

SUMMARY
The decline of tissue regenerative potential with age correlates with impaired stem cell function. However, limited strategies are available for therapeutic modulation of stem cell function during aging. Using skeletal muscle stem cells (MuSCs) as a model system, we identify cell death by mitotic catastrophe as a cause of impaired stem cell proliferative expansion in aged animals. The mitotic cell death is caused by a deficiency in Notch activators in the microenvironment. We discover that ligand-dependent stimulation of Notch activates p53 in MuSCs via inhibition of Mdm2 expression through Hey transcription factors during normal muscle regeneration and this pathway is impaired in aged animals. Pharmacologic activation of p53 promotes the expansion of aged MuSCs in vivo. Taken together, these findings illuminate a Notch-p53 signaling axis that plays an important role in MuSC survival during activation and that is dysregulated during aging, contributing to the age-related decline in muscle regenerative potential.
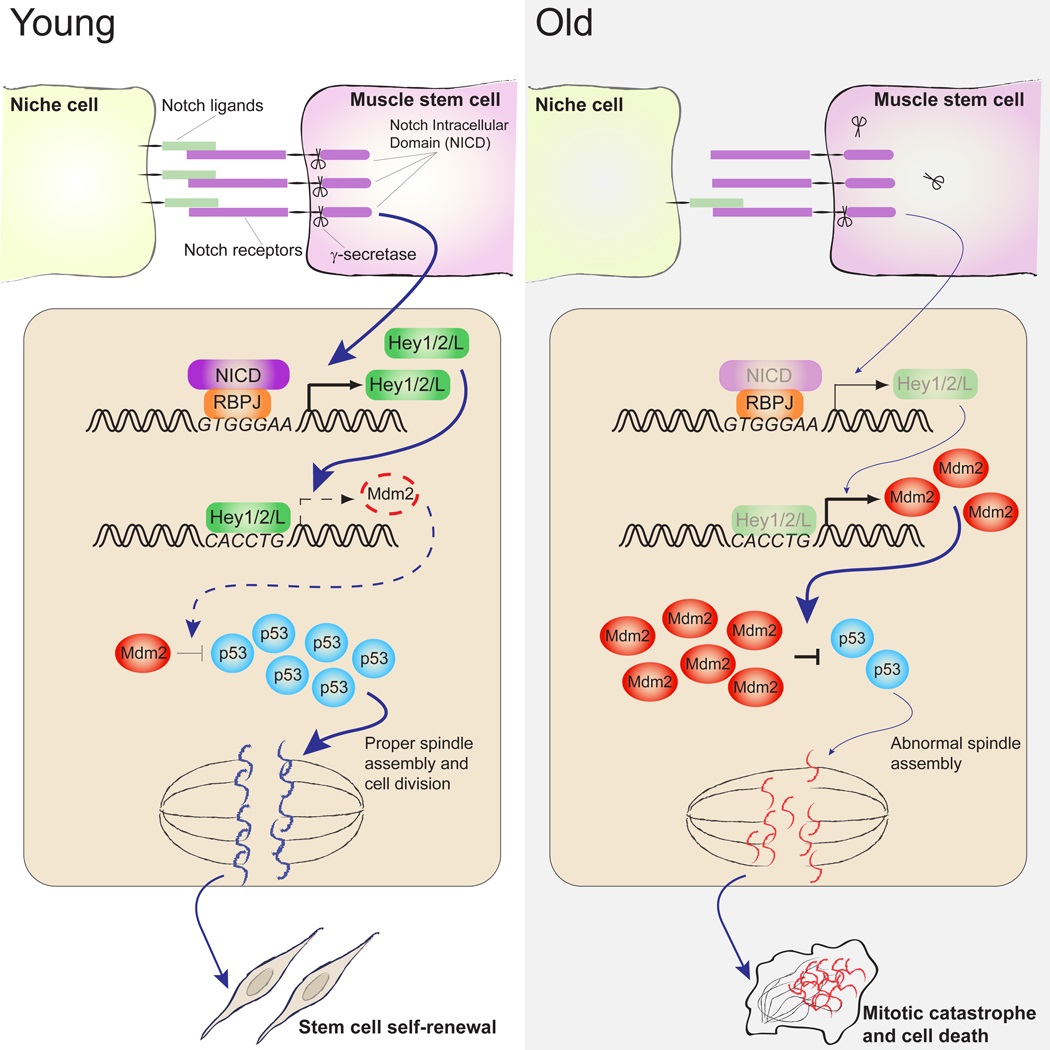
eTOC Blurb
Skeletal muscle stem cells (MuSCs) in aged animals exhibit higher incidence of cell death via mitotic catastrophe upon activation, limiting their survival and self-renewal during muscle regeneration. MuSC mitotic catastrophe is regulated by a Notch-p53 axis. Pharmacologic enhancement of p53 levels promotes the survival of aged MuSCs.
INTRODUCTION
One of the hallmarks of mammalian aging is a general decline in tissue regenerative potential (Rando, 2006). Altered stem cell function has been found to correlate with the age-dependent impairment in tissue homeostasis and/or repair in a number of adult tissues (Conboy et al., 2003; Liu et al., 2013; Molofsky et al., 2006; Nishimura et al., 2005; Rossi et al., 2005). In adult skeletal muscle, muscle stem cells (MuSCs) reside in a quiescent state between the basal lamina and the muscle fiber sarcolemma and thus termed satellite cells (Mauro, 1961). In response to injury or disease, MuSCs undergo a process of activation in which they reenter the cell cycle and proliferate to yield a pool of progenitor cells marked by the expression of the myogenic transcription factor MyoD1 (Zammit et al., 2006). In young, healthy muscle, the peak of proliferation of myogenic progenitors occurs 48–60 hours post-injury (Liu et al., 2013). Progenitor cell proliferation diminishes markedly by five days post-injury, at which point most of the cells express Myogenin and have begun to differentiate to form newly regenerated muscle fibers. While the majority of cells in the pool of activated MuSCs will differentiate and fuse to generate functional muscle, a subset will self-renew to replenish the quiescent MuSC population (Collins et al., 2005).
MuSC-mediated muscle regeneration relies on both the cell-autonomous myogenic program and niche signals. The Notch pathway plays an essential role in regulating both MuSC homeostasis in resting muscle and MuSC expansion during muscle regeneration. Genetic ablation of Notch signaling in quiescent MuSCs leads to spontaneous activation of MuSCs and depletion of the stem cell pool in adult animals (Bjornson et al., 2012). Active Notch signaling is also required for MuSC activation and the early phase of the expansion of their progeny (Conboy and Rando, 2002). In response to injury, the Notch ligand Delta is upregulated in muscle to support the expansion of myogenic progenitors. Inhibition of Notch signaling in MuSCs in injured young animals results in impairment in muscle regeneration (Conboy and Rando, 2002). Isolated MuSCs are more prone to cell death in typical in vitro culture conditions, and providing the Notch activator Delta promotes the expansion of myogenic progeny in vitro (Parker and Tapscott, 2013). While these studies clearly demonstrate the requirement of Notch signaling for MuSC activation and the subsequent expansion of myogenic progeny during normal muscle regeneration, the downstream effectors that mediate these effects of Notch in MuSCs remain largely unexplored.
Aging is associated with a decline in the regenerative capacity of multiple tissues and organs. In aging animals, muscle regeneration is often delayed and accompanied by increased fibrosis and adipogenesis (Conboy et al., 2005; Mann et al., 2011). The effects of aging on muscle regeneration are due in a large part to changes in the MuSC niche and systemic milieu (Conboy et al., 2005; Conboy and Rando, 2012). In aged muscle, the upregulation of the Notch ligand Delta is limited, resulting in an impaired regeneration (Conboy et al., 2003). Enhancing Notch signaling in aging muscle pharmacologically or by heterochronic parabiosis largely restores the regenerative potential of MuSCs (Conboy et al., 2003; Conboy et al., 2005). On the contrary, some signaling pathways, including the Wnt, TGFβ, JAK-STAT and p38 kinase signaling pathways, appear to be hyperactive in MuSCs in old animals, and suppressing these pathways markedly improves muscle regeneration (Bernet et al., 2014; Brack et al., 2007; Carlson et al., 2008; Cosgrove et al., 2014; Price et al., 2014; Tierney et al., 2014).
In this study, we demonstrate that in the absence of sufficient niche support, activated MuSCs are susceptible to mitotic catastrophe, a form of cell death that is characterized by its temporal association with mitosis. Activating Notch signaling or stabilizing p53 prevents MuSC death and promotes their expansion. We further delineate a Notch-p53 axis in which the canonical Notch target, Hey1, directly binds to a consensus E-box sequence in the promoter of Mdm2 gene and suppresses Mdm2 expression, which leads to the stabilization of p53 and the induction of its transcriptional activity. Consistent with our previous discovery that Notch signaling is impaired during muscle regeneration in aging animals, we found an elevated level of Mdm2 and a decreased level of p53 in MuSCs isolated from old mice. Stabilizing p53 in old MuSCs reduces mitotic catastrophe and markedly improved their self-renewal and regenerative potential. To our knowledge, this is the first study that delineates an axis of Notch signaling and p53 activity and a role of this axis in regulating the function of stem cells. These findings provide mechanistic insight into the role of the niche in maintaining MuSC function and a mechanism by which MuSC function deteriorates during aging.
RESULTS
Impaired Self-Renewal and Increased Cell Death of Aged MuSCs
To study the efficiency of MuSC self-renewal in young (2–3-month-old) and aged (24–25-month-old) mice, we analyzed MuSC-mediated muscle repair during the course of sequential muscle injuries. In young mice, muscle was regenerated effectively following both the first and second injury with little unrepaired tissue (Figure 1A and 1B). On the contrary, unrepaired non-muscle tissue was readily detectable in aged mice after a single injury, and increased dramatically after a second injury (Figures 1A and1B). This increase in unrepaired tissue in aged muscle upon repeated injury coincided with a decrease in the average diameter of the regenerated muscle fibers in sequentially injured, aged muscle in comparison to young muscle (Figure 1C). These observations confirmed a self-renewal defect of MuSCs from aged animals as previously reported in transplantation studies (Bernet et al., 2014; Cosgrove et al., 2014).
Figure 1. Impaired self-renewal and increased cell death of aged MuSCs.
