

Abstract
We conducted a prospective study in a large, multiethnic cohort of obese adolescents to characterize clinical and genetic features associated with pediatric nonalcoholic fatty liver (NAFL), the most common cause of chronic liver disease in youth. A total of 503 obese adolescents were enrolled, including 191 (38.0%) whites, 134 (26.6%) blacks, and 178 (35.4%) Hispanics. Participants underwent abdominal magnetic resonance imaging (MRI) to quantify hepatic fat fraction (HFF), an oral glucose tolerance test (OGTT) to assess glucose tolerance and insulin sensitivity, and the genotyping of three single‐nucleotide polymorphisms (SNPs) associated with nonalcoholic fatty liver disease (NAFLD) (patatin‐like phospholipase domain‐containing protein 3 [PNPLA3] rs738409, glucokinase regulatory protein [GCKR] rs1260326, and transmembrane 6 superfamily member 2 [TM6SF2] rs58542926). Assessments were repeated in 133 subjects after a 2‐year follow‐up. Prevalence of nonalcoholic fatty liver (NAFL) was 41.6% (209 patients) and ranged widely among ethnicities, being 42.9% in whites, 15.7% in blacks, and 59.6% in Hispanics (P < 0.0001). Among adolescents with NAFL, blacks showed the highest prevalence of altered glucose homeostasis (66%; P = 0.0003). Risk factors for NAFL incidence were white or Hispanic ethnicity (P = 0.021), high fasting C‐peptide levels (P = 0.0006), and weight gain (P = 0.0006), whereas baseline HFF (P = 0.004) and weight loss (P = 0.032) predicted resolution of NAFL at follow‐up. Adding either gene variant to these variables improved significantly the model predictive performance. Conclusion: Black obese adolescents are relatively protected from liver steatosis, but are more susceptible to the deleterious effects of NAFL on glucose metabolism. The combination of ethnicity/race with markers of insulin resistance and genetic factors might help identify obese youth at risk for developing NAFL.
Abbreviations
- 2PD
2‐point Dixon
- ALT
alanine aminotransferase
- AST
aspartate transaminase
- AUC
area under the curve
- BMI
body mass index
- BP
blood pressure
- DBP
diastolic blood pressure
- DI
disposition index
- DNL
de novo lipogenesis
- GCKR
glucokinase regulatory protein
- GGT
γ‐glutamyl transferase
- GLM
general linear model
- HbA1c
hemoglobin A1C
- HDL
high‐density lipoprotein
- HFF
hepatic fat fraction
- IGI
insulinogenic index
- IQR
interquartile range
- IR
insulin resistance
- LDL
low‐density lipoprotein
- MRI
magnetic resonance imaging
- NAFL
nonalcoholic fatty liver
- NAFLD
nonalcoholic fatty liver disease
- NAS
NAFLD activity score
- OGTT
oral glucose tolerance test
- PNPLA3
patatin‐like phospholipase domain‐containing protein 3
- ROC
receiver operating characteristic
- SBP
systolic blood pressure
- SNPs
single‐nucleotide polymorphisms
- T2D
type 2 diabetes
- TG
triglycerides
- TM6SF2
transmembrane 6 superfamily member 2
- WBISI
whole‐body insulin sensitivity index
Paralleling the growing epidemic of childhood obesity, nonalcoholic fatty liver disease (NAFLD) has become the most common cause of chronic liver disease in pediatrics, with an estimate of 7 million children and adolescents affected in the United States.1 NAFLD is characterized by excess fat accumulation in hepatocytes and encompasses a wide spectrum of disease severity, from simple nonalcoholic fatty liver (NAFL) to nonalcoholic steatohepatitis (NASH),2, 3 which can progress to cirrhosis and end‐stage liver disease (ESLD) even at a young age.4, 5, 6 ) A landmark study by Feldstein et al.(4) has shown that adolescents with NAFLD have approximately 16 times higher risk of developing ESLD by the age of 20 compared to a group of American youth with similar age and sex. Moreover, studies assessing insulin resistance (IR) with state‐of‐the‐art techniques (e.g., euglycemic clamp) have demonstrated that NAFL is a major risk factor for IR and type 2 diabetes (T2D), independent from visceral and intramyocellular lipid accumulation.7
Ethnic differences in the prevalence of NAFLD have been reported in adults8, 9 and children,1, 10 with Hispanics showing the highest prevalence and blacks showing a relative protection from hepatic fat accumulation, even in the presence of morbid obesity and severe IR.11 So far, no data in pediatrics are available about putative differences among the three major ethnic groups in the United States in the metabolic profile associated with NAFL and about the predictors of changes in hepatic fat fraction (HFF) over time.6
To gain a deeper knowledge about pediatric NAFL and identify risk factors of changes in magnetic resonance imaging (MRI)‐measured intrahepatic fat, we analyzed metabolic and imaging data obtained from a multiethnic cohort of 503 overweight and obese adolescents enrolled in an ongoing study on the pathogenesis of youth‐onset NAFL. Subjects enrolled in the study were carefully phenotyped with respect to quantification of HFF and abdominal fat distribution using MRI. Glucose tolerance and markers of IR were derived from the oral glucose tolerance test (OGTT). Genotyping of patatin‐like phospholipase domain‐containing protein 3 (PNPLA3) rs738409, glucokinase regulatory protein (GCKR) rs1260326, and transmembrane 6 superfamily member 2 (TM6SF2) rs58542926 single‐nucleotide polymorphisms (SNPs) previously associated with NAFL was also performed.12, 13, 14 Moreover, after a median follow‐up of 2 years, the abdominal MRI and all metabolic assessments were repeated in a subset of 133 obese adolescents.
1. Patients and Methods
1.1. The Yale Pediatric NAFLD Cohort
A total of 503 obese and overweight adolescents were recruited from the Yale Pediatric Obesity Clinic, including 191 (38.0%) whites, 134 (26.6%) blacks, and 178 (35.4%) Hispanics (Fig. 1). Ethnic distribution was comparable to the prevalence of the different races and ethnicities in the New Haven area.15 A detailed medical and family history was obtained from all participants, and a physical examination was performed. Blood pressure (BP) was measured three times, and the last two measurements were averaged for analysis. Tanner stage was determined by a pediatrician and was based on breast stage and pubic hair development in girls16 and genitalia development in boys.17 Clinical and metabolic characteristics of the study cohort are shown in Supporting Table S1. Participants were 206 boys (41.0%) and 297 girls (59.0%), with an average age of 13.7 ± 2.8 years (median IQR [interquartile range], 13.7 [11.7‐15.7]; age range, 8.2‐18.9); Tanner stage 3.6 ± 1.4; z‐score body mass index (BMI) 2.2 ± 0.4; systolic (SBP) and diastolic BP (DBP) 118.2 ± 11.1 and 67.8 ± 8.0 mm Hg, respectively; high‐density lipoprotein (HDL) and low‐density lipoprotein (LDL) cholesterol 43.5 ± 10.2 and 89.8 ± 25.4 mg/dL, respectively; and fasting triglycerides (TG) 104.5 ± 62.7 mg/dL. Exclusion criteria were known hepatic diseases (except for NAFLD), alcohol consumption, and the use of medications that alter BP or glucose, lipid, or amino acid metabolism. At baseline, 24 boys and 14 girls (7.6% of study cohort) had alanine aminotransferase (ALT) levels 3 times higher than the normal upper limit (25 UI/L for boys and 22 UI/L for girls18). Subjects with persistent elevation of ALT levels for more than 6 months underwent appropriate blood tests to exclude autoimmune hepatitis, Wilson’s disease, alpha‐1‐antitrypsin deficiency, hepatitis B and C, and iron overload.
Figure 1.
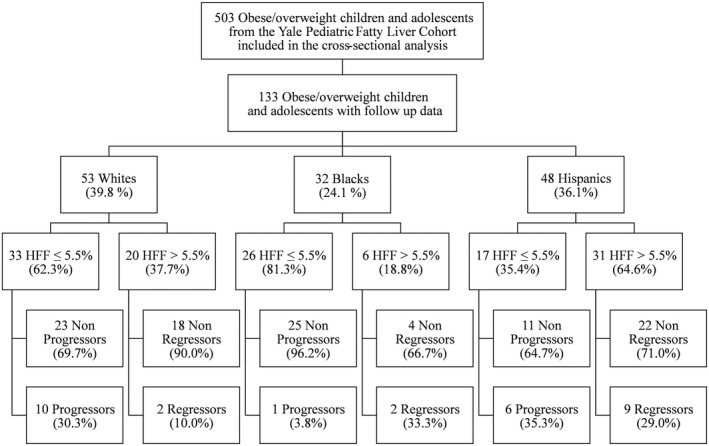
Study flowchart. Study participants are stratified according to ethnicity, presence of NAFL at presentation (HFF >5.5%), and stability or progression/regression of NAFL at follow‐up.
