

Summary
Successful cloning of monkeys, the first non-human primate species, by somatic cell nuclear transfer (SCNT) attracted world-wide attention earlier this year. Remarkably, it has taken more than 20 years since the cloning of Dolly the Sheep in 1997 to achieve this feat. This success was largely due to recent understanding of epigenetic barriers that impede SCNT-mediated reprogramming and the establishment of key methods to overcome these barriers, which also allowed efficient derivation of human pluripotent stem cells for cell therapy. Here we summarize recent advances in SCNT technology and its potential applications for both reproductive and therapeutic cloning.
Keywords: somatic cell nuclear transfer, epigenetic reprogramming, therapeutic cloning, reproductive cloning, iPSC
Introduction
Totipotency is defined as the ability of a cell to give rise to all cell types of an entire organism (Lu and Zhang, 2015). In normal mammalian development, totipotency is limited to zygotes or blastomeres of early stage preimplantation embryos. After fertilization, oocytes are capable of reprograming terminally differentiated sperm into a totipotent state. Totipotency is then gradually lost during preimplantation development to give rise to inner cell mass (ICM) and trophectoderm (TE) cells.
Beside fertilization, an artificial method named somatic cell nuclear transfer (SCNT), or cloning, can also confer totipotency. Dr. John Gurdon was the first to demonstrate that animals could be cloned from differentiated frog somatic cells by SCNT (Gurdon, 1962) (Figure 1). After three decades of efforts, Dolly the Sheep, the first cloned mammal, was born (Wilmut et al., 1997). Since then, successful cloning of more than 20 mammalian species has been reported (Table 1).
Figure 1.
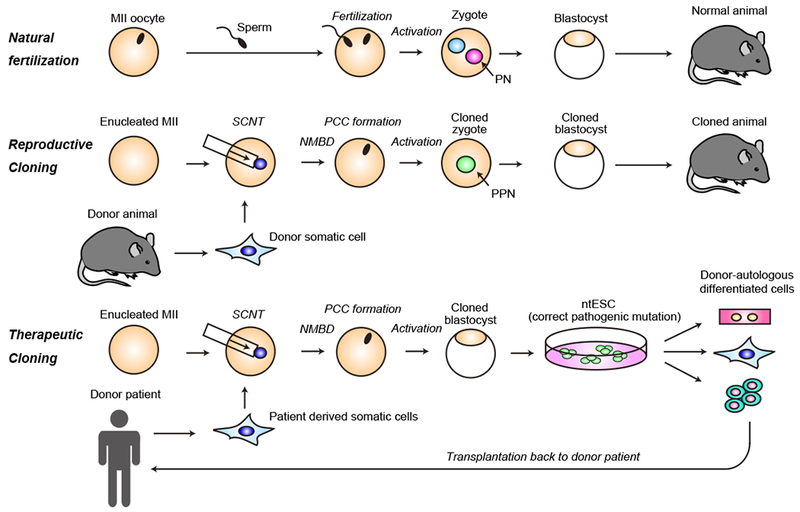
Diagram showing the major steps of reproductive and therapeutic cloning compared to natural fertilization. (Top) Natural fertilization; metaphase II (MII stage) oocytes are activated by fertilized sperm and form paternal and maternal pronuclei (PN), and continue preimplantation cleavages until they reach the blastocyst stage. (Middle) Reproductive cloning; donor somatic cell nuclei introduced into the enucleated oocytes quickly undergoes nuclear membrane break down (NMBD) to form metaphase chromosome in a process called premature chromosome condensation (PCC). The reconstructed SCNT oocytes are artificially activated to initiate developmental program to form blastocysts. (Bottom) Therapeutic cloning; patient derived somatic cells are introduced to enucleated oocytes similar to reproductive cloning. Pluripotent embryonic stem cells can be derived from the blastocysts of nuclear transferred embryos (ntESCs), and the causative mutation can be corrected in vitro if desired.
Table 1.
List of cloned mammalian species
| Type of cloning | Year of Publication | Donor cell |
Recipient oocyte |
References | |
|---|---|---|---|---|---|
| Species | cell type | Species | |||
| Intraspecies cloning | 1996 | Sheep | Differentiated embryonic cell line | Sheep | (Campbell et al., 1996) |
| 1997 | Sheep | Adult mammary epithelium | Sheep | (Wilmut et al., 1997) | |
| 1998 | Cow | Fetal fibroblasts (transgenic) | Cow | (Cibelli et al., 1998) | |
| 1998 | Cow | Adult cumulus and oviductal cells | Cow | (Kato et al., 1998) | |
| 1998 | Mouse | Adult cumulus cells | mouse | (Wakayama et al., 1998) | |
| 1999 | Goat | Fetal fibroblasts (transgenic) | Goat | (Baguisi et al., 1999) | |
| 2000 | Pig | Fetal fibroblasts | Pig | (Onishi et al., 2000) | |
| 2000 | Pig | Cultured adult granulosa cells | Pig | (Polejaeva et al., 2000) | |
| 2002 | Rabbit | Adult transgenic cumulus cells | Rabbit | (Chesné et al., 2002) | |
| 2002 | Cat | Adult cumulus cells | Cat | (Shin et al., 2002) | |
| 2003 | Mule | Fetal fibroblasts | Horse | (Woods et al., 2003) | |
| 2003 | Horse | Adult skin fibroblasts | Horse | (Galli et al., 2003) | |
| 2003 | Rat | Fetal fibroblasts | Rat | (Zhou et al., 2003) | |
| 2005 | Dog (Afghan hound) | Adult skin fibroblasts | Dog (golden retriever) | (Lee et al., 2005) | |
| 2006 | Ferret | Adult cumulus cells | Ferret | (Li et al., 2006) | |
| 2007 | Red deer | Adult antlerogenic cells | Deer | (Berg et al., 2007) | |
| 2007 | Buffalo | Fetal fibroblasts and adult granulosa cells | Buffalo | (Shi et al., 2007) | |
| 2010 | Camel | Adult cumulus cells | Camel | (Wani et al., 2010) | |
| 2018 | Cynomolgus monkey | Fetal fibroblast | Cynomolgus monkey | (Liu et al., 2018) | |
| Interspecies cloning | 2000 | Gaur (Bos gaur) | Cryopreserved adult skin cells | Cow (Bos taurus) | (Lanza et al., 2000) |
| 2001 | Mouflon (Ovis orientalis musimon) | Adult granulosa cells | Sheep (Ovis aries) | (Loi et al., 2001) | |
| 2001 | Zebu (Bos indicus) | Morula stage blastomere | Cow (Bos taurus) | (Meirelles et al., 2001) | |
| 2004 | African Wildcat (Felis lybica) | Adult skin fibroblasts | Cat (Felis catus) | (Gómez et al., 2004) | |
| 2004 | Banteng (Bos javanicus) | Cryopreserved adult fibroblasts | Cow (Bos taurus) | (Janssen et al., 2004) | |
| 2007 | Gray wolf (Canis lupus) | Adult ear fibroblasts | Dog (Canis familiaris) | (Kim et al., 2007) | |
| 2009 | Pyrenean ibex (Capra pyrenaica pyrenaica) | Cryopreserved skin fibroblasts | Goat (Capra aegagrus hircus) | (Folch et al., 2009) | |
| 2013 | Coyote (Canis latrans) | Neonatal/adult fibroblasts | Dog (Canis familiaris) | (Hwang et al., 2013) | |
| 2017 | Bactrian camel (Camelus bactrianus) | Adult skin fibroblasts | Dromedary camel (Camelus dromedaries) | (Wani et al., 2017) | |
Despite the success, several technical hurdles have limited the practical use of SCNT technology. First, the cloning efficiency is extremely low in essentially all species. Second, abnormalities are frequently observed in the extraembryonic tissues, such as placenta, of the cloned animals (Ogura et al., 2013). Moreover, some abnormalities are observed in cloned animals even after their birth, including obesity, immunodeficiency, respiratory defects and early death (Loi et al., 2016; Ogura et al., 2013), although these phenotypes are not transmitted to the offspring (Fulka et al., 2004; Tamashiro et al., 2002; Wakayama et al., 2013). These observations suggest the existence of barriers that prevent normal development of cloned embryos and animals.
In the past decade, great effort has been placed into improving cloning efficiency (Meissner and Jaenisch, 2006). However, the lack of understanding of reprogramming barriers has impeded improvements on cloning efficiency. Recent technical advances, particularly low-input sequencing techniques, have enabled analysis of transcriptome and epigenetic changes during SCNT reprogramming. These analyses have revealed molecular defects as well as provided clues about how to overcome the defects (Liu et al., 2016a; Matoba et al., 2014, 2018). Indeed, the recent success in monkey cloning (Liu et al., 2018b) has been largely attributed to this understanding and the establishment of approaches to overcome critical barriers of epigenetic reprogramming (Cibelli and Gurdon, 2018).
In addition to animal cloning, SCNT technology holds great potential for stem cell biology and human therapeutics. Similar to the derivation of embryonic stem cells (ESCs) from blastocysts of fertilized eggs, SCNT-generated blastocysts could be used to derive pluripotent stem cells (PSC), also termed, nuclear transfer ESCs (ntESCs). Because patient-derived ntESCs are isogenic to the donor patients, they could be used for therapeutic purposes including cell transplantation and disease modeling. Thus, the ntESC-derivation process is also called ‘therapeutic cloning’ (Figure 1). Although derivation of human ntESCs had been very difficult due to the extremely low efficiency of cloned human embryos to reach the blastocyst stage, derivation of human ntESCs was finally achieved after decades of struggle in optimizing SCNT conditions (Tachibana et al., 2013) and further improved (Chung et al., 2014, 2015; Yamada et al., 2014) thus making therapeutic cloning a reality.
In this review, we summarize our current understanding of the cellular and molecular events associated with SCNT reprogramming. We discuss the epigenetic barriers and potential ways to overcome them for efficient cloning with a focus on the mouse model. We also discuss the advantages and disadvantages of ntESC compared with iPSCs in human regenerative medicine. Finally, we discuss how this revamped technology could contribute to modern medicine when combined with CRISPR/Cas9-mediated genome editing.
Cellular events following SCNT
The ooplasm has a remarkable ability to reprogram a differentiated cell nucleus. However, the cellular and molecular events underlying the reprogramming process are poorly understood. Regardless of the species, the SCNT procedure involves three major steps: enucleation, injection/fusion, and activation. After removing the oocyte nucleus, the donor cell nucleus is injected or fused with the enucleated oocytes before the reconstructed embryos are activated. Below, we briefly summarize the cellular events following SCNT.
Nuclear membrane breakdown and PCC formation
Following introduction into an enucleated oocyte cytoplasm, the donor nucleus quickly undergoes nuclear membrane breakdown to form condensed metaphase-like chromosomes. This process is called premature chromosome condensation (PCC), and is trigged by the M-phase promoting factors (MPFs) present in the ooplasm (Campbell et al., 1996a). Although G0/G1 arrested quiescent cells are ideal donor cells, G2 or M phase cells can also be reprogrammed as long as the cell cycle stage of donor cells and recipient oocytes are carefully coordinated (Ono et al., 2001). PCC appears to be required for reprogramming as subsequent development of SCNT embryos is severely compromised without PCC (Kim et al., 2002). During PCC, most chromatin bound proteins including transcription factors (TFs) dissociate from the genome. PCC can be stably maintained for hours until reconstructed oocytes are activated.
Activation
Upon fertilization, sperm-borne phospholipase C zeta (PLCζ) induces oocyte activation through calcium oscillation and MPF breakdown, which triggers oocytes to exit M phase and initiate the developmental program (Saunders et al., 2002). However, since PLCζ is absent in somatic cells, SCNT reconstructed oocytes needs to be artificially activated to initiate the developmental program. The most popular method for activation of mouse SCNT reconstructed oocytes is strontium chloride (SrCl2) treatment as addition of SrCl2 to the culture medium recapitulates the signals induced by fertilization. In some species, including human and monkey, that are less sensitive to SrCl2 and easily restore MPF activity after activation (Liu et al., 2014), electropulse or calcium-ionophore treatment is widely used, while cyclohexamide or 6-Dimethylaminopurine (6-DMAP) is added to the medium during/after activation to inhibit the recovery of MPF activity for efficient metaphase exit.
