

Abstract
Toxic protein aggregates are key features of progressive neurodegenerative diseases. In addition to “seed” proteins diagnostic for each neuropathy (e.g., Aβ1–42 and tau in Alzheimer’s disease), aggregates contain numerous other proteins, many of which are common to aggregates from diverse diseases. We reported that CRAM-1, discovered in insoluble aggregates of C. elegans expressing Q40::YFP, blocks proteasomal degradation of ubiquitinated proteins and thus promotes aggregation. We now show that CRAM-1 contains three α-helical segments forming a UBA-like domain, structurally similar to those of mammalian adaptor proteins (e.g. RAD23, SQSTM1/p62) that shuttle ubiquitinated cargos to proteasomes or autophagosomes for degradation. Molecular modeling indicates that CRAM-1, through this UBA-like domain, can form tight complexes with mono- and di-ubiquitin and may thus prevent tagged proteins from interacting with adaptor/shuttle proteins required for degradation. A human ortholog of CRAM-1, SERF2 (also largely disordered), promotes aggregation in SH-SY5Y-APPSw human neuroblastoma cells, since SERF2 knockdown protects these cells from amyloid formation. Atomistic molecular-dynamic simulations predict spontaneous unfolding of SERF2, and computational large-scale protein-protein interactions predict its stable binding to ubiquitins. SERF2 is also predicted to bind to most proteins screened at random, although with lower average stability than to ubiquitins, suggesting roles in aggregation initiation and/or progression.
Introduction
Neurotoxic protein aggregation is a hallmark of neurodegenerative diseases including Alzheimer’s disease (AD), Huntington’s disease (HD), and Amyotrophic Lateral Sclerosis (ALS). Aggregates in these neuropathies contain, in addition to disease-specific proteins that are diagnostic for each condition, many common proteins found in insoluble aggregates from multiple progressive diseases and which may play roles in aggregate growth and/or toxicity1–4. Failure of protein homeostasis precedes other evidence of neurotoxicity5–7, suggesting that it may contribute causally by allowing persistence of misfolded proteins8,9. Inherently disordered proteins are predisposed to misfolding, and diverse post-translational modifications (PTMs) — including oxidation, phosphorylation, and acetylation10,11 — can introduce structural instability, exposing hydrophobic regions that may interact with those of other transiently or irreversibly denatured proteins8,11–14. Although numerous explanations have been offered for the failure of proteostasis in neurodegenerative diseases, the underlying mechanisms remain elusive.
The canonical pathway for ubiquitin-mediated targeting of misfolded proteins to proteasomes is as follows: E3 ubiquitin ligases recognize misfolded proteins and add an initial ubiquitin moiety. Extension of polyubiquitin chains marks proteins for degradation by proteasomes and/or autophagosomes. Shuttle proteins (e.g. RAD23A) bind polyubiquitin tags via a ubiquitin-binding (UBA) domain and escort their cargos to proteasomes15,16. Genetic disruption of RAD23A and/or RPN10, a regulatory/docking subunit of the 26S proteasome, leads to accumulation of ubiquitin-tagged proteins16,17.
Caenorhabditis elegans transgenic strains that simulate neurodegeneration-associated aggregation have proven invaluable in assessing mechanisms of age-dependent proteostasis failure1,5,18. For example, nematode strain AM141 expresses Q40::YFP (approximating the glutamine-array-length threshold for penetrance of Huntington’s disease) in body-wall muscle, leading to age-progressive protein aggregation and paralysis1,19. Strain CL4176 [myo-3p::Aβ1–42] expresses the human amyloid peptide Aβ1–42 in body-wall muscle, whereas CL2355 expresses Aβ1–42 in all neurons, and both show age-progressive behavioral disruptions. These nematode strains model human amyloidopathies, such as β-amyloid deposition in Alzheimer’s disease, and have provided insights into the composition of aggregates, gene activities that favor or oppose aggregation, and pharmacologic or genetic interventions that are protective1,20–22.
We recently reported that knockdown of Cytotoxicity-Related Aggregation Mediator-1 (CRAM-1) protects against aggregation in several C. elegans models1. In the present work, we extended our characterization of CRAM-1 and its human orthologs through both molecular-biology and computational approaches. We found that CRAM-1 contains a UBA-like domain, structurally similar to that of human RAD23A, which interacts with polyubiquitin and thus could compete with RAD23A for ubiquitin binding. We hypothesized that CRAM-1 and its orthologs, despite considerable sequence divergence, may still conserve key properties such as ubiquitin binding, allowing them to block degradation of ubiquitinated proteins. In support of this conjecture, knockdowns of CRAM-1 and SERF2, its closest human ortholog, appear to promote aggregation by very similar mechanisms.
Results
CRAM-1 has a UBA-like domain structurally similar to RAD-23
In previous work, we identified CRAM-1 as a minor component of insoluble aggregates from aged adults of C. elegans strain AM141 (expressing Q40::YFP in muscle). CRAM-1 is predominantly an unstructured or disordered protein, and preferentially interacts with polyubiquitins1. In this work, we further characterize structural features of CRAM-1. First, we modeled its C-terminal region (residues 54 to 96) by an ab-initio approach (Fig. 1a). Consistent with the full-length structure (Fig. 1b), which was also predicted by ab-initio methods, C-terminal modeling predicted CRAM-1 to have three alpha helices (α1, α2, and α3) of moderate stability, connected by loops. Similar structures are characteristic of UBA-like superfamily domains23,24, although full-length CRAM-1 lacks any sequence similarity to other UBA-domain proteins including RAD23, Sequestosome-1/p62, DSK2, and MUD1. UBA-domain proteins show little similarity at the sequence level except for moderately conserved hydrophobic residues, but instead display structural similarity with respect to the three-helix bundle25. We assessed structural congruity of the predicted C-terminal region of CRAM-1 with well-established UBA-domain proteins including MUD1 (yeast), RAD23A (human) and SQSTM1/p62 (human) using the Swiss-PDB viewer26 and the “multiseq” plugin for VMD27. The C-terminal region of CRAM-1 aligns quite closely with the UBA domain (UBA2) of human RAD23A28, indicating high structural homology (the two ribbon models are superimposed in Fig. 1c; RMSD ≈ 1.14 Å), but deviates substantially more from the SQSTM1/p62 UBA domain (not shown; RMSD ≈ 3.03 Å).
Figure 1.

Predicted 3-dimensional structure of CRAM-1 indicates a UBA-like domain. Structures of the CRAM-1 C-terminal region (a) and the full-length model (b) show the same bundle of three helices connected by loops. (c) Structural comparison of the CRAM-1 C-terminal region (red) with the UBA domain of human RAD23A (green). Superimposed structures illustrate structural agreement, with RMSD (root-mean-square deviation) = 1.14 Å. (d) C-terminal region of CRAM-1 (red) superimposed on the UBA domain of ceRAD-23 (green), RMSD = 1.02 Å. (e) Sequence alignment, based on superimposition of 3-dimensional structures, showing 3 α helices with conserved hydrophobic residues (highlighted in gold) and nearby exposed hydrophobic residues (boxes).
Since CRAM-1 is a C. elegans protein, we modeled the UBA domain of C. elegans RAD-23 (hereafter denoted as ceRAD-23) and compared it to CRAM-1. The CRAM-1 UBA-like domain showed even closer structural resemblance to ceRAD-23 (ribbon models are superimposed in Fig. 1d; RMSD ≈ 1.02 Å) than to human RAD23A. The interactions of UBA-domain proteins with ubiquitins are largely mediated by hydrophobic interactions, involving conserved core residues (chiefly in UBA alpha-helices) supplemented by additional, unstructured UBA-domain residues, interacting with conserved hydrophobic residues in ubiquitin24,28. We examined the CRAM-1 UBA-like (C-terminal) region for hydrophobic residues conserved with known UBA proteins. CRAM-1 possesses many of the hydrophobic residues conserved among known UBA-domain proteins including the 3-helix bundles of DSK2, SWA2p, DDI1, MUD1 and RAD2324, which are considered crucial for ubiquitin interactions (Fig. 1e, highlighted in gold), and a few additional exposed hydrophobic residues nearby (Fig. 1e, boxes). Together, these findings support the hypothesis of structural conservation between the CRAM-1 UBA-like domain and previously identified UBA domains, including those of human and nematode RAD23.
