

SUMMARY
Acetate is a major nutrient that supports acetyl-coenzyme A (Ac-CoA) metabolism and thus lipogenesis and protein acetylation. Its source however has been unclear. Here we report that pyruvate, the end product of glycolysis and key node in central carbon metabolism, quantitatively generates acetate in mammals. This phenomenon becomes more pronounced in contexts of nutritional excess such as during hyperactive glucose metabolism. Conversion of pyruvate to acetate occurs through two mechanisms: 1) coupling to reactive oxygen species (ROS), and 2) neomorphic enzyme activity from keto acid dehydrogenases that enable function as pyruvate decarboxylases. Further, we demonstrate that de novo acetate production sustains Ac-CoA pools and cell proliferation in limited metabolic environments such as during mitochondrial dysfunction or ATP citrate lyase (ACLY) deficiency. De novo acetate production occurs in mammals and is further coupled to mitochondrial metabolism providing possible regulatory mechanisms and links to pathophysiology.
In brief
Cells produce acetate directly from pyruvate through two distinct mechanisms, providing back up support for acetyl-CoA pools during times of metabolic deficiency
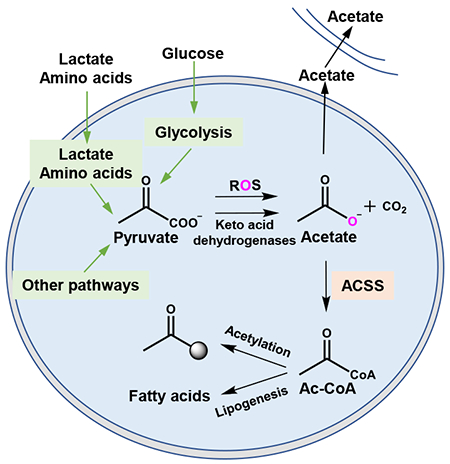
INTRODUCTION
In conditions of hyperactive cellular metabolism, excessive cellular nutrient uptake results in incomplete metabolism and excretion of intermediates. The carbon source for catabolic and anabolic processes is often incompletely catabolized and excreted into the extracellular space (Meiser et al., 2018; Reaves et al., 2013). During the Warburg effect, a phenotype characterized by increased glucose uptake, glycolysis rate (near 100 mM/h), is 10 to 100 times faster than the rate at which complete oxidation of glucose occurs in the mitochondria (Shestov et al., 2014). As a result, the excess carbon from glycolysis is secreted as lactate. Other examples are seen in the cells in diabetic tissues where excess fatty acid oxidation leads to incomplete lipid catabolism and excretion of acyl-carnitines and ketone bodies (Muoio, 2014). Numerous other examples are also apparent such as in the case of excess anabolic substrates released in nucleotide synthesis or the excretion of formic acid during excess one carbon metabolism (Meiser et al., 2016). In each case, the overflow metabolism results from imbalanced metabolic supply and demand that results in limits to enzyme activity.
The products of these overflow pathways provide a valuable resource during conditions of nutrient limitation. Ketone bodies become fuel sources during fasting conditions, while lactate and alanine are readily metabolized in local microenvironments (Faubert et al., 2017; Sousa et al., 2016). These unconventional fuel sources satisfy metabolic demands during nutrient scarcity. Interestingly, acetate is also a nutrient that has been found to be a major carbon source for central carbon metabolism in nutrient limited conditions. Acetate metabolism provides a parallel pathway for acetyl-CoA production separate from conversion of citrate to acetyl-CoA by ATP citrate lyase (ACLY) and thus acetate allows for protein acetylation and lipogenesis independent of citrate conversion to acetyl-CoA. This pathway is essential in nutrient deprived tumor microenvironments and other diverse contexts but the origin of acetate has been unclear (Balmer et al., 2016; Comerford et al., 2014; Mashimo et al., 2014; Mews et al., 2017; Perry et al., 2016; Schug et al., 2015; Schug et al., 2016). It has been postulated that acetate may be synthesized de novo in cells (Andrae et al., 1985; Asmus et al., 2015; Bunton, 1949; Salahudeen et al., 1991; Vysochan et al., 2017) but the pathways and quantitative reaction mechanisms through which this may occur are unknown. Given that alternative carbon sources often arise from overflow metabolism, we suspected that such a mechanism may allow for acetate generation. This hypothesis led us to conduct a re-evaluation of mammalian central carbon metabolism.
RESULTS
To monitor glycolysis in the setting of acetate metabolism, we incubated exponentially growing human colorectal HCT116 cells with uniformly labeled [13C6]-glucose, harvested metabolites over time, subjected the extracts to chemical derivatization with 2-hydrazinoquinoline (HQ) to capture acetate and other organic acids, and analyzed the products by liquid chromatography coupled to high resolution mass spectrometry (LC-HRMS) (Figure 1A, STAR methods). Monitoring the kinetics of [13C3]-pyruvate, [13C2]-acetate, and [13C3]-lactate, unexpectedly revealed that the rates of generation of pyruvate and lactate, the end products of glycolysis, were commensurate with the rate of acetate generation (Figure 1B). Strikingly, the concentration of [13C2]-acetate was comparable to the concentration of [13C3]-pyruvate and on the order of the amount of [13C3]-lactate generated from [13C6]-glucose in these glycolytic cells. De novo acetate production from glycolysis was also observed in several other cancer cell lines of diverse origins (Figure 1C). Indeed, there are other sources of pyruvate, such as alanine or lactate (Faubert et al., 2017; Sousa et al., 2016). Thus, the presence of exogenous pyruvate or lactate also increased acetate production (Figure 1D). Meanwhile, switching the 13C source from [13C6]-glucose to [13C5]-glutamine, which labels [l3C3]-pyruvate to a far lesser extent, diminished the formation of [13C2]-acetate (Figures S1A and S1B). Together these findings suggest that acetate can be synthesized endogenously in quantitative amounts and with kinetics comparable to pyruvate and lactate production from glycolysis.
Figure 1. Acetate is synthesized from glucose independent of acetyl-CoA related reactions.
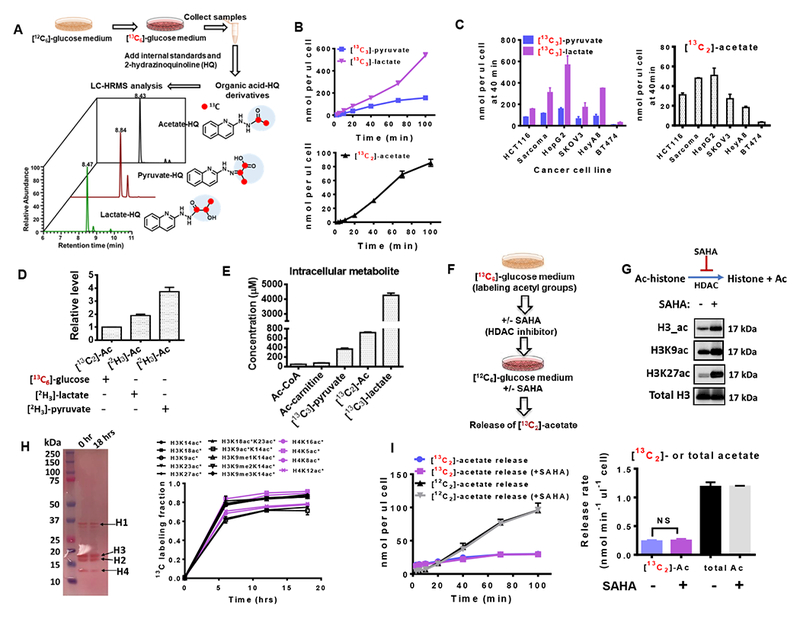
(A) Scheme of experimental setup for measuring glucose-derived acetate, pyruvate and lactate. (B) Medium concentration of [13C3]-pyruvate, [13C2]-acetate, and [13C3]-lactate secreted from HCT116 cells cultured in RPMI 1640 medium containing [13C6]-glucose. Time 0 is when medium was switched to [13C6]-glucose medium. (C) The amount of [13C2]-pyruvate, [13C3]-lactate and [13C2]-acetate released from various cultured cells at time 40 min after switching to [13C6]-glucose medium. (D) The relative levels of acetate production in HCT116 cells cultured in glucose free medium containing different tracers: [13C6]-glucose (11.1 mM), [2C3]-lactate (5 mM) or [2C3]-pyruvate (5 mM). (E) Intracellular concentrations of metabolites in HCT116 cells after incubation in [13C6]-glucose medium for 5 hrs. (F) Schematic depicting the workflow for HCT116 cells with [13C6]-glucose and or treatment with the histone deacetylation inhibitor, SAHA. (G) Western blots of histone acetylation sites from HCT116 cells in the absence or presence of SAHA (5 μM) for 1 hr. (H) Ponceau s staining (left panel) and quantitative proteomic analysis (right panel) of histone extracts from HCT116 cells cultured in the presence of 13C-glucose for 0, 6, 12 and 18 hrs. * denotes 13C labeled acetyl groups on histones. (I) Release of acetate from HCT116 cells with 6 hours pretreatment of [13C6]-glucose in the presence or absence of histone deacetylation inhibitor (SAHA, 5 μM) for 1 hr (left panel). The release rate (right panel) is obtained by calculating the slope of the acetate release curve. Abbreviations: Ac, Acetate; Ac-histone, acetylated histone. The acetylated histone sites include lysine 14 (H3K14ac), lysine 18 (H3K18ac), lysine 18 and lysine 23 (H3K18acK23ac), lysine 23 (H3K23ac), lysine 27 (H3K27ac), lysine 9 (H3K9ac), lysine 9 and lysine 14 (H3K9acK14ac), lysine 14 containing mono-methylation at lysine 9 (H3K9me1K14ac), lysine 14 containing di-methylation at lysine 9 (H3K9me2K14ac), lysine 14 containing tri-methylation at lysine 9 (H3K9me3K14ac) of histone H3, and acetylation on lysine 16 (H4Kac), lysine 5 (H4K5ac), lysine 8 (H4K8ac), and lysine 12 (H4K12ac). Values are expressed as mean ± SD of n=3 independent measurements. NS: p>0.05 in Student’s t test. See also Figure S1.
Acetate can be generated by the removal of acetyl groups from histones by histone deacetylases (Inoue and Fujimoto, 1969) and by hydrolysis of Ac-CoA (Knowles et al., 1974). We thus measured the concentrations of the metabolites involved in these processes and observed orders of magnitude lower concentrations of Ac-CoA than glucose-derived pyruvate, lactate, and acetate (Figure 1E), suggesting that de novo acetate production likely is uncoupled from these acetyl-CoA dependent reactions. Moreover, the acetate HQ derivative was not spontaneously formed by incubating Ac-CoA or acetyl (Ac)-carnitine with the HQ derivatization reagents indicating this potential artifact is sufficiently controlled (Figure S1C). To further evaluate the contribution of deacetylation reactions, including histone deacetylation, to acetate production, we cultured HCT116 cells in [13C6]-glucose containing media for 6 hrs, followed by incubation with the histone deacetylase inhibitor SAHA for an additional 1 hr (Figure 1F). Treatment with SAHA suppressed histone deacetylation (Figure 1G), indicated by the increase of total Histone 3 acetylation (H3_ac) as well as acetylation of H3 on lysines 9 (H3K9ac) and 27 (H3K27ac) of Histone H3. Furthermore, a quantitative proteomics analysis across a diverse set of acetylation sites revealed a consistent 60-80 % incorporation of [13C6]-glucose at 6 hrs that reached steady state (Figure 1H). [13C6]-glucose medium was then replaced with 12C glucose medium, and the release of [13C2]-acetate would represent the contribution from acetyl groups to acetate production. Histone deacetylase inhibition didn’t alter the [13C2]-acetate release rate (Figure 1I), and the measured [13C2]-acetate production rate from this assay was found to be nearly fivefold less than the total amount (12C and 13C acetate) of acetate (Figure 1I), indicating that hydrolysis of the acetyl group from histones, is not a major source of acetate in these experiments. Together these experiments allow us to conclude that in these conditions, more than 80% of the acetate measured is rapidly generated de novo through glycolysis.
It is generally thought that Ac-CoA formation requires glucose to first enter the mitochondria, be exported as citrate and then metabolized to cytosolic Ac-CoA (Figure S1D). We thus tested whether the pyruvate carrier inhibitor (UK5099) (Bricker et al., 2012; Divakaruni et al., 2013) that perturbs entry of pyruvate into the mitochondria would affect acetate generation. As expected, the 13C labeled Ac-CoA, Ac-carnitine (reversibly converted from Ac-CoA) and citrate (the first intermediate in the TCA cycle) were decreased by UK5099 treatment (Figures S1E-1G). Notably, no difference in the generation of [13C2]-acetate from [13C6]-glucose was observed (Figure S1H). These data thus indicate that acetate production is independent of Ac-CoA synthesis and breakdown.