Nuclear expansion
After activation, the donor cell genome enters G1 phase and forms the nuclear membrane. In a fertilized zygote, the two nuclei derived from sperm and oocyte are called paternal and maternal pronucleus (PN), respectively. The PN in SCNT embryos is called pseudo-pronucleus (PPN). The number of PPN varies between embryos depending on the random distribution of PCC chromosomes, but normally one or two PPNs are formed. One unique feature of PN in fertilized embryos is its large size. Like PN, SCNT PPN is also much larger than the original donor somatic cells (Prather et al., 1990). This is achieved through nuclear expansion, during which PPN incorporates a large amount of maternal proteins (Prather et al., 2000). Consequently, drastic changes in chromatin structure and protein association take place during this process.
DNA replication
Mouse zygotes initiate DNA replication 5–6 hours after fertilization, and the replication continues for 6–7 hours (Yamauchi et al., 2009). Although SCNT embryos have similar dynamics, the timing of replication initiation appears to be variable among SCNT embryos. In naturally fertilized embryos, since proteins involved in DNA methylation maintenance, such as DNMT1 and UHRF1, are exported out of the nucleus at this stage (Hirasawa et al., 2008; Maenohara et al., 2017), newly synthesized DNA, with the exception of imprinting genes, are mostly unmethylated, resulting in a passive dilution of DNA methylation and its oxidized derivatives in a replication-dependent manner (Inoue and Zhang, 2011; Inoue et al., 2011).
Zygotic genome activation (ZGA)
Mammalian oocytes and sperm are transcriptionally silent. Following fertilization, the zygote gradually restores transcription from its newly organized genome, which is called zygotic genome activation (ZGA). The timing of ZGA differs between species (at 2-cell stage for mice and 8-cell stage for human). As ZGA initiates, maternally stored RNAs are quickly degraded and replaced by newly synthesized zygotic RNAs. A similar mechanism is likely used in ZGA of SCNT embryos.
Chromatin, epigenetics, and transcriptional reprogramming upon SCNT
Given that most cell types of an organism have the same genetic materials, SCNT reprogramming is likely achieved mainly through epigenetic reprogramming. Currently, there is no standard definition for SCNT reprogramming. Thus, we propose SCNT reprogramming as the cellular and molecular events taking place in the transferred somatic nucleus before the onset of the major developmental event, ZGA. Below, we summarize our current knowledge on the chromatin, epigenetics, and transcriptome changes (Figure 2A) during this short time window with a focus on the best characterized mouse model. Overall, SCNT can reprogram the epigenetic identity of donor somatic cells within a very short period of time although some regions are resistant to this reprogramming in a genomic context-dependent manner.
Figure 2.

Molecular mechanisms of SCNT reprogramming and its associated barriers in reproductive cloning.
(A) Diagram showing the cellular and molecular events taking place in the SCNT reprograming process. PPN; pseudo pronuclei. ZGA; zygotic genome activation. DHSs; DNase I hypersensitive sites. TF; transcription factor. CGI; CpG island.
(B) Diagram illustration of the various epigenetic barriers in reproductive cloning. XCI; X chromosome inactivation. LOI; loss-of-imprint.
Chromatin structure reprogramming
In a eukaryotic cell, genomic DNA is packaged by histones to form chromatin in the nucleus. The nucleosome, the basic repeating unit of chromatin, is comprised of ~147bp DNA wrapped around a core histone octamer (2 copies of H2A, H2B, H3 and H4). The position of nucleosomes in the genome is dynamically regulated by chromatin remodeling factors and plays an important role in restricting the DNA access of transcription factors. Thus, nucleosome positioning and associated chromatin accessibility are expected to be globally reprogrammed to accommodate the transition from somatic cell to totipotent cell upon SCNT. Recent technological improvements have enabled the profiling of accessible chromatin sites of mouse and human preimplantation embryos using low input DNase I hypersensitive sites sequencing (liDNase-seq) (Gao et al., 2018; Inoue et al., 2017a; Lu et al., 2016) or Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq) (Wu et al., 2016).
Using liDNase-seq, chromatin accessibility profile of 1-cell SCNT embryos was recently analyzed, which revealed that global chromatin accessibility of donor somatic cells is quickly reprogramed to the pattern of a totipotent zygote within 12 hours post activation (hpa) (Djekidel et al., 2018). The SCNT chromatin showed drastic loss of DNase I hypersensitive sites (DHSs) of the somatic donor cells concurrent with the appearance of some zygote-specific DHSs. Interestingly, the change in chromatin accessibility is independent of DNA-replication. Based on the high correlation between each cell type’s specific TFs and DHSs, SCNT reprogramming may involve global loss of somatic chromatin organization and gain of zygotic chromatin landscape through global TF network switching (Figure 2A).
Despite global chromatin landscape reprogramming, some regions are resistant to this reprogramming when compared to in vitro fertilized (IVF) counterparts. Interestingly, these regions are enriched for the heterochromatin marker H3K9me3 in both donor somatic cells and 2-cell SCNT embryos (Djekidel et al., 2018). This observation is consistent with the fact that H3K9me3 in donor cells functions as an epigenetic barrier for SCNT reprogramming (Chung et al., 2015; Liu et al., 2016a; Matoba et al., 2014) (discussed below).
Incorporation of histone variants
In addition to canonical histones (H2A, H2B, H3 and H4), several histone variants can also be incorporated into nucleosomes. Upon fertilization, the sperm genome, originally packaged with protamine undergoes global remodeling so that maternally stored histones, such as H3.3 (Akiyama et al., 2011; Inoue and Zhang, 2014) and H2AX (Nashun et al., 2010), can repackage the sperm DNA. Although the chromatin of somatic cells is packaged with histones, similar drastic histone variant exchanges also occur in SCNT embryos (Nashun et al., 2011; Wen et al., 2014a, 2014b) (Figure 2A). By using ESCs that stably express flag-tagged histone variants as donor cells, Nashun et al. observed that most histone variants are eliminated within 5 hours after activation (Nashun et al., 2011). Similarly, macroH2A, which is enriched in repressive chromatin, is also quickly eliminated from the donor somatic nucleus after SCNT (Chang et al., 2010). Concomitant with global histone removal, all three H3 variants (H3.1, H3.2 and H3.3) as well as H2AX are efficiently incorporated into the donor nucleus upon SCNT (Nashun et al., 2011). These results indicate that donor cell histones are rapidly replaced by maternally stored histones upon SCNT (Figure 2A). This histone replacement appears to be critical for successful reprogramming as H3.3 knockdown (KD) prior to SCNT compromised pluripotent gene activation and SCNT embryo development (Wen et al., 2014a, 2014b). The role of other histone variants in SCNT reprogramming remains to be shown.
In addition to the histone variants mentioned above, oocytes have unique core histone variants, TH2A and TH2B. These oocyte-specific histone variants are quickly incorporated into the PNs in IVF embryos and play critical roles in paternal genome activation and embryonic development (Shinagawa et al., 2014). Since overexpression of TH2A and TH2B in somatic cells induces chromatin opening and facilitates iPSC reprogramming, they may also contribute to SCNT reprogramming. Similarly, the canonical linker histone H1 in somatic cells is also globally replaced by the oocyte specific H1FOO after SCNT (Gao et al., 2004; Teranishi et al., 2004). Addressing the functional importance of these variant histones in SCNT reprogramming and determining their genomic distribution before and after SCNT will contribute to our understanding of SCNT reprogramming.
Histone modification reprogramming
In addition to histone variants, covalent histone modifications, such as acetylation, methylation, ubiquitination, and phosphorylation, can also regulate gene transcription (Klose et al., 2006; Martin and Zhang, 2005). Thus, successful SCNT reprogramming should include reprogramming of histone modification patterns from the donor cell to those of the zygotes (Figure 2A). Earlier immunostaining studies revealed global differences in the acetylation and methylation patterns of SCNT embryos when compared to those of their IVF counterparts (Wang et al., 2007; Zhang et al., 2009). However, higher resolution of histone modification dynamics requires chromatin immunoprecipitation coupled with high-throughput sequencing (ChIP-seq).
Since it is difficult to obtain a sufficient number of SCNT embryos, particularly at the 1-cell and 2-cell stages, ChIP-seq analysis of SCNT embryos has been technically challenging. A recent study attempted to compare H3K9me3 distribution in donor cumulus cells and the resulting 2-cell SCNT embryos in mice (Liu et al., 2016a). The study revealed that the majority of H3K9me3-enriched promoters in donor cells become demethylated in 2-cell SCNT embryos, indicating a global H3K9me3 reprogramming. This demethylation might be mediated by an endogenous H3K9me3 demethylase Kdm4b, as its expression level correlates with the developmental potential of SCNT embryos (Liu et al., 2016a). Similarly, other H3K9me3 demethylases, Kdm4d and Kdm4e, are required for successful bovine SCNT reprogramming (Liu et al., 2018a). These observations suggest that H3K9me3 demethylation is generally required for successful SCNT reprogramming. Interestingly, some regions were not efficiently demethylated in the mouse SCNT 2-cell embryos (Liu et al., 2016a), suggesting that H3K9me3 demethylation might be a limiting factor for efficient SCNT reprogramming.
Very recently, our group successfully mapped H3K27me3 distribution in mouse SCNT morula embryos (Matoba et al., 2018). In contrast to the predominant enrichment of H3K27me3 in CpG island (CGI)-associated promoters in somatic cells, H3K27me3 is broadly distributed in the maternal genome but absent at promoter regions in IVF preimplantation embryos (Zheng et al., 2016). Interestingly, H3K27me3 is also absent at promoters in SCNT morulae, suggesting successful global H3K27me3 reprogramming upon SCNT. However, the maternal broad H3K27m3 domains that regulate a group of newly discovered imprinting genes (Inoue et al., 2017a, 2017b) are not established in SCNT embryos (Matoba et al., 2018), suggesting incomplete reprogramming of H3K27me3 in SCNT embryos.
DNA methylation reprogramming
In addition to histone modifications, DNA methylation at the 5-position of cytosine (5mC) is another major epigenetic modification that plays a pivotal role in mammalian development. DNA methylation is established and maintained by DNA methyltransferases (DNMTs) and can be demethylated through Ten eleven translocation (TET) protein-mediated oxidation followed by TDG-mediated base excision repair (Wu and Zhang, 2017). During mouse preimplantation development, DNA undergoes extensive demethylation through a combination of active and passive processes to reach their lowest level at the blastocyst stage (Guo et al., 2014; Shen et al., 2014). Given that the somatic cell genome is heavily methylated at most CpGs, global demethylation might be a necessary step of SCNT reprogramming. Although several earlier studies using 5mC antibody-based immunostaining suggested that DNA methylation level is relatively high in bovine SCNT embryos throughout the preimplantation stages (Dean et al., 2001; Santos et al., 2003), whether global DNA demethylation takes place during SCNT reprogramming has been elusive for many years.
After identifying TET3 as a factor mediating active DNA demethylation in paternal PN of IVF embryos, several groups showed that oocyte-stored TET3 could localize to PPN in mouse SCNT embryos to induce 5mC to 5hmC conversion (Gu et al., 2011; Iqbal et al., 2011; Wossidlo et al., 2010, 2011), indicating active DNA demethylation indeed occurs in SCNT embryos. Gu et al. further showed that TET3 depletion in recipient oocyte abolished 5hmC generation in mouse SCNT PPN, compromised DNA demethylation of the endogenous Oct4 promoter, and delayed the activation of a silenced Oct4-EGFP allele in donor cells. However, the DNA methylation analysis was only limited to the Oct4 locus and more importantly, the effect of TET3 depletion on the developmental potential of SCNT embryos was not analyzed. Unexpectedly, we found that maternal TET3 is dispensable for embryonic development in mice (Inoue et al., 2015a). Thus, more analysis on the effect of TET3 depletion on SCNT reprogramming is warranted.