Interaction of CRAM-1 with mono- and oligo-ubiquitins
Proteasome-trafficking adaptor proteins, including RAD23A, DSK2, and MUD1, bind ubiquitin chains via UBA domains29. We predicted stable interaction of the CRAM-1 UBA-like domain with ubiquitins1; we now estimate its docking affinity for mono-, di-, and tetra-ubiquitin (Ub1, Ub2, and Ub4). As positive controls for ubiquitin binding, we modeled docking for MUD1 and RAD23A, previously reported UBA-domain proteins24,28,30. Docking simulations predict stable interaction of MUD1 and RAD23A with Ub1 and Ub2 similar to those determined by NMR chemical-shift assays24,28,30, supporting the validity of our modeling conditions (Supplementary Fig. 1a,b).
Using the same parameters, we calculated docking orientations and energies for the CRAM-1 UBA-like domain and ceRAD-23 UBA, each interacting separately with Ub1, Ub2 and Ub4. The results (Fig. 2) indicate that interactions of the CRAM-1 UBA-like domain with mono-, di- and tetra-ubiquitins are quite similar to MUD1-ubiquitin interactions (Supplementary Fig. 1). These docking results predict that CRAM-1 and ceRAD-23 can bind to the same region, largely via the same residues in ubiquitin (Ub1, Ub2; Fig. 2c–e). It is noteworthy that human RAD23A is predicted to interface with a ubiquitin surface (Supplementary Fig. 1) that is supported by NMR chemical-shift data28,30 but differs from the ubiquitin aspects predicted to be bound by C. elegans RAD-23, which instead coincide with the interface of yeast MUD1 binding to ubiquitin. For this reason, in subsequent studies we considered only comparisons between proteins that co-evolved — C. elegans CRAM-1 with ceRAD-23, and human SERF2 with human RAD23A.
Figure 2.

CRAM-1 and RAD-23 have similar interactions with ubiquitins. (a) The CRAM-1 UBA-like domain (red) interacts with Ub1 (mono-ubiquitin, gray); interacting amino acids of ubiquitin are labeled. (b) The full-length structure of CRAM-1 (red) binds the same region of ubiquitin, contacting most of the same ubiquitin residues that were predicted to interact with the CRAM-1 C-terminus alone. (c) Structural superimposition of ceRAD-23 (green) and CRAM-1 (red), showing their predicted interactions with mono-ubiquitin via the same ubiquitin aspects. (d,e) Binding of CRAM-1 (d, red) or ceRAD-23 (e, green) ribbon models to di-ubiquitin (space-filling model) is facilitated by hydrophobic interactions (brown; see scale at left). (f) Predicted interaction energies (ΔEinteraction) for CRAM-1 (UBA-like) and 3 other UBA proteins, each with mono-, di-, and tetra-ubiquitin. (g,h) Simulated structures of complexes between the CRAM-1 UBA domain (cyan) and either mono-ubiquitin (light green; g) or di-ubiquitin (pink and light green; h) appear stable over a 200-ns simulation. (i,j) Western blot analyses for CRAM-1 interaction to ubiquitin: lysates from wild-type N2 worms at days 1 and 5 were immuno-precipitated with biotinylated CRAM-1 antibody (i) or antibody to ubiquitin (j), resolved in polyacrylamide gradient gel lanes (10% w/v, BioRad), electroblotted to nylon membranes, and probed with antibody to CRAM-1 (i) or ubiquitin (j), followed by peroxidase-tagged antibody to IgG and chemiluminescence imaging (Western ECL kit, Bio-Rad).
RAD23A and MUD1 belong to a subset of UBA-domain proteins that recognize K48-linked polyubiquitin chains, and preferentially target their cargos to proteasomes24,31. The ubiquitin structures we employed were reported in NMR and X-ray-crystallographic studies of K48-linked di- and tetra-ubiquitins, respectively32,33. Empirically, UBA:ubiquitin binding affinity increases with ubiquitin chain length24, so we asked whether this trend is predicted via computational docking and whether it extends to CRAM-1. UBA or UBA-like domains of MUD1, ceRAD-23, RAD23A, and CRAM-1 were docked to Ub1, Ub2 (K48-linked open conformer), or Ub4 (K48-linked). The predicted free-energy drop on ubiquitin docking (ΔEinteraction) agrees with published observations, indicating comparable gains in affinity as ubiquitin chain length increases, for established UBA domains (MUD1, ceRAD-23, and RAD23A), and for the CRAM-1 UBA-like domain (Fig. 2f). Full-length CRAM-1 is predicted to have even greater binding affinity for each ubiquitin target (−180 to −235 kcal/mol; Supplementary Fig. 1c), than the isolated UBA-like domain alone (−140 to −210 kcal/mol).
We then analyzed the stability of CRAM-1 binding to Ub1 or Ub2 by atomistic molecular-dynamic simulation. The structural integrity (stability) of complexes, comprising the CRAM-1 UBA-like domain bound to mono- or di-ubiquitin, was assessed in 200-ns simulations. Both complexes, CRAM-1/mono-ubiquitin (Fig. 2g) and CRAM-1/di-ubiquitin (Fig. 2h), remained stable throughout the simulations.
To test our computational predictions that CRAM-1 could bind to oligo-ubiquitins, we assessed interactions between CRAM-1 and ubiquitin in wild-type (Bristol-N2) adults at two ages, day 1 (pre-gravid) and day 5 (post-gravid) of adulthood. First, ubiquitinated and ubiquitin-interacting proteins were isolated from lysates of wild-type (Bristol-N2) worms by immuno-pulldown (IP) on magnetic beads coated with antibody to ubiquitin. Bound proteins were eluted, concentrated, electrophoresed, and electroblotted onto nylon membranes, which were then probed with antibody to CRAM-1 (Fig. 2i). Prominent, discrete proteins are seen at SDS-gel mobilities (marked by double arrows) corresponding to native CRAM-1 (band i, ~10.8 kDa), mono-ubiquitinated CRAM-1 (ii, ~19.3 kDa), and tetra-ubiquitinated CRAM-1 (iii, ~45 kDa). Of these bands, only mono-ubiquitinated CRAM-1 (ii) appeared more abundant in older worms.
In parallel, lysates from the same wild-type worms were used to recover CRAM-1 and associated proteins, by IP with biotin-tagged antibody against CRAM-1 and capture on streptavidin-coated magnetic beads. IP-recovered proteins were rinsed, electrophoresed, blotted, and probed with antibody to ubiquitin. CRAM-1 appears to bind a wide variety of ubiquitinated proteins that increase in abundance with age (e.g., double arrows in Fig. 2j) in agreement with previous reports of age-associated aggregation in wild-type C. elegans1,34 and our observation of CRAM-1 in insoluble protein aggregates from aged worms1. Ubiquitinated CRAM-1 must make up a rather small fraction of this signal, since only relatively minor bands are seen at mobilities observed in the preceding IP blot (Fig. 2i).
CRAM-1 could compete with ceRAD-23 for binding to polyubiquitin, impairing substrate delivery to C. elegans proteasomes
In previous studies, we found that cram-1 knockdown (KD) by RNA interference (RNAi) protects C. elegans against aggregation and its toxicity, utilizing transgenic strains that form aggregates modeling diverse neurodegenerative diseases1. If CRAM-1 functions as a UBA-like shuttle protein, then its knockdown should have been detrimental to the worms. Instead, CRAM-1 RNAi was strikingly protective in each model, and reduced aggregate burden in wild-type worms as well as in neuropathy models1. In each protein-aggregation model, integrity of the ubiquitin-proteasome system (UPS) was essential for cram-1 KD-mediated protection1.
We now predict by computational modeling, and corroborate by immuno-pulldowns, that the CRAM-1 UBA-like domain could interact with ubiquitin and its oligomers, analogous to ubiquitin binding by the ceRAD-23 UBA. These combined results led us to hypothesize that, due to structural similarity and conservation of contact residues, CRAM-1 could act as a decoy mimic that competes with ceRAD-23 for oligo-ubiquitin binding, but lacks a second recognition domain to convey its cargo to proteasomes or autophagosomes. To test this prediction in vivo, we knocked down the expression of ceRAD-23 in AM141 worms expressing Q40::YFP in muscle. As in previous experiments1, worms were transferred to RNAi plates at 48 h post-hatch, and imaged on day 5 post-hatch (d5PH), for quantitation of aggregates. Although rad-23 knockdown alone had no effect on aggregate counts per worm, relative to controls, cram-1 knockdown reduced both the number and intensity of aggregates by 23‒25% (Fig. 3a,b; t-test P < 3E-05) as reported previously1. All protection conferred by cram-1 KD was negated, however, by concurrent knockdown of rad-23 (using a 1:1 mixture of RNAi constructs, t-test P < E-4).