One considerable possibility of a substrate for acetate production is pyruvate, a keto acid, that contains an electrophilic moiety. Keto acids have been reported to be chemical scavengers in both bacterial and mammalian cells (Kim et al., 2016; Wang et al., 2007). A plausible reaction pathway for the generation of acetate could involve the nucleophilic attack of pyruvate by the reactive oxygen species generated from hydrogen peroxide (H2O2) (Figure 2A top), and the reaction would involve incorporation of the oxygen into H2O2 via production of superoxides and then into acetate. H2O2 thus obtains oxygen from molecular O2 and therefore, culturing cells in the presence of 18O2 and monitoring incorporation of 18O into acetate would enable quantitation of the endogenous contribution of ROS to acetate production. In this experimental design, other potential acetate production routes (deacetylation or aldehyde oxidation) would involve transferring the oxygen in a water molecule to acetate. 18O labeled water is negligible in this setup, and thus these two possibilities could be resolved with this experiment (Figure 2A bottom). We cultured cells in the presence of 20% O2 (18O2 and 16O2) and 80% N2 (Figure 2B). We then employed LC-HRMS to enable spectral resolution of [18O1]-acetate from other isotopically labeled species. We used labeled kynurenine (produced through oxygenation) in SKOV3 cells with high indoleamine-2,3-dioxygenase (IDO) expression (Litzenburger et al., 2014) to quantify the amount of heavy 18O2 experienced by the cells which was found to be about 40% in this setup (Figure 2C). We then identified (Figure 2D) and quantified (Figure 2E) [18O1]-acetate providing a direct observation in intact cells of the formation of [18O2]-H2O2 and oxidative decarboxylation of pyruvate by H2O2. Inhibiting the mitochondrial pyruvate carrier with UK5099 or addition of pyruvate to the culture medium substantially increased the contribution of H2O2 to acetate production from pyruvate (Figure 2E). To further test this mechanism, inhibition of superoxide dismutase (SOD), which converts superoxides to H2O2 (Figure 2F), with tetrathiomolybdate (TTM), was done to decrease endogenous H2O2 levels. Indeed, the presence of TTM decreased 18O incorporation into H2O2-coupled reactions, including methionine sulfoxide (Figure 2G) and acetate (Figures 2H and 2I). These findings together demonstrate that endogenous H2O2 contributes to acetate formation from pyruvate in cellular conditions.
Figure 2. ROS catalyzes the oxidative decarboxylation of pyruvate to generate acetate in mammalian cells.
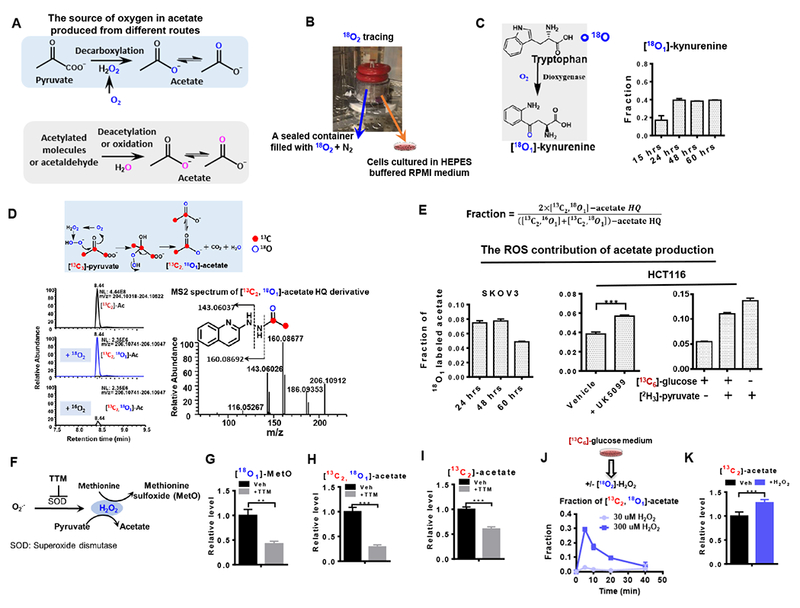
(A) Schematic of the potential sources of elemental oxygen in acetate produced from different routes. (B) Experimental setup of 18O2 tracing assay as described in STAR Methods. (C) Incorporation of 18O into metabolites in tryptophan metabolism. (D) Schematic of 18O and 13C incorporation into acetate produced from H2O2-mediated pyruvate decarboxylation, and representative chromatogram and tandem mass spectrum of [13C2, 18O1]-acetate derivative. Blue open circles and red solid circles denote O and C, respectively. (E) Fraction of [13C2, 18O1]-acetate out of the glucose-derived acetate pool. (F) Schematic of endogenous H2O2 generated from superoxides and superoxide dismutase (SOD) and H2O2-mediated methionine oxidation and pyruvate decarboxylation. TTM, Ammonium tetrathiomolybdate, a SOD inhibitor. The effect of SOD inhibitor TTM on the relative levels of [18O1]-MetO (G), [13C2, 18O1]-acetate (H) in SKOV3 cells cultured in [13C6]-glucose and 18O2 for 48 hrs. (I) Secretion of [13C2]-acetate from HCT116 cells in the presence or absence of TTM. (J) Release of [13C2, 18O1]-acetate from HCT116 cells after addition of exogenous [18O2]-H2O2. (K) Relative abundance of [13C2]-acetate. The fractions in E used to represent the ROS contribution to acetate production were corrected for 18O natural abundance and the 18O2 enrichment and. The data in J and K represent the acetate from the spent media collected at 10 min after addition of 0 or 300 μM [18O2]-H2O2 (J) or H2O2 (K) to HCT116 cells which were pre-incubated in [13C6]-glucose medium for 1 hour. Values are expressed as mean ± SD of n=3 independent measurements. ** p<0.01, *** p<0.001 in Student’s t test. See also Figure S2.
To further investigate the properties of this reaction, we performed in vitro assays by incubating pyruvate with hydrogen peroxide at 37 °C and found the major product to be acetate as measured by 1H Nuclear Magnetic Resonance Spectroscopy (Figure S2A). Thus, in the presence of hydrogen peroxide, pyruvate is converted to acetate non-enzymatically with kinetics commensurate with values needed for the reaction to occur at appreciable amounts in cells (Figure S2B). The reaction followed second order kinetics (k = 0.19 +/− 0.05 mM−1min−1) and could be accelerated with catalysts present in high concentrations in cells such as Cu2+, as Cu2+ stabilizes the intermediate of pyruvate decarboxylation (Figures S2B and C).
As a further evaluation, we tested whether exogenous H2O2 affects acetate production by adding [18O2]-H2O2 to cultured HCT116 cells (Figure 2J). Titrating concentrations of [18O2]-H2O2 revealed a dose-dependent increase in acetate levels and the acetate detected had 18O incorporation as measured by LC-HRMS (Figure 2J). Additionally, HCT116 cells pre-incubated with [13C6]-glucose and then treated with [18O2]-H2O2 showed a transient increase in the amount of [18O1]-acetate peaking around five minutes post induction of ROS with subsequent decay kinetics corresponding to the clearance of the ROS (Figure 2J) (Winterbourn, 2008). Importantly, it was observed that up to 30% of the total [13C2]-acetate pool (Figures 2J and K) could be derived from a transient increase in ROS from exogenous [18O2]-H2O2. This analysis further revealed that acetate is synthesized from pyruvate in intact cells and the activity of this reaction is mediated by ROS.
The presence of acetate unaccounted by isotopic labeling of oxygen suggests ROS-mediated pyruvate decarboxylation is not the only mechanism for de novo acetate production. Given that pyruvate is a keto acid, we next tested whether acetate can be released from keto acid dehydrogenases, considering that one function of this enzyme family is to catalyze the decarboxylation of the keto acid group. We first considered pyruvate dehydrogenase (PDH) (Figure 3A), which uses pyruvate as the substrate. Mammalian PDH mediates oxidative decarboxylation of pyruvate to acetyl-CoA. PDH is a multienzyme complex, containing three subunits (E1, E2, and E3). E1 catalyzes pyruvate decarboxylation using the cofactor thiamine pyrophosphate (TPP) and subsequent reductive acetylation of the lipoyl groups. E2 transfers the acetyl group to the thiol group in CoA and generates acetyl-CoA, and E3 regenerates the cofactor lipoate by coupling the oxidation to NAD+ reduction. Therefore, the cofactors CoA and NAD+ are required for PDH to achieve full activity. However, when CoA levels are lower than pyruvate levels, possibly, other thiol-containing molecules, such as GSH could serve as the acetyl group acceptor, considering that the cellular CoA is around 10 μM while GSH is 1 to 7 mM (Figure 3B), and similar concentration values in different types of cells or tissues were also reported by others (Chen et al., 2016; Lee et al., 2014; Sadhukhan et al., 2016). Low levels of CoA and high levels of GSH were also observed in autochthonous mouse tumors providing a physiological context for this occurrence (Figure 3C). Meanwhile, pyruvate was at much higher concentrations (Figure 1E). To study how these imbalanced substrate and cofactor concentrations affect PDH function, we incubated PDH isolated from porcine heart with [13C3]-pyruvate in the presence or absence of cofactors (TPP, CoA and NAD+) (Figure 3D). Indeed, with CoA at saturated concentrations (400 μM), more than 99% of the pyruvate (200 μM) was consumed in less than 2 mins (Figure 3D). Surprisingly, in the absence of CoA and NAD+, pyruvate was still consumed by PDH in the presence of TPP, but instead converted to [13C2]-acetate and [13C2]-acetaldehyde (Figures 3D, 3E and S3A), with lower overall PDH activity (Figure S3B). Furthermore, in the absence of CoA, GSH was found to be an acetyl group acceptor and increased overall PDH activity (Figure 3E) that resulted in the formation of [13C2]-acetyl-GSH (Ac-GSH) (Figure 3E). Interestingly, another keto acid dehydrogenase, alpha-ketoglutarate dehydrogenase (aKGDH), that normally catalyzes the conversion from alpha-ketoglutarate to succinyl-CoA via a similar mechanism as PDH, also functions as a pyruvate decarboxylase, catalyzing the conversion of pyruvate to acetaldehyde and to acetate (Figures 3F-G). Together these data indicate that the release of intermediates as a result of incomplete oxidation, including acetate, acetaldehyde or acetyl-GSH, is regulated by the activity of keto acid dehydrogenases and the availability of cofactors CoA and NAD+ (Figure 3H).
Figure 3. Conversion from pyruvate to acetate is catalyzed by mammalian PDH in a thiamine dependent manner.
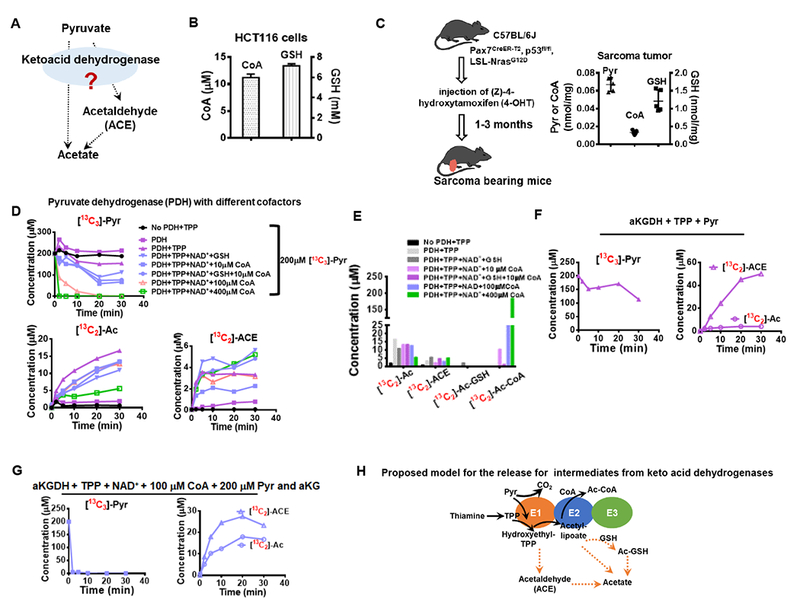
(A) Release of acetate and acetaldehyde from pyruvate is possibly catalyzed by keto acid dehydrogenases, especially pyruvate dehydrogenase (PDH). (B) Intracellular CoA and GSH concentrations in HCT116 cells. (C) CoA and GSH concentrations in mouse sarcoma tumors. (D) Conversion of pyruvate (200 μM) to acetate and acetaldehyde by PDH over 30 min in the absence or presence of cofactors. (E) Production of pyruvate-derived acetate, acetaldehyde, Ac-CoA and Ac-GSH after 30 min incubation with PDH. Consumption of pyruvate and production of acetaldehyde and acetate by alpha-ketoglutarate dehydrogenase supplemented with TPP in the absence (F) or the presence (G) of other cofactors and alpha-ketoglutarate (aKG, 200 μM). (H) Proposed model for acetate release from pyruvate catalyzed by keto acid dehydrogenase. Values are expressed as mean ± SD of n=3 (HCT116 cells) and n=5 (mouse) independent measurements. See also Figure S3.