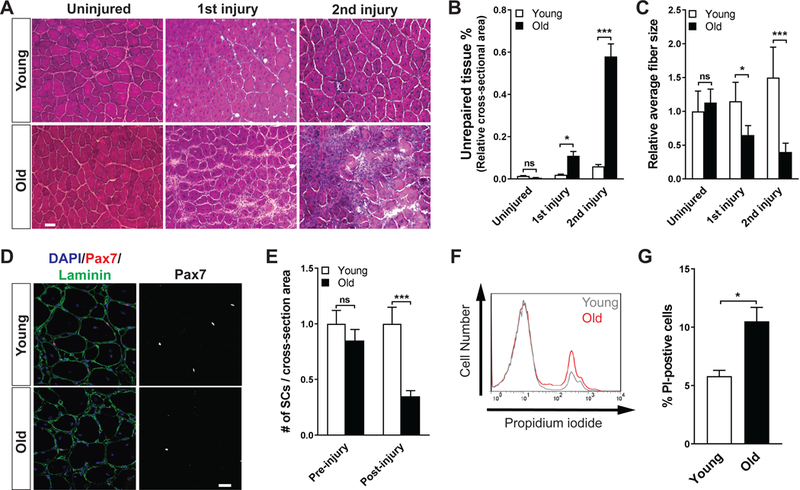
(A) Representative hematoxylin and eosin-stained frozen transverse sections of Tibialis Anterior (TA) muscles of young and old mice during the course of sequential injuries. Muscles were collected 7 days post-injury. (B) Quantification of the percentage of non- myofiber tissue over total section area of A (n = 3 per condition). (C) Quantification of fiber diameter in panel (A). The average fiber diameter of uninjured muscles from young mice set at 1. (D) Representative immunofluorescence (IF) analysis of Pax7-expressing MuSCs in regenerated TA muscles of young and old mice. Muscles were analyzed 21 days post-injury. (E) Quantification of the number of MuSCs prior to and 21 days after injury in young and old mice. The average number of Pax7-expressing cells per area of transverse section of uninjured young mice was used as standard and set at 1 (n = 4 per condition). (F) Representative flow cytometric analysis of cell death in the MuSC population from young and old animals 60 hours after injury, measured by propidium iodide (PI) incorporation. (E) Quantification of the percentage of dead (PI-positive) MuSCs from young and old animals 60 hours after injury (n = 3).
To quantify the number of MuSCs in young and old mice, we used in situ immunofluorescence to identify Pax7-expressing MuSCs in cross-sections of the tibialis anterior (TA) muscle prior to and 21 days after acute injury. It has been previously reported that MuSC number appears to decrease to various degrees, exhibit little change, or even slightly increase in mice of 22–28 months depending on the species, the specific muscle evaluated, and the detection method (Brack and Rando, 2007). In the current study, we consistently found a trend toward a decrease in MuSC number in the TA muscles of old mice in the absence of injury (Figures 1E). However, acute muscle injury led to a drastic reduction in MuSCs in old animals. In comparison to young mice, old mice exhibited far fewer activated MuSCs 2.5 days after injury (Figure S1A and S1B), and as a consequence, 60% fewer Pax7-expressing MuSCs 21 days after injury (Figure 1D and1E). These data suggest that MuSC self-renewal is compromised in old mice, leading to a gradual depletion of resident MuSCs following acute muscle injury.
One potential cause of cell depletion is that MuSC progeny in aged animals have a higher propensity to undergo cell death following activation. We thus injured the muscles of young and old mice to activate resident MuSCs and quantified dead or dying cells by various methods. We found a significant increase in the proportion of propidium iodine (PI) positive activated MuSCs in old mice 2.5 days after injury (Figures 1F and 1G). Consistently, in situ analysis of cross-sections of regenerating muscle revealed a two-fold increase in the proportion of cells positive for cleaved Caspase-3 (aCaspase-3) staining in myogenic cells in injured old animals (Figure S1C and S1D). Although activated caspase-3 has been reported in differentiating myoblasts (Fernando et al., 2002), we found the intensity of aCaspase-3 staining in Myogenin-expressing cells to be negligible (Figure S2E). Therefore, the increase in myogenic cells positive for aCaspase- 3 staining indicated MuSC progeny were more prone to die during the regenerative response in old animals. Taken together, these data suggest that the impaired self-renewal observed in old MuSCs may be due to their higher propensity to undergo cell death.
MuSCs Undergo Mitotic Catastrophe in vitro
Previous studies have demonstrated that isolated MuSCs undergo extensive cell death due to the lack of niche-derived cues and that plating MuSCs and other cells on recombinant Dll 1 (rDll1) supports their survival and proliferation (Dahlberg et al., 2015; Parker and Tapscott, 2013). We therefore reasoned that the death of purified MuSCs in vitro models that of MuSCs in vivo in the aged niche where Notch ligand expression is reduced (Conboy et al., 2003). To explore the timing of MuSC death during the activation process, we performed time-lapse microscopy of FACS-purified MuSCs from 2–3 months old mice to directly visualize their fate (Movies S1). Interestingly, we found that MuSC death always occurred during or immediately following cell division as the average time to death coincided with the average time to cell division. In the first cell- cycle following isolation, cells, on average, divided 42 ± 2 hours or died 41 ± 1 hours post-isolation (Figure 2A). In the second cell-cycle following isolation, cells, on average, divided 12.1 ± 0.5 hours or died 11.7 ± 0.2 hours after the preceding division (Figure S2A). This temporal correlation between mitosis and cell death of MuSCs suggests that they may die by mitotic catastrophe, a form of cell death that occurs either during or shortly after failed mitosis (Kroemer et al., 2009).
Figure 2. Mitotic catastrophe of MuSCs during in vitro activation.
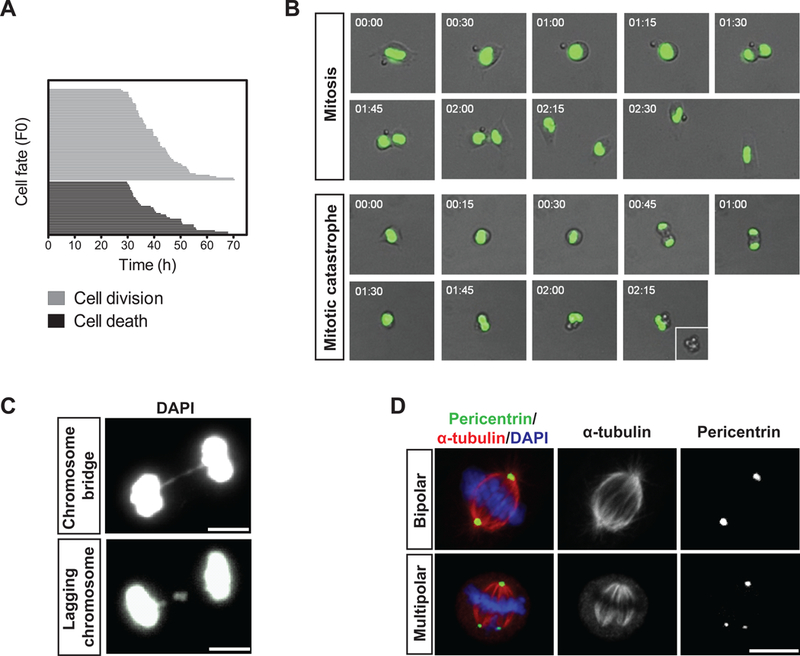
(A) Cell fate analysis by time-lapse microscopy of MuSCs following FACS isolation and plating. Each horizontal bar represents a single cell (n=100). The length of a given bar along the x-axis reflects the length of time required to reach the point of cell division (grey lines; top) or cell death (black lines; bottom). (B) Representative time-lapse images of H2B-GFP-expressing MuSCs undergoing successful mitosis or mitotic catastrophe. Merged bright field and fluorescence images are shown with the time at which the frames were captured indicated at the top left corner. (C) Representative IF images of dividing MuSCs exhibiting features of mitotic catastrophe. DNA was stained with DAPI. (D) Representative IF images of a normal bipolar (top panels) and an abnormal multipolar (bottom panels) mitotic spindle of MuSCs during in vitro activation. Pericentrin (green) and α-tubulin (red) were stained to visualize the mitotic spindle. DNA was stained with DAPI.
In addition to the timing of death, characteristics of mitotic catastrophe include aberrant segregation of chromosomes in anaphase (Kroemer et al., 2009). To test for these characteristics, we used MuSCs isolated from mice constitutively expressing a fusion protein of histone H2B and Green Fluorescent Protein (H2B-GFP) in time-lapse microscopy to directly visualize genomic DNA during cell activation. A significant portion of MuSCs underwent aberrant divisions in which cells died either prior to completing anaphase or shortly after an anaphase failure followed by the collapse of spindles (Figure 2B and Movies S2 and S3). These dying cells exhibit features of aberrant or uneven distribution of chromosomes between daughter cells (Dodson et al., 2007; Hayashi and Karlseder, 2013; Huang et al., 2005; Ichijima et al., 2010; Imreh et al., 2011), including chromosome bridges, which are formed by improperly attached chromosomes segregating to opposite daughter cells (Figure 2C, left panel), anaphase lag, in which chromatids fail to connect to the spindle apparatus or are segregated slowly (Figure 2C, right panel), and the formation of a multipolar mitotic spindle (Figure 2D). The identification of aberrant chromosome segregation in H2B-GFP-expressing MuSCs in culture provides strong evidence that mitotic catastrophe is the primary cause of cell death upon MuSC activation.
Using time-lapse microscopy, we found a two-fold increase in the frequency of cell death in MuSCs from aged animals in comparison to those from young animals (Figure S2B). As mitotic catastrophe is often induced by genomic lesions directly affecting the integrity of chromosomes, we performed phosphorylated histone H2AX (γH2AX) staining as well as comet assays to quantify the proportion of MuSCs that exhibited signs of DNA damage in young and old animals. γH2AX foci were identified in greater than 80% of MuSCs from old animals as opposed to less than 20% of MuSCs from young mice (Figure S2C and S2D). The comet assays also revealed a 3–4-fold increase in the frequency of cells with fragmented DNA from old animals (Figure S2E). Not only did a significantly higher percentage of old MuSCs exhibit signs of DNA damage, but the extent of DNA damage appeared to be also more severe in old MuSCs (Figure S2D and S2E). Taken together, these data indicated that the accumulation of DNA damage in old MuSCs is associated with increased mitotic catastrophe when the cells are activated during muscle regeneration.
p53 Regulates Mitotic Catastrophe in Activated MuSCs
Mitotic catastrophe has been previously associated with p53 deficiency (Firat et al., 2011; Fragkos and Beard, 2011; Ianzini et al., 2006). For instance, analysis of syngeneic mouse embryonic fibroblasts has demonstrated that cells expressing mutant, but not wild-type, p53 undergo mitotic catastrophe following irradiation (Ianzini et al., 2006). Intriguingly, unbiased analysis of our previously published gene expression microarray data from young and aged MuSCs (Liu et al., 2013) revealed that genes associated with p53 signaling were significantly enriched among genes showing decreased expression with age (p = 7.2×10−5, Figure S3A).
To determine whether the death of MuSCs during activation is indeed associated with a functional deficit of p53 activity, we treated MuSCs with the small pharmacological molecule Nutlin-3 (N3). N3 increases p53 levels by inhibiting the interaction between p53 and Mdm2, an E3 ubiquitin ligase that targets p53 for proteasomal degradation (Vassilev et al., 2004). We first treated freshly isolated MuSCs with increasing concentration of N3 and quantified total number of viable cells after 4 days of treatment. We found that N3 led to an increase in cell number in a dose- dependent manner (Figure 3A). Nearly all progeny of MuSCs after N3 treatment expressed MyoD1, indicating that N3 did not alter their myogenic lineage progression (Figure 3B). Given that 10 μM N3 treatment provided maximal expansion of MuSCs, we used this concentration for the rest of our studies. Time-lapse microscopy analysis of MuSCs in the presence or absence of N3 revealed that N3 did not alter the kinetics of cell division, but significantly inhibited cell death (Figure S3B and Movie S4). Consistent with this, aCaspase-3 was detected in far fewer cells when they were treated with N3 (Figure 3C). In order to directly assess the extent of DNA damage in cells that had initiated mitosis, we stained MuSCs two days after plating for γ-Η2ΑΧ, and found that in marked contrast to untreated cells whose metaphase chromosomes contained high levels of γ-Η2AX, N3-treated MuSCs exhibited little γ-Η2AX in metaphase (Figure 3D). These data suggest that N3 prevented MuSC death and mitotic catastrophe.