Subjects were phenotyped with respect to their glucose tolerance by a standard OGTT (Supporting Methods) and to HFF and abdominal fat distribution by using fast MRI.19, 20, 21 From OGTT data, insulin sensitivity and early‐phase insulin secretion were assessed by the whole‐body insulin sensitivity index (WBISI) and the insulinogenic index (IGI), respectively. The disposition index (DI) was calculated as the product of the WBISI and the IGI (Supporting Methods). Glucose tolerance was defined according to the American Diabetes Association criteria.22 NAFL was defined as liver fat content (hepatic fat fraction [HFF]%) >5.5%.8
After the first visit, all participants received standard nutritional guidance as well as recommendations for physical activity and were scheduled to be followed up every 4‐6 months. After a median follow‐up of 2 years, metabolic and imaging studies were repeated in all available participants whose parents consented to repeat those assessments (133 subjects). Baseline demographic and anthropometric characteristics of participants with follow‐up data were comparable to that of the original cohort (Supporting Table S1). During the follow‐up period, 12 subjects started new medications, including metformin (n = 7), omega‐3‐acid ethyl esters (n = 3), and angiotensin‐converting enzyme inhibitors (n = 2). Metformin was suspended at least 7 days before metabolic evaluations.
The study was approved by the Yale University Human Investigation Committee. Written parental informed consent and written child assent were obtained from all participants before enrollment.
1.2. Fast MRI: Assessment Of Liver Fat Content And Abdominal Fat Distribution
Abdominal and liver MRI studies were performed on a Siemens Sonata 1.5 Tesla system (Erlangen, Germany), as previously reported,23, 24, 25, 26 using an advanced magnitude‐based liver fat quantification MRI technique, the 2‐point Dixon (2PD) as modified by Fishbein et al.27 Briefly, this method is based on phase‐shift imaging where HFF is calculated from the signal difference between the vectors resulting from in‐phase and out‐of‐phase signals. Five regions of interest were drawn on each image, and the mean pixel signal intensity level was recorded.27 We validated the modified 2PD method against 1H‐NMR (proton nuclear magnetic resonance) in 34 lean and obese adolescents and found a very strong correlation between the two methods (r = 0.954; P < 0.0001).23 To assess its repeatability, measurements were obtained within the same day on 12 subjects. The within‐subject SD for HFF was 1.9%. This degree of reproducibility is well within the boundaries of that necessary to make this a viable method to assess the relation between HFF and metabolic outcomes. Kim et al.25 demonstrated that a 2PD HFF cutoff of 3.6%, provided good sensitivity (80%) and specificity (87%) compared to an 1H‐NMR reference. In addition, we further validated the modified 2PD method against liver biopsy—the gold standard for diagnosing NAFLD—in 15 obese adolescents. We found a very strong correlation between percent of liver fat measured by the two methods (r = 0.836; P = 0.0001; Supporting Fig. S1). Fast MRI has also been found to be able to track longitudinal changes in liver fat content in obese adolescents with NAFL.24
Abdominal MRI was also used to quantify visceral and subcutaneous fat depots, as reported.26 The pulse sequence was a T1‐weighted fast low angle shot gradient echo. Slices were acquired using a 400‐cm field of view (echo time 4.76, repetition time 100, 4 excitations, 90‐degree flip angle, matrix 256 × 128, and bandwidth 140). Visceral and subcutaneous abdominal fat distribution was determined on a single slice obtained at the level of the L4/L5 disc space, using thresholding to discriminate fat from soft tissue.26
1.3. Liver Biopsy
Liver biopsy was performed in 15 subjects (Supporting Tables S1 and S2) with persistent elevation of ALT (156.2 ± 94.5 U/L; 95% confidence interval [CI], 101.7‐210.8). Biopsies were formalin‐fixed, paraffin‐embedded and stained with hematoxylin and eosin, trichrome, and Gordon’s reticulin techniques. All biopsies were 2 cm or more in length and were reviewed by a pathologist, who established the diagnosis of steatohepatitis.28, 29 Steatosis was assessed as the percentage of hepatocytes involved within a lobule (0%‐100%, steatosis score). Staging and grading were performed according to Brunt et al.28, 29 The NAFLD activity score (NAS) was calculated according to Kleiner et al.30 The score is defined as the sum of the scores for steatosis (0, <5%; 1, 5%‐33%; 2, 33%‐66%; 3, >66%), lobular inflammation (0, none; 1, <2 foci/×200 magnification field; 2, 2‐4 foci/×200 magnification field; 3, >4 foci/×200 magnification field), and ballooning (0, none; 1, few; 2, many).
1.4. Genotyping
To test whether genetic variants previously associated with pediatric NAFLD might predict changes in HFF% over time, the PNPLA3 rs738409, GCKR rs1260326, and TM6SF2 rs58542926 variants were genotyped as previously reported.12, 13, 14 Genomic DNA was extracted from peripheral blood leukocytes in 474 subjects, including 111 individuals who underwent a follow‐up MRI (44 whites, 29 blacks, 38 Hispanics; 65 [58.6%] with HFF ≤5.5% and 46 [41.4%] with HFF >5.5%).
1.5. Biochemical Analyses
Plasma glucose was determined using a glucose analyzer by the glucose oxidase method (Beckman Instruments, Brea, CA). Plasma insulin was measured by the Linco radioimmunoassay (St. Charles, MO). Lipid levels were determined with an Auto‐Analyzer (model 747‐200, Roche Diagnostics, Indianapolis, IN). Liver enzymes were measured using standard automated kinetic enzymatic assays.
1.6. Statistical Analyses
Categorical data are presented as counts and/or percentages and were analyzed with the chi‐square test. Continuous variables are presented as means ± SDs. Distribution of continuous variables was examined for skewness using the Kolmogorov‐Smirnov test, and non‐normally distributed variables were log‐transformed before data analysis to approximate univariate normality, except for HFF% for which a square root transformation was used. A general linear model (GLM) was used to test differences among groups. Post‐hoc comparisons were performed by Tukey’s honest significant difference (HSD) tests. A GLM was used also to evaluate the association between changes in HFF% over time and changes in clinical and metabolic variables, and age, sex, z‐score BMI, and follow‐up duration were used as covariates. Correlations between variables were tested using Spearman’s rank correlations. To identify potential factors associated with changes in NAFL phenotype (progression or resolution) in adolescents without and with NAFL at baseline, a multivariable logistic regression was used. Variables included in the models were age, sex, ethnicity, z‐score BMI, changes in z‐score BMI, use of medications, and follow‐up duration, in addition to the variables associated with progression or regression of NAFL in univariate analyses (namely fasting glucose and C‐peptide for progression and baseline HFF% and subcutaneous fat for regression). The median number of visits during the follow‐up time of 2 years was 1 (range, 0‐4), and it was not different among groups at follow‐up (all P > 0.10). During the follow‐up, 12 subjects received new medications that could potentially influence the results. Hence, multivariable logistic analysis was first repeated by adding “medications” as a covariate, which did not show any significant effect in either the progression (β = 0.848; P = 0.154) or the regression (β = 0.189; P = 0.698) model. Then, a multivariable logistic analysis was repeated only in the group of subjects who did not receive any medications over time. Exclusion of individuals taking medications during the follow‐up did not affect the results. Performance of different models was assessed by the area under the receiver operating characteristic (ROC) curve. A GLM was used to assess the association between the studied SNPs and the HFF%. Because genotype was available for 94% of the cross‐sectional cohort and 83% of subjects from the initial longitudinal cohort, subjects who had not been genotyped were excluded from the genetic analysis. The subgroup excluded from the analysis did not differ from the tested cohort with respect to age, sex, and z‐score BMI (all P > 0.10). Statistical tests were performed using SAS 9.4 and JMP Pro 11.2.0 (SAS Institute Inc., Cary, NC) using a two‐sided alpha level of 0.05.
2. Results
2.1. Metabolic Phenotypes Of Obese Adolescents With NAFL According To Ethnicity
Overall prevalence of NAFL was 41.6% (209 patients) and varied significantly by ethnicity, being 42.9% in whites, 15.7% in blacks, and 59.6% in Hispanics (P < 0.0001; Table 1). Subjects with NAFL were more likely to be boys (53.6%) than girls (46.4%; P < 0.0001; Table 1). In all three ethnic groups, subjects with NAFL had higher z‐score BMI, fasting insulin, fasting C‐peptide, 2‐hour glucose, total cholesterol, TG, and visceral fat, and lower WBISI and DI, than those without NAFL (Table 1). Notably, although there was no difference in the overall HFF% (Fig. 2A) and visceral fat content (Fig. 2B) among the three ethnic groups with NAFL, stark differences emerged in their metabolic profiles, given that blacks with NAFL displayed a metabolic profile consistent with profound alterations in insulin (Fig. 2C) and glucose homeostasis (Fig. 2D). Compared to both whites and Hispanics with NAFL, black adolescents with liver steatosis had higher fasting glucose (P = 0.020 and P = 0.028, respectively), fasting insulin (P = 0.0004 and P < 0.0001), fasting C‐peptide (P = 0.038 and P = 0.002), 2‐hour glucose (P = 0.043 and P = 0.003), hemoglobin A1C (HbA1c; P < 0.0001 for both), IGI (P = 0.0008 and P = 0.007), and SBP (P = 0.005 and P = 0.008), as well as lower WBISI (P = 0.031 and P = 0.001), despite similar age (P = 0.342), sex (P = 0.373), and Tanner stage (P = 0.575) distribution (Table 1) and, more important, a similar amount of visceral fat depot (P = 0.218; Fig. 2). Blacks with NAFL also had higher z‐score BMI (P = 0.007) than Hispanics (Table 1). This pronounced reduction in insulin sensitivity observed in blacks with NAFL translated into a significantly higher prevalence (66.6%) of alterations of glucose tolerance compared to white (24.4%) and Hispanic (31.1%) obese adolescents with NAFL (P = 0.0003; Fig. 2).