We then tested whether this also occurred in intact cells and mice. Indeed, [13C2]-acetate, [13C2]-acetaldehyde and [13C2]-Ac-GSH were quickly formed in HCT116 cells within a few minutes after [13C6]-glucose media were added (Figure 4A) and further confirmed with tandem mass spectrometry (Figure S4A). The intracellular concentration of [13C2]-acetaldehyde was found to be higher than the intracellular [13C2]-acetate concentration, and also much higher than the concentration of [13C2]-acetaldehyde excreted into the media (Figure 4B). It is worth noting that the atmospheric boiling point of acetaldehyde and acetate is 20.2 and 118.1 °C, respectively. Thus acetaldehyde evaporated much faster than did acetate, and at 37 °C, some of the acetaldehyde evaporates (Figure S4B). Therefore, intracellular acetaldehyde (Figure 4B) accumulated, while medium acetaldehyde evaporated which suppresses in part the amount we are able to quantify. Production of acetate and Ac-GSH was decreased in cells engineered with CRISPR/Cas9 to lack PDH (Figures 4C and S4C). Interestingly, subcellular fractionation, followed by measurement of the levels of these dehydrogenases in the mitochondria and nucleus, indicated the quantitative presence of alpha-ketoglutarate dehydrogenase (aKGDH) in the nuclear fraction, while PDH is mostly in the mitochondria (Figure 4D). The presence of other keto acid dehydrogenases, such as aKGDH, prevented the complete depletion of de novo acetate production in PDH KO cells (Figure 4C) and in cells treated with mitochondrial pyruvate carrier inhibitor UK5099 (Figure S1H). Therefore, thiamine deprivation was performed to inactivate both PDH and aKGDH by depleting thiamine pyrophosphate. Thiamine deprivation for 4 days caused the depletion of thiamine, thiamine pyrophosphate (TPP), and alterations in the levels of TCA intermediates in HCT116 cells (Figure S4D), which is consistent with the hypothesis that thiamine depletion inactivates keto acid dehydrogenases (PDH and aKGDH). Consequently, thiamine depletion reduced acetate generation by more than 80% (Figure 4E), which is greater than the effects of PDH KO or treatment with UK5099. In addition, thiamine depletion also decreased ROS activity as evidenced by decreased ROS-mediated methionine sulfoxide formation and thus decreased the ROS-mediated acetate production (Figure S4E). However, under thiamine starvation, the addition of exogenous ROS (H2O2), especially in the presence of pyruvate in the medium greatly increased the contribution of ROS to acetate production (Figures 4F-G and S4F). In addition, CPI-613, a lipoate analogue and a PDH inhibitor, increased the formation of Ac-GSH (Fig. S4G). All together, these results link thiamine pyrophosphate-dependent keto acid dehydrogenase activity to acetaldehyde, Ac-GSH, and acetate production, consistent with our findings using purified PDH enzyme (Figure 3).
Figure 4. Acetate production from pyruvate occurs in both cultured cells and in vivo.
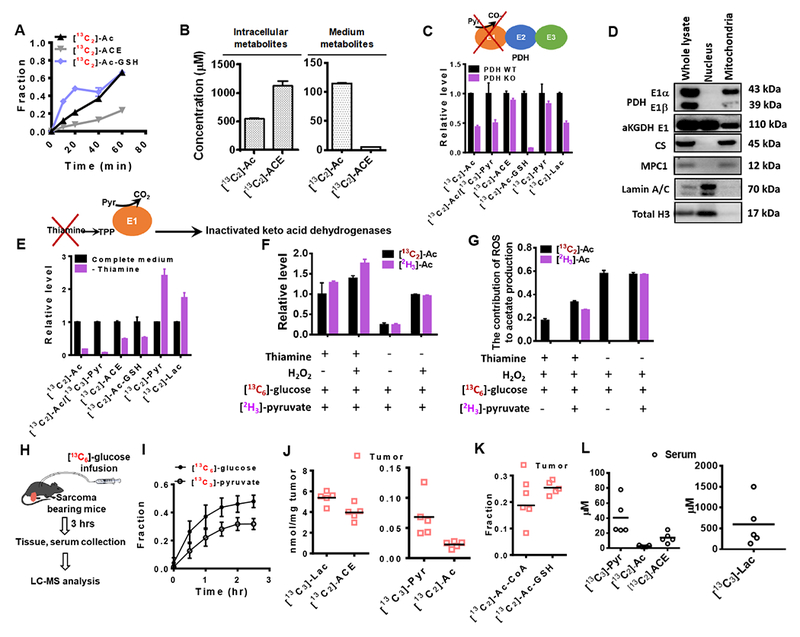
(A) 13C enrichment of acetate (Ac), acetaldehyde (ACE) and Acetyl-GSH (Ac-GSH) in HCT116 cultured in the presence of [13C6]-glucose. (B) Intracellular and medium concentrations of 13C labeled Ac and ACE in HCT116 cells after incubation in[13C6]-glucose medium for 1 hr. (C) Relative levels of 13C enriched Ac, ACE and Ac-GSH in mouse sarcoma cells with wild type PDH (PDH WT) or PDH knockout (PDH KO) using CRISPR/Cas9. (D) The presence of PDH and aKGDH proteins in the subcellular fractions of HCT116 cells. Abbreviations: E1, the E1 component of PDH or aKGDH; CS, citrate synthetase; MPC1, mitochondrial pyruvate carrier 1. (E) Relative levels of 13C enriched Ac, ACE and Ac-GSH in HCT116 cells cultured in medium with or without thiamine. (F) Relative levels of [13C6]-glucose and [2H3]-pyruvate-derived acetate after the addition of [18O2]-H2O2 (200 μM) for 10 min. (G) Contribution of ROS to acetate production (represented by the fraction of 18O labeled acetate) upon thiamine starvation. (H) Schematic of 13C glucose infusion in soft tissue sarcoma-bearing mice. (I) 13C enrichment fraction of glucose and pyruvate in the serum of sarcoma free mouse after the [13C6]-glucose infusion. (J) Concentrations of [13C3]-Pyr, [13C3]-Lac, [13C2]-ACE and [13C2]-Ac in sarcoma tumors. (K) 13C enrichment of AC-GSH and [13C2]-Ac-GSH in sarcoma. (l) Concentrations of [13C3]-Pyr, [13C3]-Lac, [13C2]-ACE and [13C2]-Ac in serum of tumor-bearing mice. J to L were obtained from sarcoma mouse samples collected 3 hrs after [13C6]-glucose infusion. Values are expressed as mean ± SD of n=3 (HCT116 cells) and n=5 (mouse) independent measurements. See also Figure S4.
To determine whether these reaction mechanisms could occur in a physiological context, we first established a euglycemic transfusion system where mice (Figure 3C) bearing autochthonous sarcoma tumors were continuously infused with [13C6]-glucose (Figure 4H, described in STAR Methods). We first conducted this experiment using tumor-free mice, and observed that over 40% of the glucose in the serum at steady state could be labeled, resulting in approximately 32% [13C3]-pyruvate labeling (Figure 4I). Next in tumor-bearing mice, both serum and tumor tissue were collected for metabolite analysis after 3 hrs of glucose infusion. 13C labeled pyruvate, lactate, acetate and acetaldehyde were detected in tumor tissue at different concentrations (Figure 4J). The enrichment of 13C labeled [13C2]-Ac-CoA and [13C2]-Ac-GSH was comparable (Figure 4K). The concentrations of [13C3]-pyruvate, [13C3]-lactate and [13C2]-acetate were much higher in the tumor than in the serum (Figure 4L), indicating that these metabolites were produced locally confirming our findings in vitro and in intact cells (Figure 3 and Figures 4A-G).
To expand the scope of the study and thus identify contexts in which this pathway is biologically necessary, we sought to explore situations in which acetate derived from some cells could interact with the metabolism of other cells. ATP citrate lyase (ACLY) is a cytosolic enzyme that converts nucleocytosolic citrate to Ac-CoA and acetate requirements in cells increase when its activity is impaired (Zhao et al., 2016). We thus considered mouse embryonic fibroblasts (MEFs) that were engineered to lack ACLY (ACLY KO) (Figures 5A and 5B) that were previously described and further confirmed to exhibit an acquired dependence on acetate supplementation (Zhao et al., 2016). Co-culturing these MEFs with acetate-producing HCT 116 cells separated by a 0.4 micron membrane allowing for free diffusion (Figure 5C) resulted in dramatically increased medium acetate concentrations in the MEF compartment (Figure 5D), and rescued deficiencies in the growth rate in the knockout cells (Figure 5E) with negligible effects on the control (ACLY WT) MEFs (Figure 5F). ACSS2 is the enzyme that metabolizes acetate for acetyl-CoA production. As shown in Figures 5G-H, ACSS2 knockdown is detrimental to the fitness of MEF cells with ACLY deficiency, so the co-culture time was shortened to 30 hrs when ACSS2 knockdown cells were used, and the co-culture with HCT116 cells ablated the rescue of cell proliferation. Consistent with prior findings (Zhao et al., 2016), [13C6]-glucose contributed minimally to fatty acid synthesis in ACLY KO cells (Figure 5I). However, increased de novo lipogenesis indicated by increased incorporation of [13C6]-glucose into palmitate in ACLY KO cells co-cultured with HCT116 cells confirmed increased cytosolic Ac-CoA production (Figure 5I). These findings demonstrate that endogenous de novo acetate produced through overflow metabolism from one cell type can be utilized to support proliferation of surrounding cells with acetyl-CoA insufficiencies (Figure 5J).
Figure 5. De novo acetate production rescues proliferation defects in cells with ACLY deficiency.
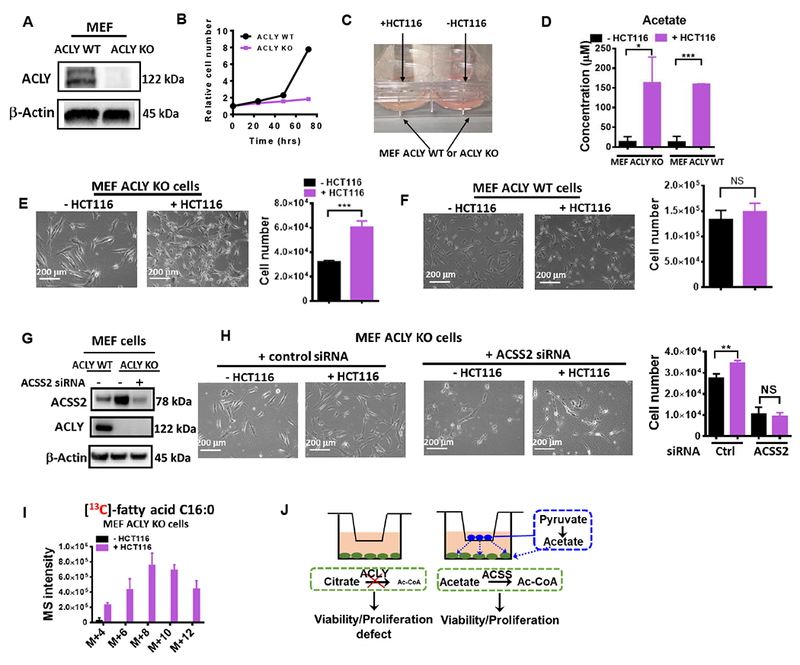
(A) Western blot of ATP citrate lyase (ACLY) in MEF (ACLY WT or KO) cells. (B) Growth curve of MEF (ACLY WT or KO) cells cultured in RPMI 1640 supplemented with 10% dialyzed FBS without acetate supplement. (C) Picture of co-culture system. (D) Medium acetate concentration in co-culture system in the absence or presence of HCT116 cells. Representative images and cell number of MEF ACLY KO (E) or WT cells (F) co-cultured with or without HCT116 for 3 days. (G) Western blot confirming the knockdown of ACLY and ACSS2 in MEF cells. (H) Representative photos and cell numbers of MEF ACSS2 WT or KD cells co-cultured with or without HCT116 for 30 hrs. (I) The incorporation of 13C into palmitate in MEF ACLY KO cells co-cultured with or without HCT116 cells in RPMI medium containing [13C6]-glucose and 10% dialyzed FBS (bottom). (J), Model of restoration proliferation in MEF ACLY KO cells by acetate released from HCT116 cells in a co-culture system. Values are expressed as mean ± SD of n=3 independent measurements. NS: p>0.05, * p<0.05, ** p<0.01, *** p<0.001 in Student’s t test.
To further explore this pathway, we examined the effects of ACLY expression in high-acetate producing HCT116 cells cultured in [13C6]-glucose medium in the absence of acetate supplementation. ACLY knockdown had minor effects on de novo lipogenesis (Figures 6A-C), while knockdown or inhibition using a small molecule inhibitor (Comerford et al., 2014) of ACSS2 had larger effects on de novo lipogenesis (Figures 6B-C) when there is no additional acetate in the medium. The knockdown of both ACLY and ACSS2 reduced de novo lipogenesis to nearly undetectable amounts (Figure 6B). Furthermore, to evaluate the contribution of ROS-mediated acetate production to intracellular Ac-CoA pools, we measured the labeling patterns of surrogate products of Ac-CoA (Figure 6D), and we saw the incorporation of 18O into Ac-carnitine (Figure 6E) and the increase in [13C2,18O2]- labeled acetylated molecules, including Ac-aspartate in the presence of UK5099 (Figure 6F). Nevertheless, the net effect was that lipogenesis is decreased by exogenous ROS and increased by the presence of catalase (Figures S5A-B), which was likely attributed to the activity of lipogenesis enzymes also being affected by oxidative stress. Separately, the oxidation of amino acids, such as methionine, was suppressed in the presence of pyruvate due to the clearance of ROS by pyruvate via oxidative decarboxylation (Figure S5C). In addition, acetaldehyde, one of the intermediates released from incomplete oxidation of pyruvate, was oxidized to acetate by aldehyde dehydrogenase (Enomoto et al., 1991), and then subsequently supplied the Ac-CoA pool (Figure 6G). Isotope tracing with deuterated acetaldehyde confirmed that ACLY knockdown cells exhibited increased acetate utilization which could be further increased by limiting nutrient availability during serum starvation (Figure 6H), which is consistent with previous findings (Bulusu et al., 2017; Schug et al., 2015). All together these experiments confirm that acetate generated from keto acid dehydrogenases or ROS-mediated pyruvate decarboxylation can have critical functional roles in supporting acetyl-CoA metabolism.