Figure 3.

Rescue by cram-1 knockdown, of both Q40::YFP and Aβ1–42 amyloid aggregation, requires RAD23. (a,b) Protection from Q40::YFP aggregation, conferred by cram-1 knockdown, is blocked by rad-23 RNAi. Dual knockdowns were conducted as described. Aggregate numbers per worm, ± SEM, were counted at day 5 post-hatch (D5PH) for 10–16 worms per group. Significance (2-tailed t-tests): ***P < 3E–5 comparing [FV + FV] to [FV + cram-1KD]; ****P < 10–4 comparing [FV + cram-1KD] to [rad-23KD + cram-1KD]. (c) CL4176 worms were treated, beginning 36–40 h after egg isolation, with dual RNAi in 3 experiments. Cram-1 RNAi reduced paralysis relative to [FV + FV] controls, but not when cram-1KD was paired with rad23KD. Shaded bars show fraction paralyzed, normalized to FV, for 3 independent experiments; open bars summarize combined data ± SEM. Significance by chi2 test within each experiment, comparing [cram-1KD + FV] to [cram-1KD + rad23KD], for 50–100 worms/group: *P < 0.05; **P < 0.01; ***P = 0.001. Treating each experiment as one point per group, ****P < 0.006 by 2-tailed paired t-test (white/open bars).
We next assessed paralysis in CL4176 [myo-3p::Aβ1–42], a model of β-amyloidosis1,21, using a similar dual-RNAi experimental design (Fig. 3c). A synchronous cohort of worms expressing Aβ1–42 in muscle was fed dsRNA that targets rad-23, cram-1, or both, starting 40 h after recovery of embryos. Worms were induced to express Aβ1–42 (by upshift to 25 °C at 48 h post-lysis), and paralysis was scored 48–52 h later. Data from 3 such experiments are presented in Fig. 3c. In all experiments, knockdown of cram-1 was highly protective against paralysis, decreasing its incidence 2- to 3-fold (P < 0.001), whereas concurrent rad-23 KD fully reversed that protection (P < 0.006 for reversal).
These experiments indicate that cram-1 KD requires rad-23 activity to rescue phenotypes resulting from aggregation of either Q40 (mimicking Huntington’s disease) or Aβ1–42 amyloid (observed in Alzheimer’s). CRAM-1 could perhaps compete with ceRAD-23 for binding to ubiquitin-tagged proteins, thereby blocking the normal role of ceRAD-23, i. e. delivery of misfolded proteins to 26S proteasomes for degradation. In principle, CRAM-1 protein might instead interact directly with ceRAD-23, with pro-aggregative effects mediated by that interaction. Our evidence does not support this alternative, however, since docking simulations do not predict a stable CRAM-1:ceRAD-23 interaction, and in vivo cross-linking studies revealed no linked peptides that indicate contact between these two proteins.
Aggregate reduction via cram-1 knockdown impedes ATG-7 inhibition
We next investigated the role of autophagy in cram-1 KD-mediated protection from aggregation. Key steps in autophagy (summarized in Fig. 4a) — nucleation (via bec-1), and protein conjugation and vesicle elongation (via atg-7 and lgg-3) — were disrupted by RNAi KD, ±RNAi to cram-1. Synchronized AM141 worms were transferred from bacteria harboring empty feeding vector (FV) to dual-RNAi combinations as indicated, beginning at the L3/L4 transition to minimize RNAi disruption of development1. Q40::YFP aggregate numbers per worm were counted at day-5 post-hatch, for ≥12 worms per group. KD of atg-7 (encoding an E1-like ubiquitin-activating enzyme involved in autophagosome targeting) blocked the anti-aggregation effects of cram-1 KD (yellow bars, Fig. 4b). In contrast, KD of lgg-3 or bec-1 did not diminish aggregation protection by cram-1 KD relative to FV controls (Fig. 4b). These results imply that protection via cram-1 KD (striped bars in Fig. 4) depends on ATG-7 but not on LGG-1 or BEC-1.
Figure 4.

Cram-1 knockdown reduces aggregation of Q40::YFP and formation of Aβ1–42 amyloid, dependent on proteasome and ATG-7 functions. (a) Schematic depiction of RNAi targets and their roles in autophagy pathways. (b) For dual RNAi, AM141 worms were fed from the L3/L4 molt on bacteria carrying empty FV, or FV expressing dsRNAs to target lgg-3, bec-1, or atg-7, each mixed 1:1 with RNAi against cram-1 (striped bars, ± SEM) or empty FV (solid bars, ± SEM). Aggregates per worm were counted on D5PH. *P < 2E–05, in a 1-tailed t-test comparing FV to [cram-1KD + FV]; **P < 0.0002, in a 2-tailed t-test comparing [cram-1KD + FV] to [cram-1KD + atg-7KD]. (c,d) LN149 worms expressing mCherry::ubiquitin in muscle were fed from the L3/L4 molt through D8PH, on dual-RNAi as described for panel b. Images of mCherry fluorescence are shown in c, and mean mCherry intensity per worm ± SEM is summarized in d. *P < 1E–04, in 1-tailed t-test between FV and [cram-1KD + FV]; **P < 6E–05, for a 2-tailed t-test of [cram-1KD + FV] vs. [cram-1KD + atg-7KD]. (e) CL4176 worms, expressing human Aβ1–42 in muscle, were fed dual RNAi as in b. Paralysis, assessed 36–40 h after induction for 50–100 worms/group, is shown as mean ± SEM, treating each experiment as one data point per group. One replicate value for [lgg-3KD + cram-1KD] was excluded as an outlier, >8.5 SDs from the mean of all other values (P < 6E–10). **Significance by 1-tailed paired t-test, P < 0.01. ***P < 1E–40, assuming a normal distribution. (f) C. elegans strain AM141 (unc54p/Q40::yfp) was fed from hatch on bacteria containing feeding vector (FV) or FV expressing dsRNA to target cram-1. Worms were treated from the L3/L4 molt onward with either 20-μM MG132 to inhibit proteasomes (cross-hatched bars), or vehicle only (solid bars). Aggregates per worm were counted on D5PH for 10–15 worms/group. *P ≈ 0.003, 1-tailed t-test between FV and cram-1KD; **P ≈ 1E–05, 2-tailed t-test for [cram-1KD + DMSO] vs. [cram-1KD + MG132].
To visualize the “backlog” of ubiquitin-tagged substrates, we employed a UPS-reporter strain (LN149) expressing both mCherry::ubiquitin and Q82::YFP in body-wall muscle. In this model, Q82::YFP rapidly forms aggregates in adolescent or mature worms, while mCherry:: ubiquitin serves as a pre-ubiquitinated reporter substrate that co-localizes with Q82::YFP. When UPS degradation functions normally, mCherry::ubiquitin is efficiently cleared, whereas residual mCherry signal indicates compromise of UPS clearance. Synchronized L3/L4 worms were transferred onto dual-RNAi plates targeting candidate genes, as in the experiment described above. Mean mCherry fluorescence per worm (a measure of uncleared, ubiquitinated substrate) was quantified on day 8 post-hatch (Fig. 4c,d). Cram-1 KD (striped bars) reduced the steady-state amount of mCherry::ubiquitin by 50–60% (each t-test P < 0.0002) whether combined with FV, lgg-3 KD, or bec-1 KD, indicating that neither LGG-3 nor BEC-1 is required for protection by cram-1 KD. Dual RNAi knockdown of atg-7 and cram-1, however, restored the mCherry::ubiquitin to at least the level of worms given either FV or atg-7 RNAi alone (N.S.), well over twice the signal seen after cram-1 KD alone [cram-1KD + FV], P < 6E–05.