Figure 3. Pharmacologic activation of p53 prevents mitotic catastrophe in MuSCs.
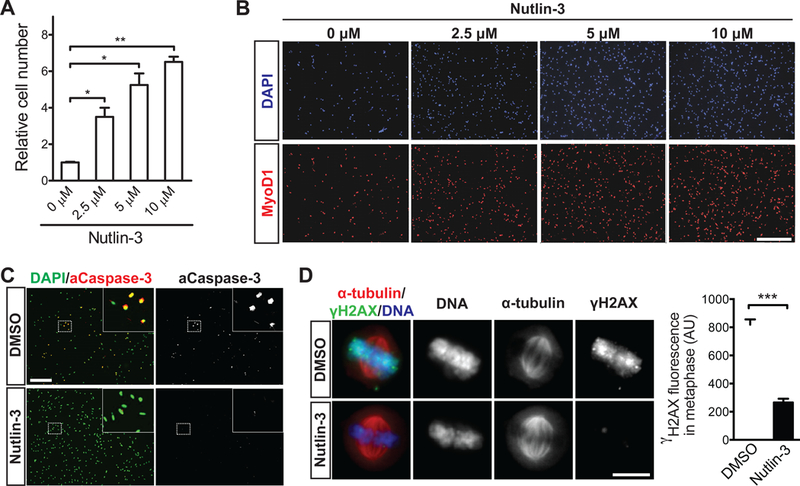
(A) Quantification of MuSC number in cultures treated without Nutlin-3a (N3) or with the given concentration of N3 for a period of four days post-isolation (n = 3 for each condition). (B) Representative IF images of activated, Myod1-expressing MuSCs following four days in culture with the given concentration of N3. DNA was stained with DAPI. (C) Representative IF images of aCaspase-3 staining in control and N3-treated (10 μM) MuSC cultures. DNA was stained with DAPI. Percentage of aCaspase-positive MuSCs is indicated at the lower right corner for each condition (n = 500 cells per condition in each of three independent experiments). (D) Representative IF images of γ- H2AX staining (green) in control and N3-treated (10 μM) MuSCs in metaphase (n = 75 cells per condition from three independent experiments). Cells were analyzed 48 hours post-isolation. The spindles were labeled with α-tubulin staining (red) and DNA was stained with DAPI. Quantitative analysis of γ-H2AX fluorescence intensity in metaphase MuSCs was shown in the right panel (AU, arbitrary units).
Activation of p53 is often associated with changes in the expression of Cyclin- dependent Kinase Inhibitors. We found that the expression of p21 and p27 was upregulated in N3-treated cells (Figure S3C). A second, chemically distinct Mdm2 inhibitor, YH239-EE (Huang et al., 2014), also led to an increase in cell number in dividing MuSCs relative to untreated cells (Figure S3D). To further confirm that it is indeed p53 that mediates the inhibitory effects of N3 on cell death, we performed time- lapse microscopy with MuSCs isolated from p53 knockout mice in the presence or absence of N3 and found that N3 did not rescue mitotic catastrophe of p53−/− cells (Figure S3E). These results confirmed that the rescue of cell death by N3 was mediated by p53.
p53 Activity Links Cell Death to Notch Signaling
We have previously demonstrated that Notch activation promotes MuSC expansion in vitro and also in vivo during muscle regeneration in aged mice (Conboy et al., 2003; Conboy and Rando, 2002). However, the downstream molecular mechanism by which Notch activation led to the expansion of MuSC progeny remained unclear. Given our finding that stabilizing p53 prevented cell death and also led to the expansion of MuSC progeny, we next sought to determine whether there could be a crosstalk between the Notch pathway and p53 activity in promoting MuSC survival and their subsequent expansion. In order to establish an in vitro assay to identify the downstream effector of Notch in MuSCs, we plated MuSCs on dishes coated with recombinant proteins of the Notch ligand Dll 1 (rDll1) and confirmed that such treatment led to the induction of canonical Notch targets HeyL and Hey1 and their transcriptional activity in a γ-secretase- dependent manner as such induction can be abrogated by the addition of the γ-secretase inhibitor DAPT (Figure S4A and S4B). Intriguingly, we found that rDll1 induced a number of known p53 target genes (Figure S4C). To understand whether p53 mediates the effect of Notch activation on MuSC survival and proliferation, we transfected MuSCs, cultured in the presence or absence of rDll1, with a p53 reporter plasmid in which the expression of a luciferase gene is driven by tandem repeats of p53 transcriptional response elements. We found that rDll1 treatment led to a significant induction of p53 transcriptional activity in these cells, and that the p53 activity could be abrogated by the addition of DAPT (Figure 4A).
Figure 4. Activation of p53 transcriptional activity by rDll1.
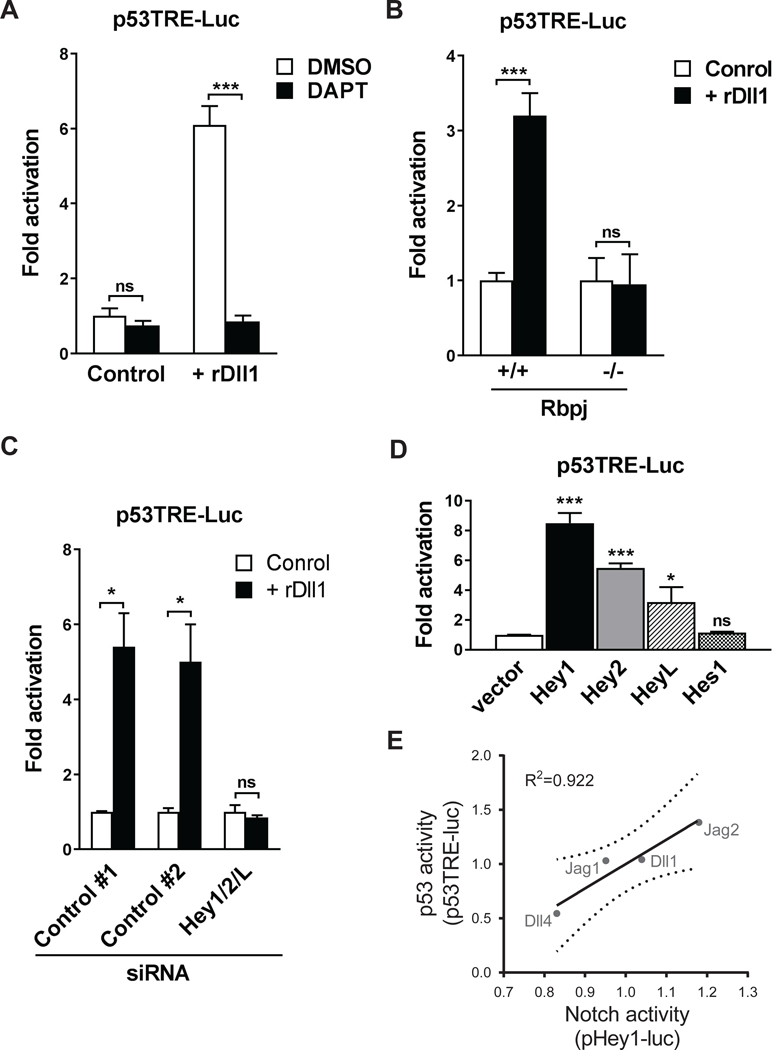
(A) γ-secretase-dependent activation of p53 transcription by rDlll. MuSCs were transfected with a p53 luciferase reporter plasmid and cultured with or without rDll1 in the presence and absence of 25 μM DAPT. Cells were collected 48 hours after transfection and assayed for luciferase activity. Normalized luciferase activity of the control culture was set at 1 (n = 3 independent cultures for each condition). (B) Rbpj- dependent activation of p53 transcription by rDll1. MuSCs isolated from conditional Rbpj knockout or control mice were transfected with a p53 luciferase reporter plasmid and cultured with or without rDll1. Cells were collected 48 hours after transfection and assayed for luciferase activity. Normalized luciferase activity of untreated cells from control mice was set at 1 (n = 3). (C) Hey1/2/L-dependent activation of p53 transcription by rDll1. MuSCs isolated from wildtype mice were co-transfected with either siRNAs specific for Hey1/2/L or control siRNAs, and with a p53 luciferase reporter plasmid. Transfected cells were cultured with or without rDll1 for 48 hours and subjected to luminescent assay. Normalized luciferase activity of untreated cells transfected with control siRNAs was set at 1 (n = 3). (D) Induction of p53 transcriptional activity by overexpression of Hey proteins. MuSCs isolated from wildtype mice were co-transfected with a p53 luciferase reporter plasmid and plasmids encoding Hey1, Hey2, HeyL or Hes1. Transfected cells were collected 48 hours after transfection and subjected to luminescent assay. Normalized luciferase activity of vector-transfected cells was set at 1 (n = 3). (E) Correlation of p53- and Notch-signaling activity in purified MuSCs treated with recombinant Notch ligands measured using p53 (p53TRE-luc) and Notch (pHey1- luc) transcriptional reporter plasmids. Linear regression (solid line) and 95% confidence intervals (dashed lines) are shown.
The Notch coactivator, Rbpj, is indispensable in activating the transcription of canonical Notch targets such as Hes and Hey genes. To determine whether the effect of Notch activation on inducing p53 transcriptional activity was mediated by Rbpj and potentially Notch target genes, we transfected the p53 reporter plasmid into MuSCs isolated from the conditional Rbpj knockout and control mice in the presence or absence of rDll1. We found that rDll1 induced p53 transcriptional activity in an Rbpj-dependent manner as the induction of the reporter gene was completely abolished in Rbpj-null cells (Figure 4B). This suggests that Rbpj-dependent Notch target genes are required for the induction of p53-mediated transcription by Notch activation. We then co-transfected MuSCs with the p53 reporter plasmid and siRNAs targeting Hey1, Hey2 and HeyL transcripts or scramble siRNAs and subjected the cells to rDll1 treatment. Knocking down the expression of these Hey genes completely ablated the induction of p53 transcriptional activity (Figure 4C and S4D). To directly investigate whether Notch target genes can induce p53 transcription, we co-transfected the p53 reporter plasmid and plasmids encoding individual Hey and Hes proteins into MuSCs. In the absence of rDll1, expression of canonical Notch target genes Hey1, Hey2 and HeyL in these cells was sufficient to induce p53 transcriptional activity (Figure 4D).
In addition to Dll1, we also tested other Notch ligands, including Jag1, Jag2, and Dll4, in similar assays for their ability to induce the p53-dependent reporter. MuSCs plated in dishes coated with these individual Notch ligands were transfected with Hey1 or p53 reporter plasmids, and collected 48 hours later to measure the reporter activity. As these ligands activated p53 to varying degrees, we found a direct positive correlation between the level of Hey1 and p53 transcriptional activity (Figure 4E), further suggesting that the induction of p53 activity relies on the Hey proteins.