Figure 6. Endogenous acetate contributes to acetyl-CoA metabolism.
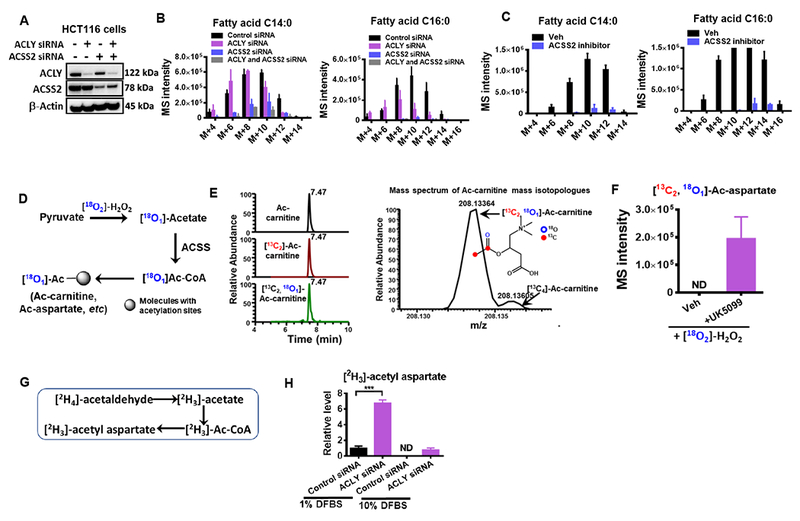
(A) Western blot confirming the knockdown of ACLY and ACSS2 in HCT116 cells. The effect of ACLY and ACSS2 knockdown (B) or ACSS2 inhibitor (20 μM) (C) on lipogenesis in HCT116 cells cultured in RPMI 1640 supplemented with 10% dialyzed FBS, without exogenous acetate. (D) Schematic of 18O incorporation to acetyl groups via ROS mediated pyruvate decarboxylation. (E) Extracted ion chromatogram (left) and mass spectra (right) of ac-carnitine mass isotopologues from HCT116 cells treated with [18O2]-H2O2 (300 μM) for 30 mins. (F) The relative levels of 18O labeled acetyl-aspartate in from HCT116 cells treated with [18O2]-H2O2 (300 μM) for 30 mins in the presence and absence of UK5099. (G-H) Carbon incorporation from deuterium labeled acetaldehyde into acetyl groups by HCT116 cells treated with control siRNA or ACLY siRNA cultured in low (1%) or regular (10%) D-FBS. Values are expressed as mean ± SD of n=3 independent measurements. ND: not detected. ** p<0.001 in Student’s t test. See also Figure S5.
DISCUSSION
While it has been demonstrated that ACLY is required for histone acetylation in cancer cells, high ACLY activity is not necessarily a universal requirement for cellular acetylation events (Wellen et al., 2009), indicating that acetate-derived acetyl groups could contribute to the cellular acetylation reactions. This is consistent with our observation (Figure 6) that endogenous acetate production can support acetyl-CoA production for lipogenesis. In addition, the dependence on acetate to supply both nucleocytosolic and mitochondrial acetyl-CoA pools has been identified in multiple cell types, including cancer cells, T cells, and neurons (Balmer et al., 2016; Comerford et al., 2014; Mashimo et al., 2014; Mews et al., 2017; Schug et al., 2015). Our study provides evidence for a pyruvate-derived overflow pathway for acetate metabolism that occurs in mammalian cells. It further identifies two mechanisms with differential regulatory capacities involving non-enzymatic and enzymatic pyruvate-derived acetate chemistry. Thus endogenous acetate production may be a fundamental component of mammalian central carbon metabolism.
One reaction mechanism found to generate acetate occurred through altered enzyme activity of thiamine-dependent keto acid dehydrogenases, which transformed their activity to keto acid decarboxylases. The availability of free CoA in the mitochondria is thought to limit the conversion of pyruvate to acetyl groups (Bremer, 1966). It has also been demonstrated (Frey et al., 1989) that the absence of either CoA or NAD+ causes the accumulation of hydroxyethyl-TPP (HETPP), which is the product of the E1 and the substrate of the E2 component. In addition, aKGDH and PDH are structurally and catalytically similar enzymes, given that the flavoprotein E3 components from PDH and aKGDH are identical (Reed, 2001). It has also been reported that pyruvate is a substrate for aKGDH (Graham et al., 1989; Steginsky et al., 1985). Interestingly, even though it has been thought that aKGDH is mainly located in mitochondria, the presence of nuclear aKGDH has been reported (Wang et al., 2017) and also supported by our data (Figure 4D). Therefore, although acetate production was depleted by thiamine starvation, neither PDH deletion nor mitochondrial pyruvate carrier inhibition alone was sufficient to completely remove acetate production. Thus, numerous pathways may exist by which acetate is coupled to other critical metabolic functions, depending on the activity and the location of keto acid dehydrogenases, the availability of cofactors.
The other reaction mechanism to generate acetate occurred by reaction with ROS, a finding which potentially links this pathway to numerous physiological and pathophysiological processes (Houstis et al., 2006; Sena et al., 2013). 18O2 tracing and LC-HRMS allowed us to directly monitor ROS-mediated acetate production in intact cells without exogeneous ROS stimulation (Figure 1), and the results suggest that oxidative stress could allow for coupling of acetate production to metabolic functions within central carbon metabolism (Dai et al., 2016; Murphy, 2009). ROS levels are dynamic and at steady state, intracellular concentrations of H2O2 vary from nM to μM (Antunes and Cadenas, 2001; Chance et al., 1979), while extracellular concentrations of H2O2 may be 10 to 100 times higher under certain circumstances, such as inflammation (Test and Weiss, 1984). Consistent with this pathway, cancer cells have been shown to produce H2O2 at a rate of around 1 nmol/104 cells/hour (Szatrowski and Nathan, 1991; Weinstain et al., 2014), while the combined pyruvate generation and secretion rate (Figure 1B) was found to be around 1 nmol/104 cells/hour.
Under typical cell culture conditions when there is active PDH and aKGDH, ROS only contributes to around 5-15% of the acetate production (Figure 2). However, thiamine deficiency is reported to be a common occurrence in cancer patients providing additional context (Isenberg-Grzeda et al., 2017; Onishi et al., 2017; Sechi et al., 2016). Thiamine starvation and the addition of exogeneous ROS greatly stimulated the ROS contribution to acetate production (Figure 4G), which can be used to replenish intracellular acetyl groups. Nevertheless, ROS can also impair the activity of certain proteins, such as those with active sites containing cysteine residues (van der Reest et al., 2018). Because the amino acid residues of proteins are targets of ROS, and proteins have large sizes, many amino acid residues, and present at high abundance (50-70% of cell dry weight), which increase the reaction efficiency by increasing the likelihood of reactive collisions, especially when these protein residues are solvent-exposed. For such reasons, exogenous ROS increased ROS-derived acetate production and the incorporation into cellular acetyl groups, but didn’t cause a net increase in lipogenesis in cells subjected to ROS. Thus, increased acetate production and release could potentially favor the neighboring cells deficient in cytosolic acetyl-CoA, as demonstrated by co-culturing ACLY KO cells with HCT116 cells (Figure 5).
In addition, increased acetate production from ROS-mediated pyruvate decarboxylation suggests that pyruvate may naturally function as a ROS scavenger. Indeed, the presence of 1-5 mM pyruvate in typical cell culture media prevents the oxidation of methionine to methionine sulfoxide (as marker of amino acid oxidation, Figure S5C). Nevertheless, under certain conditions, ROS is known to play an important role in stimulating cell proliferation such as in T cell activation (Sena et al., 2013), and we can not rule out that under such conditions acetate production may possibly be related to ROS-stimulated cell proliferation.
Overall, our study elucidates pathways and quantitative reaction mechanisms through which pyruvate-derived de novo acetate production occurs. Moreover, our results reveal a potential role for de novo acetate production to supply acetyl groups and concomitantly, maintaining lower cellular redox potentials. Although we have defined a set of biological examples which require this pathway, we have by no means defined the general biological role of these reactions. Thus, it will be of further interest to expand on the role of de novo acetate production in maintaining acetyl-CoA pools for the myriad biological settings which require acetyl groups.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the Lead Contact, Jason W. Locasale (Jason.Locasale@duke.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture.
MEF (ACLY WT (Aclyf/f) and KO) cells were prepared as described previously (Zhao et al., 2016). HCT116, SKOV3, HepG2, and BT474 cell lines were obtained from ATCC. The HeyA8 cell line was a generous gift from Dr. Ernst Lengyel’s lab, University of Chicago, IL USA (Kenny et al., 2015). Gender of the patients from whom the cell lines were derived: HCT116, male; SKOV3, female; BT474, female; HeyA8, female; HepG2, male. Mouse sarcoma cells were derived from sarcoma bearing male mice, and the procedure is described below. The original MEF cells were from male and female mixed mice. All human cell lines used in the study were authenticated at the Duke University DNA Analysis Facility using the GenePrint 10 kit (Promega) and tested to be mycoplasma-free. All cell culture related medium and reagents were sterilized using a 0.22 μm sterile filter.
All cells were first cultured in a 10 cm dish with full growth medium (RPMI 1640 supplemented with 10% FBS). The cell incubator was set at 37 °C supplemented with 5% CO2. For cell proliferation assays, cells were cultured in a 12 well plate, and at the end of treatment, cells were first harvested by trypsinization, and cell number and volume were measured using Moxi™ Z Mini Automated Cell Counter (ORFLO Technologies). For metabolite analysis, cells were seeded into 6 well plate at the density of 300 000 to 500 000 cells per well. After overnight incubation in full growth medium, for UK5099 treatment, the old medium was removed and cells were briefly washed with glucose free RPMI 1640 before 1.5 ml of RPMI 1640 (supplemented with 10 % dialyzed FBS) with 0.025 % DMSO or 5 μM UK5099 (final DMSO content 0.025 %) was added to each well. After 5 hours of incubation, medium was replaced with 1 ml of RPMI 1640 containing 11.1 mM [13C6]-glucose, 10 % dialyzed FBS, and 0.025 % DMSO or 5 mM UK5099 (final DMSO content 0.025 %). For acetate measurements, 5 μl medium from each well was collected at different time points and immediately placed on dry ice before storage in a −80 °C freezer. Intracellular metaboli tes were extracted at 10, 20 and 40 min, and the detailed extraction procedure will be described below. For [18O2]-H2O2 treatments, after incubation in [13C6]-glucose medium for 1 hr, [18O2]-H2O2 was directly added to each selected well (final concentration, 30 or 300 μM). 5 μl medium from each well was collected at 5, 10, 20 and 40 min. Intracellular metabolites were extracted at 30 min after addition of [18O2]-H2O2 (300 μM). For CPI-613 treatment, HCT116 cells were incubated in RPMI 1640 (supplemented with 10 % dialyzed FBS) in the absence or presence of 150 μM CPI-613 for 4 hours and media were replaced with 1 μl of RPMI 1640 containing 11.1 μM [13C6]-glucose, 10 % dialyzed FBS, and 0.15 % DMSO or 150 μM CPI-613 (final DMSO content 0.15 %). Media were collected after 40 min, and intracellular metabolites were analyzed after a 1 hr incubation in 13C-glucose media. For the thiamine deprivation assay, HCT116 cells were incubated in thiamine free RPMI 1640 (supplemented with 10 % dialyzed FBS) for 4 days and media were replaced with 1 ml of thiamine free RPMI 1640 containing 11.1 mM [13C6]-glucose and 10% dialyzed FBS. Media and intracellular metabolites were collected after 40 min or 1 hr incubation in 13C-glucose media.
Animal.
All animal procedures for this study were approved by the Institutional Animal Care and Use Committee (IACUC) at Duke University. 8 to 10-week old C57BL/6J male and female mixed mice (The Jackson Laboratory) were used to develop the 13C-glucose infusion workflow. Mouse models of soft tissue sarcoma were generated in a mixed 129/SVJae and C57BL/6 background (The Jackson Laboratory) using a combination of alleles: Pax7CreER-T2, p53fl/fl, LSL-NrasG12D. Primary mouse soft tissue sarcomas were generated in the mouse hind limb by intramuscular (IM) injection of (Z)-4-hydroxytamoxifen (4-OHT) (Zhang et al., 2015). A euglycemic glucose infusion was performed according to a procedure previously described (Ayala et al., 2010). Briefly, a jugular vein catheter was surgically implanted and exteriorized via a vascular access port, which allows infusion of [13C6]-glucose via the venous catheter. Blood from a tail nick in freely-moving and unanesthetized animals was serially withdrawn. Four to five days after surgery, mice were fasted for 6 hours and continuously infused with [13C6]-glucose at a rate of 20 mg/kg/min for a total of 3 hours. Blood was collected every 30 min for the duration of the infusion. Immediately following infusion, animals were sacrificed by cervical dislocation and liver tissue was excised and rapidly frozen in liquid N2 to quench metabolism. Tissue was stored at −80°C u ntil used for metabolite extractions. No data were excluded from the analysis.