We next employed a nematode model of amyloidopathy, strain CL4176 [myo-3p::Aβ1–42], that expresses the human amyloid peptide Aβ1–42 in body-wall muscle, causing paralysis 2 days post-induction. For most experiments, as in previous studies1, Aβ1–42 synthesis was induced by upshift from 20° to 25°C at the L3/L4 larval molt (~48 h after egg isolation), and paralysis was scored in young adults ~42 h later. To minimize possible developmental disruption, RNAi against target genes (candidate autophagy genes, as above) began at the L2/L3 larval transition, 8–10 h prior to upshift, and was maintained continuously thereafter. Consistent with our previous results, RNAi targeting cram-1 [FV + cram-1KD] reduced paralysis by 20–40%. In all 3 experiments conducted, this difference was significant at 36 and/or 48 h post-upshift (each Chi2 test P < 0.02; data not shown). Treating each of the three experiments as a single point per group, cram-1 KD robustly reduced paralysis (P = 0.002 by 1-tailed paired t-test, Fig. 4e, gray bars).
Protection by cram-1 KD was fully reversed by concurrent knockdown of atg-7 and was partially blocked, although not significantly, by concurrent knockdown of lgg-3 or bec-1 (Fig. 4e). That is, [cram-1KD + atg-7KD] differed significantly from [cram-1KD + FV] at 36 or 48 h post-upshift within each experiment (each Chi2 P < 0.05), and for the three experiments combined (P < 0.01, 1-tailed paired t-test, Fig. 4e). Protection against cram-1 knockdown was also fully reversed when 26 S proteasome function was inhibited by MG132 (Fig. 4f; P < 10−5). The above results imply that protection against aggregation by cram-1 knockdown requires both ATG-7 activity and active 26S proteasomes.
Protein unfolding increases the aggregation propensity of CRAM-1
Because CRAM-1 was initially identified in day-7 (post-reproductive) insoluble aggregates from a C. elegans strain that expresses huntingtin-like polyglutamine arrays1, we analyzed the aggregation propensity and disordered regions of the CRAM-1 protein structure, based on its structural dynamics in atomistic molecular-dynamic simulations. We modeled full-length CRAM-1 by the same fold recognition/ab-initio structure prediction method we had used to model its C-terminal UBA-like region. The full model predicted the same UBA-like cluster of 3 α-helices (3 turns each) joined by loops, as predicted for the C-terminal model alone. The N-terminal region contains 2 weaker α-helices (4 turns each) connected by a loop (Fig. 5a). This full-length CRAM-1 model supersedes our previous model1, which was derived by homology (with <50% template identity) and iterative loop refinement.
Figure 5.

Atomistic molecular-dynamic simulation predicts aggregation-prone regions in CRAM-1 due to structural unfolding. (a,b) MD simulation in Desmond (Desmond Molecular Dynamics System, ver. 2016.4, D.E. Shaw Research, New York, NY) showing the unfolding of protein structure from initial (a) to simulation-stability (b) conformations. (c) Helical regions (red bars) indicate unfolding of N-terminal residues, plotted against simulation time (x axis). (d–g) Predicted aggregation propensity (see scale at right) of CRAM-1 alone before simulation of structural rearrangement (d), or at the end of simulation (e) and of CRAM-1 bound to mono-ubiquitin (ub1) before simulation (f), or at the end of simulation (g). (h) Schematic depiction of a proposed mechanism, wherein CRAM-1 competes with RAD-23 for binding to ubiquitin, thus impeding clearance of ubiquitinated substrates.
Structural changes were documented during 200-ns simulations. Two of three independent simulations showed complete unfolding of the weak N-terminal helices (Fig. 5a–c), but stable retention of C-terminal helices comprising the UBA-like domain in all 3 simulations (Supplementary video 1). The aggregation propensities for initial and post-simulation (200-ns) conformations of CRAM-1 were calculated using AGGRESCAN3D. The results show that aggregation-prone regions become increasingly exposed with protein unfolding (Fig. 5d,e).
To more rigorously assess aggregation propensity, we performed computational docking of CRAM-1 with 1000 random proteins. CRAM-1 showed high affinity for a substantial majority of proteins: >85% of interactions fell between −150 and −260 kcal/mol, with −207 kcal/mol average interaction energy (Supplementary Fig. 1d), supporting the predicted exposure of aggregation-prone regions in CRAM-1. To determine whether this level of interaction is exceptional relative to other proteins, we simulated protein-protein interactions within each of 3 sets of 25 proteins (i.e. 75 total) taken at random from 1000 proteins used in Supplementary Fig. 1d. The number of pairwise interactions per set is C(25,2) = 300, or 325 including homodimers, totaling 975 interactions for 3 sets (Supplementary Fig. 1e). The average interaction energy for the 3 control sets is −96.0 ± 12.6 (SEM) kcal/mol, less than half of the CRAM-1 interaction energies (−207.3 ± 1.7 kcal/mol).
We then asked whether the aggregation propensity of CRAM-1 alters when its C-terminal UBA-like domain binds ubiquitin (Ub1) during a 200-ns atomistic simulation. The results predict stable binding of full-length CRAM-1 to ubiquitin(s), consistent with the prediction from C-terminal modeling. Remarkably, ubiquitin binding did not diminish the aggregation-propensity of any CRAM-1 regions, implying that CRAM-1 could aggregate with other proteins in addition to ubiquitin-tagged substrates (Fig. 5f,g). Based on our computational prediction and experimental data, we postulate that CRAM-1 aggregation involves both “general” interactions among disordered proteins, and annealing specific to ubiquitin-tagged proteins. This dual mechanism could account for the surprising extent of protection conferred by cram-1 knockdown (Fig. 5h).
SERF-2, a close human ortholog of CRAM-1, is disordered and aggregation-prone
By sequential tracking of the last common ancestral protein along the phylogenetic tree, we identified SERF2 as the closest human ortholog of CRAM-1, despite the absence of significant sequence similarity between these proteins1. Based on their common origin, we anticipated that SERF2 may show structural and/or functional conservation to CRAM-1 and may play a similar role in mammalian protein aggregation. Ab initio modelling of the SERF2 structure indicates 2 helices connected by a loop (Fig. 6a), with little conformational resemblance to CRAM-1 or any other UBA-domain-containing protein. Sequence-based algorithms35 indicate that 100% of SERF2 residues have high potential for disorder, and 95% are predicted to be solvent accessible. We then analyzed the structural dynamics of SERF2 by atomistic molecular-dynamic (MD) simulation for 0.5 µs. Like CRAM-1, SERF2 undergoes extensive unfolding of helices to random coils (Fig. 6b), which could cause loss of function and increased aggregation propensity.
Figure 6.

MD simulation and protein-protein interactions predict that SERF2 is aggregation-prone. (a,b) MD simulation of SERF2, in Desmond (Desmond Molecular Dynamics System, ver. 2016.4, Shaw Research, New York), indicating its initial state (a) and the unfolded state after 500 ns (b). (c) RMSD for three independent simulations of SERF2 structure monitored throughout the simulation. The inset shows the coefficient of variation for RMSD in each simulation. (d,e) Predicted aggregation-prone regions (see scale at left) for SERF2 in its initial conformation (d) and unfolded conformation after 500 ns of simulation (e). (f) Interaction energies were predicted for SERF2 in its initial (black dots) and simulated unfolded (green dots) conformations when interacting with 1000 random proteins from the PDB databank. Interaction energies are also shown for SERF2 dimer in its initial conformation (boxed red dot) and in a subsequent unfolded state (boxed yellow dot). The tinted rectangle indicates the 90% confidence interval for SERF2 interaction energies. (g,h) Predicted structures are shown for SERF2 interacting with Ub1 (g) or Ub2 (h). (i) Average H-bond number was calculated for a single SERF2 molecule interacting with Ub1 or Ub2, over a 200-ns simulation. (j) Interaction energies were predicted for RAD23A or SERF2 molecules binding to mono- and di-ubiquitin (Ub1 and Ub2, respectively).
MD simulations were performed in triplicate, allowing calculation of average SERF2 structural properties. All three simulations stabilized rapidly (<20 ns) at similar root-mean-square deviation (RMSD) values (Fig. 6c): 6.0, 6.43 and 6.05 Å (mean = 6.16 Å). These data are consistent with a preferred, relatively stable but largely unfolded conformation of SERF2, which differs substantially from its initial structure. The Coefficient of Variation (SD/mean) of RMSD beyond 35 ns was 5–7%, indicating rather little structural change after initial SERF2 unfolding. Aggregation propensities were calculated for initial and post-simulation (500-ns) SERF2 structures. Unlike CRAM-1, SERF2 did not increase its predicted aggregation propensity as it unfolded (Fig. 6e). This may reflect that, based on its high intrinsic aggregation propensity, SERF2 lacks initially-buried hydrophobic residues that could be exposed during MD simulation.