Direct Regulation of Mdm2 expression by Hey proteins
Despite the increase in p53 transcriptional activity in rDll1-treated MuSCs and their progeny, the transcription of p53 itself did not increase following Notch activation (Figure S5A). We did, however, detect a decrease in the expression of the p53 antagonist Mdm2 (Figure S5B and S5C), and this reduction can be reversed by the knock-down of the Hey proteins (Figure 5A). The Hey proteins belong to the family of basic helix-loop- helix transcriptional repressors that bind to the CANNTG consensus E-box sequence. Using SwissRegulon, a database of transcription factor biding sites (Pachkov et al.., 2007), we identified a 6-basepair sequence (CACCTG) (Chromosome 10, 117,147,266– 71, mm9) in the Mdm2 promoter which resembles a consensus E-box binding site (Figure S5C). This CACCTG sequence is conserved in orthologues of the Mdm2 gene from rat, cow, horse, opossum, orangutan, and human (Figure S5D). To confirm that this sequence is indeed the responsive element to the Hey proteins, we generated a series of luciferase reporter constructs driven by a 740-bp fragment of the Mdm2 promoter containing the CACCTG sequence or a mutated version of the Mdm2 promoter (Figure 5B). These reporter plasmids were transfected into MuSCs with or without rDll1 treatment. While rDll1 suppressed the expression of the luciferase reporter driven by the wildtype Mdm2 promoter, the reporters driven by the truncated or mutated versions of the Mdm2 promoter were unresponsive to Notch activation (Figure 5C). This provides strong evidence that the CACCTG sequence in the Mdm2 promoter is essential for the suppression of Mdm2 expression by Notch activation.
Figure 5: Activation of Mdm2 transcription by Hey1.
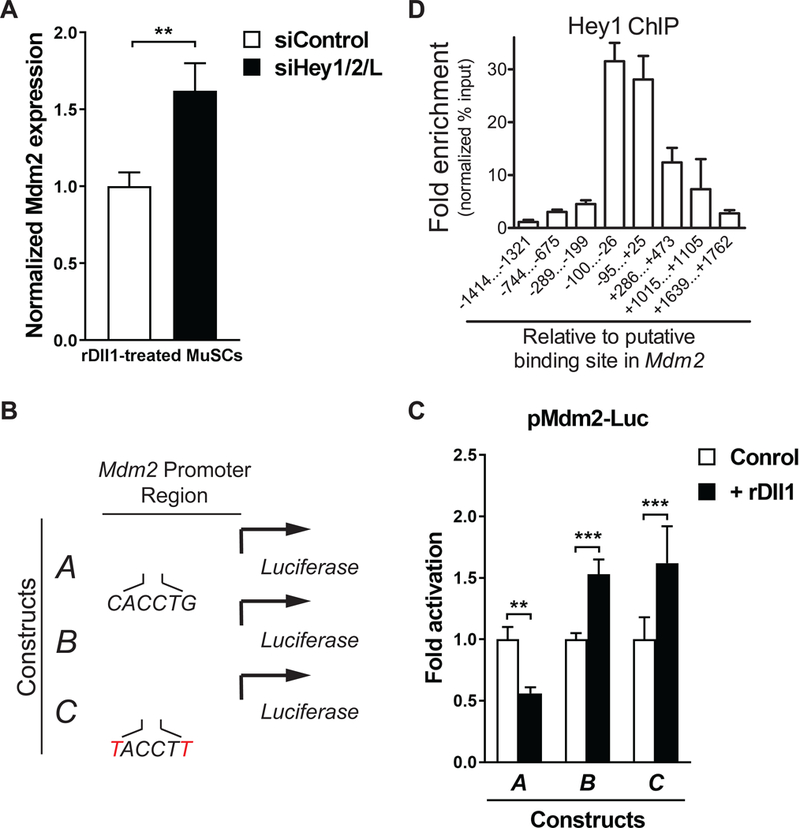
(A) Hey-dependent repression of Mdm2 expression in response to rDll1. Freshly isolated MuSCs were transfected with control siRNA or a cocktail of siRNAs targeting individual Hey genes. Transfected cells were cultured for 48 hours in the presence rDll1 and collected for RT-qPCR analysis. Mdm2 expression was normalized to GAPDH expression in each condition (n = 3 independent experiments). (B) Depiction of wild- type, truncated, and mutated versions of the Mdm2 promoter reporter. (C) Normalized activity of Mdm2 promoter luciferase reporters as depicted in (B) with and without rDll1 treatment. MuSCs were transfected with the individual reporter plasmids and cultured in the absence or presence of rDll1 for 48 hours. Cells were then harvested and subjected to luminescent assay. Normalized luciferase activity of untreated cells was set at 1. Fold change was calculated for other conditions and plotted (n = 3 independent experiments). (D) Direct binding of Hey1 to Mdm2 promoter. ChIP-qPCR analysis of Hey1 binding to various positions throughout the Mdm2 promoter using pairs of primers specific to these regions. Amplicons are identified by base-pair distance from the 5’ end of the CACCTG E-box sequence.
To directly investigate whether Hey proteins bind to the Mdm2 promoter, we performed chromatin immunoprecipitation (ChIP) followed by qPCR using cells expressing Myc-Hey1. We designed PCR primers that spanned a region from 1,500 bp downstream to 1,800 bp upstream of the CACCTG element in the Mdm2 promoter. For each pair of primers, we calculated the fold enrichment of the PCR products in equal amount of ChIP DNA over input, and found the highest fold enrichment in the 200-bp region flanking the CACCTG sequence (Figure 5D), indicating that Hey1 directly binds to the Mdm2 promoter in this region.
Abrogation of p53 Signaling Limits MuSC Survival in vivo
To study the role of p53 in regulating MuSC survival during activation, we generated a conditional knockout (cKO) mouse (Trp53fl/fl; Pax7CreER/+; ROSA26eYFP/+) in which the ablation of p53 and the expression of enhanced Yellow Fluorescent Protein (eYFP) as a reporter can be specifically induced in MuSCs upon Tamoxifen administration (Marino et al., 2000; Nishijo et al., 2009). We confirmed that YFP- expressing MuSCs isolated from cKO mice did not express p53 protein by Western Blot and immunofluorescence analysis (Figure S6A, S6B and S6C). In contrast to previous findings that germline p53 ablation leads to a reduction in MuSCs in young animals (Schwarzkopf et al., 2006), p53 ablation in the adult did not appear to affect the number of MuSCs in resting muscle even after two months (Figure 6A and6B). However, when we quantified the number of MuSC progeny by YFP expression on individual single fibers after they had been cultured for 3 days, we found a nearly 50% reduction in individual fibers isolated from p53 cKO mice compared to those from wildtype control mice (Figure S6D and 6A). This reduction in cell number was likely to result from an increase in cell death as PI staining revealed a 50% increase in the number of YFP- expressing cells that stained positive for PI in cKO mice (Figure 6C, and S6E). In addition, we performed acute muscle injury to activate MuSCs in vivo and found a 50% reduction in the number of YFP-expressing cells in cKO mice in comparison to control mice three days after muscle injury (Figure 6B).
Figure 6: Impairment of MuSCs from conditional p53 cKO mice.
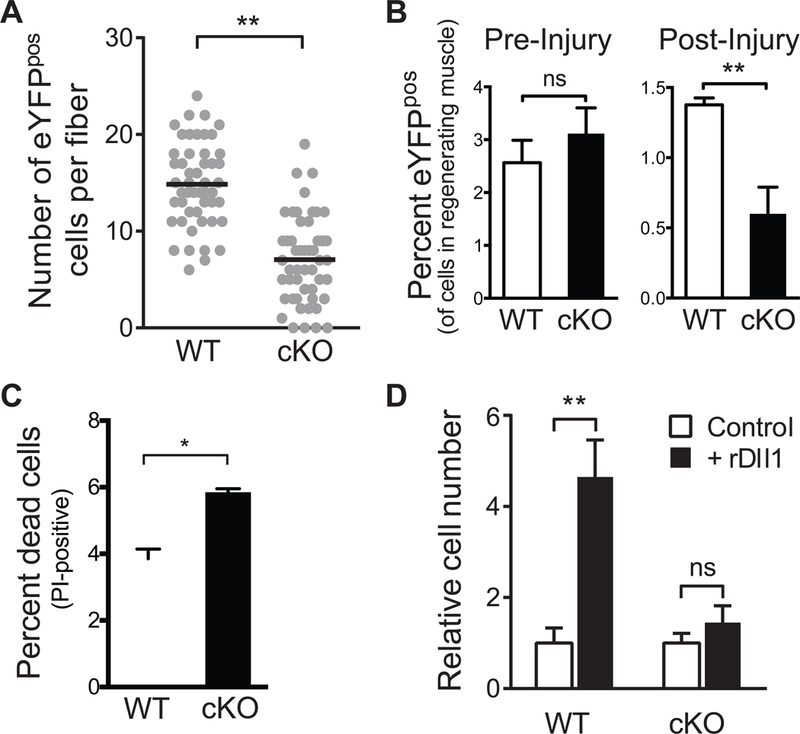
(A) Number of MuSCs on single myofibers in p53 cKO and control mice. Quantification of MuSCs that reside on single myofibers isolated from p53 cKO and control mice immediately following isolation (left) and after a period of three days in muscle fiber explant culture (right). Data were accumulated from explants in three independent cultures. Each data point represents the number of cells per fiber. The black lines represent median number of cells per fiber. (B) Quantification by FACS of MuSCs in p53 cKO and control mice pre- and three days post-injury (n = 5 mice in each condition). (C) Comparison of the percentage of dead MuSCs and their progeny in p53 cKO and control mice three days after injury. YFP-expressing cells were isolated by FACS in the presence of PI. The ratio between PI-labeled and YFP-expressing cells was calculated for each genotype (n=5). (D) Abrogation of the effect of rDll1 on MuSC expansion in the absence of p53. MuSCs were isolated from p53 cKO and control mice and cultured for 72 hours in the absence or presence of rDll1 prior to cell number quantification. The average cell number of each genotype was set at 1 (n = 3).
The survival and proliferation of MuSCs and their progeny during muscle regeneration is regulated by niche signals. The expression of the Notch ligand, Dll1, peaks 3–4 days after muscle injury and plays a critical role in promoting the expansion of MuSC progeny. Using time-lapse microscopy, we quantified the frequency of cells that died around mitosis in the presence or absence of rDll1 and confirmed that providing rDll1 in ex vivo culture conditions drastically decreased the frequency of mitotic cell death (Movie S5). To study whether the effect of Dll 1 on promoting cell survival is mediated by p53 activation, we isolated MuSCs from p53 cKO or control mice and cultured them in the presence or absence of rDll1. Consistent with our previous findings, rDll1 treatment led to a three-fold increase in live wildtype cells after three days in culture (Figure 6D). On the contrary, rDll1 failed to induce the expansion of MuSCs isolated from p53 cKO mice. Taken together, these data demonstrate the importance of the Notch-p53 axis in promoting MuSC survival in response to muscle injury.