METHOD DETAILS
siRNA transfections.
siRNA transfection was performed following manufacturer’s instructions with Lipofectamine RNAiMAX (Invitrogen), using siRNA pools targeting human ACLY (Dharmacon #L-004915-00), mouse ACSS2 (Dharmacon #L-065412-01), human ACSS2 (Dharmacon #L-010396-00) or a non-targeting control (Dharmacon #D-001810-10-20) at a concentration of 20 nM for ACLY and control, and 5 nM for ACSS2 in growth medium (RPMI 1640 with 10 % FBS). After 48 hrs, cells were split into a new 6 well plate in the absence of transfection reagents. All analysis (protein, metabolite and cell proliferation) was performed after 72 hrs of siRNA transfections.
Western blotting.
Whole cell lysates were prepared by collecting cells into ice cold RIPA buffer containing complete Protease Inhibitor Cocktail (Roche).
Protein lysate was first loaded to a NuPAGE 4-12% Bis-Tris Protein Gels (Thermo Fisher Scientific) for separation, and then transferred to a PVDF membrane. The blot was visualized using ChemiDoc (Bio-Rad). Antibodies used are described in Key Resources Table. All antibodies were used at 1:1000 dilution.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-ATP citrate lyase | Proteintech | Cat#15421-1-AP; RRID:AB_2223741 |
| Mouse monoclonal anti-β-actin | Cell Signaling Technology | Cat#3700S; RRID:AB_2242334 |
| Rabbit monoclonal anti-AceCS1 | Cell Signaling Technology | Cat#3658T |
| Rabbit monoclonal anti-pyruvate dehydrogenase | Cell Signaling Technology | Cat#3205T; RRID:AB_2162926 |
| Rabbit polyclonal anti-aKGDH | Cell Signaling Technology | Cat#26865S |
| Rabbit monoclonal anti-citrate synthase | Cell Signaling Technology | Cat#14309S |
| Rabbit monoclonal anti-MPC1 | Cell Signaling Technology | Cat#14462S |
| Rabbit polyclonal anti-Lamin A/C | Cell Signaling Technology | Cat#2032T |
| Rabbit polyclonal anti-Histone H3 | Cell Signaling Technology | Cat#9715S; RRID:AB_331563 |
| Rabbit monoclonal anti-acetyl-histone K27 | Cell Signaling Technology | Cat#8173T |
| Rabbit monoclonal anti-acetyl-histone K9 | Cell Signaling Technology | Cat#9649T;RRID:AB_823528 |
| Rabbit polyclonal anti-acetyl-histone H3 | Millipore | Cat#06-599; RRID:AB_2115283 |
| Goat anti-mouse IgG (H&L) | Rockland Immunochemicals | Cat#610-1102; RRID:AB_219646 |
| Donkey anti-rabbit IgG (H&L) peroxidase conjugated | Rockland Immunochemicals | Cat#611-7302; RRID:AB_219747 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| RPMI 1640 | Cellgro | Cat#11875-093 |
| RPMI 1640, without glucose | ThermoFisher Scientific | Cat#11879020 |
| RPMI 1640, without glucose, without sodium bicarbonate | Sigma-Aldrich | Cat#R1383-10X1L |
| RPMI 1640, without thiamine | This paper | N/A |
| Opti-MEM | ThermoFisher Scientific | Cat#11058021 |
| Fetal Bovine Serum (FBS) | Hyclone | Cat#14-486 |
| Dialyzed FBS | ThermoFisher Scientific | Cat#H00000406-P01 |
| [13C6]-D-glucose | Cambridge Isotope Laboratories | Cat#CLM-1396-1 |
| 3,3,3-[2H3]-sodium pyruvate | Cambridge Isotope Laboratories | Cat#DLM-6068-0.5 |
| [13C3]-sodium pyruvate | Cambridge Isotope Laboratories | Cat#CLM-2440-0.5 |
| 3,3,3-[2H3]-sodium lactate | Cambridge Isotope Laboratories | Cat#DLM-9071-0.1 |
| [2H3]-acetyl-carnitine hydrochloride | Santa Cruz Biotechnology | Cat#sc-480972 |
| [2H3]-acetyl-CoA | Laboratory of Hening Lin, Cornell University | (Sadhukhan et al., 2016) |
| 18O2 | Sigma-Aldrich | Cat#490474-1L |
| 2,2,2-[2H3]-sodium acetate | Sigma-Aldrich | Cat#176079-5G |
| [2H4]-acetaldehyde | Sigma-Aldrich | Cat#176567-1G |
| [2H2]-H2O | Sigma-Aldrich | Cat#151882-10G |
| [18O2]-H2O2 (2-3 % solution) | Sigma-Aldrich | Cat#609978 |
| Coenzyme A sodium salt hydrate | Sigma-Aldrich | Cat#C3144-25MG |
| β-Nicotinamide adenine dinucleotide sodium salt (NAD+) | Sigma-Aldrich | Cat#N0632-1G |
| Glutathione (reduced) | Sigma-Aldrich | Cat#G4251-1G |
| SAHA | Sigma-Aldrich | Cat#SML0061 |
| Ammonium tetrathiomolybdate (TTM) | Sigma-Aldrich | Cat#323446 |
| CPI-613 | Sigma-Aldrich | Cat#SML0404 |
| UK5099 | Sigma-Aldrich | Cat#PZ0160 |
| Copper (II) sulfate | Sigma-Aldrich | Cat#451657 |
| 2-hydrazinoquinoline | Sigma-Aldrich | Cat#CDS000062 |
| Triphenylphosphine | Sigma-Aldrich | Cat#T84409 |
| 2,2’-dipyridyl disulfide | Sigma-Aldrich | Cat#143049 |
| L-Kynurenine | Sigma-Aldrich | Cat#K8625 |
| L-Methionine sulfoxide | Sigma-Aldrich | Cat#M1126 |
| Pyruvate dehydrogenase (PDH) from porcine heart | Sigma-Aldrich | Cat#P7032-10UN |
| alpha-Ketoglutarate dehydrogenase (aKGDH) from porcine heart | Sigma-Aldrich | Cat#K1502-20UN |
| Catalase from human erythrocytes | Sigma-Aldrich | Cat#C3556 |
| Thiamine pyrophosphate chloride | Alfa Aesar | Cat#J61483 |
| Ac-CoA Synthase Inhibitor | Millipore | Cat#533756 |
| MTT powder | ThermoFisher Scientific | Cat#M6494 |
| Quick Start™ Bradford 1x Dye Reagent | Bio-Rad | Cat#5000205 |
| Lipofectamine™ RNAiMAX Transfection Reagent | ThermoFisher Scientific | Cat#13778030 |
| Ponceau S solution, 0.1% | G-Biosciences | Cat#786-576 |
| cOmplete™, EDTA-free Protease Inhibitor Cocktail | Sigma-Aldrich | Cat#11873580001 |
| IGEPAL CA-630 | Sigma-Aldrich | Cat#I8896 |
| Acetonitrile, Optima LC/MS | Fisher Scientific | Cat# A955 |
| Water, Optima LC/MS | Fisher Scientific | Cat# W6 |
| Methanol, Optima LC/MS | Fisher Scientific | Cat# A456 |
| Ammonium hydroxide, Optima LC/MS | Fisher Scientific | Cat#A470 |
| Formic acid, Optima LC/MS | Fisher Scientific | Cat#A117 |
| Acetic Acid, Glacial | Fisher Scientific | Cat#A38 |
| Ammonium hydroxide, Optima LC/MS | Fisher Scientific | Cat#A470 |
| Critical Commercial Assays | ||
| Mitochondria Isolation Kit for Cultured Cells | ThermoFisher Scientific | Cat#89874 |
| Western Lightning Plus-ECL, Enhanced Chemiluminescence Substrate | Perkin Elmer | Cat#NEL103001EA |
| Deposited Data | ||
| Histone proteomics data | This paper; Mendeley Data | http://dx.doi.org/10.17632/39rzr8xzsv.1 |
| Metabolomics data | This paper; Mendeley Data | http://dx.doi.org/10.17632/39rzr8xzsv.1 |
| NMR data | This paper; Mendeley Data | http://dx.doi.org/10.17632/39rzr8xzsv.1 |
| Experimental Models: Cell Lines | ||
| Human: HCT116 cells | ATCC | CCL-247 |
| Human: HepG2 cells | ATCC | HB-8065 |
| Human: SKOV cells | ATCC | HTB-77 |
| Human: BT474 cells | ATCC | HTB-20 |
| Human: HeyA8 cells | Laboratory of Ernst Lengyel, University of Chicago | (Kenny et al., 2015) |
| Mouse: Sarcoma cells | This paper | N/A |
| Mouse: MEF (ACLY WT and KO) cells | Laboratory of Kathryn Wellen, University of Pennsylvania | (Zhao et al., 2016) |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No. 000664 |
| Mouse: Pax7CreER-T2 | Laboratory of Chen-Ming Fan, Carnegic Institution for Science | N/A |
| Mouse: p53fl/fl | Laboratory of Anton Berns, The Netherlands Cancer Institute | N/A |
| Mouse: LSL-NrasG12D | The Jackson Laboratory | Stock No. 008304 |
| Oligonucleotides | ||
| siRNA pools for human ACLY | Dharmacon | Cat#L-004915-00 |
| siRNA pools for human ACSS2 | Dharmacon | Cat#L-010396-00 |
| siRNA pools for mouse ACSS2 | Dharmacon | Cat# L-065412-01 |
| Negative control siRNA | Dharmacon | Cat#D-001810-10-20 |
| Recombinant DNA | ||
| LentiCRISPR v2 expressing an sgRNA targeting PDH (5’-AGAACAACCGCTATGGCATG-3’) | (Sanjana et al., 2014) | Addgene Plasmid#52961 |
| Software and Algorithms | ||
| Sieve 2.0 | ThermoFisher Scientific | https://portal.thermo-brims.com/index.php/component/thermosoftwares/thermosoftware/67?Itemid=121 |
| Xcalibur 4.0 | ThermoFisher Scientific | https://www.thermofisher.com/order/catalog/product/OPTON-30487 |
| MestReNova Lite SE | Mestrelab Research | http://mestrelab.com/news/new-mnova-lite-10-special-edition/ |
| GraphPad Prism 6 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| ChemDraw 15 | PerkinElmer | http://www.cambridgesoft.com/land/ChemDraw_Q22014.aspx |
| Microsoft Office Excel | Microsoft | https://www.microsoft.com/en-us/p/excel-2016/cfq7ttc0k5f3?activetab=pivot%3aoverviewtab |
| Other | ||
| Mini vacuum desiccator | VWR | Cat#47750-242 |
Histone extraction for western blotting.
Cells were washed with ice cold PBS, supplemented with 5 mM sodium butyrate, and then collected in triton extraction buffer (TEB): PBS containing 0.5% (v/v) Triton X-100, 2mM phenylmethylsulfonyl fluoride (PMSF) and 0.02% (w/v) NaN3. After 10-min on ice with gentle stirring, the lysed cells were centrifuged at 2,000 rpm for 10 min at 4 °C. T he supernatant was removed, and the pellet was washed with half the volume of TEB and then resuspended in 0.2 N HCl. After acid extraction overnight at 4 °C, the histon e extract was centrifuged at 2,000 rpm for 10 min at 4 °C. The supernatant wa s transferred to a new Eppendorf tube and protein concentration was determined by the Bradford assay. Aliquots were stored at −80 °C until ready for blotting.
13C labeling of histones.
HCT116 cells were seeded into 10 cm dish and cultured for 24 hours. The media were then replaced with labeling medium (RPMI 1640 glucose free medium supplemented with 11.1 mM [U-13C]-glucose, and 10% dialyzed FBS). Histone extraction and sample preparation for LC-MS/MS analysis were performed as described below and previously (Lin and Garcia, 2012). At the indicated time points, cells were washed with ice cold PBS (supplemented with 5 mM sodium butyrate), and then lysed in NIB-250 buffer (15 mM TrisCl pH 7.5, 60 mM KCl, 15 mM NaCl, 5 mM MgCl2, 1 mM CaCl2, 250 mM Sucrose), to which EDTA free cOmplete Protease Inhibitor Cocktail, sodium butyrate (final 5mM), DTT (final 1mM) and IGEPAL® CA-630 (NP-40, final 0.2%, v/v) were freshly added. After allowing lysis to proceed on ice for 10 mins, nuclei were pelleted at 500 rcf for 5 mins at 4C, washed twice in NIB-250 buffer without detergent, and then extracted in 0.4 N H2SO4 for 4 hrs at 4°C with rotation. Insoluble debris was pelleted by centrifugation at 3 400 rcf for 5 mins at 4°C and proteins were precipitated overnight on ice by adding 1 volume of trichloroacetic acid to 3 volumes of supernatant. Precipitated proteins were pelleted as above, washed with acetone containing 0.1% HCl and then pure acetone, and allowed to air dry before resuspending in 100 mM NH4HCO3.