We next simulated SERF2 interactions with 1000 proteins of known structure, randomly taken from PDB (https://www.rcsb.org/). SERF2 was predicted to interact strongly with most of these proteins, in both its initial state (Fig. 6f; black dots) and in the unfolded state observed at 500 ns simulation (green dots). Of all predicted SERF2 interactions, 90% fall between −111 and −267 kcal/mol (shaded rectangle in Fig. 6f, Supplementary Fig. 1e), whereas only 34% of 1000 random protein-protein interactions fall within this range (average ΔE −96.0 kcal/mol), consistent with SERF2 being far more interaction-prone than most proteins.
We then asked whether SERF2 and CRAM-1, despite the absence of structural similarity, show functional conservation regarding interaction with ubiquitin. Protein-protein docking predicts SERF2 interaction with mono- or di-ubiquitin (Ub1, Ub2), with interaction energies even more favorable than those for RAD23A (Fig. 6g–j). We note that the ubiquitin facets that interact with SERF2 differ from those contacted by CRAM-1 or any UBA-domain protein including MUD1 or hsRAD23A (human). MD simulations of protein-protein complexes indicate stable binding of SERF2 to ubiquitin (Ub1 & Ub2). Moreover, the number of hydrogen bonds between SERF2 and ubiquitin is stable throughout each simulation, averaging 6.7 H bonds per ubiquitin moiety (Fig. 6i).
Since the binding energy of SERF2 to ubiquitin exceeds that of RAD23A (Fig. 6j), we expected that SERF2 binding to ubiquitin would block RAD23A-ubiquitin interaction. Analysis of a simulated 3-molecule interaction, comprising RAD23A, ubiquitin, and SERF2, supports this prediction (Supplementary Fig. 1f). In additional simulations, SERF2 spontaneously formed homodimers with an interaction energy (ΔEinteraction) of −302.9 kcal/mol (Fig. 6f, red dot), lying at the 0.4th percentile for simulated interactions of SERF2 with 1000 randomly chosen proteins. These data argue that SERF2 could promote aggregate progression, perhaps by the mechanisms inferred for CRAM-1: obstructing RAD23-ubiquitin interaction, and propensity to aggregation in general.
SERF2 knockdown reduces amyloid aggregation in human neuroblastoma cells
Computational studies indicate that SERF2 is disordered, aggregation-prone, and able to interact with damaged or misfolded proteins via their ubiquitin tags, or directly with disordered regions. Knockdown of CRAM-1, the C. elegans ortholog of SERF2, reduces aggregation and associated toxicity in C. elegans models of protein aggregation associated with neurodegenerative diseases1 (see also Figs 1–5). We therefore used short-hairpin RNA (shRNA) constructs to knock down SERF2 expression in a human neuroblastoma cell line expressing an aggregation-prone mutation of amyloid precursor protein (SY5Y-APPSw). These cells produce an overabundance of Aβ1–42 and continuously form extracellular amyloid aggregates. Cells were transfected with shRNA against Serf2 or Serf1a, after which cells were maintained 48 h at 37 °C and then stained for amyloid. Transfected cells, and non-transfected control cells, were incubated in 0.1% (w/v) thioflavin T followed by DAPI (see Methods) and imaged for amyloid fluorescence (thioflavin: amyloid excitation peak at 450 nm; emission at 482 nm) and for DNA (DAPI: DNA excitation at 358 nm; emission at 461 nm) as shown in Fig. 7a. ShRNA knockdown of SERF2 consistently reduced amyloid aggregation by 50–70% in four independent experiments (Fig. 7a,b), three of which showed significant shifts. Parallel experiments targeting SERF1A, a somewhat more distant ortholog of CRAM-11, also reduced amyloid aggregation by ~2-fold (2-tailed t-test P < 6E–07) in a single experiment (Supplementary Fig. 1g). These data corroborate our computational predictions that SERF2 could play a vital role in aggregate progression.
Figure 7.
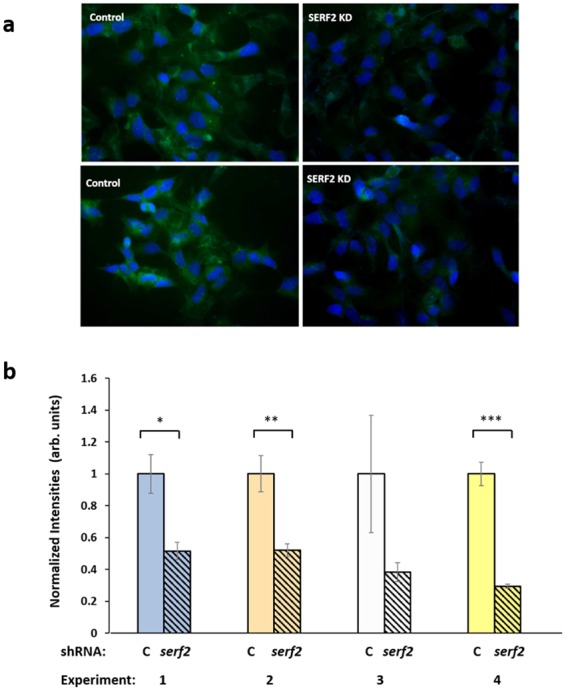
SERF2 knockdown protects against amyloid aggregation in human neural cells. (a) SH-SY5Y-APPSw neuroblastoma cells were transfected with SERF2 shRNA, and stained with thioflavin T at 48 h post-transfection. Amyloid fluorescence is displayed in green, and nuclei (counterstained with DAPI) appear blue. (b) Thioflavin-T fluorescence was quantified and divided by the number of DAPI-stained nuclei per field, to estimate amyloid deposition per cell. In 4 independent experiments, SERF-2 knockdown reduced amyloid by 50–70%, attaining statistical significance in 3 of 4 experiments. Fluorescence intensity per cell is plotted ± SEM, normalized to the control mean for each experiment. *P < 0.01 by 2-tailed t-test (initial experiment); **P < 0.01 by 1-tailed t-test; ***P < 0.001 by 1-tailed t-test.
Discussion
We identified key properties of the C. elegans CRAM-1 protein, and its closest human ortholog, SERF2, which are critical in promoting aggregation. CRAM-1 and SERF2 are predominantly-disordered proteins containing aggregation-prone regions1. The CRAM-1 C-terminal region (residues 54–96) comprises 3 moderately stable α-helices connected by loops, in a conformation that closely matches the ceRAD-23 UBA-domain structure28 (Fig. 1c,d). UBA domains primarily bind ubiquitin28,30, with increasing affinity as ubiquitin chains grow, favoring specific conveyance of poly-ubiquitinated substrates to the proteasome24,31. CRAM-1, despite lacking sequence similarity to known UBA domains, has striking structural homology to ceRAD-23 (RMSD = 1.02 Å) and its human ortholog, hsRAD23A (RMSD = 1.14 Å), suggesting that it may bind poly-ubiquitin.
We previously identified CRAM-1 as a C. elegans protein present in sarcosyl-insoluble protein aggregates and strongly favoring polyglutamine-seeded protein aggregation. Molecular modeling and atomistic dynamic simulations indicate its propensity to bind ubiquitin(s) via the UBA-like domain, interacting with the same residues of ubiquitin that ceRAD-23 binds. This interaction could prevent ceRAD-23 interaction with ubiquitin, which is a critical step in cargo conveyance to 26S proteasomes. In multiple nematode models of neurodegeneration-like protein aggregation, RNAi knockdown of cram-1 protected against aggregation and associated traits1 — protection that is lost if UPS is inhibited either by a proteasome-inactivating drug, MG132 (Fig. 3a), or by RNAi targeting pas-4, which encodes an essential α subunit of the 20S proteasome1.