Alex Meissner and colleagues analyzed the DNA methylome of late stage 1-cell mouse SCNT embryos using reduced representation bisulfite sequencing (RRBS) (Chan et al., 2012). At this stage (12–14 hpa), global DNA methylation patterns of SCNT embryos were still more similar to the donor somatic cells than to IVF embryos, suggesting that reprogramming of DNA methylation had not yet completed at this time point. Interestingly, they found that genes related to germline development were subjected to significant demethylation and that certain types of repetitive elements, including LINEs and LTRs, maintained a high methylation level even after 1-round of replication. Another study led by Shaorong Gao found that high DNA methylation levels similar to those of donor cumulus cells had been maintained until the 4-cell stage in SCNT embryos (Liu et al., 2016a). These observations suggest that, similar to IVF embryos, global demethylation in SCNT embryos may require several rounds of replication.
Our recent study confirmed this notion. By performing whole genome bisulfite sequencing (WGBS), we provided the first comprehensive DNA methylome of mouse SCNT blastocysts which revealed very low DNA methylation levels (about 20% in average), similar to that of IVF blastocysts (Matoba et al., 2018). Given that replication-dependent dilution is a driving force for DNA demethylation during preimplantation development (Shen et al., 2014), this result indicates that the same mechanism also works in mouse SCNT embryos. Although previous RRBS analysis at 1-cell SCNT embryos identified demethylation resistant CpGs at LINEs and LTRs (Chan et al., 2012), WGBS analysis revealed that the methylation levels of these regions have become similar to that of the IVF at the blastocyst stage (Matoba et al., 2018), indicating replication-dependent dilution may be able to compensate for the initial DNA methylation differences at the 1-cell embryo, at least in mice. Thus, DNA methylation reprogramming in SCNT occurs mainly through replication dependent dilution during preimplantation development of mouse SCNT embryos (Figure 2A). Given that DNA demethylation through replication-dependent dilution in preimplantation stages is likely conserved among mammals, this mechanism may be generally applicable to SCNT reprogramming in different mammalian species.
Transcriptome reprogramming
Since transcription is tightly regulated by chromatin structure and epigenetic state, the chromatin and epigenetic reprogramming described above is expected to have a transcriptional consequence (Figure 2A). Using RNA sequencing (RNA-seq), several groups analyzed the transcriptional activities of SCNT embryos in mouse (Liu et al., 2016a; Matoba et al., 2014), human (Chung et al., 2015) and bovine (Liu et al., 2018a). In mouse, the transcriptome differences between SCNT and IVF embryos become obvious at the 2-cell stage as over 1000 genes failed to activate properly in SCNT embryos (Matoba et al., 2014). Based on whether their transcriptional activities are successfully reprogrammed at 2-cell embryos, genes can be categorized into different groups. Among the genes highly expressed in donor cumulus cells, about 80% were quickly downregulated in both SCNT and IVF 2-cell embryos, indicating that the transcriptional state of this group of genes were properly reprogrammed. However, the remaining 20% of genes maintained their high expression level in the 2-cell SCNT embryos, but they were silenced in the IVF counterpart. This group of genes maintained their donor somatic transcriptional “memory,” which was not reprogramed immediately after SCNT. Another gene group is embryonic specific; they are silenced in somatic donor cells, but they are activated in 2-cell IVF embryos. About 85% of this gene group is activated in 2-cell SCNT embryos, suggesting global transcriptional reprogramming. However, the remaining 15% were resistant to reprogramming in SCNT embryos. Further studies revealed that the reprogramming resistant regions/genes (resistant to silencing or activation) possess specific epigenetic features that will be discussed below (Chung et al., 2015; Hörmanseder et al., 2017; Matoba et al., 2014).
Epigenetic barriers that impede SCNT reprogramming
Despite success in cloning more than 20 mammalian species, the rate of cloned animals reaching term is extremely low. For example, in the case of mouse, about 70% of SCNT embryos arrest development before reaching the blastocyst stage and only 1–2 % of embryos transferred to surrogate mothers can reach term (Ogura et al., 2013). The problem appears to be more severe in primates. Although Mitalipov’s group successfully generated SCNT blastocysts and derived ntESCs in rhesus monkeys more than 10 years ago (Byrne et al., 2007), the blastocyst rate remained about 16% and attempts to clone a monkey were unsuccessful until recently. Moreover, abnormalities were frequently observed in extraembryonic tissues such as placenta in almost all cloned mammalian species (Ogura et al., 2013), indicating the existence of barriers preventing normal development of cloned animals (Figures 2B).
In the 20 years following the birth of Dolly the Sheep, great efforts have been made into identifying conditions and parameters that affect cloning efficiency, including donor cell types and embryo culture conditions, but the progress has been limited. One important finding was the use of a histone deacetylase inhibitor (HDACi), trichostatin A (TSA), which led to the increase of overall cloning efficiency from 1% to 6% (Kishigami et al., 2006; Rybouchkin et al., 2006). Although the same approach appears to have also improved the cloning efficiency of pig (Zhao et al., 2010) and cow (Akagi et al., 2011), the effect was limited and more importantly, how the HDACi treatment improved SCNT reprogramming remained unclear. Nevertheless, these studies suggest that epigenetic changes might be an important aspect of SCNT reprogramming. This notion is further supported by recent transcriptome and epigenomic studies revealing that a significant proportion of genomic regions are resistant to reprogramming. By characterizing the epigenetic features of these reprogramming resistant regions, epigenetic barriers to SCNT reprogramming have been identified. Below we summarize the recent progress in identifying and overcoming these epigenetic barriers with the goal of increasing SCNT cloning efficiency (Figure 2B).
Aberrant Xist activation impedes postimplantation development
One of the most critical developmental steps where SCNT embryos exhibit developmental defects is implantation. To identify the molecular cause of implantation defects, Inoue et al compared the transcriptome of single SCNT blastocysts to those of sex-matched IVF counterparts and discovered that many X-linked genes were specifically and consistently repressed in SCNT embryos regardless of sex (Inoue et al., 2010). This observation established a link between SCNT and X-chromosome inactivation (XCI), a female-specific dosage compensation mechanism. XCI occurs in an imprinted manner at the paternal X chromosome in preimplantation embryos and extraembryonic tissues, but it occurs randomly in embryonic epiblast cells (Lee and Bartolomei, 2013). Imprinted XCI is initiated by expression of the X-linked non-coding RNA Xist from the paternal allele. Xist RNA then coats the entire X chromosome in cis and recruits Polycomb repressive complex 2 (PRC2) to deposit the repressive histone mark H3K27me3 (Cao et al., 2002), leading to heterochromatinization of the entire X chromosome (Plath et al., 2003). In contrast, maternally derived X chromosome remains active in both males and females. As such, female (XX) and male (XY) cells achieve similar gene dosage for X -linked genes.
Inoue et al postulated that X chromosome-wide gene repression observed in SCNT blastocysts might be caused by ectopic XCI. Indeed, they found that Xist RNA is transcribed from both maternal and paternal alleles and thereby induced abnormal silencing of both X chromosomes in SCNT preimplantation embryos. Importantly, they could overcome this abnormal XCI by using Xist heterozygous knockout donor cells, which resulted in an 8–9 fold increase in SCNT cloning pup rate (Inoue et al., 2010). A similar effect was observed when siRNA against Xist was injected into the male 1-cell SCNT embryos (Matoba et al., 2011), suggesting that abnormal Xist activation at the preimplantation stage has a long-term effect on the developmental capacity of SCNT embryos. Although aberrant Xist activation following SCNT has been observed in bovine (Inoue et al., 2010) and pig (Ruan et al., 2018), it is not clear whether this mechanism is conserved in human and monkey due to the diverse XCI initiation mechanism in these species (Okamoto et al., 2011). The molecular mechanism underlying ectopic Xist activation in mouse SCNT embryos is likely due to the lack of the recently discovered H3K27me3 imprinting in donor somatic cells (Inoue et al., 2017a) (see below).
H3K9me3 impedes ZGA and preimplantation development
The earliest time when SCNT embryos exhibit developmental arrest phenotype is highly correlated with ZGA in different species. Given that successful ZGA is required for embryonic development, the correlation between ZGA and SCNT embryo arrest suggest that ZGA failure might be a problem in SCNT embryos. In mice, as discussed above, about 1000 genomic regions/genes failed to activate at ZGA in SCNT embryos (Matoba et al., 2014). Interestingly, these reprogramming resistant regions (RRRs) are enriched for the transcription repressive marker H3K9me3, which raises the possibility that H3K9me3 in somatic donor cells may serve as a barrier preventing ZGA of SCNT embryos. This notion turned out to be true as injecting mRNAs encoding the H3K9me3-specific demethylase, Kdm4d, not only rescued the ZGA defect but also rescued the preimplantation developmental arrest phenotype (Matoba et al., 2014), resulting in a pup rate increase from less than 1% to more than 8%. Importantly, the H3K9me3 reprogramming barrier appears to be conserved in human as injection of mRNAs encoding a human H3K9me3 demethylase, KDM4A, not only facilitated ZGA of human SCNT embryos but also improved the development of human SCNT embryos to reach the expanded blastocyst stage from which human nuclear transfer embryonic stem cells (hntESC) were successfully derived (Chung et al., 2015). It is worth noting that the positive effect of TSA treatment might be functionally linked to H3K9me3 removal, as TSA treatment did not further improve the development of Kdm4d mRNA injected SCNT embryos (Matoba et al., 2014). In addition, genes activated by Kdm4d mRNA injection and TSA treatment overlap significantly (Inoue et al., 2015b). Besides mouse and human, recent studies indicated that KDM4 mRNA injection can also increase the cloning efficiency in pig (Ruan et al., 2018), bovine (Liu et al., 2018a), and monkey (Liu et al., 2018b). Thus, H3K9me3 in somatic cells appears to be a general barrier in mammalian SCNT reprogramming.
H3K27me3 imprinting defects impede postimplantation development
Another SCNT reprogramming barrier comes from RNA-seq analysis of cloned placentae (Okae et al., 2014). A comprehensive allelic transcriptome analysis of mouse E13.5 placentae identified three genes, Sfmbt2, Gab1, and Slc38a4, that exhibit paternal allele-specific expression in normal placenta, but become biallelic expression in SCNT placentae (Okae et al., 2014). Given that these three genes play important roles in placental development (Itoh et al., 2000; Miri et al., 2013; Matoba, unpublished), loss-of-imprint (LOI) of these genes may contribute to the enlarged placental phenotype commonly observed in SCNT embryos in a wide range of species. Interestingly, the imprinting state of these three genes is independent of maternal DNA methylation (Okae et al., 2014).
In an effort to characterize allelic differences in chromatin accessibility and expression during early embryonic development, we recently uncovered a novel genomic imprinting mechanism by which the maternally deposited H3K27me3 represses maternal allele expression of at least 76 genes during preimplantation development (Inoue et al., 2017a). Surprisingly, all three imprinted genes dysregulated in SCNT placenta belong to the newly identified H3K27me3-dependent imprinted genes. Moreover, allelic expression analysis of mouse SCNT blastocysts revealed that essentially all the H3K27me3-dependent imprinted genes detectable at that blastocyst stage lost their imprinting state to become biallelically expressed (Matoba et al., 2018). This LOI is likely due to the absence of H3K27me3 imprinting mark in the donor somatic cells because it is intrinsically lost in the embryonic lineage from which the donor cells are derived. Indeed, ChIP-seq analysis revealed lack of maternal-specific domain-like H3K27me3 in SCNT morula embryos (Matoba et al., 2018). Since at least some of the H3K27me3-imprinted genes are functionally important for postimplantation development, complete LOI on these genes likely contribute to the postimplantation developmental arrest phenotypes observed in mouse SCNT embryos. Given Xist is also regulated by maternal H3K27me3 (Inoue et al., 2017b), ectopic activation of Xist in SCNT embryos (described above) is also likely due to the lack of the H3K27m3 mark at the Xist locus in donor somatic cells. Future studies should address whether the H3K27me3-imprinting system as well as its LOI in SCNT embryos are conserved in other mammalian species.
Other epigenetic barriers, DNA methylation and H3K4me3?