Dysregulation of the Notch-p53 Signaling Axis Limits the Survival of Aged MuSCs
We have previously demonstrated that the Notch pathway is impaired in aged MuSCs and restoring Notch signaling in injured muscle of old animals promotes muscle regeneration (Conboy et al., 2003; Liu et al., 2013). We found that canonical Notch target genes were downregulated in MuSCs from old mice upon activation (Supplemental Figure 7A). Given our current findings on a Notch-p53 axis, we investigated whether the reduction in Notch target genes associates with changes in the expression levels of Mdm2 and p53. By using RT-PCR and Western Blot analyses, we found an increase in Mdm2 at both the transcript and protein levels in old MuSCs (Figure S7B and S7C). The total protein level of p53 decreased in old MuSCs while its transcript level did not change (Figure S7D and S7E). This is consistent with our finding that Hey1 transcriptionally regulates the expression of Mdm2 which in term stabilizes p53 protein.
We next determined whether restoring p53 signaling in aged MuSCs would protect them from the mitotic catastrophe that occurs during the early phase of MuSC activation and thus promote their survival and expansion in response to muscle injury. To study mitotic catastrophe in vivo, we injured the TA muscles of 24-month-old mice with or without systemic N3 treatment and collected the TA muscles three days after injury. Mitotic MuSCs were identified in situ by the stereotypical metaphase microtubule array and their expression of the myogenic marker Myod1 (Figure 7A). Our quantification revealed a greater than two-fold reduction in the number of mitotic MuSCs that exhibited γ-H2AX staining in N3-treated mice (Figure 7B). Consistent with this, FACS analysis indicated that N3 treatment led to an increase in the number of myogenic cells in the TA muscles (Figure 7C). In addition, we confirmed by time-lapse microscopy that the proportion of aged MuSCs undergoing mitotic cell death in the first division significantly decreased when cultured with N3 (Movies S6 and S7). While these data indicate that pharmacologic activation of p53 signaling ameliorated the increased cell death and limited expansion of MuSCs in old animals, they do not rule out the possibility that N3 treatment might rejuvenate the niche around MuSCs but not the MuSCs themselves. To confirm that N3 treatment enhances the function of old MuSCs in a cell-autonomous manner, we isolated YFP-expressing MuSCs from 24-month old Pax7CreER/+; ROSA26eYFP/+ mice and subjected them to N3 or vehicle treatment for two days. We then transplanted equal numbers of N3- or vehicle-treated cells into the TA muscles of wildtype 24-month old mice. Fourteen days after transplantation, we collected the TA muscles from the recipient mice and quantified the number of YFP-expressing cells. We found that N3 treatment of old MuSCs prior to transplantation led to a nearly three-fold increase in the number of engrafted YFP-expressing cells (Figure 7E and 7F). In addition, N3-treated donor cells contributed to de novo muscle fiber formation with greater efficiency than did the untreated MuSCs (Figure 7G). Taken together, these studies demonstrated that the increased frequency of cell death and impaired self-renewal of MuSCs in aged animals were at least in part due to insufficient p53 activity in these cells, and that pharmacological stabilization of p53 was able to restore the youthful function of MuSCs.
Figure 7. Rejuvenation of aged MuSC function by pharmacological activation of p53.
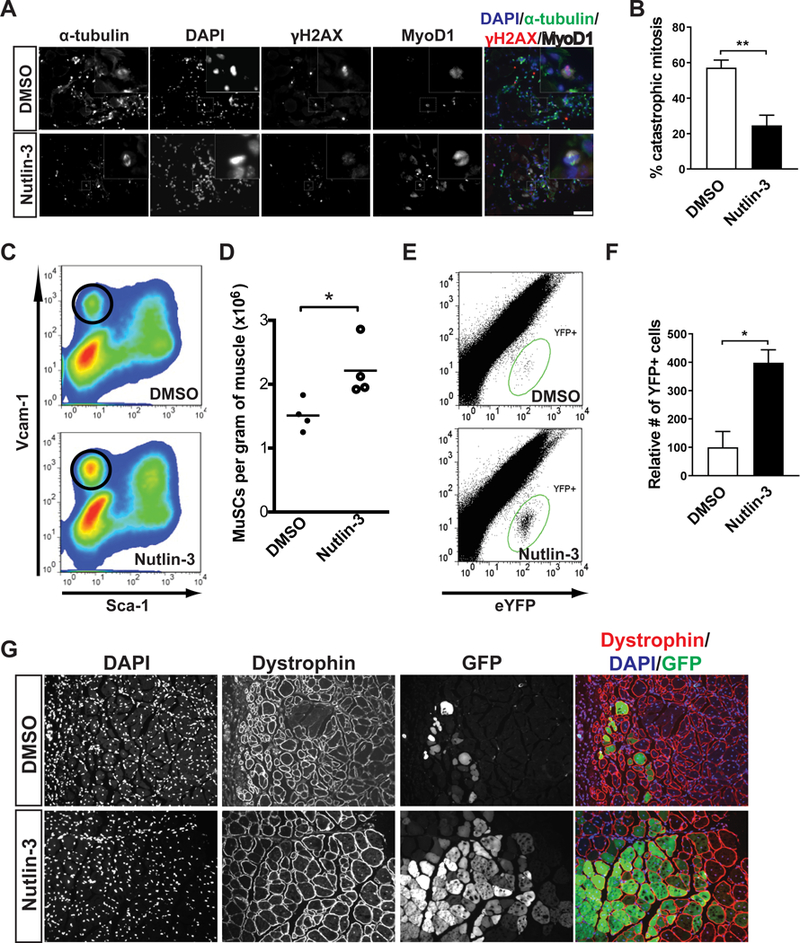
(A) Representative IF images of γ-H2AX staining (red) in mitotic myogenic cells in cross-sections of regenerating TA muscle from 24-month-old mice, three days post-injury with or without N3 treatment. Myogenic cells were labeled by MyoD1 staining (white) and spindles were labeled by α-tubulin staining (green). DNA was stained with DAPI. (B) From studies shown in panel (A), the percentages mitotic myogenic cells that were γ- H2AX-postive in each condition were calculated. (C) Representative FACS plot of MuSC progeny (circled population) in muscles of old mice, three days post-injury with or without N3 treatment. (D) The number of MuSC progeny were normalized by muscle weight from studies illustrated in panel (C) (n=4). (E) Representative FACS plots of engrafted eYFP-expressing cells 14 days following transplantation. MuSCs isolated from aged PAX7CreER/+; ROSA26eYFP/+ mice were cultured in control medium or medium supplemented with N3 for two days. From each condition, 10,000 cells were then transplanted into the TA muscles of 24-month-old mice. (F) From studies illustrated in panel (E), engrafted YFP cells were analyzed by FACS 14 days after transplantation (n = 4 mice analyzed in each condition). (G) Representative IF images of YFP-expressing muscle fibers 14 days following transplantation of YFP-expressing aged MuSCs treated or untreated with N3.
DISCUSSION
The Notch-p53 axis and stem cell function
In recent years, cross-talk between the Notch and p53 pathways has been proposed in some disease settings. Both Notch and p53 are activated in the brains of animals subjected to ischemic stroke (Balaganapathy et al., 2017). These same authors further demonstrated that, in human neuroblastoma cells, suppressing Notch signaling by DAPT inhibits p53 phosphorylation and overexpression of the active form of Notch, NICD, leads to an increase in p53 phosphorylation, stability, and transcriptional activity. The mechanism by which Notch activation induces p53-dependent transcription in these conditions remains unknown. In human sarcoma cells, expression of the canonical Notch target gene Hey1 has an inhibitory effect on Mdm2-mediated p53 degradation and sensitizes cells to chemotherapeutic drugs (Lopez-Mateo et al., 2016). A model has been put forth that Hey1 competes with p53 for binding with Mdm2 thereby suppressing the Mdm2-mediated p53 degradation. However, a physical interaction between Hey1 and Mdm2 that is strong enough to compete for the binding of p53 by Mdm2 has not been demonstrated.
Our study not only delineates a molecular mechanism by which Notch regulates p53 stability and transcriptional activity, but also demonstrates the importance of such a Notch-p53 axis in regulating normal stem cell function. We demonstrate that Hey1 directly binds to the promoter of the p53 antagonist Mdm2 and suppresses its expression. In old animals, changes in the niche environment leads to an impairment of the Notch pathway in MuSCs (Conboy et al., 2003) resulting in elevated levels of Mdm2 and reduced levels of p53 protein in aged MuSCs. The reduced p53 activity are then responsible for the increased propensity of aged MuSCs to die from mitotic catastrophe upon activation, accounting at least in part for both the impaired regenerative response and the impaired MuSC self-renewal in aged muscle. This Notch-p53 axis thus provides a link between niche and cell-autonomous changes that adversely impact MuSC function during aging.
The protective role of p53 or p53 family proteins has been documented in other stem cell compartments. Neural stem cells from p73-null mice are defective in proliferation and self-renewal (Agostini et al., 2010). Interestingly, a reduction in the expression of Notch1 and its targets Hes1 and Hes5 was found in these cells. Intestinal injury following irradiation has been found to be exacerbated by p53 deficiency in mice (Komarova et al., 2004). The frequency of non-apoptotic cell death of intestinal epithelial progenitors and their progeny in the crypts of p53- or p21-null mice is 10-fold elevated from that of wildtype mice (Leibowitz et al., 2018). These findings suggest that p53 and its target p21 are essential for productive intestinal regeneration by promoting the survival of intestinal progenitors. It remains unknown whether the protective role of p53 in these stem or progenitor cells is regulated by Notch signaling, but it is noteworthy the Notch pathway has been found to be a key regulator of many types of adult stem cells, including neural stem cells, MuSCs, intestinal stem cells, and hematopoietic stem cells (Koch et al., 2013). It will be interesting to investigate whether the Notch-p53 axis exists in these different type of stem cells and whether it can be utilized as a therapeutic target to enhance tissue regeneration in the setting of injury, disease, or aging.
Role of p53 in Aging
As a tumor suppressor, p53 promotes organismal lifespan by reducing somatic mutations that accumulate in cells or by eliminating cells that accumulate somatic mutations, thereby reducing the incidence of cancer (Rodier et al., 2007). However, in the absence of cancer, the role of p53 in tissue and cellular aging has remained elusive. Although mice harboring constitutively active p53 alleles are remarkably resistant to cancer at older age, they have a lifespan 20–30% shorter than wildtype control mice and exhibit a number of phenotypes associated with aging (Liu et al., 2010; Tyner et al., 2002). Of note, these phenotypes were linked to extensive apoptosis involving tissue progenitor cells of several organs. These mouse models with constitutively active p53 alleles imply that increased p53 activity is associated with, and potentially drives, cell and tissue aging. On the contrary, transgenic mice carrying one or two extra copies of the wildtype p53 gene under endogenous transcriptional control have a normal lifespan and show no signs of accelerated aging while being strikingly resistant to irradiation or carcinogen-induced cancers (Carrasco-Garcia et al., 2015; Garcia-Cao et al., 2002). Cells from these mice exhibit normal basal level of p53 activity which can be enhanced in response to oxidative stress and DNA damage (Garcia-Cao et al., 2002). These studies suggest that elevated but properly regulated p53 activity appears to provide protection against cellular stress and therefore to promote longevity.