Table 1.
Baseline characteristics of the study population stratified by ethnicity and by absence (HFF≤5.5%) or presence (HFF>5.5%) of NAFL
| Whites | Blacks | Hispanics | |||||||
|---|---|---|---|---|---|---|---|---|---|
| (n = 191; 38.0%) | (n = 134; 26.6%) | (n = 178; 35.4%) | |||||||
| HFF ≤ 5.5% | HFF > 5.5% | P | HFF ≤ 5.5% | HFF > 5.5% | P | HFF ≤ 5.5% | HFF > 5.5% | P | |
| (n = 109) | (n = 82) | (n = 113) | (n = 21) | (n = 72) | (n = 106) | ||||
| Clinical features | |||||||||
| Age (years) | 14.2 ± 2.7 | 13.2 ± 2.8 | 0 . 154 | 14.0 ± 3.1 | 14.1 ± 2.6 | 0 . 529 | 13.5 ± 2.6 | 13.1 ± 2.8 | 0 . 711 |
| Sex (M/F) [%] | 36/73 [33.0/67.0] | 39/43 [47.6/52.4] | 0 . 208 | 34/79 [30.1/69.9] | 12/9 [57.1/42.9] | 0.049 | 24/48 [33.3/66.7] | 61/45 [57.5/42.5] | 0.003 |
| Body mass index z‐score | 2.09 ± 0.41 | 2.34 ± 0.34 | 0.0002 | 2.24 ± 0.42 | 2.52 ± 0.39 | 0.011 | 2.11 ± 0.43 | 2.26 ± 0.37 | 0.036 |
| Body mass index (kg/m2) | 32.5 ± 6.5 | 33.9 ± 5.5 | 0 . 108 | 34.4 ± 6.6 | 38.5 ± 6.0 | 0.009 | 31.8 ± 6.3 | 32.7 ± 6.1 | 0 . 341 |
| Body fat mass (%) | 41.2 ± 7.4 | 45.4 ± 8.7 | 0.001 | 44.1 ± 7.5 | 48.4 ± 8.9 | 0.046 | 41.4 ± 7.2 | 43.9 ± 8.0 | 0.033 |
| Tanner stage (1/2/3/4/5) [%] | [5.5/11.0/18.4/21.1/44.0] | [14.6/17.1/18.3/19.5/30.5] | 0 . 616 | [7.1/15.1/16.8/15.9/45.1] | [9.5/14.3/9.5/19.1/45.6] | 0 . 781 | [12.5/11.1/16.7/19.4/40.3] | [10.4/24.5/20.8/18.9 /25.4] | 0 . 127 |
| Systolic blood pressure (mmHg) | 118.3 ± 11.6 | 117.5 ± 11.7 | 0 . 173 | 118.6 ± 9.9 | 126.2 ± 10.6 | 0.010 | 115.7 ± 11.3 | 118.3 ± 10.8 | 0 . 345 |
| Diastolic blood pressure (mmHg) | 68.0 ± 7.9 | 68.0 ± 8.8 | 0 . 967 | 67.6 ± 8.3 | 70.1 ± 6.9 | 0 . 224 | 66.7 ± 6.6 | 67.8 ± 8.1 | 0 . 100 |
| Glucose metabolism | |||||||||
| Fasting glucose (mg/dl) | 90.2 ± 7.4 | 91.9 ± 7.8 | 0 . 347 | 90.0 ± 8.7 | 98.1 ± 16.5 | 0.022 | 91.4 ± 7.3 | 92.3 ± 8.5 | 0 . 969 |
| Fasting insulin (µU/ml) | 27.5 ± 16.5 | 39.8 ± 16.6 | <0.0001 | 30.8 ± 16.8 | 60.1 ± 30.4 | <0.0001 | 32.3 ± 31.2 | 37.9 ± 22.5 | 0 . 211 |
| Fasting C peptide (pmol/l) | 1071.9 ± 430.1 | 1409.4 ± 69.4 | <0.0001 | 971.4 ± 348.0 | 1747.3 ± 602.7 | <0.0001 | 1171.9 ± 502.9 | 1274.0 ± 551.4 | 0 . 209 |
| 2 h glucose (mg/dl) | 121.3 ± 31.3 | 133.0 ± 38.0 | 0.022 | 116.3 ± 28.5 | 152.9 ± 41.7 | <0.0001 | 123.1 ± 25.0 | 126.2 ± 27.7 | 0 . 938 |
| Hemoglobin A1C (%) | 5.39 ± 0.30 | 5.46 ± 0.30 | 0 . 444 | 5.56 ± 0.30 | 6.00 ± 0.64 | 0.0001 | 5.46 ± 0.27 | 5.56 ± 0.35 | 0 . 109 |
| Whole Body Insulin Sensitivity Index (WBISI) | 2.30 ± 1.44 | 1.36 ± 0.69 | <0.0001 | 2.04 ± 1.19 | 0.89 ± 0.53 | <0.0001 | 1.80 ± 0.89 | 1.54 ± 0.82 | 0.042 |
| Insulinogenic index (IGI) | 3.77 ± 3.09 | 4.21 ± 2.83 | 0 . 949 | 5.58 ± 4.18 | 7.21 ± 5.41 | 0 . 086 | 4.70 ± 3.48 | 4.81 ± 3.06 | 0 . 883 |
| Disposition Index (DI) | 6.83 ± 4.44 | 5.15 ± 3.35 | 0.001 | 9.99 ± 7.83 | 5.70 ± 4.47 | 0.045 | 7.30 ± 5.25 | 7.21 ± 6.65 | 0 . 991 |
| Lipid profile | |||||||||
| Total cholesterol (mg/dL) | 151.1 ± 30.5 | 161.8 ± 29.6 | 0.007 | 151.9 ± 26.3 | 158.0 ± 22.5 | 0 . 205 | 148.5 ± 29.4 | 156.7 ± 35.1 | 0 . 183 |
| HDL cholesterol (mg/dL) | 45.4 ± 10.5 | 42.4 ± 11.0 | 0 . 470 | 46.7 ± 9.7 | 39.0 ± 7.7 | 0.006 | 42.2 ± 9.9 | 40.7 ± 9.1 | 0 . 745 |
| LDL cholesterol (mg/dL) | 87.6 ± 25.2 | 91.0 ± 24.0 | 0 . 365 | 91.2 ± 22.7 | 97.1 ± 21.7 | 0 . 193 | 85.6 ± 24.8 | 90.8 ± 29.8 | 0 . 414 |
| Triglycerides (mg/dL) | 91.9 ± 44.4 | 144.6 ± 80.6 | <0.0001 | 67.7 ± 36.3 | 109.2 ± 31.7 | <0.0001 | 105.8 ± 72.8 | 124.5 ± 58.0 | 0 . 079 |
| Body fat distribution | |||||||||
| Hepatic fat fraction (%) | 1.5 ± 1.8 | 19.7 ± 11.6 | <0.0001 | 0.9 ± 1.4 | 18.0 ± 10.6 | <0.0001 | 1.5 ± 1.8 | 18.9 ± 10.9 | <0.0001 |
| Visceral fat (cm2) | 55.5 ± 25.8 | 75.1 ± 27.8 | 0.001 | 47.0 ± 24.5 | 67.4 ± 28.7 | 0.043 | 50.9 ± 19.9 | 66.4 ± 29.7 | 0.006 |
| Subcutaneous fat (cm2) | 480.1 ± 203.9 | 511.2 ± 180.1 | 0 . 374 | 488.5 ± 214.5 | 641.6 ± 212.4 | 0 . 064 | 461.0 ± 177.3 | 484.1 ± 190.5 | 0 . 603 |
| Liver enzymes | |||||||||
| Alanine Transaminase (U/L) | 22.4 ± 17.5 | 42.3 ± 33.7 | <0.0001 | 17.6 ± 11.4 | 35.1 ± 27.1 | 0.0002 | 18.6 ± 12.1 | 42.8 ± 29.8 | <0.0001 |
| Aspartate transaminase (U/L) | 21.9 ± 9.7 | 31.0 ± 17.9 | 0.0004 | 21.4 ± 7.4 | 27.8 ± 14.7 | 0.026 | 20.6 ± 6.0 | 31.3 ± 15.1 | <0.0001 |
| γ‐Glutamyl transferase (U/L) | 21.0 ± 12.8 | 23.3 ± 15.0 | 0 . 271 | 20.9 ± 7.3 | 21.8 ± 8.6 | 0 . 933 | 17.1 ± 7.2 | 23.3 ± 9.0 | 0.032 |
| Alkaline phosphatase (U/L) | 154.9 ± 99.3 | 198.6 ± 93.9 | 0.002 | 181.9 ± 111.3 | 205.4 ± 104.8 | 0 . 275 | 178.9 ± 93.7 | 245.7 ± 108.2 | 0.0002 |
P values are adjusted for age, sex, and z‐score BMI when appropriate. Statistically significant P values are indicated in bold.