In addition to H3K9me3, Xist activation, as well as LOI on H3K27me3 mentioned above, other epigenetic barriers may exist. For example in the case of X-linked genes, although the use of Xist heterozygous KO cells as donors resulted in the derepression of most X-linked genes in mouse SCNT blastocysts, genes at the Magea and Xlr clusters of the X chromosomes still failed to be activated (Inoue et al., 2010), suggesting that an additional silencing mechanism independent of Xist is in play at least in mice. Our recent study analyzing DNA methylome of mouse SCNT blastocysts revealed high levels of promoter DNA methylation at most of these genes (Matoba et al., 2018), suggesting DNA methylation as a possible barrier. Moreover, this DNA methylome study also indicated that oocyte-derived DNA methylation marks that are inherited by the blastocysts via fertilization, is absent in SCNT embryos. Since some maternally-biased DNA methylation passed down to embryos play important role in trophoblast development (Branco et al., 2016), absence of oocyte-like DNA methylation might also serve as a barrier.
In addition to H3K9me3, H3K4me3 might also affect transcriptional reprogramming, and thus impair the developmental potential of SCNT embryos. By performing single cell RNA-seq of mouse preimplantation SCNT embryos, Liu et al revealed a correlation between the level of Kdm5b, an H3K4me3 demethylase, and the 4-cell to 8-cell SCNT embryo rate (Liu et al., 2016a). Importantly, knockdown of Kdm5b caused a 4-cell arrest while overexpression of Kdm5b rescued 4-cell arrest of SCNT embryos, indicating that Kdm5b level is critical for SCNT embryos to pass the 4-cell stage. Since Kdm5b is an H3K4me3-specific demethylase involved in gene repression, it is possible that H3K4me3 in donor cells serves as a barrier preventing silencing of somatic cell signature genes, which leads to developmental arrest of SCNT embryos at the 4-cell stage. This is consistent with a previous report demonstrating donor cell transcriptional memory in SCNT embryos (Gao et al., 2003). A similar observation was also reported using an Xenopus oocyte transcriptional reprogramming system (Hörmanseder et al., 2017). Thus, efficient removal of a donor cell specific H3K4me3 mark might contribute to SCNT reprogramming, although the mechanism is currently unclear.
Therapeutic cloning
Successful cloning of the Dolly the Sheep in 1997 not only made reproductive cloning possible, but it also raised the possibility of therapeutic cloning, the generation of pluripotent human embryonic stem cells from cloned blastocysts. This possibility became a realistic hope when the first human ESC line was derived in 1998 (Thomson et al., 1998). The notion of deriving human ESCs by nuclear transfer (ntESC) is exciting as the pluripotent ntESC has the same nuclear genetic materials as those of the donor and therefore could be used for regenerative medicine. The first proof-of-concept experiment was performed in a mouse model where ntESCs were derived from cloned blastocysts (Munsie et al., 2000) and had a similar differentiation capacity as those derived from a blastocyst by normal fertilization (Wakayama et al., 2001). Genetic fixation of a mutant allele in ntESCs was achieved by homologous recombination and the resultant ntESCs were used as cell source for treating immunodeficient mice (Rideout et al., 2002). Despite the success in mice, derivation of ntESCs in other animals including primates had remained difficult for many years.
Derivation of human ntESCs
A key breakthrough was made in 2007 by Mitalipov’s group when they successfully derived rhesus monkey ntESCs by inhibiting premature activation of recipient oocytes (Byrne et al., 2007). Nevertheless, the efficiency for ntESC derivation was only 0.7% per oocyte used for SCNT. Using a similar approach, coupled with TSA treatment, the same group also established the first human ntESC lines using fetal/infant fibroblast as donor cells (Tachibana et al., 2013). The next year, using the same or slightly modified conditions, two groups succeeded in generating ntESCs using adult donor cells including patients with type 1 diabetes (T1D) (Chung et al., 2014; Yamada et al., 2014). Importantly, one group showed that the ntESC lines derived from T1D patients could differentiate into insulin-secreting beta cells (Yamada et al., 2014), demonstrating a potential use of ntESCs in cell replacement therapy. Despite the success in ntESC generation, all three groups observed great variation among the egg donors on the capacity of the SCNT embryos to reach the blastocyst stage. Thus, it is necessary to establish a method to mitigate the variation for consistent ntESC generation.
Since injection of Kdm4d mRNA can improve preimplantation development of mouse SCNT embryos to reach blastocyst stage with a rate comparable to that of IVF (Matoba et al., 2014), we asked whether a similar approach could improve human SCNT embryo development reaching blastocyst stage so that ntESCs could be derived. Injection of human KDM4A mRNA, which encodes a H3K9me3 demethylase, greatly improved the developmental potential of human SCNT embryos and allowed production of at least one expanded blastocyst from each oocyte donor and subsequent establishment of multiple ntESC lines (Chung et al., 2015). In contrast, none of the SCNT embryos reached the blastocyst stage in the non-injected control groups. Thus, KDM4A mRNA injection can greatly facilitate patient-specific ntESC derivation.
SCNT and iPSC reprogramming use different mechanisms
In addition to SCNT, pluripotent stem cells can also be generated by cell fusion or transcription factor-induced reprogramming (iPSC) (Yamanaka and Blau, 2010). However, SCNT and iPSC reprogramming may use different mechanisms. In a landmark study, Yamanaka and Takahashi demonstrated that pluripotency can be induced by simply overexpressing a set of transcription factors, Oct4, Sox2, Klf4, and c-Myc, in mouse somatic cells (Takahashi and Yamanaka, 2006). One year later, two groups demonstrated that the same approach could also be used to generate human iPSCs from adult human somatic cells (Takahashi et al., 2007; Yu et al., 2007). Although both the SCNT and the iPSC technologies can reprogram differentiated somatic cells into cells of embryonic state, there are fundamental differences between these two reprogramming technologies (Table 2). First, iPSC technology reprograms cells into a pluripotent state similar to ESCs, while SCNT technology reprograms cells into totipotent state similar to zygote. Thus, although the final destination of a pluripotent state is the same in SCNT and iPSC, the paths to achieve pluripotency are likely different. Secondly, the speed of reprogramming is very different. SCNT reprogramming is very fast as evidenced by chromatin accessibility (Djekidel et al., 2018) and transcriptome (Egli et al., 2011) reprogramming within hours, probably due to the rapid histone exchanges driven by ooplasmic histone chaperones. In contrast, establishment of stable iPSC takes several days to weeks (Takahashi and Yamanaka, 2006). Furthermore, SCNT reprogramming, at least for chromatin accessibility and transcriptome reprogramming, is much more efficient than that of iPSC. These differences likely reflect their diverging reprogramming mechanisms. For example, Oct4 is a core pluripotent transcription factor for iPSC generation but is dispensable for SCNT reprogramming (Wu et al., 2013). Additionally, although H3K9me3 is a key reprogramming barrier in both systems (Chen et al., 2013; Matoba et al., 2014; Soufi et al., 2012), the critical genes impeded by this barrier are different; while it prevents activation of Sox2 or Nanog in iPSC generation, it inhibits ZGA in SCNT. Collectively, these results strongly suggest that iPSC and SCNT reprogramming are mechanistically different.
Table2.
Comparison between SCNT and iPSC reprogramming
| Reprogramming method | Endpoint | Speed | Reprogramming Factors |
|---|---|---|---|
| SCNT | Totipotency | Fast (hours) | Tet3 Kdm4b, Kdm4d, Kdm4e, Kdm5b * Oct4 is dispensable |
| iPSC | Pluripotency | Slow (days/weeks) | Tet1, Tet2 Kdm2a/2b,Utx Oct4, Sox2, Klf4, c-Myc, Lin28 Chd1, Ino80 |
Advantages and disadvantages of ntESCs compared with iPSCs
So far, three types of pluripotent stem cells (PSCs): ESCs, iPSCs and ntESCs, have been generated. With regenerative medicine in mind, traditional human ESCs generation that involves normal fertilization is unlikely suitable not only due to ethical concerns but also because they generally have different genomes from those of the patients (allogenic) and therefore could cause severe immune-rejection after transplantation. The two reprogrammed stem cell types, ntESCs and iPSCs, possess an advantage for regenerative medicine because isogenic cells can be generated directly from the patient somatic cells. Then, which cell type is better? Clearly, iPSCs have technical and ethical advantages especially during their derivation steps, as ntESC derivation involves technically difficult SCNT procedures and requires oocytes, while iPSCs can easily be derived even with commercial kits without ethical concerns. However, recent studies on the molecular characteristics of iPSC and ntESCs revealed critical differences between these two cell types that could impact on their use for therapeutic purposes. Below we compare the molecular features of these two cell types for their respective advantages and disadvantages.
Mitochondrial replacement.
One of the most critical differences between ntESC and iPSC is the composition of mitochondrial DNA (mtDNA). Although the nuclear DNA of ntESC is from donor somatic cells (the patient), the mtDNA is from the recipient oocytes. In contrast, both the nuclear and mitochondrial DNAs of iPSCs are from the starting somatic cells (the patient). Mitochondria play a major role in energy production by oxidative phosphorylation, and mutation of mtDNA can cause metabolic disorders (Gorman et al., 2016). Therefore, derivation of ntESCs followed by differentiation to the desired cell/tissue types and transplantion back to the patients can correct mitochondrial diseases. Mitalipov’s group indeed demonstrated that the mitochondrial composition of ntESCs derived from a patient with Leigh syndrome, a mitochondrial disease, was almost completely replaced with oocyte derived mitochondria (over 99%) (Ma et al., 2015). Moreover, the disorder-related defects observed in the donor fibroblasts and the fibroblast-derived iPSCs were functionally rescued in ntESCs (Ma et al., 2015). Although there were concerns regarding the potential incompatibility between nuclear DNA and different haplotypes of mitochondria from oocytes, the ntESCs derived from relatively different haplotypes (47 SNPs in mtDNA) did not show any functional abnormality, suggesting normal nuclear-mitochondrial interactions. However, another study reported that transplantation of ntESCs derived from two different mouse strains with polymorphisms in mtDNA-coded proteins to the nuclear allogenic host induced weak immune responses, although the weak immune response could be tolerated (Deuse et al., 2015). This indicates that careful studies are needed before clinical trials can be initiated. Nonetheless, the unique potential of ntESC in mitochondria replacement holds great potential for the treatment of mitochondrial diseases.
Genetic and epigenetic mutations.
For therapeutic purposes, genetic and epigenetic stability is an important consideration for the different pluripotent stem cells. Detailed genetic and epigenetic analyses have been performed comparing iPSCs to ESCs (Bock et al., 2011; Lister et al., 2011). Bock et al. analyzed DNA methylomes and transcriptomes of 12 human iPSC lines and 20 ESC lines and found that both iPSCs and ESCs contained inherent variations. Although they failed to identify common epigenetic defects in iPSC lines, they observed greater variation in iPSC lines than ESC lines, suggesting that the iPSC generation process could introduce more variables compared to that of ESCs. One factor contributing to the variation is the so called “epigenetic memory” of the donor cells, which has been shown to require long culture time to remove (Nishino et al., 2011). Similarly, Lister et al. performed DNA methylome and transcriptome analyses of 5 iPSC lines derived using different methods or different starting cell types and compared the results with ESC lines. Although the global DNA methylome of iPSCs resemble that of ESCs, hundreds of differentially methylated regions (DMRs) were identified. Some of the DMRs were common to all 5 iPSC lines, suggesting the existence of common failed epigenetic reprogramming in iPSC lines.
Following the success in deriving human ntESCs (Tachibana et al., 2013), comparison of the genetic and epigenetic mutations of isogenic iPSC and ntESCs became possible. Ma et al. compared 4 ntESC lines and 7 iPSC lines derived from the same human fetal fibroblasts, with 2 IVF lines from the same egg donor as that of ntESCs (Ma et al., 2014). While they did not detect any statistically significant difference in the frequency of copy number variation (CNV) among the samples, they observed that the DNA methylome of ntESCs was more similar to that of IVF than iPSCs. Although both ntESC and iPSC lines possess residual DNA methylation memory of donor somatic cells, iPSCs had 8 times more such memory than that of ntESCs. Moreover, iPSCs had 60 times more aberrantly methylated loci than ntESCs, and the great majority of these (90%) were likely induced during reprogramming processes. Consistently, transcriptome analysis revealed that more differentially expressed genes are present in iPSCs than in ntESCs (Ma et al., 2014). While these results show that ntESC is advantageous to iPSC in terms of the epigenetic errors introduced during reprogramming, another study reported no significant differences on the levels of genetic and epigenetic mutations when ntESCs and iPSCs were compared (Johannesson et al., 2014). Although the reasons for the discrepancy of the two studies are unknown, the passage numbers of the cells used for the analysis and the technical differences in iPSCs generation between the two studies may account for the differences. While the first study used viral expression in fetal fibroblasts for iPSC generation, the latter used modified mRNA expression in neonatal or adult fibroblasts. Regardless of the source of discrepancy, functional characterization of isogenic iPSC and ntESC showed no significant difference, indicating both methods could generate pluripotent cells capable of differentiating to functional cells (Zhao et al., 2017).