Ponceau S staining was used to assess the purity of the histone extract. Histones were then derivatized with propionic anhydride (1 volume of 25% propionic anhydride in 2-propanol added to 2 volumes of histone peptides, incubated at 37 °C for 15 mins, dried in a speed vac, and repeated) to block unmodified lysines and digested with trypsin. After trypsinization, histone peptides were derivatized with propionic anhydride again to add a propionyl group to the newly formed n-termini, desalted with C18 stage tips (Empore, 3M), and analyzed using LC-MS/MS as described below and previously (Lin and Garcia, 2012). Peptides were separated online using an EASY-nLC (ThermoFisher Scientific) nano liquid chromatography system fitted with in-house pulled and packed C18 analytical columns (75 °m i.d. × 15-20 cm fused silica, 3 μm ReproSil-Pur 120 C18-AQ from Dr. Maisch GmbH). The gradient consisted of 5 to 33% B from 0 to 45 mins and 33 to 98% B from 45 to 50 mins, holding at 98% B until 60 mins with a flow rate of 300 nl/min, 0.1% formic acid in water serving as solvent A, and 80% acetonitrile with 0.1% formic acid serving as solvent B. Peptides were analyzed with an Orbitrap Fusion mass spectrometer (ThermFisher Scientific) operating in positive profile mode with the following full scan settings: 60 000 resolution, mass range 300-1100 m/z, AGC target 2e5, maximum injection time 100 ms. After one full scan in the orbitrap, eight data-independent acquisition (DIA) MS/MS scans of isolation width 50 m/z centered on 325.4125, 375.4375, 425.4625, 475.4875, 525.5125, 575.5375, 625.5625, and 675.5875 m/z were acquired in the ion trap in centroid mode, followed by another full scan, followed by another eight DIA MS/MS scans centered on 725.6125, 775.6375, 825.6625, 875.6875, 925.7125, 975.7375, 1025.7625, and 1075.7875 m/z. Mass range was set to 200-1500 m/z and quadrupole isolation set to on for DIA scans. HCD with NCE set to 30 was used for fragmentation with AGC target set at 1e4 and a maximum injection time of 50 ms. Mass spectra were analyzed by EpiProfile (Yuan et al., 2015), which uses MS/MS spectra and chromatographic peak areas to quantify the abundance uniquely modified peptide species relative to sum of the modified and unmodified forms of a given peptide sequence.
Subcellular fractionation.
For nuclei collection, around 107 cells were scraped into 1ml NIB-250 buffer containing protease inhibitor cocktail and 0.2% (v/v) NP-40, and kept on ice for 10 min with gentle pipetting. The lysates were centrifuged at 700 rcf for 5 min at 4 °C. The pellet (nuclei) was washed with 0.5 ml NIB-250 buffer without NP-40 and centrifuged. Intact mitochondria from HCT116 cells were obtained by using a Mitochondria Isolation Kit for Cultured Cells (ThermoFisher Scientific). Briefly, around 107 cells were scraped into 1ml hypotonic buffer provided by the manufacturer, and kept on ice for 6 min. A 2ml Dounce tissue grinder was then used to increase the lysis efficiency (around 80 strokes). The remaining steps were performed following the manufacturer’s guidelines. The isolated nuclei and mitochondria were then lysed on ice for 30 min in RIPA buffer containing protease inhibitor cocktail with rigorous vortexing every 5 min. The lysates were centrifuged at 20 000 rcf for 10 min at 4 °C. The supernatant was used for protein concentration measurement by the Bradford assay. Aliquots were stored at −80 °C until the time of th e experiment.
HPLC method.
Ultimate 3000 UHPLC (Dionex) was used for metabolite separation and detection. For polar metabolite analysis, a hydrophilic interaction chromatography method (HILIC) with an Xbridge amide column (100 × 2.1 mm i.d., 3.5 °m; Waters) was used for compound separation at room temperature. The mobile phase A: water with 5 mM ammonium acetate (pH 6.8); B: acetonitrile. Linear gradient: 0-1.5 min, 85% B; 5.5-10.5 min, 35% B; 10.6-12.5 min, 10% B; 13.5-19 min, 85% B. The flow rate was 0.15 ml/min from 0 to 5.5 min; 0.17 ml/min from 6.9 to 10.5 min; 0.3 ml/min from 10.6 to 17.9 min; and 0.15 ml/min from 18 to 19 min. 2-hydrazinoquinoline (HQ) derivatives were measured using a reversed phase LC method, which employed an Acclaim RSLC 120 C8 reversed phase column (150 × 2.1 mm i.d., 2.2 μm; Dionex) with mobile phase A: water with 0.05 % formic acid, and mobile phase B: acetonitrile. Linear gradient: 0 min, 2% B; 3 min, 2% B; 8 min, 85% B; 9.5-14 min, 98% B; and 14.5-19 min, 2% B. Flow rate: 0.2 ml/min. Column temperature: 25 °C.
Mass Spectrometry.
The Q Exactive Plus mass spectrometer is equipped with a HESI probe, and the relevant parameters are as listed: heater temperature, 120 °C; sheath gas, 30; auxiliary gas, 10; sweep gas, 3; spray voltage, 3.6 kV for positive mode and 2.5 kV for negative mode. Capillary temperature was set at 320 °C, and S-lens was 55. When coupled with the HILIC method, a full scan range was set at 70 to 900 (m/z) with positive/negative switching except from 6.6 to 12.5 min, 100 to 1000 (m/z) in negative mode, and the resolution was set at 70 000 (at m/z 200). When coupled with reversed phase LC method, a full scan range was set at 170 to 800 (m/z) in positive mode, and the resolution was set at 140 000 (at m/z 200). The maximum injection time (max IT) was 200 ms at resolution of 70 000 and 450 ms at resolution of 140 000. High resolution mass spectrometry allowed for spectral resolution of isotopologues, such as [13C2,18O2]-Ac-carnitine from [13C4]-Ac-carnitine. Automated gain control (AGC) was targeted at 3 × 106 ions in full scan mode. For targeted MS2 analysis, the isolation width of the precursor ion was set at 1.0 (m/z), high energy collision dissociation (HCD) was 35%, and max IT is 100 ms. The resolution and AGC were 35 000 and 200 000, respectively.
Metabolite extraction from cultured cells.
For intracellular metabolite measurements, at different time points, the medium was removed, cells were briefly washed twice with 1 ml ice cold saline solution (0.9 % NaCl in water) before placed on dry ice, followed by the addition of 1 ml 80 % methanol/water (pre-cooled in −80 °C freezer) to each well. After incubation in −80 °C freezer for 15 min, cell s were scraped into 80 % methanol on dry ice and then transferred to Eppendorf tube. Samples were centrifuged at 20 000 g for 10 min at 4 °C, and the supernatant was split i nto two Eppendorf tubes before drying in speed vacuum concentrator (Labcono). For absolute quantitation of GSH, CoA, Ac-CoA and Ac-carnitine, 13C labeled E. coli metabolite extract (Liu et al., 2015) or Ac-carnitine standard was added to the cell metabolite extract before drying. The dry pellets were reconstituted into 30 μl sample solvent (water:methanol:acetonitrile, 2:1:1, v/v) and 3 μl was injected into the LC-HRMS. The sample sequence of data acquisition was randomized.
Metabolite extraction from tissue.
Briefly, the tumor sample was first homogenized in liquid nitrogen and then 15 to 25 mg was weighed in a 2 ml vial pre-filled with Zirconia/Silica beads (1.0mm diameter). Extraction solvent 80% methanol/water (pre-cooled in −80 °C freezer) was added to each sample at the ratio of 100 μl/15mg on dry ice, and a Beadbeater (Biospec) was used to facilitate the metabolite extraction at a shaking speed of 3400 rpm for 1 min at room temperature. The metabolite extract was then immediately centrifuged at 20 000 g at 4 μC fo r 10 min. The supernatant of the tumor extract collected from sarcoma bearing mice which received 13C-glucose infusion were used for polar metabolite measurements. For absolute quantitation of GSH and CoA, 13C labeled E.coli metabolite extract (Liu et al., 2015) was added to the supernatant of tumor extract collected from sarcoma bearing mice without 13C-glucose infusion and 3 μl was directly injected to LC-HRMS.
2-hydrazinoquinoline derivatization.
To measure acetate, pyruvate, lactate and acetaldehyde, we developed a method adapted from a previous publication (Lu et al., 2013). 20 mM of 2-hydrazinoquinoline (HQ), triphenylphosphine, and 2,2′-dipyridyl disulfide (DPDS) were freshly prepared in acetonitrile solution, except that for DPDS, methanol (final concentration, 2.5 %) was added to facilitate dissolution. The HQ derivatization reagent cocktail in acetonitrile (0.5 mM of HQ, Triphenylphosphine, and DPDS) was then prepared from a 20 mM stock solution. To make HQ derivatives, without metabolite extraction, 2 μl of cell culture medium or mouse serum were directly added to a 1.5 ml Eppendorf tube containing 200 μl of 0.5 mM HQ derivatization reagent cocktail in acetonitrile. Without drying, 2 μl intracellular metabolite extract in 80 % methanol was used for HQ derivatization. 3,3,3-[2H3]-pyruvate, 2,2,2-[2H3]-acetate, and 3,3,3-[2H3]-lactate were used as internal standards to enable absolute quantification. After vigorous vortexing, the samples were incubated in water bath at 37 °C for 70 min. After derivatization, the samples were then centrifuged at 4 °C for 10 min at the speed of 20 000 g. The supernatant was then added formic acid (final concentration, 1%), transferred to a new LC vial, and 2 μl was injected for LC-HRMS analysis. It is worth noting that at high concentrations, the addition of 3,3,3-[2H3]-pyruvate or 3,3,3-[2H3]-lactate could suppress the MS signals of [13C3]-pyruvate or [13C3]-lactate after HQ derivatization, under which situation, underivatized samples were analyzed using HILIC LC-HRMS method as described previously. To check for potential artifacts, 2 μl 100 μM [2H3]-acetyl-CoA or acetyl-carnitine was incubated with the HQ derivatization reagents (Figure S1C). In addition, LC-MS grade water, 80% MeOH, and cell free medium were always included to assess the baseline level of acetate, pyruvate, lactate and acetaldehyde. For acetate and acetaldehyde measurements, the evaporation process was avoided due to the low boiling point of acetaldehyde. [2H3]-acetyl-CoA was a generous gift from Dr. Hening Lin’s lab, Cornell University, NY (Sadhukhan et al., 2016).
18O2 tracing assay.
Cells were seeded into a 35 mm dish at a density of 80 000 (for 48 or 60 hrs tracing) or 150 000 (for 15 or 24 hrs tracing) of SKOV3 or 120 000 (for 48 or 60 hrs tracing) or 300 000 (for 15 or 24 hrs tracing) of HCT116 in growth medium (RPMI 1640 supplemented with 10 % FBS). Immediately before the 18O2 tracing assay, the medium was replaced with 2 ml RPMI 1640 (without NaHCO3), containing 11.1 mM [13C6]-glucose, 20 mM HEPES and 10 % dialyzed FBS (DFBS). For drug treatments, TTM (final: 25 μM) or UK5099 (final: 5 μM) was added to the medium. Cells in 35 mm dishes were then transferred to a 500 ml container (desiccator) containing 1 ml sterile water to maintain the humidity inside the container. The container was subjected to vacuum for 15 seconds and then immediately filled with 100 ml 18O2 and 400 ml N2. The container was then sealed with a 3-way valve and large paper clips were also employed to ensure air tightness. Balloons were employed for gas transfer and to balance the gas pressure in the container. The sealed container was then placed in a non-CO2 incubator set at 37 °C. Here, HEPES-buffered medium was used to replace CO2/HCO3− buffer system to prevent the medium pH fluctuation during the 15- sec vacuum treatment. The unlabeled O2 may come from the air trapped in the connecting system when we deliver gases into the container in which 18O2 tracing was conducted. In the end of treatment (15, 24, 48, or 60 hrs), the spent media were collected and immediately placed on dry ice. Intracellular metabolites were harvested as described previously.
Co-culture assay.
Transwell® cell culture inserts (Corning, Polycarbonate, pore size: 0.4 μm) were used to set up the co-culture system. Briefly, MEF (ACLY WT and KO) cells were seed into 6 well plate at the density of 25 000/well, and HCT116 cells were seeded into the corresponding insert at the density of 50 000/insert. After overnight incubation in growth medium, the medium was replaced with 2.5 ml RPMI 1640 supplemented with 10 % DFBS. The co-culture plates were then placed back to CO2 incubator. After 30 or 72 hrs, cell number was counted using (Moxi Z Mini Automated Cell Counter, ORFLO Technologies). Media were collected for acetate measurements, and intracellular metabolites were collected for palmitate measurements.
Purified keto acid dehydrogenase assay.
Pyruvate dehydrogenase (PDH) from porcine heart (Sigma, P7032-10UN) and alpha-ketoglutarate dehydrogenase (aKGDH) from porcine heart (Sigma, K1502-20UN) were supplied as a 50% glycerol solution containing ~9 mg/mL bovine serum albumin, 30% sucrose, 1.5 mM EDTA, 1.5 mM EGTA, 1.5 mM 2-mercaptoethanol, 0.3 TRITON® X-100, 0.003% sodium azide, and 15 mM potassium phosphate, pH 6.8. PDH or aKGDH solution was diluted to 1 unit/ml with buffer containing 20 mM sodium phosphate and 1 mM MgCl2, pH7.2. A mixture of thiamine pyrophosphate (TPP, 50 μM), NAD+ (5 mM), glutathione (GSH, 40 mM) and Coenzyme A (CoA, 0.1, 1 or 4 mM) was freshly prepared in the same buffer and diluted by 10 times before use, so that the final concentrations were 5 μM (TPP), 0.5 mM (NAD+), 4 mM (GSH), 0.01, 0.1 or 0.4 mM (CoA). Reaction buffer in Eppendorf tube (without [13C3]-pyruvate) was incubated in 37 °C water bath for 1 min, and the reaction was initiated by adding [13C3]-pyruvate (final concentration: 200 μM) or the mixture of [13C3]-pyruvate and alpha ketoglutarate (final concentration: 200 μM). For acetate, acetaldehyde, and pyruvate measurements, at 0, 2, 5, 10, 20 and 30 min, a 2 μl reaction mixture was collected and immediately added to the 200 μl HQ solution in acetonitrile. HQ derivatization was performed as described previously. At 30 min, the reaction was quenched by adding 4 volumes of ice cold methanol. Acetyl-CoA was also added as an internal standard to quantify [13C2]-acetyl-CoA generated from PDH reaction. After vigorous vortexing, the mixture was centrifuged at 20 000 rcf for 10 min at 4 °C. The supernatant was directly used for acetyl-CoA and acetyl-GSH analysis by LC-HRMS.