Figure 2.
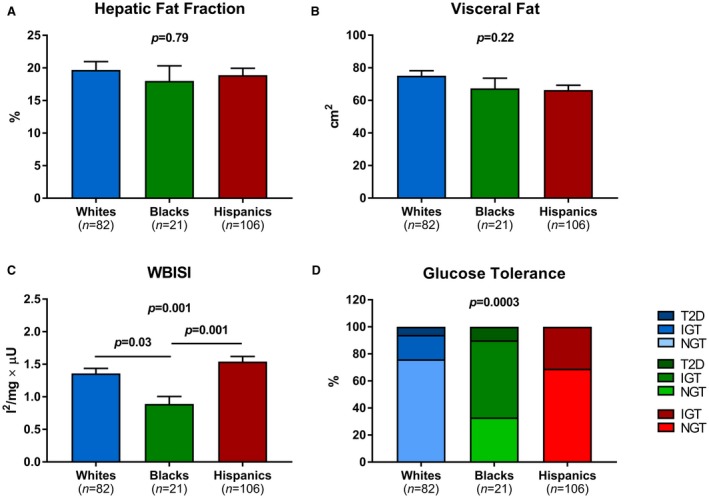
(A) HFF, (B) visceral fat, (C) WBISI, and (D) glucose tolerance of white, black, and Hispanic obese youth with NAFL. Statistical comparisons among continuous variables were made using one‐way ANOVA followed by post‐hoc pair‐wise comparisons by Tukey HSD tests. Differences in prevalence of impaired glucose control were assessed using Fisher’s test. Abbreviations: ANOVA, analysis of variance; IGT, impaired glucose tolerance; NGT, normal glucose tolerance.
2.2. Association Between Gene Variants And HFF% According To Ethnicity
The allele frequency of the PNPLA3 rs738409 minor allele (G) was 0.291 in whites, 0.158 in blacks, and 0.439 in the Hispanics. The GCKR rs1260326 minor allele (T) frequency was 0.404 in whites, 0.106 in blacks, and 0.352 in the Hispanics. The frequency of the TM6SF2 rs58542926 T allele was 0.076 in whites, 0.013 in blacks, and 0.053 in Hispanics. The allele frequencies were consistent with those shown in similar ethnic groups in the Allele Frequency Database (ALFRED; https://alfred.med.yale.edu) as well as in HAPMAP (https://hapmap.ncbi.nlm.nih.gov/). Within each ethnic group, there was no evidence against the null hypothesis that the genotype distribution was in Hardy‐Weinberg equilibrium for all the variants (all P > 0.05). The association of PNPLA3 rs738409 with HFF% was statistically significant in all the ethnic groups (P < 0.001 in whites, P = 0.0003 in blacks, and P = 0.02 in Hispanics; Fig. 3A). The association between GCKR rs1260326 and HFF% was statistically significant in whites (P = 0.003) and Hispanics (P = 0.008), and a similar trend was observed in blacks (P = 0.08; Fig. 3B). The association between the TM6SF2 rs58542926 and HFF% was statistically significant in whites (P = 0.04) and blacks (P = 0.03), but not in Hispanics (P = 0.52; Fig. 3C). These data were partially shown in our previous studies.12, 13, 14
Figure 3.
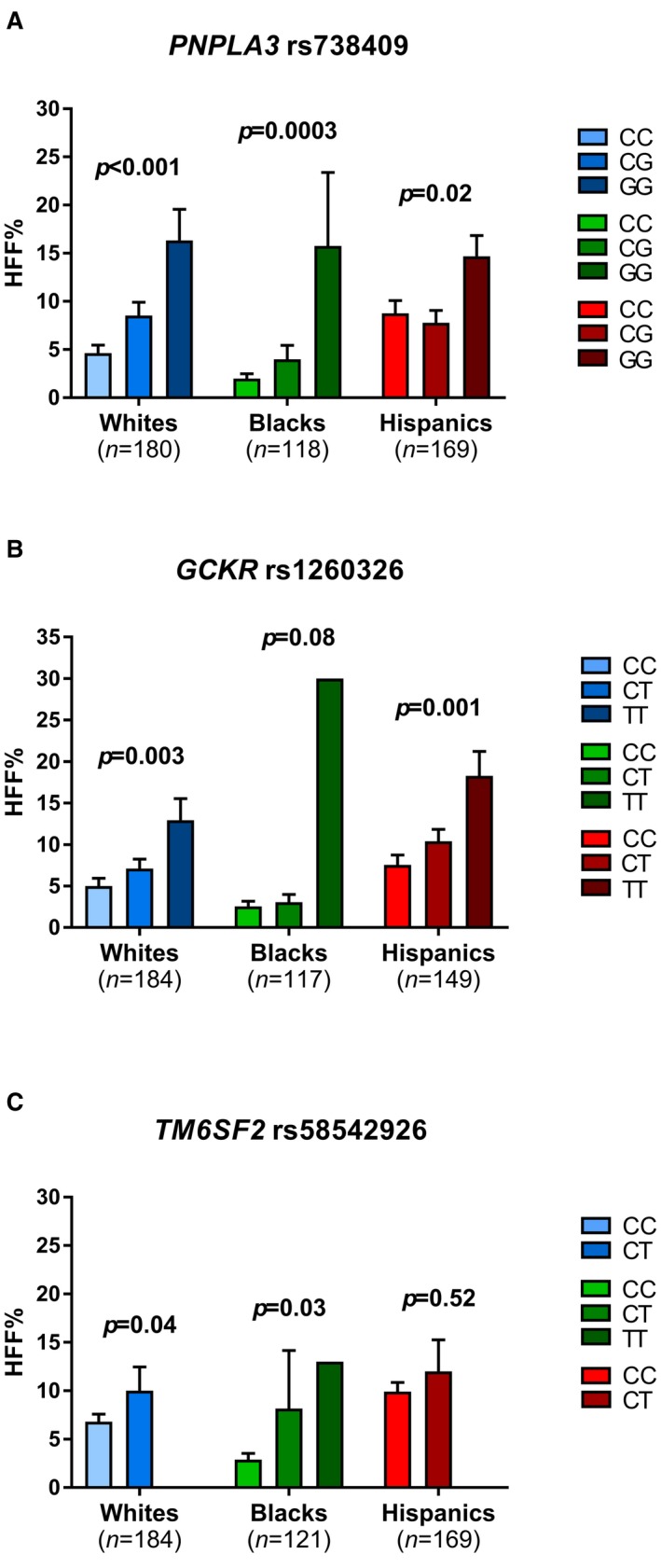
Associations between (A) PNPLA3 rs738409, (B) GCKR rs1260326, and (C) TM6SF2 rs58542926 SNPs and HFF % in obese/overweight adolescents. Statistical comparisons between groups were made using one‐way analysis of variance (ANOVA). Data are shown as mean ± SEM.
2.3. Longitudinal Assessment Of The Yale Pediatric NAFLD Cohort
One hundred thirty‐three subjects representative of the initial cross‐sectional cohort (Supporting Table S1) were followed up for an average of 2.27 ± 1.44 years (median, 1.88; IQR, 1.27‐2.80). During this period, which was similar among the three ethnicities/races (whites 2.21 ± 1.52 years, blacks 2.46 ± 1.51 years, and Hispanics 2.21 ± 1.31 years; P = 0.691), all subjects received nonpharmacological standard‐of‐care management of obesity, including nutritional consulting.
The main demographic and clinical features of these study subjects stratified by presence of NAFL are summarized in Table 2. Among them, 76 subjects (57.1%) did not have NAFL at baseline (33 whites, 26 blacks, and 17 Hispanics), whereas 57 subjects had NAFL (20 whites, 6 blacks, and 31 Hispanics; Table 2). Prevalence of NAFL in the longitudinal cohort varied largely among the three ethnic groups (37.7% in whites, 18.8% in blacks, and 64.6% in Hispanics; P = 0.0002), reflecting the difference observed in the cross‐sectional analysis.
Table 2.