We propose several routes through which CRAM-1 could impede protein aggregation. RAD23A interacts with poly-ubiquitin chains through its UBA domain, and delivers cargo to proteasomes by docking its UBL domain to the RPN10 proteasome regulatory subunit. A deletion mutant of RAD23A, lacking a UBL domain, causes a severe protein-degradation deficiency15,17,36, conjectured to involve a “decoy” mechanism in which truncated RAD23A competes with intact RAD23A for protein cargos to be degraded. On the other hand, RAD23A overexpression also blocks ubiquitin chain elongation, resulting in poor UPS clearance37; and likewise, a UBA domain termed p47-RAT inhibits chain elongation upon ubiquitin binding38. CRAM-1 could compromise proteostasis by either of these mechanisms: competition with a UBA-domain-containing shuttle protein, such as ceRAD-23, for binding to poly-ubiquitin, or binding to “immature” mono- and oligo-ubiquitin chains and disrupting their elongation. Both mechanisms are consistent with our in vivo results indicating that cram-1 KD relieves protein aggregation, but only the decoy model unambiguously predicts that ceRAD-23 KD would negate all beneficial effects of simultaneous CRAM-1 KD. Previous C. elegans studies had shown that Q40::YFP aggregates are processed chiefly by the UPS system1,39.
Autophagy is another critical protein-degradation pathway important for handling bulky cargos, including protein aggregates and damaged mitochondria40–42. We assessed several genes that play essential roles in autophagy pathways. ATG7 (an E1-like ubiquitin-activating enzyme) is a key player in autophagy; RNAi against atg-7 was reported to block autophagy in C. elegans, and thereby increase sequestosome1/p62 levels, which in turn impairs 26S proteasomes by exceeding their capacity43. Cram-1 knockdown relieves protein aggregation, but this rescue vanishes when autophagy is disrupted by atg-7 KD, whereas RNAi targeting lgg-3 and bec-1 appeared to be less effective (Fig. 4e). These results led us to postulate that, in dual KDs targeting atg-7 and cram-1, impairment of autophagy by atg-7 RNAi indirectly suppresses UPS as well, preventing any benefit of cram-1 knockdown. Like siRNA targeting just rad-23 (Fig. 3b), knockdown of atg-7 alone (Fig. 4b–e) did not elevate aggregation relative to controls — in reporter strains AM141 (Q40::yfp), LN149 (ubq::mCherry, Q82::yfp), or CL4176 (Aβ1–42) — but nevertheless fully blocked the protective effects of cram-1 knockdown.
Molecular-dynamic simulations of either CRAM-1 alone, or CRAM-1 in complex with ubiquitin(s), identified structural fluctuations in the N-terminal α-helical domains of CRAM-1, increasing the exposure of aggregation-prone regions (Fig. 5). Comparing aggregation propensities of initial and MD-simulation conformations of CRAM-1 alone (Fig. 5d,e), vs. CRAM-1 complexed with ubiquitin (Fig. 5f,g), indicates that aggregation-prone regions are exposed by unfolding of the CRAM-1. In silico modeling of interactions with random proteins implies that CRAM-1 could stably bind many other proteins (Supplementary Fig. 1d).
The above results suggest an explanation for the surprisingly large effect of CRAM-1 on the aggregate burden. In addition to acting as a decoy competing for ubiquitin sites with UPS shuttle proteins, and perhaps compromising ubiquitin chain elongation, the ubiquitin-bound form of CRAM-1 unveils its own aggregation-inclined aspects and also those of its binding partner (ubiquitin in Fig. 5g), enhancing the likelihood that both will adhere to a growing aggregate.
An RNAi screen for modifiers of protein aggregation in C. elegans identified only one gene, moag-4 (the nematode ortholog of human SERF1), for which knockdown protected nematodes against Q40::YFP aggregation44,45. Knockdown of moag-4 was thought to confer protection by mechanisms independent of UPS or autophagy, but involving direct interaction with amyloid44. CRAM-1 retains no sequence similarity to SERF2, indicating that their genes diverged long ago, and/or experienced very little selective pressure for protein-sequence conservation; likewise, CRAM-1 shows no homology to MOAG-4 or SERF1.
SERF2 (small EDRK-rich factor 2) is a small (59 residue), ubiquitously-expressed protein rich in basic residues lysine (~20%) and arginine (11%). Structural and dynamic modeling of SERF2 predicted two α-helices linked by a short random coil, which departs from the typical UBA-domain bundle of three α-helices but instead resembles the helix-turn-helix structure of the ubiquitin binding motif (UBM)46. Despite their differences, both SERF2 and CRAM-1 are predicted to interact stably with ubiquitin chains, and KDs of both protect against aggregation. In SY5Y-APPSw neuroblastoma cells that form extracellular amyloid, SERF2 knockdown reduced those aggregates by 50–70%. While SERF2 may employ a decoy mechanism as proposed for CRAM-1, it is also predicted to form stable interactions with many partners (Fig. 6f; Supplementary Fig. 1e), conferring the potential to form branch points critical to aggregate growth. We note that multiple protein interactions were recently reported for SERF2 in a yeast two-hybrid system, including the PA28γ subunit of a 20S proteasome-activating complex, and several RNA-binding and RNA-processing proteins47.
Based on in silico interaction with randomly selected proteins, we predict that SERF2 could interact with many (perhaps most) proteins. Together, these findings suggest that SERF2 is exceptionally interactive, due to its unstructured and flexible nature — properties that are consistent with roles in aggregate progression and (through ubiquitin binding) disruption of aggregate-clearance pathways. Further studies assessing the mechanistic role of SERF2 in aggregate accrual should shed light on the balance between aggregate formation and clearance, while at the same time suggesting new therapeutic targets for the many age-progressive diseases that feature protein aggregation.
Methods
Strains and maintenance
C. elegans strains AM141 [unc-54p::Q40::yfp] and CL4176 [smg-1ts; myo-3p::Aβ1–42::let-851 [3′-UTR]; rol-6(su1006)] were obtained from the Caenorhabditis Genetics Center (CGC). The LN149 strain [unc-54p::Q82::yfp; unc-54p:: mCherry::ubiquitin] was kindly provided by Lynn Boyd and Gregory Skibinski (Univ. Alabama, Huntsville, AL).
The strains listed above were routinely maintained on regular solid-agar nematode growth medium (NGM) seeded with E. coli (strain OP50) bacteria, at 20 °C except for CL4176 paralysis experiments in which worms were induced by upshift from 20° to 25° C at the L3/L4 transition (~48 hours after eggs hatched). For knockdown experiments, well-fed, gravid day-1 adults were lysed in alkaline hypochlorite to release unlaid eggs, which were allowed to hatch on plates with E. coli (strain HT115) RNAi sublines, each carrying a plasmid expressing an RNAi construct to target a gene of interest48. If RNAi exposures caused developmental delays or defects, eggs were instead hatched on HT115 bacteria carrying empty plasmid vector, and transferred only at the L3/L4 molt to RNAi-expressing bacteria, on which they were subsequently maintained. Dual-RNAi experiments used 1:1 mixtures of HT115 bacteria carrying two distinct RNAi-expressing plasmids, or a subline carrying an RNAi-expressing plasmid mixed 1:1 with empty-vector control bacteria.
Visualization of reporter strains
AM141 worms were imaged on a Nikon Eclipse E600 fluorescence microscope fitted with a Nikon CoolSnap ES camera, and Q40::YFP aggregates were counted (DotCount, http://reuter.mit.edu/software/dotcount/). Incident and emitted light were filtered to 490 ± 20 nm and 535 ± 30 nm, respectively. Similarly, red and yellow fluorescence were imaged in LN149 adults (expressing unc54p/mCherry::ubiquitin and unc54p/Q82::yfp in body wall muscle), and the average fluorescence intensity of mCherry::ubiquitin foci was quantified with FIJI (ImageJ).
Paralysis assay
The CL4176 strain, expressing Aβ1–42 in muscle, was synchronized by lysis as above, and eggs were transferred onto 60-mm agar plates seeded with bacteria expressing dsRNAs against targeted genes (or to plates seeded with dual dsRNA vectors in 1:1 ratio). Worms in experimental groups were upshifted from 20° to 25° C at the L3/L4 transition (47–49 h post-lysis) to induce expression of Aβ1–42. Paralysis of worms (defined as loss of motility in response to touch) was scored from 18-h post-upshift until motility fell below 60% in the FV (control) group. To slow development of progeny in synchronized populations, 5-fluoro-2′-deoxyuridine (FUdR) was added at a final concentration of 2 μM to RNAi plates and to control (FV) plates, each containing worms from pre-gravid (L4/adult molt, day 2.5 post-hatch) through post-gravid ages (beyond 6–7 days post-hatch).