On the basis of our observations of decreased p53 signaling in aged myogenic progenitors and the association of p53 inhibition with mitotic catastrophe, we were able to promote progenitor cell expansion in aged animals by pharmacologic stabilization of p53. This result suggests that p53 activation is beneficial for cell survival and expansion of the progenitor cell pool. We did note during the course of our studies that increasing the dose of N3 beyond those sufficient to prevent cell death caused a dose-dependent decline in cell number, presumably due to the cytotoxic or cytostatic effects of supraphysiological levels of p53. This observation, along with the aging-related phenotypes of the mice bearing constitutive active isoforms of p53 noted above, suggest that the regulation of p53 on cell survival and aging may follow a model more complex than the simple absence versus presence of the protein. There may be a threshold dose of cellular p53 over which the function of p53 switches from promoting cell survival to driving cell death.
Consistent with previous publications (Liu et al., 2013; Sousa-Victor et al., 2014), we did not observe changes in the expression of inhibitory cell cycle regulators such as p16INK4a, p15INK4b, or p19ARF in MuSCs from mice used in our studies at the age of 22–24 months old. Toward the very end of life (i.e. in mice that are 28–32 months of age), the expression of p16INK4a becomes elevated (Sousa-Victor et al., 2014). Although MuSCs do not acquire a senescent phenotype in resting muscle, they acquire a propensity to undergo senescence in response to activation following muscle injury. It remains to be determined whether the chronic loss of p53 expression in MuSCs with age will eventually lead to their senescent phenotype upon activation. It has been reported that p53 is necessary but not sufficient to suppress the expression of p16INK4a in different cell types (Leong et al., 2009). Future studies using genetic and pharmacologic manipulations of p53 levels will be required to define thresholds at which p53 exerts an inhibitory effect on p16INK4a expression. Tuning p53 levels according to these thresholds may allow for targeted treatments that promote healthy skeletal muscle regeneration in old age.
MuSC Death by Mitotic Catastrophe
The term “mitotic catastrophe” was first used to describe the phenotype of a Schizosaccharomycespombe strain that prematurely enters mitosis due to an activating Cdc2 mutation (Russell and Nurse, 1986). These yeast cells are unable to complete mitosis properly and therefore fail to produce viable progeny. In cancer biology, mitotic catastrophe is often used to explain the process by which cancer cells undergo mitosis-linked cell death triggered by physical or chemical stress (Castedo et al., 2004a). It can be induced by ionizing irradiation which damages DNA and various anti-cancer agents that destabilize microtubules or inactivate cell cycle checkpoint proteins. Of note, impairments in the G1 and/or G2 checkpoint controls are essential for mitotic catastrophe to take place. Cells with functional cell cycle checkpoint controls are often capable of activating apoptosis pathways in interphase and therefore have little chance to exhibit signs of mitotic catastrophe in response to DNA damage. In this two-hit model, central checkpoint proteins are often considered “negative regulators” of mitotic catastrophe. For example, deletion of the cell cycle checkpoint kinases Chk1 or Chk2 triggers mitotic catastrophe of cancer cells treated with chemotherapeutic drugs (Castedo et al., 2004b; Niida et al., 2005). It has also been shown that functional p53 proteins inhibits the induction of mitotic catastrophe of cancer cells following irradiation proteins (Ianzini et al., 2006; Reinhardt et al., 2007).
Our studies reveal that MuSCs from aged animals have accumulated DNA damage, and that, when activated in response to acute muscle injury, they exhibit a lower level of p53 protein due to the lack of niche support. Consequently, aged MuSCs undergo mitotic catastrophe and exhibit an increase in the frequency of cell death in the transient expansion phase of muscle regeneration. More importantly, restoring p53 activity in aged MuSCs inhibited mitotic catastrophe and promoted their expansion and self-renewal. To our knowledge, this is the first study that demonstrates mitotic catastrophe as a manifestation of cellular aging outside of the context of cancer. Whereas in the cancer setting, mitotic catastrophe provides a means to eliminate cancer cells and potentially has a beneficial effect, during aging it limits the production of functional stem cell progeny and therefore has a detrimental effect on tissue regeneration. Nevertheless, the two-hit model of mitotic catastrophe in cancer appears to be applicable to cells undergoing mitotic catastrophe regardless of the setting in which it occurs. In MuSCs, as in cancer, the co-occurrence of DNA damage and impairment in cell cycle checkpoint control appears to be sufficient for mitotic catastrophe to take place.
The accumulation of DNA damage with age has been documented in a number of adult stem cells, including hematopoietic and epidermal stem cells (Rossi et al., 2005; Sotiropoulou et al., 2010). Although a reduction in p53 protein and activity has been found in multiple tissues in aged animals (Feng et al., 2007), it remains to be determined whether the stem or progenitor cells that reside in these tissues exhibit a decrease in p53 or undergo mitotic catastrophe during activation and proliferation. As such, it will be interesting to explore the extent to which p53-dependent mitotic catastrophe of stem or progenitor cells is a general mechanism underlying the decline in tissue regeneration with age. To this end, drugs that restore p53 activity or targets components in this pathway may hold the potential to treat age-related regenerative decline.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| Anti-mouse CD31-FITC, Clone MEC13.3 | BioLegend | Catalog # 102506; RRID:AB 312913 |
| Anti-mouse CD31-APC, Clone MEC13.3 | BioLegend | Catalog # 102510; RRID:AB 312917 |
| Anti-mouse CD45-FITC, Clone 30-F11 | BioLegend | Catalog # 103108; RRID:AB 312973 |
| Anti-mouse CD45-APC, Clone 30-F11 | BioLegend | Catalog # 103112; RRID:AB 312977 |
| Anti-mouse Ly-6A/E(Sca1)-Pacific Blue, Clone D7 |
BioLegend | Catalog # 108120; RRID:AB 493273 |
| Anti-mouse CD106 (VCAM1)-PE/Cy7, Clone 429(MVCAM.A) | BioLegend | Catalog # 105720; RRID:AB 2214046 |
| Anti-mouse Pax7 | Developmental Studies Hybridoma Bank | RRID: AB_528428 |
| Anti-laminin 2α | Abcam | Catalog # 11576; RRID:AB 298180 |
| Anti-tubulin DM1A-FITC | Sigma | Catalog # F2168; RRID:AB 476967 |
| Anti-pericentrin | Covance | Catalog # PRB-432C; RRID:AB_2313709 |
| Anti-MyoD1 | BD Pharmingen | Clone 5.8A; Catalog # 554130; RRID:AB_395255 |
| Anti-cleaved caspase 3 (Asp 175) | Cell Signaling Technologies | Catalog # 9661; RRID:AB_2341188 |
| Anti-phospho-H2A.X (Ser 139) | Cell Signaling Technologies | Catalog # 9718; RRID:AB 2118009 |
| Anti-Myc, clone 4A6 | Millipore | Catalog # 05–724; RRID:AB 309938 |
| Anti-GFP | Abcam | Catalog # 13970; RRID:AB_300798 |
| Anti-Dystrophin, clone 1C7 | Developmental Studies Hybridoma Bank | Catalog #: MANEX1011B (1C7); RRID:AB 1157876 |
| Anti-mouse Myogenin, clone F5D | BD Pharmingen | Catalog # 556358; RRID:AB 396383 |
| Anti-mouse p53 | Leica | NCL-L-p53-CM5p; RRID:AB_563933 |
| Anti-Mdm2, clone 2A10 | Abcam | Catalog # 16895; RRID:AB_2143534 |
| Anti-β-actin-peroxidase, Clone AC15 | Sigma | Catalog #A3854; RRID:AB_262011 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 4’,6-diamidino-2-phenylindole (DAPI) | Thermo Fisher Scientific | Catalog # D1306 |
| Propidium iodine | Thermo Fisher Scientific | Catalog # P3566 |
| DMSO | Sigma | Catalog # 41639 |
| Nutlin-3 | Selleck Chemicals | Catalog # S1061 |
| DAPT | Selleck Chemicals | Catalog # S2215 |
| Recombinant mouse Dll-1 Fc Chimera | R&D Systems | Catalog # 5026-DL |
| Recombinant mouse Dll-4 | R&D Systems | Catalog # 1389- D4ICF |
| Recombinant human Jag1 Fc Chimera | Enzo Life Sciences | ALX-201–390 |
| Recombinant mouse Jag2 Fc Chimera | R&D Systems | Catalog # 4748-JG |
| YH239-EE | Selleck Chemicals | Catalog # S7489 |
| Tamoxifen | Sigma | Catalog # T5648 |
| Critical Commercial Assays | ||
| Dual Luciferase Reporter Assay System | Promega | Catalog # E1910 |
| Experimental Models: Cell Lines | ||
| MuSC culture | In this paper | NIA |
| Single fiber culture | In this paper | NIA |
| C2C12 | ATCC | RRID:CVCL_0188 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | JAX or NIA | RRID:IMSR JAX:000 664 |
| Mouse: B6.129P2-Trp53tm1Bm/J (or p53LoxP) | JAX | Catalog # 008462; RRID:IMSR JAX:008 462 |
| Mouse: Pax7tm1(cre/Esr1*)Cklr (or Pax7CreER/CreER) | (Nishijo et al., 2009) | RRID:IMSR NCIMR: 01XBS |
| Mouse: Pax7tm1(cre/Esr1*)Cklr (or Pax7CreER/CreER) | JAX | Catalog # 006148; RRID:IMSR JAX:006 148 |
| Oligonucleotides | ||
| non-targeting control siRNA | Dharmacon | D-001810–10-05 |
| Hey1 siRNA | Dharmacon | L-040475–01-0005 |
| Hey2 siRNA | Dharmacon | L-043647–01-0005 |
| HeyL siRNA | Dharmacon | L-060622–01-0005 |
| See Method S1 for PCR primers | NIA | |
| Recombinant DNA | ||
| p53TRE-Luc | cloned in a Stratagene vector | NIA |
| pMdm2-Luc A | (Chang et al., 2004) | NIA |
| pMdm2-Luc B | (Chang et al., 2004) | NIA |
| pMdm2-Luc C | In this paper | NIA |
| pCMV6-Myc-Hey1 | Cloned in an Origene vector | NIA |
| pHey1-Luc | (Maier and Gessler, 2000) | NIA |
| Software and Algorithms | ||
| Improvision Volocity | Perkin Elmer | |
| ZEN 2010 software | Carl Zeiss | |
| Image Lab | Bio-Rad | |
| OpenComet | (Gyori et al., 2014) | |
| DAVID | (Dennis et al., 2003) | |
| Prism GraphPad | GraphPad Software | |
Contact for Reagents and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Thomas A. Rando (rando@stanford.edu)
Experimental Model Details
Animals and Treatments
Two to four month-old C57BL/6 male mice were used to collect young MuSCs and twenty-two to twenty-four month-old mice were used to collect old MuSCs. Animals were housed and maintained in the Veterinary Medical Unit at the Veterans Affairs Palo Alto Health Care System. Animal protocols were approved by the Institutional Animal Care and Use Committee. To activate Cre in mice, Tamoxifen (Sigma-Aldrich) injection was performed as previously described (Nishijo et al., 2009). Mice received 5 mg of Tamoxifen dissolved in a mixture of corn oil and 7% ethanol delivered via intraperitoneal injection for 5 consecutive days. To activate MuSCs in vivo, mice were injured by injecting 50 μL of 1.2% BaCl2 (w/v in H2O) into the lower hind-limb muscles (below the knee). For sequential regeneration studies, injuries and analyses were performed at 7-day intervals. Nutlin-3a (Selleck Chemicals) was dissolved in 30% DMSO (v/v in normal saline) and injected into the intraperitoneal space at a dose of 4 mg/kg/day. Mice received once daily treatment for three days, beginning at the time of injury. Analyses following Nutlin treatment were performed three days post-injury.