Characteristics of the study population stratified by absence (HFF≤5.5%) or presence (HFF>5.5%) of NAFL at baseline and by stability or progression/regression of NAFL at follow up
| Baseline HFF ≤ 5.5% | Baseline HFF > 5.5% | |||||
|---|---|---|---|---|---|---|
| (n = 76) | (n = 57) | |||||
| Non Progressors | Progressors | p | Non Regressors | Regressors | p | |
| (n = 59) | (n=17) | (n=44) | (n = 13) | |||
| Clinical features | ||||||
| Age (years) | 14.2 ± 2.6 | 13.8 ± 3.1 | 0 . 696 | 13.3 ± 2.5 | 13.9 ± 2.3 | 0 . 754 |
| Follow up duration (years) | 2.59 ± 1.61 | 2.15 ± 1.46 | 0 . 382 | 2.39 ± 1.42 | 2.00 ± 1.19 | 0 . 333 |
| Sex (M/F) [%] | 19/40 [32.2/67.8] | 4/13 [23.5/76.5] | 0 . 267 | 26/18 [59.1/40.9] | 9/4 [69.2/30.8] | 0 . 722 |
| Race (Caucasian/African American/Hispanic) [%] | 23/25/11 [39.0/42.4/18.6] | 10/1/6 [58.8/5.9/35.3] | 0.006 | 18/4/22 [40.9/9.1/50.0] | 2/2/9 [15.4/15.4/69.2] | 0 . 324 |
| z‐score body mass index | 2.12 ± 0.51 | 2.33 ± 2.21 | 0 . 053 | 2.40 ± 0.27 | 2.27 ± 0.40 | 0 . 199 |
| Changes in body mass index z‐score at follow up | ‐0.07 ± 0.36 | 0.13 ± 0.23 | 0.012 | 0.02 ± 0.22 | ‐0.11 ± 0.23 | 0.030 |
| Body mass index (kg/m2) | 33.4 ± 6.9 | 35.0 ± 5.8 | 0 . 357 | 34.8 ± 5.0 | 33.6 ± 5.9 | 0 . 528 |
| Body fat mass (%) | 41.3 ± 9.3 | 44.4 ± 5.6 | 0 . 104 | 46.9 ± 8.7 | 42.7 ± 7.5 | 0 . 101 |
| Tanner stage (1/2/3/4/5) [%] | [5.1/8.5/20.3/22.0/44.1] | [11.8/5.9/29.4/17.6/35.3] | 0 . 538 | [9.1/15.9/15.9/20.5/38.6] | [7.7/7.7/38.5/0.0/46.1] | 0 . 140 |
| Changes in tanner stage at follow up (0/+1/+2) [%] | 43/12/4 [72.9/20.3/6.8] | 11/5/1 [64.7/29.4/5.9] | 0 . 795 | 28/11/5 [63.6/25.0/11.4] | 10/3/0 [76.9/23.1/0.0] | 0 . 618 |
| Systolic blood pressure (mmHg) | 117.2 ± 9.7 | 119.3 ± 12.1 | 0 . 281 | 119.7 ± 8.9 | 121.5 ± 13.0 | 0 . 609 |
| Diastolic blood pressure (mmHg) | 68.8 ± 9.0 | 68.3 ± 8.8 | 0 . 819 | 69.3 ± 4.8 | 69.3 ± 6.9 | 0 . 810 |
| Glucose metabolism | ||||||
| Fasting glucose (mg/dl) | 92.5 ± 8.9 | 98.7 ± 8.0 | 0.028 | 96.1 ± 8.5 | 92.4 ± 7.8 | 0 . 186 |
| Fasting insulin (µU/ml) | 30.3 ± 17.6 | 30.6 ± 9.8 | 0 . 421 | 43.2 ± 22.3 | 43.7 ± 24.3 | 0 . 720 |
| Fasting C peptide (pmol/l) | 1002.0 ± 349.7 | 1312.0 ± 345.4 | 0.005 | 1376.6 ± 427.0 | 1261.0 ± 359.7 | 0 . 394 |
| 2 h glucose (mg/dl) | 125.3 ± 25.3 | 135.1 ± 23.2 | 0 . 867 | 133.3 ± 27.4 | 124.6 ± 24.0 | 0 . 212 |
| Hemoglobin A1C (%) | 5.51 ± 0.40 | 5.55 ± 0.32 | 0 . 308 | 5.51 ± 0.38 | 5.57 ± 0.33 | 0 . 873 |
| Whole Body Insulin Sensitivity Index (WBISI) | 2.05 ± 1.06 | 1.63 ± 0.56 | 0 . 497 | 1.27 ± 0.67 | 1.20 ± 0.38 | 0 . 638 |
| Insulinogenic index (IGI) | 4.06 ± 3.19 | 3.07 ± 1.41 | 0 . 178 | 5.25 ± 3.70 | 7.00 ± 7.89 | 0 . 219 |
| Disposition index (DI) | 6.85 ± 4.39 | 4.76 ± 2.17 | 0 . 170 | 5.74 ± 5.48 | 8.00 ± 9.81 | 0 . 387 |
| Lipid Profile | ||||||
| Total cholesterol (mg/dL) | 153.0 ± 29.7 | 150.2 ± 23.4 | 0 . 961 | 158.8 ± 36.9 | 162.7 ± 35.9 | 0 . 674 |
| HDL cholesterol (mg/dL) | 46.7 ± 12.9 | 42.7 ± 11.5 | 0 . 606 | 43.0 ± 10.4 | 37.7 ± 5.9 | 0 . 138 |
| LDL cholesterol (mg/dL) | 90.2 ± 24.1 | 89.4 ± 21.7 | 0 . 849 | 89.3 ± 33.5 | 96.3 ± 27.3 | 0 . 457 |
| Triglycerides (mg/dL) | 80.5 ± 41.7 | 91.7 ± 32.1 | 0 . 893 | 147.8 ± 111.2 | 143.5 ± 69.0 | 0 . 875 |
| Body fat distribution | ||||||
| Hepatic fat fraction (%) | 0.98 ± 1.36 | 1.56 ± 1.63 | 0 . 841 | 21.27 ± 10.34 | 12.30 ± 6.18 | 0.005 |
| Changes in hepatic fat fraction at follow up (%) | ‐0.11 ± 1.47 | 9.21 ± 9.01 | <0.001 | 1.19 ± 11.13 | ‐10.34 ± 6.55 | <0.001 |
| Visceral fat (cm2) | 49.9 ± 23.7 | 68.2 ± 26.4 | 0 . 200 | 79.1 ± 23.6 | 68.9 ± 30.1 | 0 . 677 |
| Subcutaneous Fat (cm2) | 496.0 ± 196.7 | 537.7 ± 217.9 | 0 . 360 | 525.1 ± 145.0 | 564.4 ± 236.1 | 0.011 |
| Liver enzymes | ||||||
| Alanine transaminase (U/L) | 17.1 ± 13.6 | 25.0 ± 29.9 | 0 . 849 | 45.2 ± 34.2 | 32.4 ± 27.8 | 0 . 369 |
| Aspartate transaminase (U/L) | 20.0 ± 7.4 | 21.3 ± 12.9 | 0 . 756 | 32.5 ± 17.1 | 26.8 ± 10.3 | 0 . 462 |
| γ‐Glutamyl transferase (U/L) | 21.7 ± 19.7 | 23.8 ± 15.4 | 0 . 250 | 26.3 ± 19.8 | 16.6 ± 6.1 | 0 . 217 |
| Alkaline phosphatase (U/L) | 166.5 ± 92.0 | 218.6 ± 128.9 | 0 . 165 | 182.1 ± 89.8 | 144.0 ± 105.7 | 0 . 075 |
P values adjusted for age, sex, ethnicity, and z‐score BMI when appropriate. Statistically significant p values are indicated in bold.
2.4. Longitudinal Dynamic Changes In Hepatic Fat Content
The absolute changes in HFF% ranged from –21.4 to +40 (median, 0; IQR, –2.25 to 3.55) and were associated with the individual changes in z‐score BMI (r = 0.193; P = 0.032), fasting insulin (r = 0.256; P = 0.005), WBISI (r = –0.263; P = 0.006), visceral fat (r = 0.284; P = 0.001), subcutaneous fat (r = 0.397; P < 0.0001), ALT (r = 0.369; P = 0.0001), and aspartate transaminase (AST) levels (r = 0.262; P = 0.009; Table 3). After adjustment for age, sex, race, baseline z‐score BMI, and follow‐up duration, the association between changes in HFF% and in z‐score BMI (P = 0.006), fasting insulin (P = 0.004), WBISI (P = 0.015), visceral fat (P = 0.0002), subcutaneous fat (P < 0.0001), ALT (P = 0.0001), and AST (P = 0.041) remained statistically significant.
Table 3.