Structure generation
Three-dimensional structures of full length CRAM-1 and its UBA-like domain, and of ceRAD23, were modelled using the I-TASSER server, which performs fold recognition followed by ab initio prediction of structure49. Structures retrieved from the Protein DataBase (PDB; https://www.rcsb.org/) were human RAD23A (1F4I; C-terminal UBA(2) domain), MUD1 (1Z96), di-ubiquitin open conformer (2PE9), plus 1000 randomly chosen proteins. The full-length structure of CRAM-1 was simulated briefly (10 ns) in implicit solvent (GBSA) using the GROMACS simulation package50, prior to docking studies.
Protein-protein docking
To model protein-protein interactions, the HEX 6.1 program1,51 was used with default parameters. Interactions from each run were ranked by ΔEinteraction energies, and the lowest-energy (most stable) models were chosen for dynamic simulations. Automated Linux shell scripts were written to automate large-scale computational docking studies in Hex.
Molecular-dynamic simulation
Atomistic molecular dynamics of individual proteins and complexes were simulated using Desmond software (Desmond Molecular Dynamics System, version 2016.4, D.E. Shaw Research, New York, NY). Proteins were initially immersed in an orthorhombic box containing SPC water, pH neutralized, and salt set to 0.15-M NaCl. For MD runs, the ensemble class was set to NPT, and temperature and pressure were set to 300 °K (Nose-Hoover chain method) and 1.013 bar (Martyna-Tobias-Klein method) respectively. Velocities were randomized every 25 ps to minimize sampling bias. Each MD run was performed for 200–500 ns in duplicate or triplicate, and trajectories were analyzed using built-in packages from Desmond-Maestro, VMD and Discovery Studio (BIOVIA Discovery Studio [Ver. 17.2.0], Dassault Systèmes, San Diego [2017]).
Cell culture and maintenance
SH-SY5Y-APPSw cells, overexpressing an aggregation-prone familial-AD mutation of amyloid precursor protein, APPSw, were kindly provided by Dr. Steven Barger. These cells model Alzheimer’s-like amyloid formation to study the contribution of SERF2 to amyloidopathy. Cells were maintained at 37 °C with 5% CO2 in a tissue-culture incubator, in culture dishes containing DMEM-F12 (1:1) nutrient mixture (Ham’s medium) with 10% v/v fetal bovine serum.
Lipofection and thioflavin-T assay
To knock down expression of SERF1A or SERF2, target-specific shRNAs were introduced into SH-SY5Y-APPSw cells by lipofection. Well-maintained cells were grown to confluence, then trypsinized and subcultured in 12-well plates (at 15,000–20,000 cells/well) containing antibiotic-free DMEM medium. After 24 h, shRNA against each candidate gene was introduced with Lipofectamine 2000 (Invitrogen). Transfected cells, along with control cells (mock treatment), were maintained at 37 °C for 48 hours and then incubated with 0.1% (w/v) thioflavin T in phosphate-buffered saline to stain amyloid aggregates. Total aggregate fluorescence was quantified from images using FIJI (ImageJ), with background subtraction at a rolling-ball radius of 50 for all images. To obtain the average thioflavin T fluorescence per cell, the total aggregate fluorescence in each captured image was divided by the number of cells (nuclei stained with DAPI) counted in the same image.
Western-blot detection of CRAM-1/ubiquitin binding
Wild type N2 (Bristol) worms were fed with regular E. coli (OP50) bacteria and maintained at 20 °C. Adult day-1 (D1) and day-5 (D5) worms were flash frozen and homogenized. Lysates were then incubated with either biotin-tagged antibody to CRAM-1 (captured on streptavidin-coated magnetic beads), or magnetic beads coated with antibody to ubiquitin. Eluted proteins were resuspended in 2x Laemmli buffer with β-mercaptoethanol at 95 °C, and equal worm equivalents loaded on a 1% SDS, 10% (w/v) polyacrylamide gel, electrophoresed, and transferred to nylon membranes. Blots were incubated with murine antibodies to ubiquitin (Abcam) or rabbit antibodies to CRAM-1 (Genscript). Membranes were imaged using FluorChem Q (Cell Biosciences) after incubation with horseradish peroxidase (HRP)-coupled antibody to either mouse IgG or rabbit IgG.
Statistical tests
Groups are generally compared by 2-tailed Behrens-Fisher t tests (conservative t tests for samples of unequal or unknown variances), unless the direction of the difference is known or strongly predicted, in which case a 1-tailed t test is used. Differences in proportions are assessed by Chi-squared (Chi2) or Fisher Exact tests. Control samples used for normalization across multiple experiments (e.g., bars at left of Fig. 3c) cannot be evaluated by t tests due to zero variance of normalized control values. The null hypothesis is instead tested by calculating the area under a normal-distribution tail for control values ≥1.00, given a normal distribution with the observed mean and SEM (0.47 ± 0.015 in Fig. 3c, for [cram-1KD + FV]).
Electronic supplementary material
Acknowledgements
This work was supported by grants (Merit 2 I01 BX001655 and Senior Research Career Scientist Award) to R.J.S.R. from the U.S. Dept. of Veteran Affairs, to M.B. from the Inglewood Scholars Program, and by Program Project grant 2P01AG012411-17A1 (W.S.T. Griffin, P.I.) from the National Institute on Aging (NIA/NIH). The authors thank the Windgate Foundation and the Philip R. Jonsson Foundation for additional support, and Ramani Alla for superb technical assistance.
Author Contributions
M.B. performed all computer modeling and molecular simulations, and calculations based on those. All in vivo (nematode and human cell-culture) experiments were designed by R.J.S.R. and S.A. and executed by M.B. with guidance from S.A. R.J.S.R. oversaw data analysis and manuscript preparation. M.B. and R.J.S.R. wrote the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Srinivas Ayyadevara, Email: AyyadevaraSrinivas@uams.edu.
Robert J. Shmookler Reis, Email: rjsr@uams.edu
Electronic supplementary material
Supplementary information accompanies this paper at 10.1038/s41598-018-33143-1.