MuSC Isolation
Isolation of muscle-resident cells by sequential enzymatic and physical dissociation was performed as previously described (Liu et al., 2015). Sorted cells were routinely analyzed by flow cytometry immediately after sorting to ensure high sorting efficiency. Also, a fraction of the sorted cells were routinely plated and stained for Pax7 to determine the purity of the sorted population. For analysis of cell death by flow cytometry, sorted cells were treated immediately after sorting with propidium iodide (Invitrogen), incubated for 5 minutes at room temperature, and re-analyzed by flow cytometry. For transplantation, cells were rinsed and resuspended with sterile PBS immediately following FACS isolation and injected into the TA muscle of the recipient mouse.
MuSC Culture
Purified MuSCs were immediately plated on extracellular matrix (ECM)-coated plastic dishes or glass slides. Cells were routinely cultured at a density of 3×103 −4×103 cells per cm2. Vessels were coated with ECM (Sigma-Aldrich) by incubating with ECM in DMEM (1:500, V:V) with 1% penicillin-streptomycin for 12 hours at 4°C with gentle rocking. Vessels were used within 2 days of coating. Cells were cultured in Ham’s F10 supplemented with 20% FBS (Omega Scientific) and 1% penicillin-streptomycin. Medium was changed every 48 hours unless noted otherwise. Nutlin-3a (Selleck Chemicals) and DAPT (Sigma-Aldrich) were dissolved in DMSO, stored at −20°C, and used at the concentrations noted in the text and figures. Drugs were also refreshed at each medium change. Recombinant mouse Dll1, Dll4, and Jag2 ligands fused to the IgG Fc domain (R&D Systems) and Jag1 fused to Fc (Enzo Life Sciences) were reconstituted in sterile PBS and stored at −20°C before use. Ligands were absorbed to the cell culture surface by coating for 12 hours at 4°C in a 1:500 mix of ECM:DMEM (prepared as above). The concentration of ligand in the coating medium was 5 μg/mL for all experiments. The coating medium was removed prior to plating cells in growth medium. siRNA (Dharmacon) and plasmid transfection was performed with Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions.
Single Muscle Fiber Isolation
Single muscle fibers were prepared essentially as described (Rosenblatt et al., 1995). Briefly, the extensor digitorum longus (EDL) muscles were carefully dissected from mice following euthanization and digested in 2 mg/ml collagenase II prepared in Ham’s F10 with 10% horse serum at 37°C for 80 min with gentle agitation. The digested EDL muscles were then triturated with a wide bore glass pipet in 20 ml 10% horse serum in Ham’s F10 in a 10-cm tissue culture dish. Individual fibers were washed four times in medium before they were fixed or cultured to allow SC activation. To culture fibers ex vivo, approximately 100 fibers were maintained in suspension in 10 ml medium (20% FBS in Ham’s F10) in 10-cm dishes for 3 days.
Method Details
Immunofluorescence (IF) and Quantitative Microscopy
Fixed and permeabilized cells/fibers were blocked in 10% goat serum (in PBS) and incubated with primary antibodies for 12 hours at 4°C. After washing out primary antibodies with PBS, cells were incubated with secondary antibodies for one hour at room temperature. Goat anti-mouse antibodies conjugated to Alexa Fluor 594 or Alexa Fluor 488 were used to detect mouse primary antibodies. Cells were next washed with PBS and mounted with Fluoro-Gel (Electron Microscopy Sciences). IF imaging was performed with an AxioObserver Z1 epifluorescence microscope (Carl Zeiss) equipped with an Orca-R2 CCD camera (Hamamatsu Photonics) or an LSM 710 confocal system (Carl Zeiss). Image analysis was performed using Improvision Volocity software (Perkin Elmer) or ZEN 2010 software (Carl Zeiss). For fiber analyses, the number of eYFP- expressing cells was quantified for a representative sample of fibers from three animals of each genotype. For γ-H2AX fluorescence intensity measurements, a region of interest (ROI) consisting of the DAPI-stained DNA of a metaphase cell was identified using the Improvision Volocity software. The average intensity of γ-H2AX fluorescence within the ROI was recorded. A background fluorescence level, calculated by defining a background ROI near the metaphase cell, was calculated for each cell and subtracted from the fluorescence intensity measurement.
Time-Lapse Microscopy
Prospectively isolated MuSCs were plated at a density of 4×103 cells per 0.7 cm2 and cultured as above. Cultures were analyzed using an Axiovert 200M inverted microscope (Carl Zeiss) equipped with an environmental control chamber (CTI controller, Tempcontrol; Carl Zeiss; humidified 5% CO2 at 37°C). Brightfield images were obtained every 15 min for 36–48 hours with a Zeiss camera controlled with the Axiovision software (Carl Zeiss). For analyses of cell fate, individual cells were selected at random at the beginning of the analysis, and were followed throughout the course of each time- lapse video.
Luciferase Reporter Assays
Cells plated for luciferase reporter assays were transfected after one day in culture with firefly luciferase reporter constructs, a constitutive Renilla luciferase construct, and other protein-coding plasmids when applicable using Lipofectamine 2000 (Invitrogen). Two days post-transfection, cells were harvested and luciferase expression was analyzed using the Dual Luciferase Assay System (Promega) and a luminometer (Turner Biosystems). All luciferase assays were performed in triplicate in each of a minimum of three independent experiments. For each well, firefly luciferase activity was normalized to Renilla luciferase activity.
Histology
For immunofluorescence (IF) and hematoxylin/eosin staining, dissected muscles were mounted on tragacanth gum and snap frozen in liquid nitrogen-cooled isopentane. Acutely injured muscles (3 days post-injury) were fixed in 0.5% (v/v in PBS) electron microscopy-grade paraformaldehyde at room temperature for 5 hours prior to dehydration in 20% sucrose (w/v in PBS) at 4°C for 8 hours and subsequently embedded in OCT medium (Tissue-Tek) and snap frozen. Hematoxylin/eosin staining was performed on 20- μm-thick sections. IF staining was performed on 6-μm-thick sections. Following rehydration of sections in PBS and post-fixation for 5 min in 2% paraformaldehyde, IF staining was carried out essentially as described above for cells. Detection of mouse antigens was performed using a M.O.M. kit (Vector Labs) or Zenon labelling kit (Invitrogen). For quantification of MuSC self-renewal, 25 fields from each of 4 animals per condition were analyzed. For histomorphometric analysis of tissue regeneration, 25 independent fields representative of poorest regeneration were analyzed from each of 3 animals per condition.
Western Blotting
Two hundred thousand MuSCs pooled from two animals were used for each analyzed lane. Cells were purified from lower hind-limb muscles three days post-injury. Protein extracts were separated by electrophoresis on 4–15% polyacrylamide gradient gels and then transferred to PVDF membranes. Protein levels were quantified in Adobe Photoshop using pixel analysis of protein bands in scanned images. Background pixel intensity was calculated individually for each blot and subtracted from band intensity values.
RT-qPCR
Cells were rinsed in PBS and then lysed. RNA was isolated using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. Total RNA (0.2–1.0 micrograms) was reverse-transcribed using the High Capacity cDNA Reverse Transcription Kit (Invitrogen). Quantitative PCR was performed on an ABI 7900HT Fast Real-Time PCR system using custom synthesized oligonucleotide primers (Invitrogen) designed to amplify the cDNA of selected target genes. Relative quantification of gene expression normalized to Gapdh was carried-out using the comparative CT method (Pfaffl, 2001). Each measurement was performed in triplicate in three independent experiments.
Chromatin Immunoprecipitation
Mouse C2C12 myoblasts were cultured in DMEM supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin. Myoblasts were transiently transfected with pCMV6-entry bearing an insert encoding the myc-tagged Hey l open reading frame (Origene) using Lipofectamine 2000 (Invitrogen). Analysis of Hey1 binding to the Mdm2 promoter was carried out using enzymatic shearing (Active Motif) according to the manufacturer’s instructions. IP was performed using a purified anti-myc antibody (clone 4A6, EMD Millipore). Mouse IgG (Santa Cruz Biotechnologies) was used as a control. Fold enrichment was calculated as a percentage relative to input DNA for each set of primers. qPCR analysis was performed in triplicate for each of two independent pull- downs.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 5 (GraphPad Software). Significance was calculated using two-tailed, unpaired Student’s t-tests. Differences were considered to be statistically significant at the P < 0.05 level (*p < 0.05, **p<0.01, ***p < 0.001, ns: not significant). Unless otherwise noted, all error bars represent SEM.
Supplementary Material
HIGHLIGHTS.
Muscle stem cells (MuSCs) tend to die by mitotic catastrophe without niche support.
MuSC mitotic catastrophe is exacerbated by a decline in p53 associated with aging.
Notch signaling represses Mdm2 expression thereby increasing p53 levels in MuSCs.
Pharmacologic enhancement of p53 levels promotes the survival of aged MuSCs.
ACKNOWLEDGEMENTS
We thank all members of the Rando Laboratory for helpful discussions. We gratefully acknowledge input from Dr. Laura Attardi. We also acknowledge Dr. Charles Keller and Dr. Manfred Gessler for generously providing reagents. This work was supported by the Glenn Foundation for Medical Research, by fellowships from the NIH (F30 AG035521) and Stanford University School of Medicine Medical Scientist Training Program to G.W.C., and by grants from the Department of Veterans Affairs (Merit Review) and the NIH (P01 AG036695, R37 AG023806, and TR01 AG047820) to T.A.R.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
REFERENCES
- Agostini M, Tucci P, Chen H, Knight RA, Bano D, Nicotera P, McKeon F, and Melino G (2010). p73 regulates maintenance of neural stem cell. Biochem Biophys Res Commun 403, 13–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balaganapathy P, Baik SH, Mallilankaraman K, Sobey CG, Jo DG, and Arumugam TV (2017). Interplay between Notch and p53 promotes neuronal cell death in ischemic stroke. J Cereb Blood Flow Metab, 271678X17715956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernet JD, Doles JD, Hall JK, Kelly Tanaka K, Carter TA, and Olwin BB (2014). p38 MAPK signaling underlies a cell-autonomous loss of stem cell self-renewal in skeletal muscle of aged mice. Nat Med 20, 265–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornson CR, Cheung TH, Liu L, Tripathi PV, Steeper KM, and Rando TA (2012). Notch signaling is necessary to maintain quiescence in adult muscle stem cells. Stem cells 30, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brack AS, Conboy MJ, Roy S, Lee M, Kuo CJ, Keller C, and Rando TA (2007). Increased Wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 317, 807–810. [DOI] [PubMed] [Google Scholar]
- Brack AS, and Rando TA (2007). Intrinsic changes and extrinsic influences of myogenic stem cell function during aging. Stem Cell Rev 3, 226–237. [DOI] [PubMed] [Google Scholar]
- Carlson ME, Hsu M, and Conboy IM (2008). Imbalance between pSmad3 and Notch induces CDK inhibitors in old muscle stem cells. Nature 454, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco-Garcia E, Arrizabalaga O, Serrano M, Lovell-Badge R, and Matheu A (2015). Increased gene dosage of Ink4/Arf and p53 delays age-associated central nervous system functional decline. Aging Cell 14, 710–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Andreau K, Medema R, and Kroemer G (2004a). Cell death by mitotic catastrophe: a molecular definition. Oncogene 23, 2825–2837. [DOI] [PubMed] [Google Scholar]
- Castedo M, Perfettini JL, Roumier T, Valent A, Raslova H, Yakushijin K, Horne D, Feunteun J, Lenoir G, Medema R, et al. (2004b). Mitotic catastrophe constitutes a special case of apoptosis whose suppression entails aneuploidy. Oncogene 23, 4362–4370. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Freeman DJ, and Wu H (2004). PTEN regulates Mdm2 expression through the P1 promoter. J Biol Chem 279, 29841–29848. [DOI] [PubMed] [Google Scholar]
- Collins CA, Olsen I, Zammit PS, Heslop L, Petrie A, Partridge TA, and Morgan JE (2005). Stem cell function, self-renewal, and behavioral heterogeneity of cells from the adult muscle satellite cell niche. Cell 122, 289–301. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Smythe GM, and Rando TA (2003). Notch-mediated restoration of regenerative potential to aged muscle. Science 302, 1575–1577. [DOI] [PubMed] [Google Scholar]
- Conboy IM, Conboy MJ, Wagers AJ, Girma ER, Weissman IL, and Rando TA (2005). Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764. [DOI] [PubMed] [Google Scholar]
- Conboy IM, and Rando TA (2002). The regulation of Notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Developmental cell 3, 397–409. [DOI] [PubMed] [Google Scholar]
- Conboy IM, and Rando TA (2012). Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches. Cell cycle 11, 2260–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosgrove BD, Gilbert PM, Porpiglia E, Mourkioti F, Lee SP, Corbel SY, Llewellyn ME, Delp SL, and Blau HM (2014). Rejuvenation of the muscle stem cell population restores strength to injured aged muscles. Nat Med 20, 255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlberg A, Woo S, Delaney C, Boyle P, Gnirke A, Bock C, Bernstein BE, Meissner A, Gottardo R, and Bernstein ID (2015). Notch-mediated expansion of cord blood progenitors: maintenance of transcriptional and epigenetic fidelity. Leukemia 29, 1948–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis G Jr., Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, and Lempicki RA (2003). DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol 4, P3. [PubMed] [Google Scholar]
- Dodson H, Wheatley SP, and Morrison CG (2007). Involvement of centrosome amplification in radiation-induced mitotic catastrophe. Cell cycle 6, 364–370. [DOI] [PubMed] [Google Scholar]
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, and Levine AJ (2007). Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci U S A 104, 16633–16638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernando P, Kelly JF, Balazsi K, Slack RS, and Megeney LA (2002). Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci U S A 99, 11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firat E, Gaedicke S, Tsurumi C, Esser N, Weyerbrock A, and Niedermann G (2011). Delayed cell death associated with mitotic catastrophe in gamma-irradiated stem-like glioma cells. Radiat Oncol 6, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragkos M, and Beard P (2011). Mitotic catastrophe occurs in the absence of apoptosis in p53-null cells with a defective G1 checkpoint. PLoS One 6, e22946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Cao I, Garcia-Cao M, Martin-Caballero J, Criado LM, Klatt P, Flores JM, Weill JC, Blasco MA, and Serrano M (2002). “Super p53” mice exhibit enhanced DNA damage response, are tumor resistant and age normally. EMBO J 21, 6225–6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyori BM, Venkatachalam G, Thiagarajan PS, Hsu D, and Clement MV (2014). OpenComet: an automated tool for comet assay image analysis. Redox Biol 2, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi MT, and Karlseder J (2013). DNA damage associated with mitosis and cytokinesis failure. Oncogene 32, 4593–4601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Tran T, Zhang L, Hatcher R, and Zhang P (2005). DNA damage-induced mitotic catastrophe is mediated by the Chk1-dependent mitotic exit DNA damage checkpoint. Proc Natl Acad Sci U S A 102, 1065–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wolf S, Beck B, Kohler LM, Khoury K, Popowicz GM, Goda SK, Subklewe M, Twarda A, Holak TA, et al. (2014). Discovery of highly potent p53- MDM2 antagonists and structural basis for anti-acute myeloid leukemia activities. ACS Chem Biol 9, 802–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ianzini F, Bertoldo A, Kosmacek EA, Phillips SL, and Mackey MA (2006). Lack of p53 function promotes radiation-induced mitotic catastrophe in mouse embryonic fibroblast cells. Cancer Cell Int 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichijima Y, Yoshioka K, Yoshioka Y, Shinohe K, Fujimori H, Unno J, Takagi M, Goto H, Inagaki M, Mizutani S, et al. (2010). DNA lesions induced by replication stress trigger mitotic aberration and tetraploidy development. PLoS One 5, e8821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imreh G, Norberg HV, Imreh S, and Zhivotovsky B (2011). Chromosomal breaks during mitotic catastrophe trigger gammaH2AX-ATM-p53-mediated apoptosis. J Cell Sci 124, 2951–2963. [DOI] [PubMed] [Google Scholar]
- Koch U, Lehal R, and Radtke F (2013). Stem cells living with a Notch. Development 140, 689–704. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Kondratov RV, Wang K, Christov K, Golovkina TV, Goldblum JR, and Gudkov AV (2004). Dual effect of p53 on radiation sensitivity in vivo: p53 promotes hematopoietic injury, but protects from gastro-intestinal syndrome in mice. Oncogene 23, 3265–3271. [DOI] [PubMed] [Google Scholar]
- Kroemer G, Galluzzi L, Vandenabeele P, Abrams J, Alnemri ES, Baehrecke EH, Blagosklonny MV, El-Deiry WS, Golstein P, Green DR, et al. (2009). Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ 16, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibowitz BJ, Yang L, Wei L, Buchanan ME, Rachid M, Parise RA, Beumer JH, Eiseman JL, Schoen RE, Zhang L, et al. (2018). Targeting p53-dependent stem cell loss for intestinal chemoprotection. Sci Transl Med 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong WF, Chau JF, and Li B (2009). p53 Deficiency leads to compensatory up- regulation of p16INK4a. Mol Cancer Res 7, 354–360. [DOI] [PubMed] [Google Scholar]
- Liu DP, Song H, and Xu Y (2010). A common gain of function of p53 cancer mutants in inducing genetic instability. Oncogene 29, 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, Hurgo BM, Leavitt T, Shih J, Brunet A, and Rando TA (2013). Chromatin modifications as determinants of muscle stem cell quiescence and chronological aging. Cell reports 4, 189–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Cheung TH, Charville GW, and Rando TA (2015). Isolation of skeletal muscle stem cells by fluorescence-activated cell sorting. Nat Protoc 10, 1612–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Mateo I, Arruabarrena-Aristorena A, Artaza-Irigaray C, Lopez JA, Calvo E, and Belandia B (2016). HEY1 functions are regulated by its phosphorylation at Ser- 68. Biosci Rep 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier MM, and Gessler M (2000). Comparative analysis of the human and mouse Hey1 promoter: Hey genes are new Notch target genes. Biochem Biophys Res Commun 275, 652–660. [DOI] [PubMed] [Google Scholar]
- Mann CJ, Perdiguero E, Kharraz Y, Aguilar S, Pessina P, Serrano AL, and Munoz-Canoves P (2011). Aberrant repair and fibrosis development in skeletal muscle. Skelet Muscle 1, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, and Berns A (2000). Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 14, 994–1004. [PMC free article] [PubMed] [Google Scholar]
- Mauro A (1961). Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol 9, 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, and Morrison SJ (2006). Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature 443, 448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Tsuge S, Katsuno Y, Konishi A, Takeda N, and Nakanishi M (2005). Depletion of Chk1 leads to premature activation of Cdc2-cyclin B and mitotic catastrophe. J Biol Chem 280, 39246–39252. [DOI] [PubMed] [Google Scholar]
- Nishijo K, Hosoyama T, Bjornson CR, Schaffer BS, Prajapati SI, Bahadur AN, Hansen MS, Blandford MC, McCleish AT, Rubin BP, et al. (2009). Biomarker system for studying muscle, stem cells, and cancer in vivo. FASEB J 23, 2681–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura EK, Granter SR, and Fisher DE (2005). Mechanisms of hair graying: incomplete melanocyte stem cell maintenance in the niche. Science 307, 720–724. [DOI] [PubMed] [Google Scholar]
- Pachkov M, Erb I, Molina N, and van Nimwegen E (2007). SwissRegulon: a database of genome-wide annotations of regulatory sites. Nucleic Acids Res 35, D127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker MH, and Tapscott SJ (2013). Expanding donor muscle-derived cells for transplantation. Curr Protoc Stem Cell Biol Chapter 2, Unit 2C 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffl MW (2001). A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res 29, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price FD, von Maltzahn J, Bentzinger CF, Dumont NA, Yin H, Chang NC, Wilson DH, Frenette J, and Rudnicki MA (2014). Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat Med 20, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando TA (2006). Stem cells, ageing and the quest for immortality. Nature 441, 1080–1086. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Aslanian AS, Lees JA, and Yaffe MB (2007). p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer Cell 11, 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Campisi J, and Bhaumik D (2007). Two faces of p53: aging and tumor suppression. Nucleic Acids Res 35, 7475–7484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt JD, Lunt AI, Parry DJ, and Partridge TA (1995). Culturing satellite cells from living single muscle fiber explants. In Vitro Cell Dev Biol Anim 31, 773–779. [DOI] [PubMed] [Google Scholar]
- Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, and Weissman IL (2005). Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A 102, 9194–9199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell P, and Nurse P (1986). cdc25+ functions as an inducer in the mitotic control of fission yeast. Cell 45, 145–153. [DOI] [PubMed] [Google Scholar]
- Schwarzkopf M, Coletti D, Sassoon D, and Marazzi G (2006). Muscle cachexia is regulated by a p53-PW1/Peg3-dependent pathway. Genes Dev 20, 3440–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotiropoulou PA, Candi A, Mascre G, De Clercq S, Youssef KK, Lapouge G, Dahl E, Semeraro C, Denecker G, Marine JC, et al. (2010). Bcl-2 and accelerated DNA repair mediates resistance of hair follicle bulge stem cells to DNA-damage-induced cell death. Nat Cell Biol 12, 572–582. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P, Gutarra S, Garcia-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz- Bonilla V, Jardi M, Ballestar E, Gonzalez S, Serrano AL, et al. (2014). Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506, 316–321. [DOI] [PubMed] [Google Scholar]
- Tierney MT, Aydogdu T, Sala D, Malecova B, Gatto S, Puri PL, Latella L, and Sacco A (2014). STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat Med 20, 1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyner SD, Venkatachalam S, Choi J, Jones S, Ghebranious N, Igelmann H, Lu X, Soron G, Cooper B, Brayton C, et al. (2002). p53 mutant mice that display early ageing-associated phenotypes. Nature 415, 45–53. [DOI] [PubMed] [Google Scholar]
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, et al. (2004). In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Zammit PS, Relaix F, Nagata Y, Ruiz AP, Collins CA, Partridge TA, and Beauchamp JR (2006). Pax7 and myogenic progression in skeletal muscle satellite cells. J Cell Sci 119, 1824–1832. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.