Correlations between changes in HFF and modifications in clinical and metabolic parameters at follow up
| HFF | BMIZ | SBP | DBP | Glu0 | Ins | CPEP | Glu120 | HBA1C | WBISI | IGI | DI | CholT | HDL | LDL | TG | VFAT | SFAT | ALT | AST | GGT | ALP | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HFF | 1.000 | 0.193a | ‐0.149 | ‐0.071 | ‐0.055 | 0.256b | 0.168 | 0.069 | 0.139 | ‐0.263b | 0.160 | ‐0.165 | 0.126 | ‐0.197 | 0.153 | 0.117 | 0.284b | 0.397d | 0.369c | 0.262b | 0.183 | 0.232a |
| BMIZ | 1.000 | 0.115 | 0.120 | 0.342 | 0.325c | 0.183 | 0.149 | 0.438 | ‐0.399d | 0.285b | ‐0.127 | 0.123 | ‐0.147 | 0.186 | 0.028 | 0.210a | 0.421d | 0.230a | 0.120 | ‐0.094 | 0.145 | |
| SBP | 1.000 | 0.563d | 0.242b | ‐0.017 | 0.320b | 0.180 | 0.255a | 0.050 | 0.084 | 0.092 | ‐0.001 | ‐0.073 | 0.007 | 0.045 | ‐0.052 | ‐0.071 | 0.070 | 0.235a | 0.190 | 0.021 | ||
| DBP | 1.000 | 0.172 | 0.106 | 0.391c | 0.159 | 0.035 | ‐0.048 | 0.108 | ‐0.027 | ‐0.048 | ‐0.193 | 0.016 | 0.014 | ‐0.042 | ‐0.079 | 0.061 | 0.068 | 0.416 | 0.058 | |||
| GLU0 | 1.000 | 0.217a | 0.213a | 0.463d | 0.178 | ‐0.227a | ‐0.077 | ‐0.343c | 0.161 | ‐0.068 | 0.147 | 0.147 | 0.255b | ‐0.052 | 0.046 | 0.094 | 0.525 | 0.018 | ||||
| INS | 1.000 | 0.560d | 0.367d | 0.053 | ‐0.811d | 0.398d | ‐0.463d | 0.171 | ‐0.029 | 0.146 | 0.121 | 0.070 | 0.264 b | 0.248a | 0.063 | ‐0.265a | 0.262a | |||||
| CPEP | 1.000 | 0.217a | 0.059 | ‐0.492d | 0.391c | ‐0.261a | ‐0.044 | ‐0.167 | ‐0.074 | 0.104 | ‐0.020 | 0.169 | 0.100 | ‐0.022 | ‐0.122 | 0.093 | ||||||
| GLU120 | 1.000 | 0.200 | ‐0.435d | ‐0.052 | ‐0.481d | 0.098 | ‐0.096 | 0.074 | 0.112 | 0.055 | 0.059 | 0.149 | 0.095 | 0.320 | 0.154 | |||||||
| HBA1C | 1.000 | ‐0.036 | 0.091 | ‐0.046 | ‐0.070 | ‐0.014 | ‐0.104 | 0.064 | ‐0.132 | 0.096 | 0.103 | 0.206 | 0.238 | 0.079 | ||||||||
| WBISI | 1.000 | ‐0.319c | 0.566d | ‐0.266b | 0.183 | ‐0.272b | ‐0.229a | ‐0.208a | ‐0.377c | ‐0.066 | 0.056 | 0.088 | ‐0.272a | |||||||||
| IGI | 1.000 | 0.448d | ‐0.094 | ‐0.047 | ‐0.003 | ‐0.216a | ‐0.120 | 0.128 | ‐0.104 | ‐0.176 | ‐0.193 | 0.168 | ||||||||||
| DI | 1.000 | ‐0.281b | 0.044 | ‐0.184 | ‐0.309b | ‐0.199a | ‐0.193 | ‐0.165 | ‐0.055 | ‐0.260 | ‐0.148 | |||||||||||
| CHOLT | 1.000 | 0.352c | 0.851 | 0.251a | 0.156 | 0.137 | 0.234a | 0.275b | ‐0.004 | 0.057 | ||||||||||||
| HDL | 1.000 | 0.135d | ‐0.201a | ‐0.343c | ‐0.179 | 0.044 | 0.134 | 0.303 | ‐0.062 | |||||||||||||
| LDL | 1.000 | ‐0.037 | 0.229a | 0.255 a | 0.174 | 0.229a | 0.374 | 0.035 | ||||||||||||||
| TG | 1.000 | 0.210a | ‐0.093 | 0.139 | 0.152 | 0.028 | 0.099 | |||||||||||||||
| VFAT | 1.000 | 0.432 d | 0.071 | 0.108 | ‐0.171 | 0.110 | ||||||||||||||||
| SFAT | 1.000 | 0.219a | 0.165 | ‐0.141 | 0.097 | |||||||||||||||||
| ALT | 1.000 | 0.760d | 0.000 | ‐0.038 | ||||||||||||||||||
| AST | 1.000 | ‐0.018 | 0.017a | |||||||||||||||||||
| GGT | 1.000 | ‐0.284 | ||||||||||||||||||||
| ALP | 1.000 |
Correlations between variables were assessed by Spearman correlations. a P<0.05; b P<0.01; c P<0.001; d P≤0.0001.
HFF%, hepatic fat fraction; BMIZ, body mass index z‐score; WC, waist circumference; SBP, systolic blood pressure; DBP, diastolic blood pressure; GLU0, plasma glucose at fasting; INS, plasma insulin at fasting; CPEP, plasma C peptide at fasting; GLU120, plasma glucose at 120 min (end of OGTT); HBA1C, hemoglobin A1c; WBISI, whole‐body insulin sensitivity index; IGI, insulinogenic index; DI, disposition index; CHOLT, total cholesterol; HDL, high‐density lipoprotein cholesterol; LDL, low‐density lipoprotein cholesterol; TG, triglycerides; VFAT, visceral fat; SFAT, subcutaneous fat; ALT, alanine transaminase; AST, aspartate transaminase; GGT, gamma‐glutamyl transferase; ALP, alkaline phosphatase.
2.5. Risk Factors Of Progression And Regression OF NAFL
Among subjects without NAFL at presentation, obese adolescents who developed NAFL over time were more likely whites (58.8%) and Hispanics (35.3%) than blacks (5.9%; P = 0.006) and had higher fasting glucose (P = 0.028) and C‐peptide levels (P = 0.005; Table 2). Among subjects presenting with NAFL, a lower baseline HFF% (P = 0.005) and a higher subcutaneous fat mass, despite similar total body fat mass (P = 0.101), characterized obese adolescents whose HFF% decreased at follow‐up reaching values below 5.5% (Table 2). Notably, small increments (P = 0.012) or decrements (P = 0.030) in z‐score BMI over time were significantly associated with progression or resolution of NAFL at follow‐up, respectively (Table 2).
Accordingly, ethnicity strongly predicted the onset of NAFL in obese adolescent who did not have NAFL at baseline (P = 0.021 at multivariate logistic regression analysis). Other significant predictors were changes in z‐score BMI (P = 0.0006) and baseline fasting C‐peptide levels (P = 0.0006). The area under the ROC curve for the prediction of NAFL by race, z‐score BMI change and baseline fasting C peptide levels was 0.887 (Fig. 4A).
Figure 4.
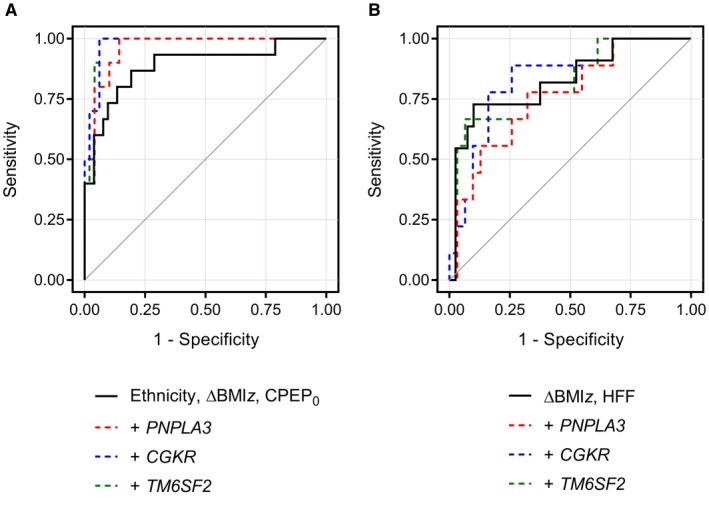
ROC curves for predicting (A) NAFL progression (n = 76) or (B) regression (n = 57) at follow‐up in obese/overweight adolescents. In (A), the AUC was 0.887 when using ethnicity, z‐score BMI change (ΔBMIz), and fasting C peptide (CPEP0) at baseline as predictors and increased to 0.959, 0.978, and 0.976 when adding either the PNPLA3 rs738409, GCKR rs1260326, or TM6SF2 rs58542926 variant to the model, respectively. In (B), the AUC was 0.827 when using ΔBMIz and HFF% at baseline as predictors and did not improve by adding either the PNPLA3 rs738409, GCKR rs1260326, or TM6SF2 rs58542926 variant (0.763, 0.828, and 0.814, respectively).
Adding either the PNPLA3 rs738409, the GCKR rs1260326, or the TM6SF2 rs58542926 variant to our model of progression to NAFL improved the area under the curve (AUC) of the ROC curve from 0.887 to 0.959, 0.978, and 0.976, respectively (Fig. 4A).
Among subjects with NAFL at baseline, the reduction of HFF% below 5.5% was more likely to follow a reduction of z‐score BMI over time (P = 0.032) and to occur in obese subjects with a lower HFF at baseline (P = 0.004). The area under the ROC curve for the prediction of NAFL regression was 0.827, including z‐score BMI change and baseline HFF as factors (Fig. 4B). Adding the PNPLA3 rs738409, the GCKR rs1260326, or the TM6SF2 rs58542926 variant to our predictive model of NAFL regression decreased or only mildly affected the AUC of the ROC curve (from 0.827 to 0.763, 0.828, and 0.814, respectively; Fig. 4B).
2.6. Histological Phenotypes Of Obese Adolescents With NAFLD
A total of 15 obese adolescents had biopsy‐proven NAFLD, including 4 whites and 11 Hispanics. Their clinical and metabolic characteristics are shown in Supporting Table S1. The two ethnicity groups showed similar sex distribution (P = 0.28), age (P = 0.61), Tanner stage (P = 0.57), adiposity (z‐score BMI, P = 0.28), and IR (WBISI, P = 0.19). Compared to the overall study cohort, adolescents with liver biopsy showed significantly higher HFF%, fasting insulin, fasting C‐peptide, 2‐hour glucose, fasting TG, visceral fat, ALT, AST, and γ‐glutamyl transferase (GGT), as well as lower WBISI, DI, and HDL cholesterol level (Supporting Table S1).