A recent study revealed that long-time culture of pluripotent stem cells is a source of oncogenic mutation in the TP53 gene (Merkle et al., 2017). Kevin Eggan and colleagues performed an extensive genomic and transcriptomic sequencing analyses on 140 human ESC lines, including 26 lines prepared for potential clinical use, and found that TP53, a well-known tumor suppressor, is frequently mutated during in vitro culture and cells harboring a mutant allele have growth advantages during ESC passage. Given that the establishment of epigenetically stable iPSC lines requires long-time culture to get rid of somatic cell “epigenetic memory”, the genetic mutations introduced during such long time culture has raised a potential safety concern. Indeed, a comprehensive analysis of 711 human iPSC lines derived from 301 health individuals revealed many iPSC lines possess recurrent copy number alterations (CNAs) at certain genomic regions on chromosomes X, 17 and 20 (Kilpinen et al., 2017). These observations suggest that iPSCs may have genetic mutations at specific genomic regions due to the long-time derivation and maintenance processes. The fact that the ntESC derivation process is relatively shorter may give ntESC an advantage, at least in terms of culturing-induced mutations. To demonstrate this is indeed the case, careful comparison of the frequency of genetic mutations in ESC, iPSC, and ntESC should be performed using large number of lines under comparable experimental settings, including donor somatic cell source, recipient eggs, culturing conditions, and passage numbers. However, the number of currently existing ntESC lines is limited due to the extremely low efficiency in ntESC derivation. In this regard, more efficient human ntESC derivation involving KDM4A mRNA injection (Chung et al., 2015) using healthy donor somatic cells will facilitate such comparative study.
Future directions in SCNT research
With technological advancement, particularly the development of more sensitive sequencing related methods, great progress has made in understanding SCNT reprogramming barriers that lead to improved cloning efficiency. In addition, work focusing on the molecular events immediately after SCNT is beginning to reveal reprogramming mechanisms. Below, we discuss the main remaining questions and where we will likely see great progress in the years to come.
Further understanding of the reprogramming mechanisms
Five decades have lapsed since the first successful frog cloning, yet our understanding of SCNT reprogramming at the molecular level is still limited. Toward this goal, a systematic and detailed analysis of the chromatin and epigenomic changes during the reprogramming process is needed. Although it is still technically challenging to obtain sufficient SCNT samples for such analyses, recent studies have demonstrated the feasibility of performing such studies using preimplantation embryos (Ke et al., 2017; Lu et al., 2016; Zheng et al., 2016). Thus, further improvement of the current techniques may make the analysis of SCNT samples possible. Such analysis may reveal new epigenetic abnormalities in SCNT embryos, which can serve as the basis for improving SCNT cloning efficiency. In addition, comparative analysis of SCNT reprogramming with other reprogramming systems, such as iPSC and cell fusion, may reveal novel insights as different reprogramming systems may share some common features. Indeed, H3K9me3 has also been shown to be a barrier to iPSC reprogramming (Soufi et al., 2012; Sridharan et al., 2013). Similarly, CAF1 and HP1 can serve as barriers to iPSC reprogramming (Cheloufi et al., 2015; Sridharan et al., 2013). Testing whether these barriers also function in SCNT reprogramming would be of potential interest.
Improving efficiency and quality of cloning by targeted epigenetic modification
As a natural extension to the previous findings that both ectopic activation of Xist and H3K9me3 in somatic donor cells serve as barriers impeding SCNT reprogramming, we applied a combined approach using Xist KO donor cells coupled with Kdm4d mRNA injection and achieved 24% pup rate, the highest cloning rate reported in mouse when Sertoli cells are used as donors (Matoba et al., 2018). However, this rate is still lower than that of IVF (more than 50%), and the resultant SCNT embryos still had abnormally enlarged placentae. Detailed allelic transcriptome, ChIP-seq and DNA methylome analyses revealed that embryos generated in this manner exhibit LOI on H3K27me3-imprinted genes as well as aberrant DNA methylation at many loci (Matoba et al., 2018). These results suggest both aberrant DNA methylation and H3K27me3-mediated genomic imprinting defects are likely responsible for the observed phenotypes (Matoba et al., 2018). Since these epigenetic modifications are reversible, targeted epigenetic changes might be a good strategy to overcome the defects. In this regard, the dCas9-guided epigenetic modulation system is particularly powerful as it has been recently used to achieve targeted DNA methylation and histone modification changes (Hilton et al., 2015; Kungulovski and Jeltsch, 2016; Liu et al., 2016b). However, a special consideration for epigenetic editing of imprinted genes is its allele-specific targeting. For example, in the case of H3K27me3-dependent imprinted genes, LOI in SCNT embryos is likely due to the absence of the H3K27me3-mark at the maternal allele of the donor somatic cells (Matoba et al., 2018). Therefore, to fix the LOI problem, targeted deposition of H3K27me3 in the maternal allele in the donor cells would be necessary.
Generation of novel human disease models for drug development
The recent progress in improving cloning efficiency has made the various applications of SCNT technology possible. In addition to mitochondrial replacement therapy for therapeutic cloning, reproductive cloning has tremendous potential for expansion of agricultural and economically important animal traits and rescue of near extinct animals (Beyhan et al., 2007; Loi et al., 2001) without sacrificing the donor animals (Kamimura et al., 2013; Wakayama et al., 2008). Another great potential of cloning is development of novel human disease models for disease study and drug development. To realize this potential, the first thing that needs to be done is to test whether the reprogramming barriers identified in mice are conserved in other relevant animal species. Thus, we are likely to see more studies on improving cloning efficiency in agricultural and economically important animal species, including pig, bovine, sheep, etc. Similarly, improved cloning techniques will likely be used for pets, like dogs and cats, to make this service more affordable.
Another potential application of SCNT technology that we would like to emphasize is generation of novel animal models for human diseases. Large scale sequencing efforts have established a correlation between genetic variations and pathogenic phenotypes in human (Landrum et al., 2016). However, the causal relationships between genetic variations and pathogenic phenotypes need to be addressed using in vitro and in vivo models. Indeed, many such in vivo models have been produced in mice (Birling et al., 2017), but due to the physiological differences between rodent and human, many human diseases, including psychiatric and immune diseases, cannot be modeled in rodents. Rather, primate species that are physiologically more relevant to human might be needed (Belmonte et al., 2015).
One unique feature of SCNT is that it enables direct generation of organisms from single donor cells. This feature allows quick and efficient generation of human disease models in large animals including primates, particularly when combined with genome editing techniques such as CRISPR/Cas9 (Doudna and Charpentier, 2014; Hsu et al., 2014). Although direct injection of CRSPR/Cas9 genome editing components into zygotes allows generation of such human disease models in various species, several important problems exist in such an approach, including frequent mosaicism (Yen et al., 2014), random mutations, and relatively low editing efficiency for homology dependent repair (HDR)-mediated knockin (KI) or knockout (KO). This is especially problematic when trying to establish disease models involving multiple edited genes, as the chance to obtain desired multiple gene mutations is extremely low. In small animal models with relatively short generation time and large litter sizes, these issues are not detrimental as the desired whole-body mutant line can be established by crossing offspring of the founder line. However, such an approach is not feasible for large animals with longer gestation periods, sexual maturation time, and uniparous features. For example, in the case of the cynomolgus monkey, gestation takes 164 days to produce a single offspring per pregnancy and sexual maturation takes 3–4 years. Thus, it would take more than 10 years to establish a disease model in such primate species using this approach. On the other hand, if CRISPR/Cas9 genome editing in somatic cells and screening for the desired mutations are done in vitro prior to SCNT, the desired disease model can be generated within 1–2 years.
Indeed, a similar approach has been employed in goat (Ni et al., 2014) and pig (Yan et al., 2018; Zhou et al., 2015), and has been summarized in a review (Tan et al., 2016). Nevertheless, the limiting factor of such an approach is the extremely low efficiency in animal cloning. For example, based on the summary, the cloning efficiency of genome edited somatic cells is only 0.5–1.0% in livestock animals (Tan et al., 2016). If the cloning efficiency is improved, such an approach will be the most efficient way for generating large animal models of human diseases, including non-human primate models.
Concluding remarks
Here we summarized our current understanding of the cellular and molecular mechanisms of reprogramming by SCNT. Recent technology advancement has revealed reprogramming barriers and have prompted the development of methods to overcome such barriers leading to increased cloning efficiency. Increased cloning efficiency has made both therapeutic cloning and reproductive cloning, including cloning of primates, possible. The generation of patient-specific ntESCs has provided a complementary strategy to iPSCs in pluripotent cell generation. Improved cloning technology, when combined with the state-of-art genome editing technology, will expand the potential applications, including generation of novel human disease models for drug screening and evaluation. We believe that further refinement of SCNT technology will turn many of the applications, once dreams of the past, into reality.
Figure 3.

Application of SCNT technology in human disease model generation. CRISPR/Cas9-mediated genome editing coupled with improved SCNT cloning enables rapid generation of human disease animal models. CRISPR/Cas9-mediated genome editing can induce deletion to produce gene knockout (KO) alleles, or insertion to produce knockin (KI) alleles, or replacement of specific pathogenic mutation via HDR.
Matoba and Zhang discuss recent advances in somatic cell nuclear transfer technology, particularly new methods to overcome epigenetic barriers, and highlight the potential applications of this cloning technology for both reproductive efforts and therapy.
Acknowledgements
The authors thank Drs. Luis Tsuesta, Zhiyuan Chen, Azusa Inoue, Kimiko Inoue and Atsuo Ogura for comments on the manuscript. Work on the SCNT-related work in the Zhang lab is supported by NIH (R01HD092465) and HHMI. SM is supported by JSPS KAKENHI Grant JP16H06146. Y.Z is an investigator of HHMI. The authors apologize to colleagues whose work cannot be cited due to a space limitation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Lead Contact: Yi Zhang
Declaration of Interests
YZ is a scientific founder of the NewStem Biotechnology.
YZ and SM are inventors of a patent on the role of Kdm4 in improving cloning efficiency.
