2010;8:1532–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matafonov A, Cheng Q, Geng Y, Verhamme IM, Umunakwe O, Tucker EI, Sun M-F, Serebrov V, Gruber A, Gailani D. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost 2013;11:2118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sebesty´en MG, Budker VG, Budker T, Subbotin VM, Zhang G, Monahan SD, Lewis DL, Wong SC, Hagstrom JE, Wolff JA. Mechanism of plasmid delivery by hydrodynamic tail vein injection. I. Hepatocyte uptake of various molecules. J Gene Med 2006;8:852–873. [DOI] [PubMed] [Google Scholar]
- 27.Gailani D, Wheeler AP, Neff AT. Rare Coagulation Factor Deficiencies. Hematol. Basic [Google Scholar]
- 28.Mumford AD, Ackroyd S, Alikhan R, Bowles L, Chowdary P, Grainger J, Mainwaring J, Mathias M, O’Connell N, BCSH Committee. Guideline for the diagnosis and management of the rare coagulation disorders: a United Kingdom Haemophilia Centre Doctors’ Organization guideline on behalf of the British Committee for Standards in Haematology. Br J Haematol 2014;167:304–326. [DOI] [PubMed] [Google Scholar]
- 29.Pike GN, Bolton-Maggs PH. Factor XI-related thrombosis and the role of concentrate treatment in factor XI deficiency. Haemophilia 2015;21:477–480. [DOI] [PubMed] [Google Scholar]