References
- 1.Ayyadevara S, et al. Proteins in aggregates functionally impact multiple neurodegenerative disease models by forming proteasome-blocking complexes. Aging Cell. 2015;14:35–48. doi: 10.1111/acel.12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kopito RR, Ron D. Conformational disease. Nat. Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 3.Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl) 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- 4.Youmans KL, et al. Intraneuronal Abeta detection in 5xFAD mice by a new Abeta-specific antibody. Mol. Neurodegener. 2012;7:8. doi: 10.1186/1750-1326-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kikis EA, Gidalevitz T, Morimoto RI. Protein homeostasis in models of aging and age-related conformational disease. Adv. Exp. Med. Biol. 2010;694:138–159. doi: 10.1007/978-1-4419-7002-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Labbadia J, Morimoto RI. The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 2015;84:435–464. doi: 10.1146/annurev-biochem-060614-033955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liachko Nicole F., Saxton Aleen D., McMillan Pamela J., Strovas Timothy J., Currey Heather N., Taylor Laura M., Wheeler Jeanna M., Oblak Adrian L., Ghetti Bernardino, Montine Thomas J., Keene C. Dirk, Raskind Murray A., Bird Thomas D., Kraemer Brian C. The phosphatase calcineurin regulates pathological TDP-43 phosphorylation. Acta Neuropathologica. 2016;132(4):545–561. doi: 10.1007/s00401-016-1600-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arrasate M, Finkbeiner S. Protein aggregates in Huntington’s disease. Exp. Neurol. 2012;238:1–11. doi: 10.1016/j.expneurol.2011.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DeToma AS, Salamekh S, Ramamoorthy A, Lim MH. Misfolded proteins in Alzheimer’s disease and type II diabetes. Chem. Soc. Rev. 2012;41:608–621. doi: 10.1039/C1CS15112F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ayyadevara S, et al. Proteins that mediate protein aggregation and cytotoxicity distinguish Alzheimer’s hippocampus from normal controls. Aging Cell. 2016;15:924–939. doi: 10.1111/acel.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang Q, et al. Human proteins with target sites of multiple post-translational modification types are more prone to be involved in disease. J. Proteome Res. 2014;13:2735–2748. doi: 10.1021/pr401019d. [DOI] [PubMed] [Google Scholar]
- 12.Fink AL. Protein aggregation: folding aggregates, inclusion bodies and amyloid. Fold. Des. 1998;3:R9–23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 13.Karve TM, Cheema AK. Small changes huge impact: the role of protein posttranslational modifications in cellular homeostasis and disease. J. Amino Acids. 2011;2011:207691. doi: 10.4061/2011/207691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vitalis A, Wang X, Pappu RV. Atomistic simulations of the effects of polyglutamine chain length and solvent quality on conformational equilibria and spontaneous homodimerization. J. Mol. Biol. 2008;384:279–297. doi: 10.1016/j.jmb.2008.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen L, Madura K. Rad23 promotes the targeting of proteolytic substrates to the proteasome. Mol. Cell. Biol. 2002;22:4902–4913. doi: 10.1128/MCB.22.13.4902-4913.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verma R, Oania R, Graumann J, Deshaies RJ. Multiubiquitin chain receptors define a layer of substrate selectivity in the ubiquitin-proteasome system. Cell. 2004;118:99–110. doi: 10.1016/j.cell.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 17.Liang RY, et al. Rad23 interaction with the proteasome is regulated by phosphorylation of its ubiquitin-like (UbL) domain. J. Mol. Biol. 2014;426:4049–4060. doi: 10.1016/j.jmb.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guthrie CR, Greenup L, Leverenz JB, Kraemer BC. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011;20:1989–1999. doi: 10.1093/hmg/ddr079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brignull HR, Morley JF, Garcia SM, Morimoto RI. Modeling polyglutamine pathogenesis in C. elegans. Methods Enzymol. 2006;412:256–282. doi: 10.1016/S0076-6879(06)12016-9. [DOI] [PubMed] [Google Scholar]
- 20.Ayyadevara S, et al. Aspirin inhibits oxidant stress, reduces age-associated functional declines, and extends lifespan of Caenorhabditis elegans. Antioxid. Redox Signal. 2013;18:481–490. doi: 10.1089/ars.2011.4151. [DOI] [PubMed] [Google Scholar]
- 21.Dosanjh LE, Brown MK, Rao G, Link CD, Luo Y. Behavioral phenotyping of a transgenic Caenorhabditis elegans expressing neuronal amyloid-beta. J. Alzheimers Dis. 2010;19:681–690. doi: 10.3233/JAD-2010-1267. [DOI] [PubMed] [Google Scholar]
- 22.Morley JF, Brignull HR, Weyers JJ, Morimoto RI. The threshold for polyglutamine-expansion protein aggregation and cellular toxicity is dynamic and influenced by aging in Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA. 2002;99:10417–10422. doi: 10.1073/pnas.152161099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chang YG, et al. Solution structure of the ubiquitin-associated domain of human BMSC-UbP and its complex with ubiquitin. Protein Sci. 2006;15:1248–1259. doi: 10.1110/ps.051995006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trempe JF, et al. Mechanism of Lys48-linked polyubiquitin chain recognition by the Mud1 UBA domain. EMBO J. 2005;24:3178–3189. doi: 10.1038/sj.emboj.7600797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ciani B, Layfield R, Cavey JR, Sheppard PW, Searle MS. Structure of the ubiquitin-associated domain ofp62 (SQSTM1) and implications for mutations that cause Paget’s disease of bone. J. Biol. Chem. 2003;278:37409–37412. doi: 10.1074/jbc.M307416200. [DOI] [PubMed] [Google Scholar]
- 26.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 27.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 28.Mueller TD, Feigon J. Solution structures of UBA domains reveal a conserved hydrophobic surface for protein-protein interactions. J. Mol. Biol. 2002;319:1243–1255. doi: 10.1016/S0022-2836(02)00302-9. [DOI] [PubMed] [Google Scholar]
- 29.Hurley JH, Lee S, Prag G. Ubiquitin-binding domains. Biochem. J. 2006;399:361–372. doi: 10.1042/BJ20061138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mueller TD, Kamionka M, Feigon J. Specificity of the interaction between ubiquitin-associated domains and ubiquitin. J. Biol. Chem. 2004;279:11926–11936. doi: 10.1074/jbc.M312865200. [DOI] [PubMed] [Google Scholar]
- 31.Raasi S, Orlov I, Fleming KG, Pickart CM. Binding of polyubiquitin chains to ubiquitin-associated (UBA) domains of HHR23A. J. Mol. Biol. 2004;341:1367–1379. doi: 10.1016/j.jmb.2004.06.057. [DOI] [PubMed] [Google Scholar]
- 32.Ryabov Y, Fushman D. Structural assembly of multidomain proteins and protein complexes guided by the overall rotational diffusion tensor. J. Am. Chem. Soc. 2007;129:7894–7902. doi: 10.1021/ja071185d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Satoh T, et al. Crystal structure of cyclic Lys48-linked tetraubiquitin. Biochem. Biophys. Res. Commun. 2010;400:329–333. doi: 10.1016/j.bbrc.2010.08.057. [DOI] [PubMed] [Google Scholar]
- 34.David DC, et al. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010;8:e1000450. doi: 10.1371/journal.pbio.1000450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xue B, Dunbrack RL, Williams RW, Dunker AK, Uversky VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta. 2010;1804:996–1010. doi: 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schauber C, et al. Rad23 links DNA repair to the ubiquitin/proteasome pathway. Nature. 1998;391:715–718. doi: 10.1038/35661. [DOI] [PubMed] [Google Scholar]
- 37.Hwang GW, Sasaki D, Naganuma A. Overexpression of Rad23 confers resistance to methylmercury in saccharomyces cerevisiae via inhibition of the degradation of ubiquitinated proteins. Mol. Pharmacol. 2005;68:1074–1078. doi: 10.1124/mol.105.013516. [DOI] [PubMed] [Google Scholar]
- 38.Meyer Hemmo H, Shorter James G, Seemann Joachim, Pappin Darryl, Warren Graham. A complex of mammalian Ufd1 and Npl4 links the AAA-ATPase, p97, to ubiquitin and nuclear transport pathways. The EMBO Journal. 2000;19(10):2181–2192. doi: 10.1093/emboj/19.10.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmberg CI, Staniszewski KE, Mensah KN, Matouschek A, Morimoto RI. Inefficient degradation of truncated polyglutamine proteins by the proteasome. Embo J. 2004;23:4307–4318. doi: 10.1038/sj.emboj.7600426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mizushima N. The role of the Atg1/ULK1 complex in autophagy regulation. Curr. Opin. Cell Biol. 2010;22:132–139. doi: 10.1016/j.ceb.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 41.Nah J, Yuan J, Jung YK. Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol. Cells. 2015;38:381–389. doi: 10.14348/molcells.2015.0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tanida I. Autophagy basics. Microbiol. Immunol. 2011;55:1–11. doi: 10.1111/j.1348-0421.2010.00271.x. [DOI] [PubMed] [Google Scholar]
- 43.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Falsone SF, et al. SERF protein is a direct modifier of amyloid fiber assembly. Cell Rep. 2012;2:358–371. doi: 10.1016/j.celrep.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Ham TJ, et al. Identification of MOAG-4/SERF as a regulator of age-related proteotoxicity. Cell. 2010;142:601–612. doi: 10.1016/j.cell.2010.07.020. [DOI] [PubMed] [Google Scholar]
- 46.Burschowsky D, et al. Structural analysis of the conserved ubiquitin-binding motifs (UBMs) of the translesion polymerase iota in complex with ubiquitin. J. Biol. Chem. 2011;286:1364–1373. doi: 10.1074/jbc.M110.135038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim J, et al. A protein-protein interaction network for human inherited ataxias and disorders of Purkinje cell degeneration. Cell. 2006;125:801–814. doi: 10.1016/j.cell.2006.03.032. [DOI] [PubMed] [Google Scholar]
- 48.Kamath RS, Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods. 2003;30:313–321. doi: 10.1016/S1046-2023(03)00050-1. [DOI] [PubMed] [Google Scholar]
- 49.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ritchie DW, Kozakov D, Vajda S. Accelerating and focusing protein-protein docking correlations using multi-dimensional rotational FFT generating functions. Bioinformatics. 2008;24:1865–1873. doi: 10.1093/bioinformatics/btn334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.