References
- Akagi S, Matsukawa K, Mizutani E, Fukunari K, Kaneda M, Watanabe S, and Takahashi S (2011). Treatment with a histone deacetylase inhibitor after nuclear transfer improves the preimplantation development of cloned bovine embryos. J. Reprod. Dev. 57, 120–126. [DOI] [PubMed] [Google Scholar]
- Akiyama T, Suzuki O, Matsuda J, and Aoki F (2011). Dynamic Replacement of Histone H3 Variants Reprograms Epigenetic Marks in Early Mouse Embryos. PLoS Genet 7, e1002279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baguisi A, Behboodi E, Melican DT, Pollock JS, Destrempes MM, Cammuso C, Williams JL, Nims SD, Porter CA, Midura P, et al. (1999). Production of goats by somatic cell nuclear transfer. Nat. Biotechnol 17, 456–461. [DOI] [PubMed] [Google Scholar]
- Belmonte JCI, Callaway EM, Caddick SJ, Churchland P, Feng G, Homanics GE, Lee K-F, Leopold DA, Miller CT, Mitchell JF, et al. (2015). Brains, Genes, and Primates. Neuron 86, 617–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg DK, Li C, Asher G, Wells DN, and Oback B (2007). Red Deer Cloned from Antler Stem Cells and Their Differentiated Progeny1. Biol. Reprod 77, 384–394. [DOI] [PubMed] [Google Scholar]
- Beyhan Z, Iager AE, and Cibelli JB (2007). Interspecies Nuclear Transfer: Implications for Embryonic Stem Cell Biology. Cell Stem Cell 1, 502–512. [DOI] [PubMed] [Google Scholar]
- Birling M-C, Herault Y, and Pavlovic G (2017). Modeling human disease in rodents by CRISPR/Cas9 genome editing. Mamm. Genome 28, 291–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock C, Kiskinis E, Verstappen G, Gu H, Boulting G, Smith ZD, Ziller M, Croft GF, Amoroso MW, Oakley DH, et al. (2011). Reference Maps of Human ES and iPS Cell Variation Enable High-Throughput Characterization of Pluripotent Cell Lines. Cell 144, 439–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco MR, King M, Perez-Garcia V, Bogutz AB, Caley M, Fineberg E, Lefebvre L, Cook SJ, Dean W, Hemberger M, et al. (2016). Maternal DNA Methylation Regulates Early Trophoblast Development. Dev. Cell 36, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne JA, Pedersen DA, Clepper LL, Nelson M, Sanger WG, Gokhale S, Wolf DP, and Mitalipov SM (2007). Producing primate embryonic stem cells by somatic cell nuclear transfer. Nature 450, 497–502. [DOI] [PubMed] [Google Scholar]
- Campbell KH, Loi P, Otaegui PJ, and Wilmut I (1996a). Cell cycle co-ordination in embryo cloning by nuclear transfer. Rev. Reprod 1, 40–46. [DOI] [PubMed] [Google Scholar]
- Campbell KHS, McWhir J, Ritchie WA, and Wilmut I (1996b). Sheep cloned by nuclear transfer from a cultured cell line. Nature 380, 64–66. [DOI] [PubMed] [Google Scholar]
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS, and Zhang Y (2002). Role of Histone H3 Lysine 27 Methylation in Polycomb-Group Silencing. Science 298, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Chan MM, Smith ZD, Egli D, Regev A, and Meissner A (2012). Mouse ooplasm confers context-specific reprogramming capacity. Nat. Genet 44, 978–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C-C, Gao S, Sung L-Y, Corry GN, Ma Y, Nagy ZP, Tian XC, and Rasmussen TP (2010). Rapid elimination of the histone variant MacroH2A from somatic cell heterochromatin after nuclear transfer. Cell. Reprogram 12, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheloufi S, Elling U, Hopfgartner B, Jung YL, Murn J, Ninova M, Hubmann M, Badeaux AI, Euong Ang C, Tenen D, et al. (2015). The histone chaperone CAF-1 safeguards somatic cell identity. Nature 528, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Liu H, Liu J, Qi J, Wei B, Yang J, Liang H, Chen Y, Chen J, Wu Y, et al. (2013). H3K9 methylation is a barrier during somatic cell reprogramming into iPSCs. Nat. Genet 45, 34–42. [DOI] [PubMed] [Google Scholar]
- Chesné P, Adenot PG, Viglietta C, Baratte M, Boulanger L, and Renard J-P (2002). Cloned rabbits produced by nuclear transfer from adult somatic cells. Nat. Biotechnol 20, 366–369. [DOI] [PubMed] [Google Scholar]
- Chung YG, Eum JH, Lee JE, Shim SH, Sepilian V, Hong SW, Lee Y, Treff NR, Choi YH, Kimbrel E. a, et al. (2014). Human Somatic Cell Nuclear Transfer Using Adult Cells. Cell Stem Cell 14, 777–780. [DOI] [PubMed] [Google Scholar]
- Chung YG, Matoba S, Liu Y, Eum JH, Lu F, Jiang W, Lee JE, Sepilian V, Cha KY, Lee DR, et al. (2015). Histone Demethylase Expression Enhances Human Somatic Cell Nuclear Transfer Efficiency and Promotes Derivation of Pluripotent Stem Cells. Cell Stem Cell 17, 758–766. [DOI] [PubMed] [Google Scholar]
- Cibelli JB, and Gurdon JB (2018). Custom-Made Oocytes to Clone Non-human Primates. Cell 172, 647–649. [DOI] [PubMed] [Google Scholar]
- Cibelli JB, Stice SL, Golueke PJ, Kane JJ, Jerry J, Blackwell C, Ponce de Leon FA, and Robl JM (1998). Cloned transgenic calves produced from nonquiescent fetal fibroblasts. Science 280, 1256–1258. [DOI] [PubMed] [Google Scholar]
- Dean W, Santos F, Stojkovic M, Zakhartchenko V, Walter J, Wolf E, and Reik W (2001). Conservation of methylation reprogramming in mammalian development: Aberrant reprogramming in cloned embryos. Proc Natl Acad Sci U S A 98, 13734–13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuse T, Wang D, Stubbendorff M, Itagaki R, Grabosch A, Greaves LC, Alawi M, Grunewald A, Hu X, Hua X, et al. (2015). SCNT-Derived ESCs with Mismatched Mitochondria Trigger an Immune Response in Allogeneic Hosts. Cell Stem Cell 16, 33–38. [DOI] [PubMed] [Google Scholar]
- Djekidel MN, Inoue A, Matoba S, Suzuki T, Zhang C, Lu F, Jiang L, and Zhang Y (2018). Reprogramming of chromatin accessibility in somatic cell nuclear transfer is DNA replication independent. Cell Rep [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doudna JA, and Charpentier E (2014). The new frontier of genome engineering with CRISPR-Cas9. Science 346, 1258096–1258096. [DOI] [PubMed] [Google Scholar]
- Egli D, Chen AE, Saphier G, Ichida J, Fitzgerald C, Go KJ, Acevedo N, Patel J, Baetscher M, Kearns WG, et al. (2011). Reprogramming within hours following nuclear transfer into mouse but not human zygotes. Nat. Commun 2, 488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Cocero MJ, Chesne P, Alabart JL, Dominguez V, Cognié Y, Roche A, Fernández-Árias A, Martí JI, Sánchez P, et al. (2009). First birth of an animal from an extinct subspecies (Capra pyrenaica pyrenaica) by cloning. Theriogenology 71, 1026–1034. [DOI] [PubMed] [Google Scholar]
- Fulka J, Miyashita N, Nagai T, and Ogura A (2004). Do cloned mammals skip a reprogramming step? Nat. Biotechnol 22, 25–26. [DOI] [PubMed] [Google Scholar]
- Galli C, Lagutina I, Crotti G, Colleoni S, Turini P, Ponderato N, Duchi R, and Lazzari G (2003). Pregnancy: A cloned horse born to its dam twin. Nature 424, 635–635. [DOI] [PubMed] [Google Scholar]
- Gao L, Wu K, Liu Z, Yao X, Yuan S, Tao W, Yi L, Yu G, Hou Z, Fan D, et al. (2018). Chromatin Accessibility Landscape in Human Early Embryos and Its Association with Evolution. Cell 173, 248–259.e15. [DOI] [PubMed] [Google Scholar]
- Gao S, Chung YG, Williams JW, Riley J, Moley K, and Latham KE (2003). Somatic Cell-Like Features of Cloned Mouse Embryos Prepared with Cultured Myoblast Nuclei1. Biol. Reprod 69, 48–56. [DOI] [PubMed] [Google Scholar]
- Gao S, Chung YG, Parseghian MH, King GJ, Adashi EY, and Latham KE (2004). Rapid H1 linker histone transitions following fertilization or somatic cell nuclear transfer: evidence for a uniform developmental program in mice. Dev. Biol 266, 62–75. [DOI] [PubMed] [Google Scholar]
- Gómez MC, Earle Pope C, Giraldo A, Lyons LA, Harris RF, King AL, Cole A, Godke RA, and Dresser BL (2004). Birth of African Wildcat Cloned Kittens Born from Domestic Cats. Cloning Stem Cells 6, 247–258. [DOI] [PubMed] [Google Scholar]
- Gorman GS, Chinnery PF, DiMauro S, Hirano M, Koga Y, McFarland R, Suomalainen A, Thorburn DR, Zeviani M, and Turnbull DM (2016). Mitochondrial diseases. Nat. Rev. Dis. Prim 2, 16080. [DOI] [PubMed] [Google Scholar]
- Gu T-P, Guo F, Yang H, Wu H-P, Xu G-F, Liu W, Xie Z-G, Shi L, He X, Jin S, et al. (2011). The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature 477, 606–610. [DOI] [PubMed] [Google Scholar]
- Guo F, Li X, Liang D, Li T, Zhu P, Guo H, Wu X, Wen L, Gu T-P, Hu B, et al. (2014). Active and Passive Demethylation of Male and Female Pronuclear DNA in the Mammalian Zygote. Cell Stem Cell 15, 447–458. [DOI] [PubMed] [Google Scholar]
- Gurdon JB (1962). The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol 10, 622–640. [PubMed] [Google Scholar]
- Hilton IB, D’Ippolito AM, Vockley CM, Thakore PI, Crawford GE, Reddy TE, and Gersbach CA (2015). Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat. Biotechnol 33, 510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirasawa R, Chiba H, Kaneda M, Tajima S, Li E, Jaenisch R, and Sasaki H (2008). Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev 22, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hörmanseder E, Simeone A, Allen GE, Bradshaw CR, Figlmüller M, Gurdon J, and Jullien J (2017). H3K4 Methylation-Dependent Memory of Somatic Cell Identity Inhibits Reprogramming and Development of Nuclear Transfer Embryos. Cell Stem Cell 21, 135–143.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, and Zhang F (2014). Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157, 1262–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang I, Jeong YW, Kim JJ, Lee HJ, Kang M, Park KB, Park JH, Kim YW, Kim WT, Shin T, et al. (2013). Successful cloning of coyotes through interspecies somatic cell nuclear transfer using domestic dog oocytes. Reprod. Fertil. Dev 25, 1142–1148. [DOI] [PubMed] [Google Scholar]
- Inoue A, and Zhang Y (2011). Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science 334, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, and Zhang Y (2014). Nucleosome assembly is required for nuclear pore complex assembly in mouse zygotes. Nat. Struct. Mol. Biol 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Shen L, Dai Q, He C, and Zhang Y (2011). Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res 21, 1670–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Shen L, Matoba S, and Zhang Y (2015a). Haploinsufficiency, but Not Defective Paternal 5mC Oxidation, Accounts for the Developmental Defects of Maternal Tet3 Knockouts. Cell Rep 10, 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, Suzuki T, and Zhang Y (2017a). Maternal H3K27me3 controls DNA methylation-independent imprinting. Nature 547, 419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue A, Jiang L, Lu F, and Zhang Y (2017b). Genomic imprinting of Xist by maternal H3K27me3. Genes Dev 31, 1927–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue K, Kohda T, Sugimoto M, Sado T, Ogonuki N, Matoba S, Shiura H, Ikeda R, Mochida K, Fujii T, et al. (2010). Impeding Xist expression from the active X chromosome improves mouse somatic cell nuclear transfer. Science 330, 496–499. [DOI] [PubMed] [Google Scholar]