Generation of mouse sarcoma cell lines and CRISPR/Cas9 PDH knockout cell lines.
Mouse primary sarcoma cell lines were generated from Pax7CreER-T2; p53fl/fl; LSL-NrasG12D tumors as described previously (Zhang et al., 2015). Briefly, tumor tissue was excised and digested with 5 mg/mL Collagenase Type I at 37 °C for 45 mins. Red blood cells were lysed and cell pellets washed with PBS before plating. Cells were passaged 4-5 times to deplete stromal cells. PDH knockout cell lines were generated using lentiviral transduction of primary cells with the lentiCRISPR v2 backbone, which contains mammalian Cas9, a sgRNA for PDH (5’-catgccatagcggttgttct-3’), and a puromycin resistance marker. LentiCRISPR v2 was a gift from Feng Zhang (Addgene plasmid # 52961) (Sanjana et al., 2014). 48 hours after transduction, cells were treated with 5 ug/uL puromycin to select for infected cells. Once selected, cells were diluted and plated to sort single-cell colonies. Single-cell outgrowth was confirmed by sequencing and TIDE analysis (Brinkman et al., 2014). Loss of PDH activity was confirmed by culturing PDH WT and KO cells in RPMI medium containing [13C6]-glucose for 6 hrs. 13C metabolite analysis was performed on LC-HRMS as described above. All LC-HRMS analysis was conducted in a blinded fashion.
Nuclear Magnetic Resonance kinetics assay.
An NMR tube containing 0.6 ml of D2O was pre-warmed at 37 °C and to check the background , 6 μl of freshly prepared pyruvate stock solution (50 mM or 100 mM in D2O) was added. The NMR tube was then equilibrated to 37 °C before data acquisition on 50 0 MHz Varian Inova spectrometer. After the first 8 scans, 6 μl of H2O2 stock (50, 100, or 200 mM in D2O) was added to NMR tube to initiate the reaction. A further equilibration of the reaction mixture in the NMR tube to 37 °C was required before data acquisit ion. To check the transition metal effects on this reaction, CuSO4 (final 200 μM) was added to NMR tube before the addition of H2O2. After temperature equilibration, data was then collected continuously for 24 data points (8 scans per data point, roughly 48 seconds per data point). However, it is worth mentioning that due to instrument limitations, the first data point collected had a delay from the real reaction time, and we performed the reaction at 37 °C to mimic the physiological temperature.
Data analysis.
LC-MS peak extraction and integration were performed using commercial available software Sieve 2.0 (ThermoFisher Scientific). The integrated peak area was used to calculate the fold changes between different treatments and 13C or 18O enrichment. The fraction of 18O labeled acetate was corrected for the 18O natural abundance and for the 18O2 enrichment in the container where the 18O2 tracing was conducted. All data are represented as mean ± SD. All p values were obtained from student’s t-test two-tailed using GraphPad Prism 6 unless otherwise noted. NMR data was analyzed using the software MestReNova Lite SE (Mestrelab Research), following manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise noted, all values were reported as mean ± SD with n = 3 (for cell culture assays), or n = 5 (for mouse assays) independent measurements and statistical tests resulting in p value computations were computed using a Student’s t test two tailed. All statistics were computed using Microsoft Excel 2016 or GraphPad Prism 6 (GraphPad, http://www.graphpad.com/scientific-software/prism/).
DATA AND SOFTWARE AVAILABILITY
Mass spectrometry-based metabolomics and proteomics data and NMR-based data have been deposited to Mendeley Data and are available at http://dx.doi.org/10.17632/39rzr8xzsv.1
Supplementary Material
Figure S1. Evaluation of the effects of glutamine and pyruvate carrier inhibition on acetate production and the levels of TCA metabolites, Related to Figure 1
(A) [13C6]-glucose enrichment pattern of metabolites produced by HCT116 cells for 70 mins (acetate) or 40 mins (pyruvate and Ac-CoA). (B) Same as in (A) but for [13C5]-glutamine (C) Extracted ion chromatogram of acetate or the [2H3]-acetate HQ derivative from samples prepared by incubating acetyl-carnitine, [2H3]-acetyl-CoA or [2H3]-acetate (100 μM) with HQ at 37 °C for 70 mins. (D) 13C enrichment patterns of metabolites derived from [13C6]-glucose. Red solid circle denotes 13C. Ac: Acetate; Ac-carnitine: Acetyl-carnitine; Pyr: Pyruvate. The relative levels of 13C enriched Ac-CoA (E), Ac-carnitine (F), citrate (G), and acetate (H) in HCT116 cells cultured in RPMI 1640 medium containing [13C6]-glucose with or without UK5099 treatment for 40 min. Values are expressed as mean ± SD of n=3 independent measurements. NS: p>0.05, *** p<0.001 in Student’s t test.
Figure S2. Exogenous H2O2 catalyzes pyruvate decarboxylation to acetate, Related to Figure 2
(A) Representative NMR spectra for pyruvate and acetate peaks from time 0 to 18.7 min. Time 0 min is when data acquisition began, and there is a delay from the actual reaction time due to temperature equilibration in the NMR tube. (B) Conversion rate of pyruvate (Pyr) to acetate under different conditions from 0 to 18.7 mins from the inception of the data acquisition. Conversion rate was calculated by dividing acetate peak area by the sum of acetate and pyruvate peak area. (C) Summary of reaction conditions, conversion rate (at 18.7 min), and reaction constant (mean ± SD).
Figure S3. Keto-acid dehydrogenases catalyze acetate and acetaldehyde production from pyruvate, Related to Figure 3
(A) Release of acetate from pyruvate in the presence of pyruvate dehydrogenase (PDH) supplemented with thiamine pyrophosphate (TPP). (B) Pyruvate consumption rate (blue), relative to that in the presence of TPP, NAD+ and CoA, representing relative activity of PDH; Acetate (yellow) and acetaldehyde (grey) production, relative to total pyruvate consumption.
Figure S4. Metabolites from HCT116 cells subjected to alterations in mitochondrial metabolism, Related to Figure 4
(A) Extracted ion chromatogram and tandem mass spectrum (positive ion mode) of [13C2]-Ac-GSH in HCT116 cells cultured in [13C6]-glucose medium for 40 min. (B) The decrease of acetaldehyde in cell free PBS buffer or RPMI medium in cell culture plates at 37 °C. (C) 13C enrichment of citrate in mouse sarcoma (PDH WT and KO) cells cultured in 13C glucose for 6 hrs. (D) The effect of thiamine depletion on intracellular metabolite levels and cell proliferation. (E) The effect of thiamine depletion on the formation of [13C2, 18O1]-Ac, [18O1]-methionine sulfoxide in HCT116 cells cultured in the presence of 18O2 for 48 hrs. (F) The contribution of ROS to acetate production with increasing doses of exogenous H2O2 (10 mins) in HCT116 treated with thiamine starvation. (G) Relative levels of 13C enriched Ac, ACE and Ac-GSH in HCT116 cells in the absence or presence of CPI-613, a lipoate analog. For thiamine depletion, HCT116 cells were cultured in thiamine free medium for 4 days before 18O2 or [18O2]-H2O2 treatment. Values are expressed as mean ± SD of n=3 independent measurements. ** p<0.01 in Student’s t test.
Figure S5. The effect of exogenous, endogenous ROS or catalase on lipogenesis and amino acid oxidation in HCT116 cells, Related to Figure 6
(A-B) The relative levels of 13C labeled fatty acid. HCT116 cells were first thiamine starved for 4 days, and then the old media were replaced with fresh media containing 100% [13C6]-glucose with or without catalase (600 U/ml). After incubation for 1 hr, increasing doses of H2O2 were added, and 1 hr after H2O2 addition, free fatty acids were extracted from HCT116 cells. (C) The relative levels of methionine oxidation in HCT116 cells cultured in the presence of [18O2]-H2O2 (200 μM) (left) with or without 1 mM [2H3]-pyruvate for 1 hr or 18O2 (right) with or without 5 mM [2H3]-pyruvate for 24 hrs. Values are expressed as mean ± SD of n=3 independent measurements. ** p<0.01 in Student’s t test.
Highlights.
Glucose-derived pyruvate generates acetate in mammals
Two reaction mechanisms leading acetate production are defined
Both mechanisms are coupled to mitochondrial function
Endogenous acetate can be required to maintain acetyl-CoA pools and lipogenesis
ACKNOWLEDGEMENTS
We acknowledge support from National Institutes of Health (awards R01CA193256, R00 CA16899 to JWL, T32 CA093240 to DEC, R35 CA197616 to DGK, R01CA174761 to KEW, R01GM110174 to BAG, T32CA009140 to PJL and 1F99CA222741 to SZ), the American Cancer Society (TBE434120 to JWL and TBE130927 to MAR), FAPESP 2017/15835-1 to ML, and the King Abdullah International Medical Research Center under the Ministry of National Guard Health graduate fellowship to AAC. We thank Dr. Anthony Ribeiro (NMR facility at Duke University) for help with spectroscopy measurements and Dr. Clementina Mesaros (University of Pennsylvania) for helpful advice on the 18O2 tracing assay. We also thank members of the Locasale Lab for helpful discussions. JWL is especially grateful to Eyal Gottlieb and Zach Schug for discussions about their then unpublished work on the importance of acetate that conceptualized of this study.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests at this time.