The percentage of intrahepatic fat content assessed by liver biopsy was 49.7 ± 21.6% (median, 50%; 95% CI, 37.7‐61.6; Supporting Table S2). In most subjects, biopsies showed the presence of fibrosis (stage 1‐2 in 13 subjects and stage 3 in 1 subject) and an overall NAS indicative for NASH (NAS ≥5 in 12 subjects), without significant differences between ethnicities (P > 0.50). Intrahepatic fat content assessed by liver biopsy at baseline showed a strong correlation with HFF assessed by MRI either at baseline (r = 0.836; P = 0.0001; n = 15) or after a 2.5‐ ± 1.2‐year follow‐up (r = 0.768; p = 0.016; n = 9; Supporting Fig. S1).
3. Discussion
This study provides insights into the relative importance of ethnicity, clinical risk factors, and gene variants in the development of NAFL in adolescents. Consistent with previous cross‐sectional studies,1, 8, 10, 31 we observed that black youth have a lower prevalence of NAFL and a lower tendency to accumulate intrahepatic fat over time as compared to whites and Hispanics. However, when NAFL is present, black obese adolescents show a more severely impaired metabolic phenotype than whites and Hispanics, with profound alterations in insulin and glucose homeostasis that translate into a 2‐fold higher prevalence of prediabetes and T2D (Fig. 2). We used a multiple logistic regression analysis to determine which clinical factors might help predict changes in HFF in youth. This analysis revealed that ethnicity strongly predicted the onset of NAFL in obese adolescents who did not have NAFL at baseline (P = 0.021). Other significant predictors were changes in z‐score BMI (P = 0.0006) and baseline fasting C‐peptide levels (P = 0.0006). On the other hand, basal HFF and weight loss were the major factors associated with reduction of intrahepatic fat content at follow‐up. In addition, we observed that adding the three major SNPs associated with NAFLD, such as the rs738409 in the PNPLA3 gene,13, 32 the rs1260326 in the GCKR gene,12, 33 and the rs58542926 in TM6SF2 gene,14 significantly increased the likelihood to predict changes in HFF at follow‐up.
The lower propensity of black patients to develop NAFL is certainly independent of degree of IR, given that black obese youth have a similar or even higher degree of IR than whites and Hispanics.10 Therefore, such a diversity is more likely to reflect biological and genetic differences in lipid metabolism rather than differences in IR or degree of obesity.11 Notably, the minor allele frequency of the SNP rs738409 in the PNPLA3 and rs1260326 in the GCKR varies among races, being the highest in Hispanics and the lowest in blacks.10, 34, 35 thus reflecting the different prevalence of NAFL among these ethnic groups. Fat distribution likely also plays a role in the lower propensity of blacks to develop NAFL. In fact, visceral adipose tissue is the main source of free fatty acids (FFAs) for hepatic TG synthesis (~65%)36 and is typically less represented in blacks than in whites and Hispanics. Therefore, one could speculate that differences in the degree of visceral fat accretion among races might lead to a lower flux of FFA in the liver explaining the lower HFF even in the presence of greater IR.8, 11 Finally, although hepatic de novo lipogenesis (DNL) is a major contributor of intrahepatic fat accumulation,37 whether differences in DNL exist among ethnic groups remains unexplored.
It is interesting to note that in our group of adolescents with NAFL, the volume of the visceral fat depot was similar across ethnicities/races, even in the blacks in whom the volume visceral depot is known to be very small.38 This suggests that black adolescents with NAFL may carry a genetic predisposition to accumulate fat into the visceral depot. Therefore, it would be important to explore whether genetic differences between black adolescents with low and high visceral depot exist and how they might contribute to the metabolic derangements observed in this group.
Ethnic differences were also observed in the longitudinal data. In fact, we show in a longitudinal setting that blacks were the least prone to develop NAFL over time, as compared to whites and Hispanics. Furthermore, we found that among metabolic features associated with NAFL in obese youth, high fasting glucose and C‐peptide levels at baseline were associated with development of NAFL in subjects without evidence of intrahepatic fat. This probably reflects the fact that IR in obese patients is selective for glucose metabolism, whereas the high concentration of insulin available in plasma can still enhance the synthetic rate of hepatic DNL, which, in such an environment, is already increased as a consequence of the high availability of substrates (glucose). Interestingly, the degree of IR seemed to be a strong risk factor for developing NAFL also among blacks, who overall carry the lowest risk to accumulate intrahepatic fat. This observation further supports the role of IR as a major trigger of NAFL in obese adolescents.
Although the molecular bases of the relationship between IR and NAFLD are not entirely clear, puberty per se seems to play an important role. In fact, puberty is characterized by considerable metabolic and hormonal changes.39 In particular, adolescents experience a decline in insulin sensitivity during puberty, with a nadir in midpuberty and a complete recover at the end of it, when the Tanner stage 5 is achieved.40 It is conceivable that the IR of puberty might fuel the accumulation of intrahepatic TG and lead to hepatic IR, but this hypothesis remains to be studied. Pubertal IR is more severe in girls than in boys,40 and this might be partially explained by differences in adiposity distribution. In fact, for a given BMI, girls tend to have greater triceps and subscapular skinfold thickness than boys.40
In addition, consistent with previous cross‐sectional studies,1, 8, 10, 31 we showed that the prevalence of NAFL is higher in boys than girls and associated with the degree of obesity and IR in our multiethnic cohort of adolescents. This difference might be attributed to the different levels of estrogens during puberty. In fact, it has been shown in ovariectomized female mice that estradiol protects against intrahepatic lipid accumulation.41
Along with these metabolic features, genetic biomarkers were associated with intrahepatic fat accumulation. Common gene variants previously associated with NAFLD, particularly the rs1260326 in the GCKR gene, further strengthen the association between our model factors and progression of NAFL.
Although our longitudinal analyses involved a relatively small sample, they clearly demonstrate that taking into account the ethnic background, tracking some simple clinical information (such as fasting glucose and C‐peptide and changes in BMI) and knowing the genotype of one of the major genetic risk factors for NAFLD might represent a strong tool for the clinician in the follow‐up of obese adolescents.
3.1. Strengths And Limitations
Although some cross‐sectional studies assessing racial and ethnic variation of NAFL have focused primarily on the prevalence of the disorder,8, 10 there are no studies that have delved further into metabolic associations of NAFL in different racial and ethnic groups over time. Similarly, no studies have explored whether the metabolic phenotype is different among youth with NAFL in different ethnic groups in the United States. In the present study, using an advanced magnitude‐based liver fat quantification MRI technique in a large cohort of obese adolescents that have undergone a detailed characterization of abdominal fat patterning, extensive metabolic phenotyping, and genotyping of common SNPs, we performed a detailed analysis of the associations of ethnicity with clinical and metabolic features of NAFL early in its development in a pediatric population. Limitations of our study are the lack of liver biopsy for most subjects, the lack of a larger sample size at follow‐up and of a replication cohort to validate longitudinal data, and the lack of a more detailed genetic assessment of the global ancestry markers. The lack of a larger number of liver biopsies is certainly a major limitation of the study. In fact, collecting liver biopsies in all study subjects would have provided useful additional information about the real prevalence of NASH42 and the natural history of NAFLD in youth, ethnic differences in NAFLD, and the relationship between histological changes occurring in the liver of subjects with NAFLD and glucose metabolism.
In conclusion, black adolescents are protected from NAFL, even in the presence of severe obesity. On the other hand, when NAFL is present in blacks, they show a more severe metabolic profile than whites and Hispanics, characterized by a higher prevalence of prediabetes and T2D. Moreover, taking into account ethnicity, degree of IR, and genetic risk factors allows to identify obese adolescents at highest risk for development of NAFL.
4. Potential conflict of interest
Nothing to report.
5.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgments
The authors are grateful to the patients and their families as well as to the Yale Center for Genome Analysis, the Yale Center for Clinical Investigation and Hospital Research Unit.
Supported by American Heart Association (16IRG27390002, 13SDG14640038 and 11CRP5620013), Allen Foundation (DOI: 10.13039/100008796), National Institute of Diabetes and Digestive and Kidney Diseases (R01‐HD‐40787, R01‐HD‐28016, R01DK111038‐01) , R01DK114504, DK045735, and K24‐HD01464), and National Center for Advancing Translational Sciences (UL1‐RR‐ 024139).
Trial Registration: NCT01966627.
Contributor Information
Sonia Caprio, Email: sonia.caprio@yale.edu.
Nicola Santoro, Email: nicola.santoro@yale.edu.