- Inoue K, Oikawa M, Kamimura S, Ogonuki N, Nakamura T, Nakano T, Abe K, and Ogura A (2015b). Trichostatin A specifically improves the aberrant expression of transcription factor genes in embryos produced by somatic cell nuclear transfer. Sci. Rep 5, 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K, Jin S-G, Pfeifer GP, and Szabo PE (2011). Reprogramming of the paternal genome upon fertilization involves genome-wide oxidation of 5-methylcytosine. Proc. Natl. Acad. Sci 108, 3642–3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh M, Yoshida Y, Nishida K, Narimatsu M, Hibi M, and Hirano T (2000). Role of Gab1 in heart, placenta, and skin development and growth factor- and cytokine-induced extracellular signal-regulated kinase mitogen-activated protein kinase activation. Mol. Cell. Biol 20, 3695–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen DL, Edwards ML, Koster JA, Lanza RP, and Ryder OA (2004). Postnatal management of chryptorchid Banteng calves cloned by nuclear transfer utilizing frozen fibroblast cultures and enucleated cow ova. Reprod. Fertil. Dev 16, 224–224. [Google Scholar]
- Johannesson B, Sagi I, Gore A, Paull D, Yamada M, Golan-Lev T, Li Z, LeDuc C, Shen Y, Stern S, et al. (2014). Comparable Frequencies of Coding Mutations and Loss of Imprinting in Human Pluripotent Cells Derived by Nuclear Transfer and Defined Factors. Cell Stem Cell 15, 634–642. [DOI] [PubMed] [Google Scholar]
- Kamimura S, Inoue K, Ogonuki N, Hirose M, Oikawa M, Yo M, Ohara O, Miyoshi H, and Ogura A (2013). Mouse cloning using a drop of peripheral blood. Biol. Reprod 89, 24. [DOI] [PubMed] [Google Scholar]
- Kato Y, Tani T, Sotomaru Y, Kurokawa K, Kato J, Doguchi H, Yasue H, and Tsunoda Y (1998). Eight calves cloned from somatic cells of a single adult. Science 282, 2095–2098. [DOI] [PubMed] [Google Scholar]
- Ke Y, Xu Y, Chen X, Feng S, Liu Z, Sun Y, Yao X, Li F, Zhu W, Gao L, et al. (2017). 3D Chromatin Structures of Mature Gametes and Structural Reprogramming during Mammalian Embryogenesis. Cell 170, 367–381.e20. [DOI] [PubMed] [Google Scholar]
- Kilpinen H, Goncalves A, Leha A, Afzal V, Alasoo K, Ashford S, Bala S, Bensaddek D, Casale FP, Culley OJ, et al. (2017). Common genetic variation drives molecular heterogeneity in human iPSCs. Nature 546, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-M, Ogura A, Nagata M, and Aoki F (2002). Analysis of the mechanism for chromatin remodeling in embryos reconstructed by somatic nuclear transfer. Biol. Reprod 67, 760–766. [DOI] [PubMed] [Google Scholar]
- Kim MK, Jang G, Oh HJ, Yuda F, Kim HJ, Hwang WS, Hossein MS, Kim JJ, Shin NS, Kang SK, et al. (2007). Endangered Wolves Cloned from Adult Somatic Cells. Cloning Stem Cells 9, 130–137. [DOI] [PubMed] [Google Scholar]
- Kishigami S, Mizutani E, Ohta H, Hikichi T, Thuan N. Van, Wakayama S, Bui H-T, and Wakayama T (2006). Significant improvement of mouse cloning technique by treatment with trichostatin A after somatic nuclear transfer. Biochem. Biophys. Res. Commun 340, 183–189. [DOI] [PubMed] [Google Scholar]
- Klose RJ, Kallin EM, and Zhang Y (2006). JmjC-domain-containing proteins and histone demethylation. Nat. Rev. Genet 7, 715–727. [DOI] [PubMed] [Google Scholar]
- Kungulovski G, and Jeltsch A (2016). Epigenome Editing: State of the Art, Concepts, and Perspectives. Trends Genet 32, 101–113. [DOI] [PubMed] [Google Scholar]
- Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, Gu B, Hart J, Hoffman D, Hoover J, et al. (2016). ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 44, D862–D868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanza RP, Cibelli JB, Diaz F, Moraes CT, Farin PW, Farin CE, Hammer CJ, West MD, and Damiani P (2000). Cloning of an Endangered Species (Bos gaurus) Using Interspecies Nuclear Transfer. Cloning 2, 79–90. [DOI] [PubMed] [Google Scholar]
- Lee JT, and Bartolomei MS (2013). X-Inactivation, Imprinting, and Long Noncoding RNAs in Health and Disease. Cell 152, 1308–1323. [DOI] [PubMed] [Google Scholar]
- Lee BC, Kim MK, Jang G, Oh HJ, Yuda F, Kim HJ, Shamim MH, Kim JJ, Kang SK, Schatten G, et al. (2005). Dogs cloned from adult somatic cells. Nature 436, 641–641. [DOI] [PubMed] [Google Scholar]
- Li Z, Sun X, Chen J, Liu X, Wisely SM, Zhou Q, Renard J-P, Leno GH, and Engelhardt JF (2006). Cloned ferrets produced by somatic cell nuclear transfer. Dev. Biol 293, 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Kida YS, Hawkins RD, Nery JR, Hon G, Antosiewicz-Bourget J, O’Malley R, Castanon R, Klugman S, et al. (2011). Hotspots of aberrant epigenomic reprogramming in human induced pluripotent stem cells. Nature 471, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Liu X, Wang C, Gao Y, Gao R, Kou X, Zhao Y, Li J, Wu Y, Xiu W, et al. (2016a). Identification of key factors conquering developmental arrest of somatic cell cloned embryos by combining embryo biopsy and single-cell sequencing. Cell Discov 2, 16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wang Y, Gao Y, Su J, Zhang J, Xing X, Zhou C, Yao K, An Q, and Zhang Y (2018a). H3K9 demethylase KDM4E is an epigenetic regulator for bovine embryonic development and a defective factor for nuclear reprogramming. Development 145, dev158261. [DOI] [PubMed] [Google Scholar]
- Liu XS, Wu H, Ji X, Stelzer Y, Wu X, Czauderna S, Shu J, Dadon D, Young RA, and Jaenisch R (2016b). Editing DNA Methylation in the Mammalian Genome. Cell 167, 233–247.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Han X-J, Liu M-H, Wang S-Y, Jia C-W, Yu L, Ren G, Wang L, and Li W (2014). Three-day-old human unfertilized oocytes after in vitro fertilization/intracytoplasmic sperm injection can be activated by calcium ionophore a23187 or strontium chloride and develop to blastocysts. Cell. Reprogram 16, 276–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Cai Y, Wang Y, Nie Y, Zhang C, Xu Y, Zhang X, Lu Y, Wang Z, Poo M, et al. (2018b). Cloning of Macaque Monkeys by Somatic Cell Nuclear Transfer. Cell 172, 881–887.e7. [DOI] [PubMed] [Google Scholar]
- Loi P, Ptak G, Barboni B, Fulka J, Cappai P, and Clinton M (2001). Genetic rescue of an endangered mammal by cross-species nuclear transfer using post-mortem somatic cells. Nat. Biotechnol 19, 962–964. [DOI] [PubMed] [Google Scholar]
- Loi P, Iuso D, Czernik M, and Ogura A (2016). A New, Dynamic Era for Somatic Cell Nuclear Transfer? Trends Biotechnol 34, 791–797. [DOI] [PubMed] [Google Scholar]
- Lu F, and Zhang Y (2015). Cell totipotency: molecular features, induction, and maintenance. Natl. Sci. Rev 2, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu F, Liu Y, Inoue A, Suzuki T, Zhao K, and Zhang Y (2016). Establishing Chromatin Regulatory Landscape during Mouse Preimplantation Development. Cell 165, 1375–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Morey R, O’Neil RC, He Y, Daughtry B, Schultz MD, Hariharan M, Nery JR, Castanon R, Sabatini K, et al. (2014). Abnormalities in human pluripotent cells due to reprogramming mechanisms. Nature 511, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Folmes CDL, Wu J, Morey R, Mora-Castilla S, Ocampo A, Ma L, Poulton J, Wang X, Ahmed R, et al. (2015). Metabolic rescue in pluripotent cells from patients with mtDNA disease. Nature. [DOI] [PubMed] [Google Scholar]
- Maenohara S, Unoki M, Toh H, Ohishi H, Sharif J, Koseki H, and Sasaki H (2017). Role of UHRF1 in de novo DNA methylation in oocytes and maintenance methylation in preimplantation embryos. PLOS Genet 13, e1007042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin C, and Zhang Y (2005). The diverse functions of histone lysine methylation. Nat. Rev. Mol. Cell Biol 6, 838–849. [DOI] [PubMed] [Google Scholar]
- Matoba S, Inoue K, Kohda T, Sugimoto M, Mizutani E, Ogonuki N, Nakamura T, Abe K, Nakano T, Ishino F, et al. (2011). RNAi-mediated knockdown of Xist can rescue the impaired postimplantation development of cloned mouse embryos. Proc. Natl. Acad. Sci. U. S. A 108, 20621–20626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Liu Y, Lu F, Iwabuchi KA, Shen L, Inoue A, and Zhang Y (2014). Embryonic development following somatic cell nuclear transfer impeded by persisting histone methylation. Cell 159, 884–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matoba S, Wang H, Lu F, Iwabuchi KA, Yang L, Press W, Lee JT, Ogura A, Shen L, and Zhang Y (2018). Defective H3K27me3 imprinting in somatic cell nuclear transfer embryos. Cell Stem Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles FV, Bordignon V, Watanabe Y, Watanabe M, Dayan A, Lôbo RB, Garcia JM, and Smith LC (2001). Complete replacement of the mitochondrial genotype in a Bos indicus calf reconstructed by nuclear transfer to a Bos taurus oocyte. Genetics 158, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner A, and Jaenisch R (2006). Mammalian nuclear transfer. Dev. Dyn 235, 2460–2469. [DOI] [PubMed] [Google Scholar]
- Merkle FT, Ghosh S, Kamitaki N, Mitchell J, Avior Y, Mello C, Kashin S, Mekhoubad S, Ilic D, Charlton M, et al. (2017). Human pluripotent stem cells recurrently acquire and expand dominant negative P53 mutations. Nature 545, 229–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miri K, Latham K, Panning B, Zhong Z, Andersen A, and Varmuza S (2013). The imprinted polycomb group gene Sfmbt2 is required for trophoblast maintenance and placenta development. Development 140, 4480–4489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsie MJ, Michalska AE, O’Brien CM, Trounson AO, Pera MF, and Mountford PS (2000). Isolation of pluripotent embryonic stem cells from reprogrammed adult mouse somatic cell nuclei. Curr. Biol 10, 989–992. [DOI] [PubMed] [Google Scholar]
- Nashun B, Yukawa M, Liu H, Akiyama T, and Aoki F (2010). Changes in the nuclear deposition of histone H2A variants during pre-implantation development in mice. Development 137, 3785–3794. [DOI] [PubMed] [Google Scholar]
- Nashun B, Akiyama T, Suzuki MG, and Aoki F (2011). Dramatic replacement of histone variants during genome remodeling in nuclear-transferred embryos. Epigenetics 6, 1489–1497. [DOI] [PubMed] [Google Scholar]
- Ni W, Qiao J, Hu S, Zhao X, Regouski M, Yang M, Polejaeva IA, and Chen C (2014). Efficient Gene Knockout in Goats Using CRISPR/Cas9 System. PLoS One 9, e106718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishino K, Toyoda M, Yamazaki-Inoue M, Fukawatase Y, Chikazawa E, Sakaguchi H, Akutsu H, and Umezawa A (2011). DNA Methylation Dynamics in Human Induced Pluripotent Stem Cells over Time. PLoS Genet 7, e1002085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura A, Inoue K, and Wakayama T (2013). Recent advancements in cloning by somatic cell nuclear Recent advancements in cloning by somatic cell nuclear transfer. Phil. Trans. R. Soc. B 368, 20110329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okae H, Matoba S, Nagashima T, Mizutani E, Inoue K, Ogonuki N, Chiba H, Funayama R, Tanaka S, Yaegashi N, et al. (2014). RNA sequencing-based identification of aberrant imprinting in cloned mice. Hum. Mol. Genet 23, 992–1001. [DOI] [PubMed] [Google Scholar]
- Okamoto I, Patrat C, Thepot D, Peynot N, Fauque P, Daniel N, Diabangouaya P, Wolf J-P, Renard J-P, Duranthon V, et al. (2011). Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature 472, 370–374. [DOI] [PubMed] [Google Scholar]
- Onishi A, Iwamoto M, Akita T, Mikawa S, Takeda K, Awata T, Hanada H, and Perry AC (2000). Pig cloning by microinjection of fetal fibroblast nuclei. Science 289, 1188–1190. [DOI] [PubMed] [Google Scholar]
- Ono Y, Shimozawa N, Ito M, and Kono T (2001). Cloned Mice from Fetal Fibroblast Cells Arrested at Metaphase by a Serial Nuclear Transfer. Biol. Reprod 64, 44–50. [DOI] [PubMed] [Google Scholar]
- Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, and Zhang Y (2003). Role of histone H3 lysine 27 methylation in X inactivation. Science 300, 131–135. [DOI] [PubMed] [Google Scholar]
- Polejaeva IA, Chen S-H, Vaught TD, Page RL, Mullins J, Ball S, Dai Y, Boone J, Walker S, Ayares DL, et al. (2000). Cloned pigs produced by nuclear transfer from adult somatic cells. Nature 407, 86–90. [DOI] [PubMed] [Google Scholar]
- Prather RS, Sims MM, and First NL (1990). Nuclear transplantation in the pig embryo: Nuclear swelling. J. Exp. Zool 255, 355–358. [DOI] [PubMed] [Google Scholar]
- Prather RS, Kuhholzer B, Lai L, and Park K-W (2000). Changes in the Structure of Nuclei after Transfer to Oocytes. Cloning 2, 117–122. [DOI] [PubMed] [Google Scholar]
- Rideout WM, Hochedlinger K, Kyba M, Daley GQ, and Jaenisch R (2002). Correction of a genetic defect by nuclear transplantation and combined cell and gene therapy. Cell 109, 17–27. [DOI] [PubMed] [Google Scholar]
- Ruan D, Peng J, Wang X, Ouyang Z, Zou Q, Yang Y, Chen F, Ge W, Wu H, Liu Z, et al. (2018). XIST Derepression in Active X Chromosome Hinders Pig Somatic Cell Nuclear Transfer. Stem Cell Reports 10, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybouchkin A, Kato Y, and Tsunoda Y (2006). Role of histone acetylation in reprogramming of somatic nuclei following nuclear transfer. Biol. Reprod 74, 1083–1089. [DOI] [PubMed] [Google Scholar]
- Santos F, Zakhartchenko V, Stojkovic M, Peters A, Jenuwein T, Wolf E, Reik W, and Dean W (2003). Epigenetic Marking Correlates with Developmental Potential in Cloned Bovine Preimplantation Embryos. Curr. Biol 13, 1116–1121. [DOI] [PubMed] [Google Scholar]
- Saunders CM, Larman MG, Parrington J, Cox LJ, Royse J, Blayney LM, Swann K, and Lai FA (2002). PLC zeta: a sperm-specific trigger of Ca(2+) oscillations in eggs and embryo development. Development 129, 3533–3544. [DOI] [PubMed] [Google Scholar]
- Shen L, Inoue A, He J, Liu Y, Lu F, and Zhang Y (2014). Tet3 and DNA Replication Mediate Demethylation of Both the Maternal and Paternal Genomes in Mouse Zygotes. Cell Stem Cell 15, 459–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi D, Lu F, Wei Y, Cui K, Yang S, Wei J, and Liu Q (2007). Buffalos (Bubalus bubalis) Cloned by Nuclear Transfer of Somatic Cells1. Biol. Reprod 77, 285–291. [DOI] [PubMed] [Google Scholar]
- Shin T, Kraemer D, Pryor J, Liu L, Rugila J, Howe L, Buck S, Murphy K, Lyons L, and Westhusin M (2002). A cat cloned by nuclear transplantation. Nature 415, 859–859. [DOI] [PubMed] [Google Scholar]
- Shinagawa T, Takagi T, Tsukamoto D, Tomaru C, Huynh LM, Sivaraman P, Kumarevel T, Inoue K, Nakato R, Katou Y, et al. (2014). Histone Variants Enriched in Oocytes Enhance Reprogramming to Induced Pluripotent Stem Cells. Cell Stem Cell 14, 217–227. [DOI] [PubMed] [Google Scholar]
- Soufi A, Donahue G, and Zaret KS (2012). Facilitators and impediments of the pluripotency reprogramming factors’ initial engagement with the genome. Cell 151, 994–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridharan R, Gonzales-Cope M, Chronis C, Bonora G, McKee R, Huang C, Patel S, Lopez D, Mishra N, Pellegrini M, et al. (2013). Proteomic and genomic approaches reveal critical functions of H3K9 methylation and heterochromatin protein-1γ in reprogramming to pluripotency. Nat. Cell Biol 15, 872–882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Amato P, Sparman M, Gutierrez NM, Tippner-Hedges R, Ma H, Kang E, Fulati A, Lee H-S, Sritanaudomchai H, et al. (2013). Human embryonic stem cells derived by somatic cell nuclear transfer. Cell 153, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, and Yamanaka S (2006). Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, and Yamanaka S (2007). Induction of Pluripotent Stem Cells from Adult Human Fibroblasts by Defined Factors. Cell 131, 861–872. [DOI] [PubMed] [Google Scholar]
- Tamashiro KLK, Wakayama T, Akutsu H, Yamazaki Y, Lachey JL, Wortman MD, Seeley RJ, D’Alessio DA, Woods SC, Yanagimachi R, et al. (2002). Cloned mice have an obese phenotype not transmitted to their offspring. Nat. Med 8, 262–267. [DOI] [PubMed] [Google Scholar]
- Tan W, Proudfoot C, Lillico SG, and Whitelaw CBA (2016). Gene targeting, genome editing: from Dolly to editors. Transgenic Res 25, 273–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teranishi T, Tanaka M, Kimoto S, Ono Y, Miyakoshi K, Kono T, and Yoshimura Y (2004). Rapid replacement of somatic linker histones with the oocyte-specific linker histone H1foo in nuclear transfer. Dev. Biol 266, 76–86. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, and Jones JM (1998). Embryonic stem cell lines derived from human blastocysts. Science 282, 1145–1147. [DOI] [PubMed] [Google Scholar]
- Wakayama S, Ohta H, Hikichi T, Mizutani E, Iwaki T, Kanagawa O, and Wakayama T (2008). Production of healthy cloned mice from bodies frozen at −20 degrees C for 16 years. Proc. Natl. Acad. Sci. U. S. A 105, 17318–17322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakayama S, Kohda T, Obokata H, Tokoro M, Li C, Terashita Y, Mizutani E, Nguyen VT, Kishigami S, Ishino F, et al. (2013). Successful serial recloning in the mouse over multiple generations. Cell Stem Cell 12, 293–297. [DOI] [PubMed] [Google Scholar]
- Wakayama T, Perry AC, Zuccotti M, Johnson KR, and Yanagimachi R (1998). Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 394, 369–374. [DOI] [PubMed] [Google Scholar]
- Wakayama T, Tabar V, Rodriguez I, Perry a C., Studer L, and Mombaerts P (2001). Differentiation of embryonic stem cell lines generated from adult somatic cells by nuclear transfer. Science 292, 740–743. [DOI] [PubMed] [Google Scholar]
- Wang F, Kou Z, Zhang Y, and Gao S (2007). Dynamic reprogramming of histone acetylation and methylation in the first cell cycle of cloned mouse embryos. Biol. Reprod 77, 1007–1016. [DOI] [PubMed] [Google Scholar]
- Wani NA, Wernery U, Hassan FAH, Wernery R, and Skidmore JA (2010). Production of the First Cloned Camel by Somatic Cell Nuclear Transfer1. Biol. Reprod 82, 373–379. [DOI] [PubMed] [Google Scholar]
- Wani NA, Vettical BS, and Hong SB (2017). First cloned Bactrian camel (Camelus bactrianus) calf produced by interspecies somatic cell nuclear transfer: A step towards preserving the critically endangered wild Bactrian camels. PLoS One 12, e0177800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D, Banaszynski LA, Liu Y, Geng F, Noh K-M, Xiang J, Elemento O, Rosenwaks Z, Allis CD, and Rafii S (2014a). Histone variant H3.3 is an essential maternal factor for oocyte reprogramming. Proc. Natl. Acad. Sci 111, 7325–7330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D, Banaszynski LA, Rosenwaks Z, Allis CD, and Rafii S (2014b). H3.3 replacement facilitates epigenetic reprogramming of donor nuclei in somatic cell nuclear transfer embryos. Nucleus 5, 369–375. [DOI] [PubMed] [Google Scholar]
- Wilmut I, Schnieke A, McWhir J, Kind A, and Campbell K (1997). Viable offspring derived from fetal and adult mammalian cells. Nature 385, 810–813. [DOI] [PubMed] [Google Scholar]
- Woods GL, White KL, Vanderwall DK, Li G-P, Aston KI, Bunch TD, Meerdo LN, and Pate BJ (2003). A mule cloned from fetal cells by nuclear transfer. Science 301, 1063. [DOI] [PubMed] [Google Scholar]
- Wossidlo M, Arand J, Sebastiano V, Lepikhov K, Boiani M, Reinhardt R, Schöler H, and Walter J (2010). Dynamic link of DNA demethylation, DNA strand breaks and repair in mouse zygotes. EMBO J. 29, 1877–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wossidlo M, Nakamura T, Lepikhov K, Marques CJ, Zakhartchenko V, Boiani M, Arand J, Nakano T, Reik W, and Walter J (2011). 5-Hydroxymethylcytosine in the mammalian zygote is linked with epigenetic reprogramming. Nat. Commun 2, 241. [DOI] [PubMed] [Google Scholar]
- Wu X, and Zhang Y (2017). TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet 18, 517–534. [DOI] [PubMed] [Google Scholar]
- Wu G, Han D, Gong Y, Sebastiano V, Gentile L, Singhal N, Adachi K, Fischedick G, Ortmeier C, Sinn M, et al. (2013). Establishment of totipotency does not depend on Oct4A. Nat. Cell Biol 15, 1089–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Huang B, Chen H, Yin Q, Liu Y, Xiang Y, Zhang B, Liu B, Wang Q, Xia W, et al. (2016). The landscape of accessible chromatin in mammalian preimplantation embryos. Nature 534, 652–657. [DOI] [PubMed] [Google Scholar]
- Yamada M, Johannesson B, Sagi I, Burnett LC, Kort DH, Prosser RW, Paull D, Nestor MW, Freeby M, Greenberg E, et al. (2014). Human oocytes reprogram adult somatic nuclei of a type 1 diabetic to diploid pluripotent stem cells. Nature 510, 533–536. [DOI] [PubMed] [Google Scholar]
- Yamanaka S, and Blau HM (2010). Nuclear reprogramming to a pluripotent state by three approaches. Nature 465, 704–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Ward MA, and Ward WS (2009). Asynchronous DNA replication and origin licensing in the mouse one-cell embryo. J. Cell. Biochem 107, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S, Tu Z, Liu Z, Fan N, Yang H, Yang S, Yang W, Zhao Y, Ouyang Z, Lai C, et al. (2018). A Huntingtin Knockin Pig Model Recapitulates Features of Selective Neurodegeneration in Huntington’s Disease. Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen S-T, Zhang M, Deng JM, Usman SJ, Smith CN, Parker-Thornburg J, Swinton PG, Martin JF, and Behringer RR (2014). Somatic mosaicism and allele complexity induced by CRISPR/Cas9 RNA injections in mouse zygotes. Dev. Biol 393, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, Nie J, Jonsdottir GA, Ruotti V, Stewart R, et al. (2007). Induced Pluripotent Stem Cell Lines Derived from Human Somatic Cells. Science 318, 1917–1920. [DOI] [PubMed] [Google Scholar]
- Zhang M, Wang F, Kou Z, Zhang Y, and Gao S (2009). Defective chromatin structure in somatic cell cloned mouse embryos. J. Biol. Chem 284, 24981–24987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hao Y, Ross JW, Spate LD, Walters EM, Samuel MS, Rieke A, Murphy CN, and Prather RS (2010). Histone deacetylase inhibitors improve in vitro and in vivo developmental competence of somatic cell nuclear transfer porcine embryos. Cell. Reprogram 12, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M-T, Chen H, Liu Q, Shao N-Y, Sayed N, Wo H-T, Zhang JZ, Ong S-G, Liu C, Kim Y, et al. (2017). Molecular and functional resemblance of differentiated cells derived from isogenic human iPSCs and SCNT-derived ESCs. Proc. Natl. Acad. Sci 201708991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Huang B, Zhang B, Xiang Y, Du Z, Xu Q, Li Y, Wang Q, Ma J, Peng X, et al. (2016). Resetting Epigenetic Memory by Reprogramming of Histone Modifications in Mammals. Mol. Cell 63, 1066–1079. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Renard J-P, Le Friec G, Brochard V, Beaujean N, Cherifi Y, Fraichard A, and Cozzi J (2003). Generation of fertile cloned rats by regulating oocyte activation. Science 302, 1179. [DOI] [PubMed] [Google Scholar]
- Zhou X, Xin J, Fan N, Zou Q, Huang J, Ouyang Z, Zhao Y, Zhao B, Liu Z, Lai S, et al. (2015). Generation of CRISPR/Cas9-mediated gene-targeted pigs via somatic cell nuclear transfer. Cell. Mol. Life Sci 72, 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]