REFERENCES
- Andrae U, Singh J, and Ziegler-Skylakakis K (1985). Pyruvate and related alpha-ketoacids protect mammalian cells in culture against hydrogen peroxide-induced cytotoxicity. Toxicol Lett 28, 93–98. [DOI] [PubMed] [Google Scholar]
- Antunes F, and Cadenas E (2001). Cellular titration of apoptosis with steady state concentrations of H(2)O(2): submicromolar levels of H(2)O(2) induce apoptosis through Fenton chemistry independent of the cellular thiol state. Free Radic Biol Med 30, 1008–1018. [DOI] [PubMed] [Google Scholar]
- Asmus C, Mozziconacci O, and Schoneich C (2015). Low-temperature NMR characterization of reaction of sodium pyruvate with hydrogen peroxide. J Phys Chem A 119, 966–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Samuel VT, Morton GJ, Obici S, Croniger CM, Shulman GI, Wasserman DH, McGuinness OP, and Consortium NIHMMPC (2010). Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech 3, 525–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmer ML, Ma EH, Bantug GR, Grahlert J, Pfister S, Glatter T, Jauch A, Dimeloe S, Slack E, Dehio P, et al. (2016). Memory CD8(+) T Cells Require Increased Concentrations of Acetate Induced by Stress for Optimal Function. Immunity 44, 1312–1324. [DOI] [PubMed] [Google Scholar]
- Bremer J (1966). Comparison of acylcarnitines and pyruvate as substrates for rat-liver mitochondria. Biochim Biophys Acta 116, 1–11. [DOI] [PubMed] [Google Scholar]
- Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, Cox JE, Cardon CM, Van Vranken JG, Dephoure N, et al. (2012). A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science 337, 96–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulusu V, Tumanov S, Michalopoulou E, van den Broek NJ, MacKay G, Nixon C, Dhayade S, Schug ZT, Vande Voorde J, Blyth K, et al. (2017). Acetate Recapturing by Nuclear Acetyl-CoA Synthetase 2 Prevents Loss of Histone Acetylation during Oxygen and Serum Limitation. Cell Rep 18, 647–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunton CA (1949). Oxidation of α-Diketones and α-Keto-Acids by Hydrogen Peroxide. Nature 163, 444.18115097 [Google Scholar]
- Chance B, Sies H, and Boveris A (1979). Hydroperoxide metabolism in mammalian organs. Physiol Rev 59, 527–605. [DOI] [PubMed] [Google Scholar]
- Chen WW, Freinkman E, Wang T, Birsoy K, and Sabatini DM (2016). Absolute Quantification of Matrix Metabolites Reveals the Dynamics of Mitochondrial Metabolism. Cell 166, 1324–1337 e1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comerford SA, Huang Z, Du X, Wang Y, Cai L, Witkiewicz AK, Walters H, Tantawy MN, Fu A, Manning HC, et al. (2014). Acetate dependence of tumors. Cell 159, 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Z, Shestov AA, Lai L, and Locasale JW (2016). A Flux Balance of Glucose Metabolism Clarifies the Requirements of the Warburg Effect. Biophysical journal 111, 1088–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divakaruni AS, Wiley SE, Rogers GW, Andreyev AY, Petrosyan S, Loviscach M, Wall EA, Yadava N, Heuck AP, Ferrick DA, et al. (2013). Thiazolidinediones are acute, specific inhibitors of the mitochondrial pyruvate carrier. Proceedings of the National Academy of Sciences of the United States of America 110, 5422–5427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto N, Takase S, Yasuhara M, and Takada A (1991). Acetaldehyde metabolism in different aldehyde dehydrogenase-2 genotypes. Alcohol Clin Exp Res 15, 141–144. [DOI] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey PA, Flournoy DS, Gruys K, and Yang YS (1989). Intermediates in reductive transacetylation catalyzed by pyruvate dehydrogenase complex. Ann N Y Acad Sci 573, 21–35. [DOI] [PubMed] [Google Scholar]
- Graham LD, Packman LC, and Perham RN (1989). Kinetics and specificity of reductive acylation of lipoyl domains from 2-oxo acid dehydrogenase multienzyme complexes. Biochemistry 28, 1574–1581. [DOI] [PubMed] [Google Scholar]
- Houstis N, Rosen ED, and Lander ES (2006). Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948. [DOI] [PubMed] [Google Scholar]
- Inoue A, and Fujimoto D (1969). Enzymatic deacetylation of histone. Biochem Biophys Res Commun 36, 146–150. [DOI] [PubMed] [Google Scholar]
- Isenberg-Grzeda E, Shen MJ, Alici Y, Wills J, Nelson C, and Breitbart W (2017). High rate of thiamine deficiency among inpatients with cancer referred for psychiatric consultation: results of a single site prevalence study. Psychooncology 26, 1384–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Lal-Nag M, White EA, Shen M, Chiang CY, Mitra AK, Zhang Y, Curtis M, Schryver EM, Bettis S, et al. (2015). Quantitative high throughput screening using a primary human three-dimensional organotypic culture predicts in vivo efficacy. Nat Commun 6, 6220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JG, Park SJ, Sinninghe Damste JS, Schouten S, Rijpstra WI, Jung MY, Kim SJ, Gwak JH, Hong H, Si OJ, et al. (2016). Hydrogen peroxide detoxification is a key mechanism for growth of ammonia-oxidizing archaea. Proceedings of the National Academy of Sciences of the United States of America 113, 7888–7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles SE, Jarrett IG, Filsell OH, and Ballard FJ (1974). Production and utilization of acetate in mammals. Biochem J 142, 401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JV, Carrer A, Shah S, Snyder NW, Wei S, Venneti S, Worth AJ, Yuan ZF, Lim HW, Liu S, et al. (2014). Akt-dependent metabolic reprogramming regulates tumor cell histone acetylation. Cell Metab 20, 306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, and Garcia BA (2012). Examining histone posttranslational modification patterns by high-resolution mass spectrometry. Methods Enzymol 512, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litzenburger UM, Opitz CA, Sahm F, Rauschenbach KJ, Trump S, Winter M, Ott M, Ochs K, Lutz C, Liu X, et al. (2014). Constitutive IDO expression in human cancer is sustained by an autocrine signaling loop involving IL-6, STAT3 and the AHR. Oncotarget 5, 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sadhukhan S, Sun S, Wagner GR, Hirschey MD, Qi L, Lin H, and Locasale JW (2015). High-Resolution Metabolomics with Acyl-CoA Profiling Reveals Widespread Remodeling in Response to Diet. Mol Cell Proteomics 14, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Yao D, and Chen C (2013). 2-Hydrazinoquinoline as a Derivatization Agent for LC-MS-Based Metabolomic Investigation of Diabetic Ketoacidosis. Metabolites 3, 993–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashimo T, Pichumani K, Vemireddy V, Hatanpaa KJ, Singh DK, Sirasanagandla S, Nannepaga S, Piccirillo SG, Kovacs Z, Foong C, et al. (2014). Acetate is a bioenergetic substrate for human glioblastoma and brain metastases. Cell 159, 1603–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser J, Schuster A, Pietzke M, Vande Voorde J, Athineos D, Oizel K, Burgos-Barragan G, Wit N, Dhayade S, Morton JP, et al. (2018). Increased formate overflow is a hallmark of oxidative cancer. Nat Commun 9, 1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meiser J, Tumanov S, Maddocks O, Labuschagne CF, Athineos D, Van Den Broek N, Mackay GM, Gottlieb E, Blyth K, Vousden K, et al. (2016). Serine one-carbon catabolism with formate overflow. Sci Adv 2, e1601273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mews P, Donahue G, Drake AM, Luczak V, Abel T, and Berger SL (2017). Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature 546, 381–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM (2014). Metabolic inflexibility: when mitochondrial indecision leads to metabolic gridlock. Cell 159, 1253–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP (2009). How mitochondria produce reactive oxygen species. Biochem J 417, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H, Ishida M, Tanahashi I, Takahashi T, Taji Y, Ikebuchi K, Furuya D, and Akechi T (2017). Subclinical thiamine deficiency in patients with abdominal cancer. Palliat Support Care, 1–3. [DOI] [PubMed] [Google Scholar]
- Perry RJ, Peng L, Barry NA, Cline GW, Zhang D, Cardone RL, Petersen KF, Kibbey RG, Goodman AL, and Shulman GI (2016). Acetate mediates a microbiome-brain-beta-cell axis to promote metabolic syndrome. Nature 534, 213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reaves ML, Young BD, Hosios AM, Xu YF, and Rabinowitz JD (2013). Pyrimidine homeostasis is accomplished by directed overflow metabolism. Nature 500, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ (2001). A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J Biol Chem 276, 38329–38336. [DOI] [PubMed] [Google Scholar]
- Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, et al. (2016). Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci U S A 113, 4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salahudeen AK, Clark EC, and Nath KA (1991). Hydrogen peroxide-induced renal injury. A protective role for pyruvate in vitro and in vivo. J Clin Invest 88, 1886–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Peck B, Jones DT, Zhang Q, Grosskurth S, Alam IS, Goodwin LM, Smethurst E, Mason S, Blyth K, et al. (2015). Acetyl-CoA synthetase 2 promotes acetate utilization and maintains cancer cell growth under metabolic stress. Cancer Cell 27, 57–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schug ZT, Vande Voorde J, and Gottlieb E (2016). The metabolic fate of acetate in cancer. Nat Rev Cancer. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sechi G, Batzu L, Agro L, and Fois C (2016). Cancer-related Wernicke-Korsakoff syndrome. Lancet Oncol 17, e221–e222. [DOI] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shestov AA, Liu X, Ser Z, Cluntun AA, Hung YP, Huang L, Kim D, Le A, Yellen G, Albeck JG, et al. (2014). Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steginsky CA, Gruys KJ, and Frey PA (1985). alpha-Ketoglutarate dehydrogenase complex of Escherichia coli. A hybrid complex containing pyruvate dehydrogenase subunits from pyruvate dehydrogenase complex. J Biol Chem 260, 13690–13693. [PubMed] [Google Scholar]
- Szatrowski TP, and Nathan CF (1991). Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 51, 794–798. [PubMed] [Google Scholar]
- Test ST, and Weiss SJ (1984). Quantitative and temporal characterization of the extracellular H2O2 pool generated by human neutrophils. J Biol Chem 259, 399–405. [PubMed] [Google Scholar]
- van der Reest J, Lilla S, Zheng L, Zanivan S, and Gottlieb E (2018). Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat Commun 9, 1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vysochan A, Sengupta A, Weljie AM, Alwine JC, and Yu Y (2017). ACSS2-mediated acetyl-CoA synthesis from acetate is necessary for human cytomegalovirus infection. Proc Natl Acad Sci U S A 114, E1528–E1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Perez E, Liu R, Yan LJ, Mallet RT, and Yang SH (2007). Pyruvate protects mitochondria from oxidative stress in human neuroblastoma SK-N-SH cells. Brain research 1132, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Guo YR, Liu K, Yin Z, Liu R, Xia Y, Tan L, Yang P, Lee JH, Li XJ, et al. (2017). KAT2A coupled with the alpha-KGDH complex acts as a histone H3 succinyltransferase. Nature 552, 273–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstain R, Savariar EN, Felsen CN, and Tsien RY (2014). In vivo targeting of hydrogen peroxide by activatable cell-penetrating peptides. J Am Chem Soc 136, 874–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterbourn CC (2008). Reconciling the chemistry and biology of reactive oxygen species. Nat Chem Biol 4, 278–286. [DOI] [PubMed] [Google Scholar]
- Yuan ZF, Lin S, Molden RC, Cao XJ, Bhanu NV, Wang X, Sidoli S, Liu S, and Garcia BA (2015). EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Mol Cell Proteomics 14, 1696–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Qiu Q, Li Z, Sachdeva M, Min H, Cardona DM, DeLaney TF, Han T, Ma Y, Luo L, et al. (2015). HIF-1 Alpha Regulates the Response of Primary Sarcomas to Radiation Therapy through a Cell Autonomous Mechanism. Radiat Res 183, 594–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Torres A, Henry RA, Trefely S, Wallace M, Lee JV, Carrer A, Sengupta A, Campbell SL, Kuo YM, et al. (2016). ATP-Citrate Lyase Controls a Glucose-to-Acetate Metabolic Switch. Cell Rep 17, 1037–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Evaluation of the effects of glutamine and pyruvate carrier inhibition on acetate production and the levels of TCA metabolites, Related to Figure 1
(A) [13C6]-glucose enrichment pattern of metabolites produced by HCT116 cells for 70 mins (acetate) or 40 mins (pyruvate and Ac-CoA). (B) Same as in (A) but for [13C5]-glutamine (C) Extracted ion chromatogram of acetate or the [2H3]-acetate HQ derivative from samples prepared by incubating acetyl-carnitine, [2H3]-acetyl-CoA or [2H3]-acetate (100 μM) with HQ at 37 °C for 70 mins. (D) 13C enrichment patterns of metabolites derived from [13C6]-glucose. Red solid circle denotes 13C. Ac: Acetate; Ac-carnitine: Acetyl-carnitine; Pyr: Pyruvate. The relative levels of 13C enriched Ac-CoA (E), Ac-carnitine (F), citrate (G), and acetate (H) in HCT116 cells cultured in RPMI 1640 medium containing [13C6]-glucose with or without UK5099 treatment for 40 min. Values are expressed as mean ± SD of n=3 independent measurements. NS: p>0.05, *** p<0.001 in Student’s t test.
Figure S2. Exogenous H2O2 catalyzes pyruvate decarboxylation to acetate, Related to Figure 2
(A) Representative NMR spectra for pyruvate and acetate peaks from time 0 to 18.7 min. Time 0 min is when data acquisition began, and there is a delay from the actual reaction time due to temperature equilibration in the NMR tube. (B) Conversion rate of pyruvate (Pyr) to acetate under different conditions from 0 to 18.7 mins from the inception of the data acquisition. Conversion rate was calculated by dividing acetate peak area by the sum of acetate and pyruvate peak area. (C) Summary of reaction conditions, conversion rate (at 18.7 min), and reaction constant (mean ± SD).
Figure S3. Keto-acid dehydrogenases catalyze acetate and acetaldehyde production from pyruvate, Related to Figure 3
(A) Release of acetate from pyruvate in the presence of pyruvate dehydrogenase (PDH) supplemented with thiamine pyrophosphate (TPP). (B) Pyruvate consumption rate (blue), relative to that in the presence of TPP, NAD+ and CoA, representing relative activity of PDH; Acetate (yellow) and acetaldehyde (grey) production, relative to total pyruvate consumption.
Figure S4. Metabolites from HCT116 cells subjected to alterations in mitochondrial metabolism, Related to Figure 4
(A) Extracted ion chromatogram and tandem mass spectrum (positive ion mode) of [13C2]-Ac-GSH in HCT116 cells cultured in [13C6]-glucose medium for 40 min. (B) The decrease of acetaldehyde in cell free PBS buffer or RPMI medium in cell culture plates at 37 °C. (C) 13C enrichment of citrate in mouse sarcoma (PDH WT and KO) cells cultured in 13C glucose for 6 hrs. (D) The effect of thiamine depletion on intracellular metabolite levels and cell proliferation. (E) The effect of thiamine depletion on the formation of [13C2, 18O1]-Ac, [18O1]-methionine sulfoxide in HCT116 cells cultured in the presence of 18O2 for 48 hrs. (F) The contribution of ROS to acetate production with increasing doses of exogenous H2O2 (10 mins) in HCT116 treated with thiamine starvation. (G) Relative levels of 13C enriched Ac, ACE and Ac-GSH in HCT116 cells in the absence or presence of CPI-613, a lipoate analog. For thiamine depletion, HCT116 cells were cultured in thiamine free medium for 4 days before 18O2 or [18O2]-H2O2 treatment. Values are expressed as mean ± SD of n=3 independent measurements. ** p<0.01 in Student’s t test.
Figure S5. The effect of exogenous, endogenous ROS or catalase on lipogenesis and amino acid oxidation in HCT116 cells, Related to Figure 6
(A-B) The relative levels of 13C labeled fatty acid. HCT116 cells were first thiamine starved for 4 days, and then the old media were replaced with fresh media containing 100% [13C6]-glucose with or without catalase (600 U/ml). After incubation for 1 hr, increasing doses of H2O2 were added, and 1 hr after H2O2 addition, free fatty acids were extracted from HCT116 cells. (C) The relative levels of methionine oxidation in HCT116 cells cultured in the presence of [18O2]-H2O2 (200 μM) (left) with or without 1 mM [2H3]-pyruvate for 1 hr or 18O2 (right) with or without 5 mM [2H3]-pyruvate for 24 hrs. Values are expressed as mean ± SD of n=3 independent measurements. ** p<0.01 in Student’s t test.