References
- 1. Schwimmer JB, Deutsch R, Kahen T, Lavine JE, Stanley C, Behling C. Prevalence of fatty liver in children and adolescents. Pediatrics 2006;118:1388‐1393. [DOI] [PubMed] [Google Scholar]
- 2. Brunt EM, Wong VW, Nobili V, Day CP, Sookoian S, Maher JJ, et al. Nonalcoholic fatty liver disease. Nat Rev Dis Primers 2015;1:15080. [DOI] [PubMed] [Google Scholar]
- 3. Spengler EK, Loomba R. Recommendations for diagnosis, referral for liver biopsy, and treatment of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Mayo Clin Proc 2015;90:1233‐1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Feldstein AE, Charatcharoenwitthaya P, Treeprasertsuk S, Benson JT, Enders FB, Angulo P. The natural history of non‐alcoholic fatty liver disease in children: a follow‐up study for up to 20 years. Gut 2009;58:1538‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nobili V, Alisi A, Grimaldi C, Liccardo D, Francalanci P, Monti L, et al. Non‐alcoholic fatty liver disease and hepatocellular carcinoma in a 7‐year‐old obese boy: coincidence or comorbidity? Pediatr Obes 2014;9:e99‐e102. [DOI] [PubMed] [Google Scholar]
- 6. Goyal NP, Schwimmer JB. The progression and natural history of pediatric nonalcoholic fatty liver disease. Clin Liver Dis 2016;20:325‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. D’Adamo E, Cali AM, Weiss R, Santoro N, Pierpont B, Northrup V, Caprio S. Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents. Diabetes Care 2010;33:1817‐1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387‐1395. [DOI] [PubMed] [Google Scholar]
- 9. Bril F, Portillo‐Sanchez P, Liu IC, Kalavalapalli S, Dayton K, Cusi K. Clinical and histologic characterization of nonalcoholic steatohepatitis in African American patients. Diabetes Care 2018;41:187‐192. [DOI] [PubMed] [Google Scholar]
- 10. Santoro N, Feldstein AE, Enoksson E, Pierpont B, Kursawe R, Kim G, Caprio S. The association between hepatic fat content and liver injury in obese children and adolescents: effects of ethnicity, insulin resistance, and common gene variants. Diabetes Care 2013;36:1353‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guerrero R, Vega GL, Grundy SM, Browning JD. Ethnic differences in hepatic steatosis: an insulin resistance paradox? Hepatology 2009;49:791‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Santoro N, Zhang CK, Zhao H, Pakstis AJ, Kim G, Kursawe R, et al. Variant in the glucokinase regulatory protein (GCKR) gene is associated with fatty liver in obese children and adolescents. Hepatology 2012;55:781‐789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Santoro N, Kursawe R, D’Adamo E, Dykas DJ, Zhang CK, Bale AE, et al. A common variant in the patatin‐like phospholipase 3 gene (PNPLA3) is associated with fatty liver disease in obese children and adolescents. Hepatology 2010;52:1281‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goffredo M, Caprio S, Feldstein AE, D’Adamo E, Shaw MM, Pierpont B, et al. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: a multiethnic study. Hepatology 2016;63:117‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. U.S. Census Bureau 2010 . Profile of general population and housing characteristics. https://factfinder.census.gov/bkmk/table/1.0/en/DEC/10_DP/DPDP1/ 1600000US0952000. Accessed November 28, 2017.
- 16. Marshall WA, Tanner JM. Variations in pattern of pubertal changes in girls. Arch Dis Child 1969;44:291‐303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marshall WA, Tanner JM. Variations in the pattern of pubertal changes in boys. Arch Dis Child 1970;45:13‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schwimmer JB, Dunn W, Norman GJ, Pardee PE, Middleton MS, Kerkar N, Sirlin CB. SAFETY study: alanine aminotransferase cutoff values are set too high for reliable detection of pediatric chronic liver disease. Gastroenterology 2010;138(1357‐1364):1364.e1‐e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tricò D, Di Sessa A, Caprio S, Chalasani N, Liu W, Liang T, et al. Oxidized derivatives of linoleic acid in pediatric metabolic syndrome: is their pathogenic role modulated by the genetic background and the gut microbiota? Antioxid Redox Signal 2017. Apr 7. doi: 10.1089/ars.2017.7049. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Goffredo M, Santoro N, Tricò D, Giannini C, D’Adamo E, Zhao H, Peng G et al. A branched‐chain amino acid‐related metabolic signature characterizes obese adolescents with non‐alcoholic fatty liver disease. Nutrients 2017;9:E642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tricò D, Prinsen H, Giannini C, de Graaf R, Juchem C, Li F, et al. Elevated alpha‐hydroxybutyrate and branched‐chain amino acid levels predict deterioration of glycemic control in adolescents. J Clin Endocrinol Metab 2017;102:2473‐2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. American Diabetes Association . Classification and diagnosis of diabetes. Diabetes Care 2017;40(Suppl 1):S11‐S24. [DOI] [PubMed] [Google Scholar]
- 23. Cali AM, De Oliveira AM, Kim H, Chen S, Reyes‐Mugica M, Escalera S, et al. Glucose dysregulation and hepatic steatosis in obese adolescents: is there a link? Hepatology 2009;49:1896‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kim G, Giannini C, Pierpont B, Feldstein AE, Santoro N, Kursawe R, et al. Longitudinal effects of MRI‐measured hepatic steatosis on biomarkers of glucose homeostasis and hepatic apoptosis in obese youth. Diabetes Care 2013;36:130‐136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kim H, Taksali SE, Dufour S, Befroy D, Goodman TR, Petersen KF, et al. Comparative MR study of hepatic fat quantification using single‐voxel proton spectroscopy, two‐point dixon and three‐point IDEAL. Magn Reson Med 2008;59:521‐527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Burgert TS, Taksali SE, Dziura J, Goodman TR, Yeckel CW, Papademetris X, et al. Alanine aminotransferase levels and fatty liver in childhood obesity: associations with insulin resistance, adiponectin, and visceral fat. J Clin Endocrinol Metab 2006;91:4287‐4294. [DOI] [PubMed] [Google Scholar]
- 27. Fishbein MH, Gardner KG, Potter CJ, Schmalbrock P, Smith MA. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging 1997;15:287‐293. [DOI] [PubMed] [Google Scholar]
- 28. Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander‐Tetri BA, Bacon BR. Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 1999;94:2467‐2474. [DOI] [PubMed] [Google Scholar]
- 29. Brunt EM, Kleiner DE, Wilson LA, Belt P, Neuschwander‐Tetri BA, NASH,. Clinical Research Network (CRN). Nonalcoholic fatty liver disease (NAFLD) activity score and the histopathologic diagnosis in NAFLD: distinct clinicopathologic meanings. Hepatology 2011;53:810‐820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 31. Newton KP, Hou J, Crimmins NA, Lavine JE, Barlow SE, Xanthakos SA, et al. Prevalence of prediabetes and type 2 diabetes in children with nonalcoholic fatty liver disease. JAMA Pediatr 2016;170:e161971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Santoro N, Savoye M, Kim G, Marotto K, Shaw MM, Pierpont B, Caprio S. Hepatic fat accumulation is modulated by the interaction between the rs738409 variant in the PNPLA3 gene and the dietary omega6/omega3 PUFA intake. PLoS One 2012;7:e37827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Santoro N, Caprio S, Pierpont B, Van Name M, Savoye M, Parks EJ. Hepatic de novo lipogenesis in obese youth is modulated by a common variant in the GCKR gene. J Clin Endocrinol Metab 2015;100:E1125‐E1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Palmer ND, Musani SK, Yerges‐Armstrong LM, Feitosa MF, Bielak LF, Hernaez R, et al. Characterization of European ancestry nonalcoholic fatty liver disease‐associated variants in individuals of African and Hispanic descent. Hepatology 2013;58:966‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 2005;115:1343‐1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lambert JE, Ramos‐Roman MA, Browning JD, Parks EJ. Increased de novo lipogenesis is a distinct characteristic of individuals with nonalcoholic fatty liver disease. Gastroenterology 2014;146:726‐735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goran MI, Nagy TR, Treuth MS, Trowbridge C, Dezenberg C, McGloin A, Gower BA. Visceral fat in white and African American prepubertal children. Am J Clin Nutr 1997;65:1703‐1708. [DOI] [PubMed] [Google Scholar]
- 39. Amiel SA, Sherwin RS, Simonson DC, Lauritano AA, Tamborlane WV. Impaired insulin action in puberty. A contributing factor to poor glycemic control in adolescents with diabetes. N Engl J Med 1986;315:215‐219. [DOI] [PubMed] [Google Scholar]
- 40. Moran A, Jacobs DR Jr, Steinberger J, Hong CP, Prineas R, Luepker R, Sinaiko AR. Insulin resistance during puberty: results from clamp studies in 357 children. Diabetes 1999;48:2039‐2044. [DOI] [PubMed] [Google Scholar]
- 41. Camporez JP, Jornayvaz FR, Lee HY, Kanda S, Guigni BA, Kahn M, et al. Cellular mechanism by which estradiol protects female ovariectomized mice from high‐fat diet‐induced hepatic and muscle insulin resistance. Endocrinology 2013;154:1021‐1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schwimmer JB, Newton KP, Awai HI, Choi LJ, Garcia MA, Ellis LL. Paediatric gastroenterology evaluation of overweight and obese children referred from primary care for suspected non‐alcoholic fatty liver disease. Aliment Pharmacol Ther 2013;38:1267‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials