

SUMMARY
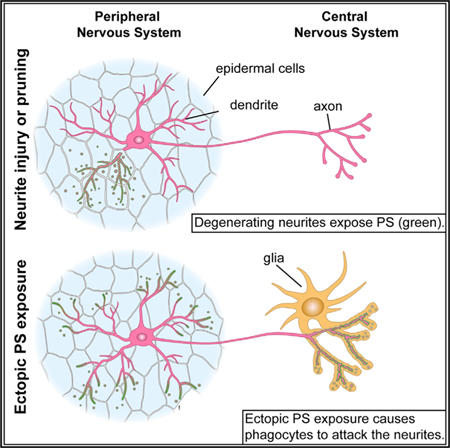
Phagocytic clearance of degenerating dendrites or axons is critical for maintaining tissue homeostasis and preventing neuroinflammation. Externalized phosphatidylserine (PS) has been postulated to be an ‘‘eat-me’’ signal allowing recognition of degenerating neurites by phagocytes. Here we show that in Drosophila, PS is dynamically exposed on degenerating dendrites during developmental pruning and after physical injury, but PS exposure is suppressed when dendrite degeneration is genetically blocked. Ectopic PS exposure via phospholipid flippase knockout and scramblase overexpression induced PS exposure preferentially at distal dendrites and caused distinct modes of neurite loss that differ in larval sensory dendrites and in adult olfactory axons. Surprisingly, extracellular lactadherin that lacks the integrin-interaction domain induced phagocyte-dependent degeneration of PS-exposing dendrites, revealing an unidentified bridging function that potentiates phagocytes. Our findings establish a direct causal relationship between PS exposure and neurite degeneration in vivo.
Graphical Abstract
In Brief
Using in vivo phosphatidylserine (PS) sensors, Sapar et al. reveal dynamic patterns of PS exposure on degenerating dendrites in Drosophila. Flippase knockout and scramblase overexpression lead to ectopic PS exposure on distal dendrites and context-dependent neurite degeneration. Lactadherin potentiates phagocytes to destruct PS-exposing dendrites, independent of its integrin-interaction domain.
INTRODUCTION
The nervous system requires active monitoring and maintenance. During development, erroneous neuronal processes and excessive connections between neurons are trimmed (Chung et al., 2015; Luo and O’Leary, 2005). Animals undergoing metamorphosis prune existing neural circuits before establishing new ones (Emoto, 2011; Schuldiner and Yaron, 2015). Further-more, physical insults can cause neuronal processes to degenerate. Consequently, efficient clearance of pruned or damaged neurites by phagocytes is critical for maintaining tissue homeostasis and preventing inflammation. Failure of clearance may cause secondary neurodegeneration and autoimmune disease (Nagata, 2010; Salter and Stevens, 2017).
How phagocytes recognize and clear neurites destined to be removed, while sparing other parts of the same neuron, is unclear in vivo. In comparison, apoptotic cell recognition by phagocytes is known to rely on ‘‘eat-me’’ signals exposed on the surface of dying cells (Ravichandran, 2010). A well-defined eat-me signal is phosphatidylserine (PS), a phospholipid found exclusively in the cytoplasmic leaflet of most healthy cells (Segawa and Nagata, 2015). During apoptosis, PS is externalized and flags the cell for engulfment by directly or indirectly interacting with engulfment receptors on phagocytes. The asymmetric PS distribution across the plasma membrane is established and maintained by aminophospholipid flippases encoded by the P4-ATPase family (Natarajan et al., 2004; Tang et al., 1996), whereas PS externalization is promoted by phospholipid scramblases. TMEM16F is a calcium-dependent scramblase required for PS exposure in platelet activation (Fujii et al., 2015; Suzuki et al., 2010). XKR family members, including mammalian XKR8, are caspase-activated scramblases that stimulate PS exposure in apoptotic cells (Suzuki et al., 2013).
Recent evidence links PS exposure to neurite degeneration. Dorsal root ganglion neurons induced to degenerate in vitro expose PS on their axons (Kim et al., 2010; Wakatsuki et al., 2017). Knockdown of the scramblase XKR8 or a lipid translocase ABC1 reduces PS exposure during degeneration (Wakatsuki and Araki, 2017). Moreover, PS recognition pathways are implicated in the phagocytosis of neurons. The mammalian TAM receptors (Tyro3, Axl, and Mertk) mediate phagocytosis of apoptotic cells by interacting with two PS binding proteins, Growth arrest-specific protein 6 (Gas6) and Protein S (Pros1) (Lemke, 2013; Lew et al., 2014). Loss of Axl and Mertk in the adult mouse CNS results in impaired cell clearance after viral infection (Tufail et al., 2017) and accumulation of apoptotic cells in the CNS (Fourgeaud et al., 2016). Another secreted PS-binding protein lactadherin (alternatively called MFG-E8), which interacts with the engulfment receptor avb3 integrin (Andersen et al., 2000; Hanayama et al., 2002), stimulates the phagocytosis of PS-exposing photoreceptor outer segments (POSs) in the mouse retina (Nandrot et al., 2007) and that of viable neurons during neuroinflammation induced by lipopolysaccharide treatment (Fricker et al., 2012). In addition, exogenous PS treatment has been shown to be neuroprotective in a degenerative disease model (Naftelberg et al., 2016).
Despite these evidences, several important questions regarding the roles of PS exposure in neurodegeneration remain unanswered. PS exposure induced on live neurons can lead to phagocytosis by co-cultured microglia (Brown and Neher, 2014). However, PS exposure on live cells does not necessarily lead to engulfment in vivo (Appelt et al., 2005; Heemskerk et al., 2002; Segawa et al., 2011). Therefore, direct in vivo evidence is required to demonstrate the sufficiency of PS exposure in driving neuronal degeneration. Mutations in mouse Atp8a2,a P4-ATPase gene, are associated with axon degeneration (Zhu et al., 2012), but the mechanism is unknown. Because PS exposure is associated with cell-autonomous membrane vesicle shedding in many contexts (Lima et al., 2009; Morel et al., 2004; Muralidharan-Chari et al., 2009), it is unclear whether PS exposure can by itself induce cell-autonomous loss of neurite membranes or whether degeneration is a consequence of phagocyte activity. Additionally, both flippase inactivation and scramblase activation can result in PS exposure in non-apoptotic cells (Segawa and Nagata, 2015), but whether they produce similar effects in neurons remains to be determined. Lastly, it is unknown whether different spatial domains of the same neuron are equally sensitive to disruptions of PS asymmetry under pathological conditions.
To address these questions, we utilized the Drosophila dendritic arborization (da) neurons that extend dendritic arbors underneath the larval epidermis (Grueber et al., 2002; Han et al., 2012; Kim et al., 2012). During dendrite remodeling and after dendrite injury, epidermal cells function as the primary phagocytes that break down and engulf da dendrites (Han et al., 2014) in a process similar to the clearance of injured sensory axons by zebrafish epidermal cells (Rasmussen et al., 2015). In this study, we examined the spatiotemporal pattern of PS exposure on degenerating dendrites using genetically coded GFP-tagged PS binding proteins (Mapes et al., 2012) and investigated the roles of nicotinamide adenine dinucleotide (NAD+) depletion and caspase activity in injury-induced PS exposure. Furthermore, we studied the effects of ectopically induced PS exposure in uninjured neurons in the larval peripheral nervous system (PNS) and the adult CNS. Lastly, we found that extracellular lactadherin potentiates Drosophila epidermal cells to destruct PS-exposing dendrites through an unidentified bridging domain. Together, our study reveals in vivo dynamics of PS exposure on degenerating neurites and establishes a direct functional association between PS exposure and phagocytosis-dependent neurite loss in Drosophila.
RESULTS
PS Is Dynamically Exposed on Degenerating Dendrites after Injury and during Dendrite Remodeling
Building on two PS-binding proteins, Annexin V (AV) (Koopman et al., 1994) and the lactadherin C1C2 domain (LactC1C2) (Andersen et al., 2000), we developed an in vivo system in Drosophila larvae to visualize surface PS externalization on cells exposed to the hemolymph. The GFP-tagged, secreted forms of these proteins (Mapes et al., 2012) are referred to as AV-GFP and GFP-Lact. LactC1C2 specifically binds to PS but lacks the integrin-interacting motif of the full-length protein (Hanayama et al., 2002). AV-GFP and GFP-Lact secreted by the fat body diffuse to da neuron dendrites labeled by CD4-tdTom (Han et al., 2011) via the hemolymph and decorate the outer surface of the branches exposing PS (Figure 1A).
Figure 1. Phosphatidylserine Is Dynamically Exposed on Degenerating Dendrites after Injury and during Dendrite Remodeling.
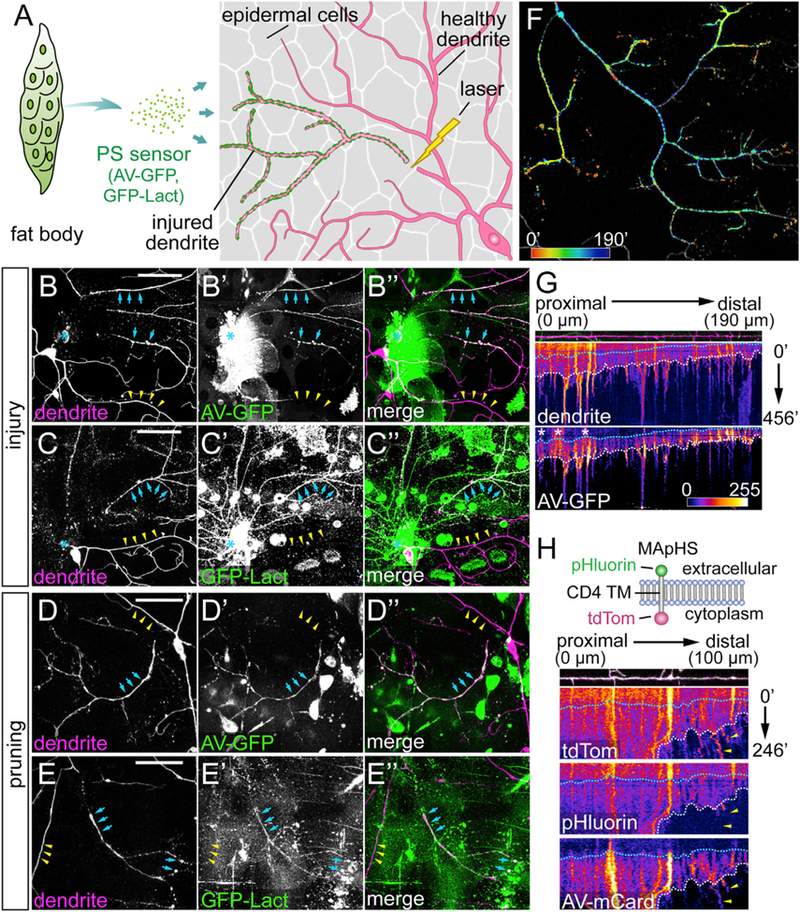
(A) Experimental design for visualizing PS exposure on injured dendrites of C4 da neurons.
(B–C”) Labeling of injured C4 da dendrites by AV-GFP (B–B”) and GFP-Lact (C–C”) at 4–5 hr after injury (AI). Blue arrows: injured dendrites; yellow arrowheads: uninjured dendrites; blue asterisk: injury sites.
(D–E”) Labeling of severed C4 da dendrites by AV-GFP (D–D”) and GFP-Lact (E–E”) during dendrite pruning between 6 and 7 hr after puparium formation (APF). Blue arrows: severed dendrites; yellow arrowheads: not-yet-severed dendrites. Some hemocytes are visible in the GFP channel in (B)–(D”) because Dcg-Gal4 also drives expression in hemocytes.
(F) Temporal sequence of AV-GFP binding to injured dendrites (Video S3). Red: early binding; blue: late binding. Indicated times are relative to the first frame.
(G) Kymographs of morphology and AV-GFP binding of an injured dendrite. The top panel shows the straightened dendrite with indicated distances relative to the proximal (left) end. The middle and bottom panels show kymographs of tdTom and AV-GFP signals, respectively, with indicated times relative to the first frame. White dotted lines: timing of dendrite fragmentation based on the continuity of the dendrite signal; blue dotted lines: timing of AV-GFP initial binding to the dendrite; white asterisks: interfering AV-GFP signals from injured dendrites of other da neurons.
(H) Kymographs of engulfment and AV-mCard binding of an injured dendrite. Top panel: diagram of the MApHS marker (adapted from Han et al., 2011). Dendrite and kymograph panels are arranged similar to (G). White dotted lines: timing of dendrite fragmentation; blue dotted lines: timing of AV-mCard initial binding; yellow arrowheads: engulfed dendrite debris (based on the loss of pHluorin) still associated with AV-mCard.
Scale bars: 50 μm (B–E). See also Figure S1 and Videos S1, S2, S3, and S4.
We examined PS exposure of class IV da (C4 da) neurons both after dendrite injury and during dendrite pruning. At 4–5 hr after laser injury, severed dendrites actively underwent blebbing and fragmentation (Han et al., 2014; Tao and Rolls, 2011) and exhibited robust labeling by both AV-GFP (Figures 1B–1B”, blue arrows, n = 29 neurons) and GFP-Lact (Figures 1C–1C″, blue arrows, n = 22 neurons). In contrast, uninjured dendrites of ablated neurons (Figures 1B–1C″, yellow arrowheads) or healthy neurons (Figure S1A–S1A″) did not show any labeling. PS sensor labeling correlated with the stage of dendrite degeneration: smooth and healthy-looking branches often did not show obvious labeling, whereas branches with roughened surfaces or blebs usually showed strong labeling. As controls, AV(mut)-GFP (Figures S1B–S1B″; n = 24 neurons) and GFP-Lact(mut) (Figures S1C–S1C″; n = 17 neurons), which carry mutations that abolish their ability to interact with PS (Dubois et al., 1998; Lin et al., 2007; Mapes et al., 2012), did not label any injured dendrites.
For dendrite pruning, we used time-lapse imaging to examine PS exposure on C4 da dendrites, which exhibit asynchronized severing and fragmentation (Kanamori et al., 2013). AV-GFP consistently labeled the branches that were degenerating, but not before branches showed any sign of degeneration (Figures 1D–1D″; Video S1). GFP-Lact labeled pruned dendrites (Figures 1E–1E”; Video S2) less consistently than AV-GFP. During metamorphosis, many larval tissues underwent large-scale apoptosis, possibly sequestering GFP-Lact away from pruned dendrites. Nevertheless, these results demonstrate that PS is specifically exposed on degenerating dendrites both after injury and during pruning.
To further investigate the timing of PS exposure on degenerating dendrites, we recorded AV-GFP labeling of injured dendrites in larvae using long-term time-lapse imaging (Poe et al., 2017), which began at 1.5–2 hr after injury (AI) and lasted 5–7 hr, long enough to capture most of the dendrite degeneration process (Han et al., 2014). Uninjured dendrites exhibit normal growth dynamics (e.g., extension, retraction, and turning) and no AV-GFP binding, signifying the health of the larvae (Video S3). In contrast, severed dendritic arbors exhibit specific spatiotemporal patterns of AV-GFP labeling (Video S3). The peak of AV-GFP binding usually started at terminal branches and then spread to low-order branches, as apparent in color-coded timing of AV-GFP peak signal on the dendritic arbor (Figure 1F), suggesting that the PS membrane asymmetry is disrupted earlier at high-order branches than at lower-order branches. Kymographs for visualizing changes of dendrite morphology and AV-GFP binding over time (Figure 1G) further show that the appearance of AV-GFP signals (indicated by the blue dotted line) correlated with roughening of the dendritic membrane and occurred before dendrite fragmentation (indicated by the white dotted line). Both AV-GFP labeling and dendrite fragmentation proceeded from the distal end of the dendrite to the proximal end. These data suggest that PS exposure is among the early events of dendrite degeneration AI.
To serve as an eat-me signal for degenerating dendrites, PS should be externalized before debris engulfment. Using MApHS, a pH sensitive dendritic marker consisting of extracellular pHluorin and intracellular tdTom (Han et al., 2014), to monitor engulfment and AV-mCardinal (AV-mCard) (Chu et al., 2014) to visualize PS exposure, we found that AV-mCard labeling (blue dotted line) appeared much earlier than engulfment (Figure 1H), which is signified by the loss of pHluorin signal due to the drop of pH in early phagosomes (Botelho and Grinstein, 2011) and mostly correlated with dendrite fragmentation (white dotted line). AV-mCard labeling can persist on dendrite debris even after engulfment (yellow arrowheads).
Together, our results demonstrate that PS is specifically and dynamically exposed on degenerating dendrites both in injury and during dendrite remodeling. The timing of PS exposure is consistent with its potential role as an eat-me signal.
PS Exposure on Injured Dendrites Depends on NAD+ Depletion but Does Not Require Caspase Activity
Injured neurites degenerate following a sequence of signaling events triggered by NAD+ depletion (Gerdts et al., 2016). Expression of Wlds, a chimeric protein containing nicotinamide mononucleotide adenyltransferase (NMNAT1), which synthesizes NAD+, in neurons prevents neurite degeneration AI (Mack et al., 2001). Interestingly, transected axons of retinal ganglion cells isolated from transgenic Wlds rats showed a delay of PS exposure and halved the speed of PS exposure spread (Almasieh et al., 2017). To test whether PS exposure on injured dendrites is also regulated by NAD+ depletion in vivo, we examined Wlds-overexpressing C4 da neurons. At 10 hr AI, the majority of wild-type neurons showed complete fragmentation of injured dendrites, and the remaining dendrite segments were consistently labeled by GFP-Lact (Figures 2A–2A” and 2E). In contrast, injured dendrites of Wlds-overexpressing neurons showed little sign of fragmentation at 10 hr AI (Figures 2B–2B” and 2E) or 24 hr AI. Neither did they show any GFP-Lact labeling (yellow arrowheads, Figure 2F), even though injured dendrites of other da neurons lacking Wlds showed strong GFP-Lact labeling in the same segment (blue arrows). Occasionally (18%, n = 17), injured Wlds-expressing dendrites broke into large segments that exhibited local enlargement and thinning without GFP-Lact labeling (Figures 2C–2C”, yellow arrowheads).
Figure 2. PS Exposure in Injured Dendrites Depends on NAD+ Depletion but Does Not Require Caspase Activity.
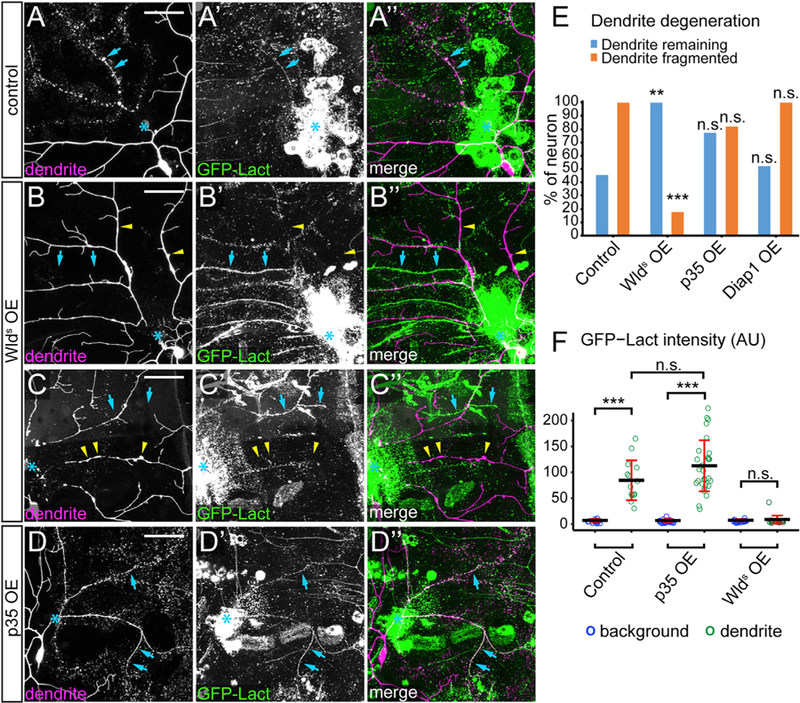
(A–A”) A control C4 da neuron at 8–10 hr AI. Blue arrows: fragmenting dendrites.
(B–C”) Wlds-overexpressing (OE) C4 da neuron at 8–10 hr AI showing unfragmented dendrites (B–B”) and fragmented dendrites (C–C”). Yellow arrowheads: injured dendrites of C4 da neurons; blue arrows: injured dendrites of other classes of neurons that were labeled by GFP-Lact.
(D) A p35 OE neuron at 8–10 hr AI. Blue arrows: fragmenting dendrites.
(E) Quantification of dendrite degeneration showing percentages of neurons with uncleared dendrites and fragmented dendrites. n = number of neurons: control (n = 11, 7 animals); Wlds OE (n = 17, 10 animals); p35 OE (n = 22, 11 animals); Diap1 OE (n = 21, 11 animals). Fisher’s exact test, **p < 0.01; ***p < 0.001, n.s., not significant.
(F) Quantification of GFP-Lact binding on dendrites. GFP-Lact intensity is shown for both background epidermal regions and GFP-Lact-labeled dendrites. n = number of measurements: control (n = 15, 5 animals); p35 OE (n = 30, 9 animals), Wlds OE (n = 24, 4 animals). ***p % 0.001; n.s., not significant; Kruskal-Wallis (one-way ANOVA on ranks) and Dunn’s test; p values adjusted with the Benjamini-Hochberg method. Black bar, mean; red bars, SD.
Scale bars represent 50 μm (A–D). Blue asterisks indicate injury sites (A–D). See also Figure S2.
Caspase activity is involved in neurite pruning (Kuo et al., 2006; Schoenmann et al., 2010; Simon et al., 2012; Williams et al., 2006). During apoptosis, caspases inhibit P4-ATPases and activate XKR scramblases (Segawa and Nagata, 2015), and consequentially promote PS exposure. To test whether caspase activity is also required for PS exposure after dendrite injury, we expressed the caspase inhibitor p35 (Williams et al., 2006) in C4 da neurons. We did not observe statistically significant delay of dendrite degeneration (Figure 2E) or weaker GFP-Lact labeling on injured dendrites (Figures 2D–2F). Neurons overexpressing Diap1, a Drosophila inhibitor of caspases (Hay and Guo, 2006), showed normal degeneration and GFP-Lact labeling of injured dendrites (Figure 2E). Consistent with these results, caspase activity was not detected in injured dendrites of wild-type or p35-expressing neurons using a caspase activity reporter CD8::PARP::Venus (Williams et al., 2006) (Figure S2). Together, our results suggest that PS exposure on injured dendrites requires NAD+ depletion, but not caspase activity.
CDC50 Knockout and TMEM16F Overexpression Disrupt PS Asymmetry Preferentially at Distal Dendrites
To understand the functional consequences of PS exposure, we sought to ectopically induce PS exposure on otherwise healthy neurons by removing P4-ATPases or overexpressing scramblases. The Drosophila genome encodes six P4-ATPases (Tanaka et al., 2011), five of which are predicted to require the chaperone CDC50 for proper subcellular localization and function (Paulusma et al., 2008; Saito et al., 2004; Tanaka et al., 2011). To maximize the chance of causing ectopic PS exposure, we knocked out the only Drosophila CDC50 gene, CG9947 (referred to as CDC50 hereafter), in C4 da neurons using a C4 da-specific ppk-Cas9 and two ubiquitously expressed guide RNAs (gRNAs) targeting CDC50. These CDC50 mutant neurons were labeled by AV-GFP in bright puncta along high-order branches of distal dendrites (yellow arrowheads) and also smoothly on the shafts of low-order branches (blue arrows, Figures 3A–3A”). AV-GFP puncta appeared to be intracellular endosomes (Figures S3A–S3C”) and were also found in the cell body (Figures S3A–S3A”), possibly because of retrograde trafficking from dendrites. In contrast with AV-GFP, which did not seem to affect the morphology of CDC50 knockout neurons, constitutive GFP-Lact expression in the fat body caused drastic dendrite degeneration of CDC50 mutant neurons (more details later). To alleviate this degeneration, we transiently expressed GFP-Lact for 24 hr using temperature-sensitive Gal80 (McGuire et al., 2003), which resulted in varying degrees of GFP-Lact labeling on CDC50 knockout neurons, with the strongest labeling typically seen at the most distal dendrites (Figure S3F). The GFP-Lact labeling was weak and smooth on dendrites without obvious sign of degeneration (Figures 3B–3B”, inset 1), whereas laser-injured dendrites of the same neurons exhibited much stronger GFP-Lact labeling (Figures 3B–3B”, inset 2), suggesting that loss of flippases causes low PS exposure preferentially at the distal dendritic arbor, and mechanisms other than P4-ATPase inactivation must exist to boost PS exposure AI.
Figure 3. CDC50 Knockout and TMEM16F Overexpression Disrupt PS Asymmetry Preferentially at Distal Dendrites.
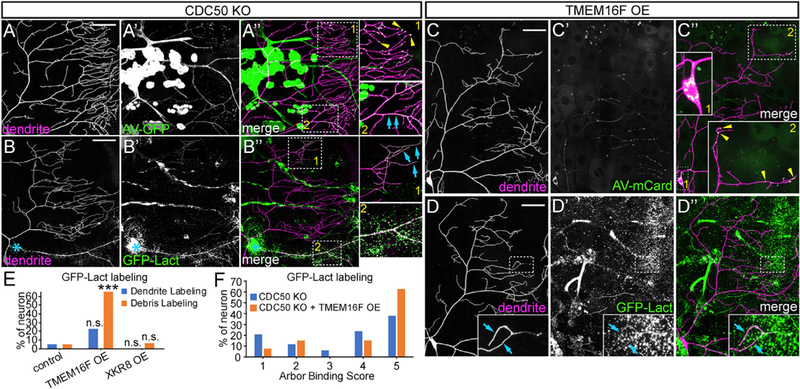
(A–A”) CDC50 knockout (KO) neuron labeled by AV-GFP. Yellow arrowheads: bright AV-GFP puncta; blue arrows: smooth AV-GFP labeling on dendrite shafts.
(B–B”) CDC50 KO neuron labeled by transiently induced GFP-Lact at 3–4 hr AI. Inset 1: weak, smooth labeling (blue arrows) on uninjured distal dendrites; inset 2: strong labeling on the injured dendrite; blue asterisks: injury sites.
(C–C”) TMEM16F OE neuron labeled by AV-mCard. Inset 1: AV-mCard in the cell body; inset 2: AV-mCard puncta along distal dendrites (yellow arrowheads).
(D–D”) TMEM16F OE neuron labeled by constitutively expressed GFP-Lact. Inset: smooth GFP-Lact labeling on distal dendrites (blue arrows).
(E)Percentage of neurons showing dendrite and debris labeling by constitutively expressed GFP-Lact. n = number of neurons: control (n = 20, 5 animals); TMEM16F OE (n = 31, 8 animals); XKR8 OE (n = 16, 4 animals). Fisher’s exact test, ***p < 0.001. n.s., not significant.
(F)Arbor binding scores of GFP-Lact-labeled neurons. n = number of neurons: CDC50 KO (n = 34, 10 animals, total neurons = 41); CDC50 KO + TMEM16F OE (n = 27, 10 animals, total neurons = 45). Scoring system is illustrated in Figures S3F and S3H. Scale bars: 50 μm (A–D).
See also Figure S3.
We next overexpressed mammalian scramblases TMEM16F and XKR8 in da neurons. A hyperactive TMEM16F mutant, TMEM16F D430G-L (abbreviated as TMEM16F), causes a high level of ectopic PS exposure on mouse lymphoma cells (Segawa et al., 2011). TMEM16F overexpression in da neurons caused AV-mCard labeling in half of the neurons (50%, n = 12, 6 animals) (Figures 3C–3C”) in puncta in the cell body (inset 1) and on distal dendrites (inset 2). Constitutive GFP-Lact expression did not cause large-scale degeneration of TMEM16F-expressing neurons but labeled distal high-order branches infrequently (Figures 3D and 3E) and debris near distal dendrites frequently (Figures S3D and 3E). XKR8 overexpression did not cause obvious labeling by AV-GFP (data not shown) or GFP-Lact (Figures 3E and S3E).
We then asked whether combining CDC50 knockout (KO) and TMEM16F overexpression (OE) (referred to as CDC50 KO + TMEM16F OE) would enhance PS exposure by scoring GFP-Lact labeling (Figures S3F and S3H). Indeed, among neurons that showed GFP-Lact labeling, higher percentages of CDC50 KO + TMEM16F OE neurons (Figure S3G) were strongly labeled than CDC50 KO alone (Figures 3F and S3F). Together, these data show that CDC50 KO and TMEM16F OE induce different degrees of PS exposure preferentially at distal dendrites.
Ectopic PS Exposure in Neurons Causes Membrane Loss at Distal Dendrites
We next examined whether the low-level PS exposure on uninjured dendrites induced by flippase KO or scramblase OE would cause recognition and engulfment of dendrites by phagocytic epidermal cells, which would result in tdTom-positive phagosomes resembling dendrite debris (Han et al., 2014). When CDC50 was knocked out in C4 da neurons, some dendrite debris was detected in epidermal cells underneath high-order dendrite branches (Figure 4B). To confirm that this phenotype is indeed caused by P4-ATPase loss of function (LOF), we knocked out CG42321, the closest Drosophila homolog for human 8A class of P4-ATPases (Tanaka et al., 2011) (referred to as ATP8A hereafter), in C4 da neurons and also detected dendrite debris in epidermal cells (Figures 4C and 4J). Knocking out both CDC50 and ATP8A did not enhance debris formation (Figures 4D and 4J), consistent with the notion that ATP8A and CDC50 function in the same complex.
Figure 4. Ectopic PS Exposure in Neurons Causes Membrane Loss at Distal Dendrites.
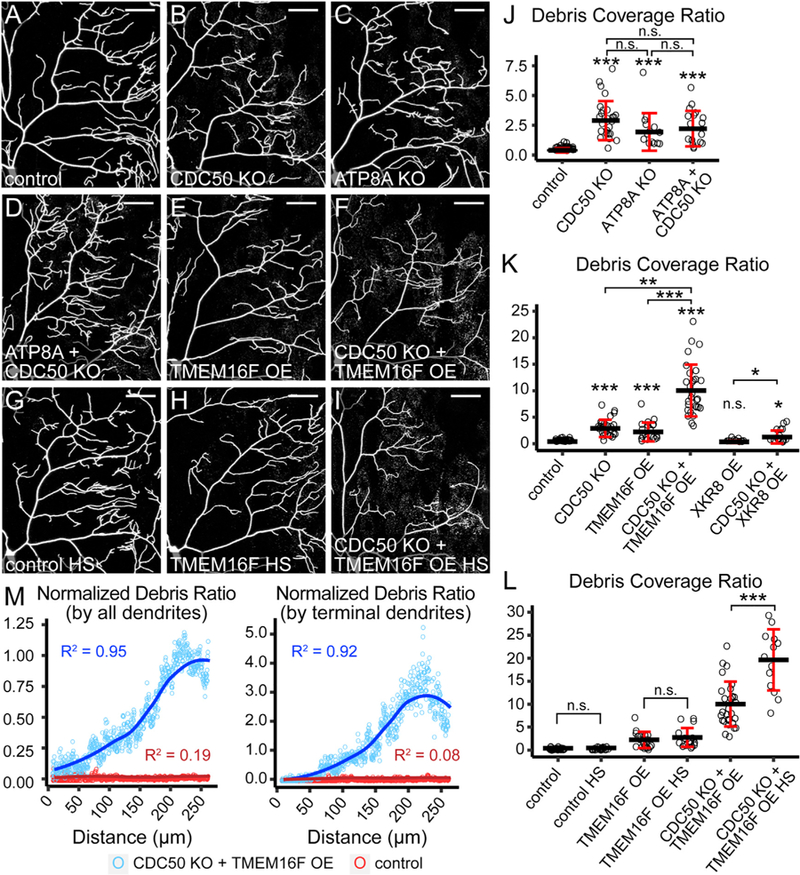
(A–F) Partial dendritic fields of control (A), CDC50 KO (B), ATP8A KO (C), ATP8A and CDC50 double KO (D), TMEM16F OE (E), and CDC50 KO + TMEM16F OE (F) C4 da neurons.
(G–I) Partial dendritic fields of control (G), TMEM16F OE (H), and CDC50 KO + TMEM16F OE (I) C4 da neurons after heat shock (HS) treatments.
(J and K) Quantification of debris coverage showing effects of flippase KO (J) and scramblase OE (K). The debris coverage ratio is debris area ratio normalized by dendrite area ratio. n = number of neurons: control (n = 35, 16 animals); CDC50 KO (n = 25, 11 animals); ATP8A KO (n = 14, 9 animals); ATP8A + CDC50 KO (n = 17, 9 animals); TMEM16F OE (n = 19, 9 animals); CDC50 KO + TMEM16F OE (n = 29, 12 animals); XKR8 OE (n = 10, 8 animals); CDC50 KO + XKR8 OE (n = 16, 9 animals).
*p % 0.05, **p % 0.01, ***p % 0.001, Kruskal-Wallis (one-way ANOVA on ranks) and Dunn’s test; p values adjusted with the Benjamini-Hochberg method.
(L)Quantification of debris coverage with or without heat shocks. n = number of neurons: control (n = 35, 16 animals); control HS (n = 23, 12 animals); TMEM16F OE (n = 19, 9 animals); TMEM16F OE HS (n = 18, 10 animals); CDC50 KO + TMEM16F OE (n = 29, 12 animals); CDC50 KO + TMEM16F OE HS (n = 13, 7 animals).
***p % 0.001, pairwise t test; p values adjusted with the Bonferroni method. n.s., not significant.
(M)Correlation between the debris ratio and the distance from the cell body in control (red) and CDC50 KO + TMEM16F OE (blue). The average debris ratio at a given distance is average debris particle number divided by either total dendrite number (left panel) or terminal dendrite number (right panel). Circle: average debris ratio; solid line: polynomial fit of the average debris ratio; R2: coefficient of determination of the linear regression. n = number of neurons: control (n = 35, 16 animals); CDC50 KO + TMEM16F OE (n = 29, 12 animals).
Scale bars: 50 mm. Black bar: mean; red bars: SD. The control in (J)–(L) is the same dataset; CDC50 KO in (J) and (K) is the same dataset; TMEM16F OE in (K) and (L) is the same dataset; CDC50 KO + TMEM16F OE in (K) and (L) is the same dataset. See also Figure S4. n.s., not significant.
Whereas knocking out CDC50 and ATP8A individually or in combination did not affect C4 da dendrite morphology (Figures S4A–S4D), TMEM16F OE caused a mild dendrite reduction (Figures S4E, S4J, and S4K). Consistent with their weaker PS-exposure phenotypes, TMEM16F-overexpressing neurons also produced a lower level of dendrite debris (Figures 4E and 4K). In contrast, XKR8 OE caused strong dendrite reduction (Figures S4H, S4J, and S4K) without generating dendrite debris (Figures 4K and S4N), suggesting that the dendrite reduction is due to dendrite growth defects rather than epidermal engulfment. Interestingly, CDC50 KO + TMEM16F OE neurons showed a much greater level of debris than what would be expected from an additive effect of CDC50 KO and TMEM16F overexpression (Figures 4F and 4K), suggesting that these two manipulations synergistically boost the loss of dendrite membranes. As a result, compared with TMEM16F OE alone, CDC50 KO + TMEM16F OE neurons had fewer high-order dendrite branches (Figures S4F and S4J), even though arbor sizes were not affected (Figure S4K). In contrast, CDC50 KO and XKR8 OE did not show synergistic effects on debris formation (Figures 4K and S4O) or dendrite reduction (Figure S4I, S4J, and S4K).
The scramblase activity of TMEM16F is activated by calcium. We asked whether increasing intracellular calcium levels by activating C4 da neurons would further enhance the membrane loss of CDC50 KO + TMEM16F OE neurons. Because C4 da neurons respond to a variety of noxious stimuli including high temperatures (Hwang et al., 2007), we subjected larvae to brief heat shocks and examined C4 da neurons 30 min later. The heat shock treatment greatly increased the debris level (Figures 4I and 4L) and further reduced high-order dendrites (Figures S4G, S4J, and S4K) of CDC50 KO + TMEM16F OE neurons but had no effect on wild-type neurons (Figures 4G and 4L). Interestingly, heat shocks did not affect TMEM16F OE alone (Figures 4H and 4L), suggesting that flippase loss is required for the enhancement effect of TMEM16F. Together, these data demonstrate that loss of P4-ATPase activity and TMEM16F activation in C4 da neurons synergistically causes engulfment of dendrite membrane by resident phagocytes.
Consistent with the preferential PS exposure at distal dendrites of CDC50 KO and TMEM16F OE (Figure 3), dendrite debris also correlated with the distance from the cell body: the debris level is greater the farther away from the cell body and peaks near the edge of the dendritic field when normalized by either the number of all dendrite branches or that of terminal dendrites (Figure 4M). This spatial preference of membrane loss is not due to TMEM16F enrichment at distal dendrites, because immunostaining showed that the TMEM16F protein was mostly found at the cell body and proximal dendrites (Figures S4L–S4L”) and was below detection at distal high-order branches (Figures S4M–S4M”). Together, these data suggested that distal dendrites of C4 da neurons are most susceptible to membrane loss due to perturbation of PS asymmetry caused by LOF of P4-ATPases or gain of function (GOF) of calcium-activated scramblases.
PS-Exposing Dendrites Shed Membrane Vesicles in a Phagocyte-Dependent Manner
To investigate how PS-exposing dendrites lose membranes, we conducted time-lapse imaging of CDC50 KO + TMEM16F OE neurons and observed two ways by which dendrites shed membrane vesicles. More often, terminal dendrites shed vesicles from branch ends during retraction (Figure 5A; Video S5). Sometimes vesicles were shed from dendritic shafts (Figure 5B; Video S6), resembling the ‘‘shedosomes’’ emanated from dendritic shafts during dendrite pruning (Han et al., 2011). Both kinds of membrane shedding were rarely observed on wild-type neurons (Figure 5C), indicating that they are unique to PS-exposing dendrites. To ask whether PS exposure is sufficient to drive cellautonomous membrane shedding, we examined animals mutant for drpr, which encodes an engulfment receptor necessary for larval epidermal cells to engulf dendrite debris (Han et al., 2014). We generated drprindel, a null allele, using CRISPR/ Cas9. Strikingly, neither CDC50 KO neurons (Figure 5E) nor CDC50 KO + TMEM16F OE neurons (Figure 5F) produced detectable debris in the drprindel background, similar to control neurons in the drprindel background (Figure 5D) or in wild-type animals (Figure 5G). These data suggest that vesicle shedding of PS-exposing dendrites results from Drpr-mediated attack and engulfment by epidermal cells on still live dendrites.
Figure 5. PS-Exposing Dendrites Shed Membrane Vesicles in a Phagocyte-Dependent Manner.
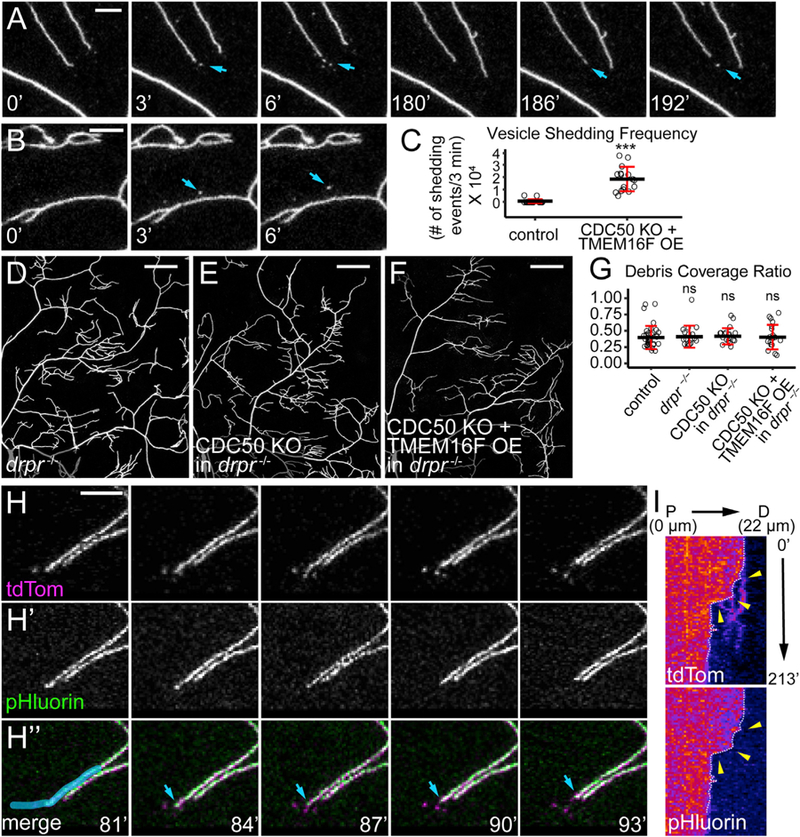
(A and B) Time series of CDC50 KO + TMEM16F OE dendrites showing vesicle shedding from a terminal branch (A) and a dendritic shaft (B). Indicated times are relative to the first frame.
(C) Vesicle shedding frequency defined as the number of shedding events per terminal dendrite in 3 min multiplied by 104. n = number of neurons: control (n = 21, 7 animals); CDC50 KO +TMEM OE (n = 16, 5 animals). ***p % 0.001, Kruskal-Wallis (one-way ANOVA on ranks) and Dunn’s tests.
(D–F) Partial dendritic fields of control (D), CDC50 KO (E), and CDC50 KO + TMEM16F OE (F) neurons all in the drpr—/— background.
(G) Quantification of debris coverage. n = number of neurons: control (n = 35, 16 animals); drpr—/— (n = 17, 9 animals); CDC50 KO in drpr—/— (n = 19, 11 animals); CDC50 KO + TMEM16F OE in drpr—/— (n = 19, 9 animals). ANOVA, H0 is true. n.s., not significant.
(H) A time series of CDC50 KO + TMEM16F OE dendrites labeled by MApHS. Blue arrows: the tip of a vesicle-shedding terminal dendrite.
(I) Kymographs of membrane shedding and engulfment of the dendrite overlaid by blue color in (H”). White dotted lines: the tip of the dendrite in each frame; yellow arrowheads: vesicle shedding events.
Scale bars represent 10 μm (A, B, and H); 50 μm (D–F). Black bar: mean; red bars: SD (C and G). See also Videos S5, S6, and S7.
Mammalian macrophages have been reported to degrade unengulfed apoptotic cells by exocytosing lysosomal contents in a process called exophagy (Haka et al., 2016). To investigate whether epidermal cells also use similar mechanisms to acidify and degrade dendrite tips before separating them from the branches, we examined dendrite vesicle shedding with the MApHS marker. In all cases of vesicle shedding from dendrite tips (n = 77), the pHluorin signal persisted on the tips before shedding, and pHluorin quenching occurred at or shortly after the separation of vesicles from the branches (Figures 5H–5H and 5I; Video S7), suggesting that epidermal cells do not acidify tips of terminal dendrites, and that epidermal cells may break dendrite tips by physical force (Han et al., 2014).
Flippase Loss and Scramblase OE Cause Distinct Modes of Axonal Degeneration in the Adult Fly Brain
We next asked whether flippase loss or scramblase OE could also cause neurons to lose axonal membrane in the CNS by examining adult Or22a neurons, which are a well-established model for studying axon degeneration and clearance in the brain (MacDonald et al., 2006). Or22a neurons are olfactory receptor neurons (ORNs) on the antenna that respond to ethyl butyrate and project axons to the DM2 glomeruli in the antennal lobe of the adult brain (Dobritsa et al., 2003). We knocked out CDC50 in mature Or22a neurons labeled by Or22a-Gal4 UAS-mCD8-GFP to avoid potential early developmental defects. In 3-day-old adults, wild-type (Figure 6A) and CDC50 KO (Figure 6B) neurons showed no differences in axonal GFP patterns and intensities (Figure 6E), whereas in 25-day-old adults, CDC50 KO axons (Figures 6D, 6E, and S5F) showed much lower GFP than the wild-type (Figure 6C) and contained many GFP aggregates (inset of Figure 6D), indicating axonal degeneration.
Figure 6. Flippase Loss and Scramblase Overexpression Cause Distinct Modes of Axonal Degeneration in the Adult Fly Brain.
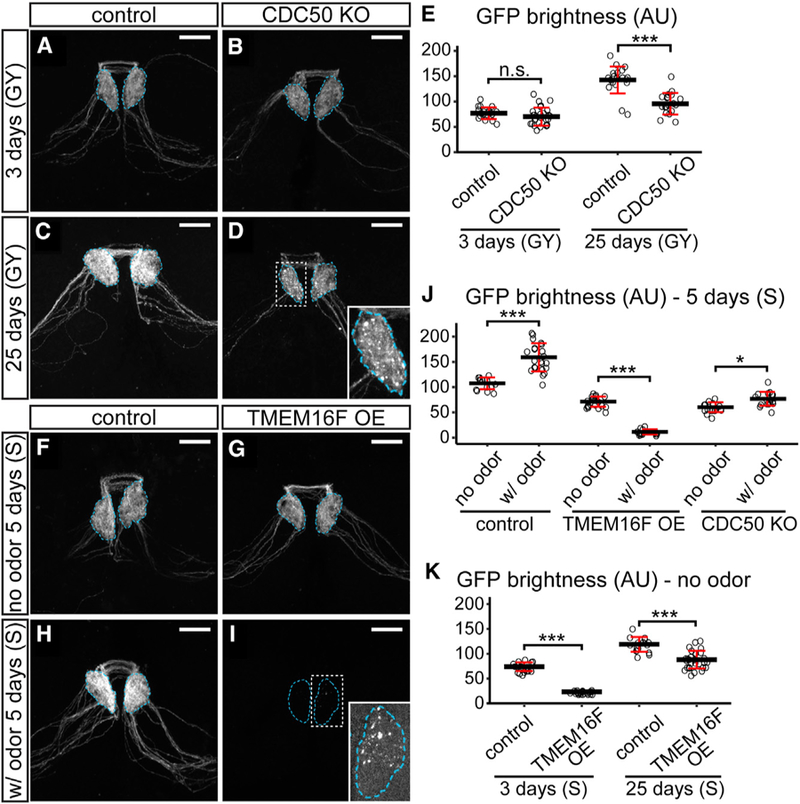
(A–D) Axons of control (A) and CDC50 KO (B) Or22a neurons in 3-day-old adult brains and axons of control (C) and CDC50 KO (D) OR22a neurons in 25-day-old brains. The inset in (D) is an enlarged glomerulus.
(E) Quantification of GFP brightness of Or22a glomeruli in age tests. n = number of brains: control 3 days (n = 19); CDC50 KO 3 days (n = 26); control 25 days (n = 21); CDC50 KO 25 days (n = 19). ***p % 0.001, one-way ANOVA and Tukey’s HSD. n.s., not significant.
(F–I) Axons of control (F) and TMEM16F OE (G) Or22a neurons in 5-day-old adult flies that were not exposed to the odor and control (H) and TMEM16F OE (I) axons in flies that were exposed to the odor. The inset in (I) is an enlarged glomerulus imaged at a much brighter setting.
(J) Quantification of GFP brightness in odor tests. n = number of brains: control no odor (n = 14); control with (w/) odor (n = 22); TMEM16F OE no odor (n = 21); TMEM16F OE w/ odor (n = 16); CDC50 KO no odor (n = 16); CDC50 KO w/ odor (n = 18). *p % 0.05; ***p % 0.001, one-way ANOVA and Tukey’s HSD test.
(K) Quantification of GFP brightness in age tests. n = number of brains: control 3 days (n = 20); TMEM16F OE3 days (n = 21); control 25 days (n = 16); TMEM16F OE 25 days (n = 29). ***p % 0.001, one-way ANOVA and Tukey’s HSD.
In all image panels, Or22a glomeruli are outlined. The types of food used are indicated for each panel: GY, glucose yeast medium; S, sucrose agar. Scale bars: 20 μm. Black bars: mean; red bars: SD. See also Figure S5.
To examine the effects of overexpressing TMEM16F in Or22a neurons, we compared 5-day-old flies exposed to ethyl butyrate with those not exposed to any odorant, all kept on sucrose agar to avoid potential exposure to odorants from the food, expecting that ethyl butyrate exposure would activate Or22a neurons and hence promote TMEM16F activity. In the absence of the odorant, TMEM16F OE slightly reduced axonal and somatic GFP signals (Figures 6G and S5A) compared with wild-type neurons (Figures 6F, 6J, and S5A). In the presence of the odorant, whereas wild-type neurons showed increased axonal and somatic GFP signals (Figures 6H, 6J, and S5B), TMEM16F-expressing neurons became fewer (Figures S5B) and their axons were largely missing except for few weak GFP-positive membrane blebs visible only under a much higher detection setting or by GFP staining (Figure 6I and S5C). The DM2 glomeruli in these TMEM16-expressing brains appeared to be much smaller and express much less the synaptic marker Brp (Figure S5C). These data suggest that activation of TMEM16F scramblase in adult ORNs causes strong axonal degeneration.
In comparison, odor exposure did not decrease axonal signals of CDC50 KO neurons in 5-day-old flies (Figures 6J and S5D). Without odor exposure, TMEM16F-expressing axons did not degenerate with age: although they were much dimmer than controls in 3-day-old flies (Figures S5E and 6K), the signals increased rapidly in 5-day-old flies (Figures 6G and 6J) and further increased in 25-day-old flies (Figures S5E, S5F, and 6K). Interestingly, the somatic signals of TMEM16F-overex-pressing neurons were comparable with those of the control in 3-day-old flies (Figure S5A), indicating a delay of axonal growth in young animals. Together, our results suggest that loss of P4-ATPase activity and TMEM16F OE causes distinct modes of axonal degeneration in ORNs: the former results in gradual and age-dependent axon degeneration, whereas the latter causes fast and neuronal activity-dependent axon degeneration.
The LactC1C2 Induces Engulfment-Dependent Dendritic Degeneration of PS-Exposing Neurons
Although CDC50 KO C4 da neurons shed membrane at distal dendrites, their primary dendrites did not degenerate (Figures 7A–7A”). Surprisingly, in the presence of constitutively expressed GFP-Lact, most CDC50 KO neurons showed severe degeneration (Figures 7B–7B”) and loss of high-order dendrites (Figure 7D), with membrane roughening and fragmentation on remaining branches. Strong GFP-Lact labeling was detected on dendrites and most cell bodies (14/20 neurons). ATP8A KO in C4 da neurons produced similar results in the presence of constitutively expressed GFP-Lact (Figures S6A–S6A”), confirming that these phenotypes are related to the loss of P4-ATPase activity. To test whether GFP-Lact induces degeneration of PS-exposing neurons in the absence of epidermal phagocytic activity, we conducted similar experiments in the drprindel background. Strikingly, drprindel homozygosity completely rescued the degeneration and reduction of CDC50 KO dendrites (Figures 7C and 7D), suggesting that GFP-Lact-induced degeneration is engulfment dependent. Consistent with the role of P4-ATPases, GFP-Lact was detected on distal dendrites of CDC50 KO neurons in drprindel (Figures 7E–7E”), albeit at a much lower level than that on CDC50 KO neurons in the wild-type background (Figure 7F). Together, these data argue that GFP-Lact functions as a bridging molecule and potentiates phagocyte-dependent destruction of dendrites exposing low levels of PS. Our results also suggest that the LactC1C2 domain contains an unidentified sequence that can interact with a Drosophila engulfment receptor, perhaps Drpr.
Figure 7. The Lactadherin C1C2 Domain Induces Engulfment-Dependent Dendritic Degeneration of PS-Exposing Neurons.
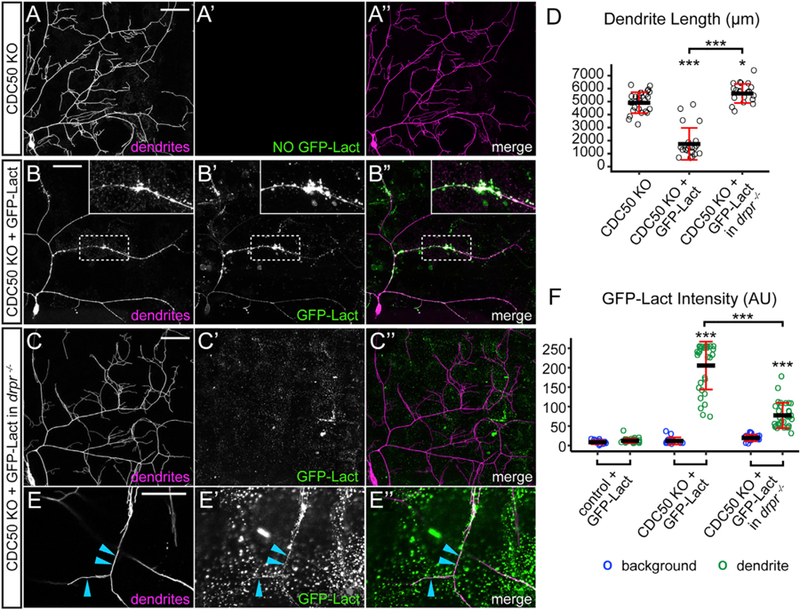
(A–C”) Partial dendritic fields of CDC50 KO neurons in the absence (A–A”) and presence (B–B”) of constitutive GFP-Lact expression. Inset in (B)–(B”): a segment of degenerating dendrite strongly labeled by GFP-Lact.
(C–C”) Partial dendritic fields of CDC50 KO neurons in drpr–/– background in the presence of constitutive GFP-Lact expression.
(D) Dendrite length in the partial ddaC dendritic field. n = number of neurons: CDC50 KO (n = 27, 9 animals); CDC50 KO + GFP-Lact (n = 20, 9 animals); CDC50 KO + GFP-Lact in drpr—/— (n = 21, 9 animals). *p % 0.05; ***p % 0.001, one-way ANOVA and Tukey’s HSD test.
(E–E”) High-resolution images of CDC50 KO distal dendrites in the presence of constitutively expressed GFP-Lact in drpr—/— background taken at a brighter GFP setting. Maximum projection of two Z slices covering 2-mm-thick volume. Blue arrows: GFP-Lact labeling on dendrites.
(F) Quantification of GFP-Lact binding on dendrites. Background: epidermal regions without dendrites. n = number of measurements: control background (n = 14) and control dendrite (n = 28), 7 animals; CDC50 KO + GFP-Lact background (n = 16) and CDC50 KO + GFP-Lact dendrite (n = 32), 8 animals; CDC50 KO + GFP-Lact in drpr—/— background (n = 16) and CDC50 KO + GFP-Lact in drpr—/— dendrite (n = 32), 5 animals. ***p % 0.001, Kruskal-Wallis (one-way ANOVA on ranks) and Dunn’s test; p values adjusted with the Benjamini-Hochberg method.
Black bars: mean; red bars: SD. See also Figure S6.
DISCUSSION
PS Exposure Is Associated with Neurite Degeneration
Our results demonstrate that externalized PS is associated with neurite degeneration in two ways. First, PS exposure results from neurite degeneration (Figure S7A). Both pruned dendrites during developmental remodeling and severed dendrites after physical injury dynamically expose PS. The onset of PS exposure coincides with early signs of dendrite degeneration such as membrane roughening and blebbing, but precedes dendrite fragmentation and engulfment. Moreover, PS exposure is blocked when degeneration of injured dendrites is inhibited by Wlds expression, suggesting that PS exposure is a downstream event of the neurite degeneration program triggered by NAD+ depletion (Almasieh et al., 2017; Gerdts et al., 2016). Interestingly, PS exposure in injured dendrites does not depend on caspase activity, consistent with previous findings that blocking caspase activity does not protect dendrites or axons from injury-induced degeneration (Simon et al., 2012; Tao and Rolls, 2011).
Second, ectopic PS exposure on neurites causes phagocytedependent membrane loss (Figure S7B). Flippase KO or TMEM16F OE led to PS exposure preferentially on distal high-order dendrites of C4 da neurons and, consequently, membrane shedding from PS-exposing dendrites. This kind of membrane loss in severe cases (such as in CDC50 KO + TMEM16F OE neurons) can lead to strong dendrite reduction. In the CNS, CDC50 KO and TMEM16F OE in adult olfactory neurons also caused age-dependent and neuronal activity-dependent axon degeneration, respectively. Although PS exposure is associated with cell-autonomous membrane vesicle shedding in apoptosis (Frey and Gaipl, 2011), necroptosis (Gong et al., 2017), tumor cell microvesicle formation (Lima et al., 2009; Muralidharan-Chari et al., 2009), and cell membrane repair (Scheffer et al., 2014), in the context of da neurons, PS-exposure by itself does not cause vesicle shedding, because membrane loss of PS-exposing dendrites is completely blocked in the drpr mutant background. Instead, epidermal cells likely latch on PS-exposing dendritic tips and cause them to separate as dendrites retract. In the CNS, glia may also actively damage PS-exposing axons through phagocytosis (Freeman, 2015). Together, our results establish a strong association between PS exposure and neurite degeneration, and argue that PS exposure is sufficient to cause phagocyte-dependent neurite degeneration.
Different Spatial Domains of Neurons Are Differentially Sensitive to the Perturbation of PS Asymmetry
Our results reveal that distal dendritic arbors of C4 da neurons are most vulnerable to disruption of PS asymmetry. Ectopically externalized PS appeared preferentially on distal high-order branches where the phagocyte-dependent neuronal membrane loss was the most severe. These results suggest that different spatial domains of the same dendritic arbor have different demands for maintaining PS asymmetry. One possibility for such spatial differences is that distal dendrites are more active in certain cellular activities that could cause PS externalization. Such activities may include calcium influx and stimulation of calcium-dependent scramblases, endocytosis, and exocytosis in inducing phospholipid scrambling (Janmey and Kinnunen, 2006), and the generation of oxidative stress (de Jong and Kuypers, 2006). Mechanistically, this differential sensitivity to PS asymmetry disruption is likely different from the diurnal and spatially restricted PS exposure on mammalian POS tips (Ruggiero et al., 2012), because the latter is a constant, physiological, and tightly regulated process that involves coordination between the neurons and their specialized phagocytes.
The Effects of Flippase LOF and Scramblase GOF Depends on the Cellular Context
Interestingly, flippase LOF and scramblase GOF produced distinct modes of degeneration that differ between larval C4 da dendrites and adult Or22a axons. These outcomes might be explained by the kinetics of PS externalization. The absence of P4-ATPase activity may lead to slow and cumulative PS externalzation, which could explain the age dependency of Or22a axon degeneration and the weak phenotype of larval C4 da neurons (considering the relatively short larval period). In contrast, the effect of TMEM16F, which can cause a more rapid PS exposure, is likely controlled by local calcium influx. In nociceptive C4 da neurons, short bursts of TMEM16F activity likely only cause local and transient PS exposure as endogenous flippases reestablish PS asymmetry. In contrast, sustained and strong TMEM16F activity, such as in activated Or22a neurons, may override the effect of endogenous flippases, leading to massive PS exposure and severe neurite degeneration. Lastly, when TMEM16F is overexpressed in CDC50 KO neurons, the lack of flippase activity may unmask the full effect of TMEM16F, which could underlie the synergistic effects of CDC50 KO and TMEM16 OE. Although other in-direct effects of CDC50 KO and TMEM16F OE cannot be ruled out, our results highlight the importance of the cellular context in determining the outcome of ectopic PS exposure.
A Potential Bridging Function of the LactC1C2 Domain
We found that prolonged exposure to GFP-Lact caused PS-exposing dendrites to degenerate in a phagocyte-dependent manner. This is a surprising result because the RGD motif required for lactadherin interaction with mammalian integrin receptors (Hanayama et al., 2002; Mapes et al., 2012) is missing in LactC1C2 used to construct GFP-Lact. LactC1C2 may contain a yet unidentified sequence that can interact with other engulfment receptors. In comparison, AV appears to be a more passive PS-binding protein: AV did not affect the clearance of the degenerating dendrites or cause degeneration of CDC50 KO neurons, but was often located in intracellular vesicles in PS-exposing neurons, likely a result of endocytosis.
Potential Roles of PS Exposure in Neurodevelopment and Neurodegenerative Diseases
Trimming of unnecessary neuronal branches or neuronal connections is an important step in the assembly of functional neural circuits (Chung et al., 2015; Schuldiner and Yaron, 2015). Our results suggest that PS exposure plays an important role in phagocytic recognition and clearance of pruned C4 da dendrites. It will be interesting to examine whether PS exposure also marks processes or synapses for elimination in other contexts of neurite and synaptic pruning. Furthermore, in neurodegenerative diseases, neurons can lose dendrites and axonal branches well before they die. Our results demonstrate that improper PS exposure in living neurons can cause neurite loss. Therefore, it is important to fully investigate whether and how spatial and temporal disruption of PS asymmetry on neurons relates to neurodegenerative pathologies.
EXPERIMENTAL PROCEDURES
Live Imaging and Dendrite Ablation
Animals were reared at 25°C in density-controlled vials. Larvae at 125 hr after legg laying (AEL) (wandering stage) were imaged using a Leica SP8 micro-scope. Unless stated otherwise, confocal images shown in all figures are maximum intensity projections of z stacks encompassing the epidermal layer and the sensory neurons beneath, which are typically 8–10 mm for larvae and 12–15 mm for pupae. For dendrite lesion, laser ablation was performed on a Zeiss LSM880 Confocal/Multiphoton Upright Microscope, using a 790-nm two-photon laser at primary dendrites of ddaC neurons in A1 and A3 segments at 90 hr AEL. Animals were recovered on grape juice agar plates following lesion for appropriate time. After recovery, the larvae were imaged using a Leica SP8 microscope. Long-term time-lapse imaging at the larval stage was done as described previously (Poe et al., 2017) with small modifications. For long-term time-lapse imaging of dendrite pruning, white pupae were directly mounted in the imaging chamber similar to larval mounting and kept on the imaging chamber for 4–6 hr before imaging.
Quantification and Statistical Analysis
Data acquisition and quantification were performed non-blinded in ImageJ and Microsoft Excel, respectively. Statistical significance was determined using one-way ANOVA and Tukey’s honest significant difference (HSD) or Kruskal-Wallis (one-way ANOVA on ranks) and Dunn’s test in R (*p < 0.05; **p < 0.01; ***p < 0.001). Statistical significance was set at p < 0.05.
The detailed descriptions for Drosophila stocks, molecular cloning and transgenic flies, generation of drprindel allele by CRISPR/Cas9, tissue-specific gene KO via CRISPR/Cas9, adult brain preparation and adult odor treatment, immunohistochemistry, heat shock conditions, live imaging and long-term time-lapse imaging, image analysis, and quantification can be found in the Supplemental Experimental Procedures.
Supplementary Material
Highlights.
PS is exposed on degenerating dendrites in a specific spatiotemporal pattern
PS exposure on injured dendrites depends on NAD depletion, not caspase activity
Ectopic PS exposure on live neurons causes phagocytes to engulf distal neurites
Lactadherin C1C2 domain potentiates phagocytes to destruct PS-exposing dendrites
ACKNOWLEDGMENTS
We thank Marc Freeman, Yang Xiang, Jon Graff, and Bloomington Stock Center for fly stocks; Ding Xue, Shigekazu Nagata, Thomas Kornberg, and Addgene for plasmids; Yalei Wu for cloning and immunochemistry reagents; Halocarbon Products Corporation for Halocarbon oils; Cornell BRC Imaging facility for access to microscopes (funded by National Institutes of Health (NIH), United States, grant S10OD018516); and Scott Emr, Mariana Wolfner, Joe Fetcho, Chris Fromme, Michael Goldberg, Fenghua Hu, and Felicity Emerson for critical reading and suggestions on the manuscript. This work was supported by a Cornell Fellowship awarded to H.J., and a Cornell start-up fund and NIH grants (R01NS099125 and R21OD023824) awarded to C.H.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and seven videos and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.095.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Almasieh M, Catrinescu MM, Binan L, Costantino S, and Levin LA (2017). Axonal degeneration in retinal ganglion cells is associated with a mem-brane polarity-sensitive redox process. J. Neurosci 37, 3824–3839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen MH, Graversen H, Fedosov SN, Petersen TE, and Rasmussen JT (2000). Functional analyses of two cellular binding domains of bovine lactadherin. Biochemistry 39, 6200–6206. [DOI] [PubMed] [Google Scholar]
- Appelt U, Sheriff A, Gaipl US, Kalden JR, Voll RE, and Herrmann M (2005). Viable, apoptotic and necrotic monocytes expose phosphatidylserine: cooperative binding of the ligand Annexin V to dying but not viable cells and implications for PS-dependent clearance. Cell Death Differ 12, 194–196. [DOI] [PubMed] [Google Scholar]
- Botelho RJ, and Grinstein S (2011). Phagocytosis. Curr. Biol 21, R533–R538. [DOI] [PubMed] [Google Scholar]
- Brown GC, and Neher JJ (2014). Microglial phagocytosis of live neurons. Nat. Rev. Neurosci 15, 209–216. [DOI] [PubMed] [Google Scholar]
- Chu J, Haynes RD, Corbel SY, Li P, Gonza´ lez-Gonza´ lez E, Burg JS, Ataie NJ, Lam AJ, Cranfill PJ, Baird MA, et al. (2014). Non-invasive intravital imaging of cellular differentiation with a bright red-excitable fluorescent protein. Nat. Methods 11, 572–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Welsh CA, Barres BA, and Stevens B (2015). Do glia drive synaptic and cognitive impairment in disease? Nat. Neurosci 18, 1539–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong K, and Kuypers FA (2006). Sulphydryl modifications alter scramblase activity in murine sickle cell disease. Br. J. Haematol 133, 427–432. [DOI] [PubMed] [Google Scholar]
- Dobritsa AA, van der Goes van Naters W, Warr CG, Steinbrecht RA, and Carlson JR(2003). Integrating the molecular and cellular basis of odor coding in the Drosophila antenna. Neuron 37, 827–841. [DOI] [PubMed] [Google Scholar]
- Dubois T, Mira JP, Feliers D, Solito E, Russo-Marie F, and Oudinet JP (1998). Annexin V inhibits protein kinase C activity via a mechanism of phospholipid sequestration. Biochem. J 330, 1277–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emoto K (2011). Dendrite remodeling in development and disease. Dev. Growth Differ 53, 277–286. [DOI] [PubMed] [Google Scholar]
- Fourgeaud L, Trave´ s PG, Tufail Y, Leal-Bailey H, Lew ED, Burrola PG, Callaway P, Zago´ rska A, Rothlin CV, Nimmerjahn A, and Lemke G (2016). TAM receptors regulate multiple features of microglial physiology. Nature 532, 240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman MR (2015). Drosophila central nervous system glia. Cold Spring Harb. Perspect. Biol 7, a020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey B, and Gaipl US (2011). The immune functions of phosphatidylserine in membranes of dying cells and microvesicles. Semin. Immunopathol 33, 497–516. [DOI] [PubMed] [Google Scholar]
- Fricker M, Neher JJ, Zhao JW, The´ ry C, Tolkovsky AM, and Brown GC (2012). MFG-E8 mediates primary phagocytosis of viable neurons during neuroinflammation. J. Neurosci 32, 2657–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii T, Sakata A, Nishimura S, Eto K, and Nagata S (2015). TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc. Natl. Acad. Sci. USA 112, 12800–12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdts J, Summers DW, Milbrandt J, and DiAntonio A (2016). Axon selfdestruction: new links among SARM1, MAPKs, and NAD+ metabolism. Neuron 89, 449–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Guy C, Olauson H, Becker JU, Yang M, Fitzgerald P, Linkermann A, and Green DR (2017). ESCRT-III acts downstream of MLKL to regulate necroptotic cell death and its consequences. Cell 169, 286–300.e216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grueber WB, Jan LY, and Jan YN (2002). Tiling of the Drosophila epidermis by multidendritic sensory neurons. Development 129, 2867–2878. [DOI] [PubMed] [Google Scholar]
- Haka AS, Barbosa-Lorenzi VC, Lee HJ, Falcone DJ, Hudis CA, Dannenberg AJ, and Maxfield FR (2016). Exocytosis of macrophage lysosomes leads to digestion of apoptotic adipocytes and foam cell formation. J. Lipid Res 57, 980–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Jan LY, and Jan YN (2011). Enhancer-driven membrane markers for analysis of nonautonomous mechanisms reveal neuron-glia interactions in Drosophila. Proc. Natl. Acad. Sci. USA 108, 9673–9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Wang D, Soba P, Zhu S, Lin X, Jan LY, and Jan YN (2012). Integrins regulate repulsion-mediated dendritic patterning of Drosophila sensory neurons by restricting dendrites in a 2D space. Neuron 73, 64–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Song Y, Xiao H, Wang D, Franc NC, Jan LY, and Jan YN (2014). Epidermal cells are the primary phagocytes in the fragmentation and clearance of degenerating dendrites in Drosophila. Neuron 81, 544–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanayama R, Tanaka M, Miwa K, Shinohara A, Iwamatsu A, and Nagata S (2002). Identification of a factor that links apoptotic cells to phagocytes. Nature 417, 182–187. [DOI] [PubMed] [Google Scholar]
- Hay BA, and Guo M (2006). Caspase-dependent cell death in Drosophila. Annu. Rev. Cell Dev. Biol 22, 623–650. [DOI] [PubMed] [Google Scholar]
- Heemskerk JW, Bevers EM, and Lindhout T (2002). Platelet activation and blood coagulation. Thromb. Haemost 88, 186–193. [PubMed] [Google Scholar]
- Hwang RY, Zhong L, Xu Y, Johnson T, Zhang F, Deisseroth K, and Tracey WD (2007). Nociceptive neurons protect Drosophila larvae from parasitoid wasps. Curr. Biol 17, 2105–2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janmey PA, and Kinnunen PK (2006). Biophysical properties of lipids and dynamic membranes. Trends Cell Biol 16, 538–546. [DOI] [PubMed] [Google Scholar]
- Kanamori T, Kanai MI, Dairyo Y, Yasunaga K, Morikawa RK, and Emoto K (2013). Compartmentalized calcium transients trigger dendrite pruning in Drosophila sensory neurons. Science 340, 1475–1478. [DOI] [PubMed] [Google Scholar]
- Kim YE, Chen J, Chan JR, and Langen R (2010). Engineering a polaritysensitive biosensor for time-lapse imaging of apoptotic processes and degeneration. Nat. Methods 7, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ME, Shrestha BR, Blazeski R, Mason CA, and Grueber WB (2012). Integrins establish dendrite-substrate relationships that promote dendritic self-avoidance and patterning in Drosophila sensory neurons. Neuron 73, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, and van Oers MH (1994). Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood 84, 1415– 1420. [PubMed] [Google Scholar]
- Kuo CT, Zhu S, Younger S, Jan LY, and Jan YN (2006). Identification of E2/E3 ubiquitinating enzymes and caspase activity regulating Drosophila sensory neuron dendrite pruning. Neuron 51, 283–290. [DOI] [PubMed] [Google Scholar]
- Lemke G (2013). Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol 5, a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew ED, Oh J, Burrola PG, Lax I, Zago´ rska A, Trave´ s PG, Schlessinger J, and Lemke G (2014). Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife 3, e03385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima LG, Chammas R, Monteiro RQ, Moreira ME, and Barcinski MA (2009). Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett 283, 168–175. [DOI] [PubMed] [Google Scholar]
- Lin L, Huai Q, Huang M, Furie B, and Furie BC (2007). Crystal structure of the bovine lactadherin C2 domain, a membrane binding motif, shows similarity to the C2 domains of factor V and factor VIII. J. Mol. Biol 371, 717–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo L, and O’Leary DD (2005). Axon retraction and degeneration in development and disease. Annu. Rev. Neurosci 28, 127–156. [DOI] [PubMed] [Google Scholar]
- MacDonald JM, Beach MG, Porpiglia E, Sheehan AE, Watts RJ, and Freeman MR (2006). The Drosophila cell corpse engulfment receptor Draper mediates glial clearance of severed axons. Neuron 50, 869–881. [DOI] [PubMed] [Google Scholar]
- Mack TG, Reiner M, Beirowski B, Mi W, Emanuelli M, Wagner D, Thomson D, Gillingwater T, Court F, Conforti L, et al. (2001). Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci 4, 1199–1206. [DOI] [PubMed] [Google Scholar]
- Mapes J, Chen YZ, Kim A, Mitani S, Kang BH, and Xue D (2012). CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr. Biol 22, 1267–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire SE, Le PT, Osborn AJ, Matsumoto K, and Davis RL (2003). Spatiotemporal rescue of memory dysfunction in Drosophila. Science 302, 1765–1768. [DOI] [PubMed] [Google Scholar]
- Morel O, Toti F, Hugel B, and Freyssinet JM (2004). Cellular microparticles: a disseminated storage pool of bioactive vascular effectors. Curr. Opin. Hematol 11, 156–164. [DOI] [PubMed] [Google Scholar]
- Muralidharan-Chari V, Clancy J, Plou C, Romao M, Chavrier P, Raposo G, and D’Souza-Schorey C (2009). ARF6-regulated shedding of tumor cellderived plasma membrane microvesicles. Curr. Biol 19, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naftelberg S, Abramovitch Z, Gluska S, Yannai S, Joshi Y, Donyo M, Ben-Yaakov K, Gradus T, Zonszain J, Farhy C, et al. (2016). Phosphatidylserine ameliorates neurodegenerative symptoms and enhances axonal transport in a mouse model of familial dysautonomia. PLoS Genet 12, e1006486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S (2010). Apoptosis and autoimmune diseases. Ann. N Y Acad. Sci 1209, 10–16. [DOI] [PubMed] [Google Scholar]
- Nandrot EF, Anand M, Almeida D, Atabai K, Sheppard D, and Finnemann SC (2007). Essential role for MFG-E8 as ligand for alphavbeta5 integrin in diurnal retinal phagocytosis. Proc. Natl. Acad. Sci. USA 104, 12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan P, Wang J, Hua Z, and Graham TR (2004). Drs2p-coupled aminophospholipid translocase activity in yeast Golgi membranes and relationship to in vivo function. Proc. Natl. Acad. Sci. USA 101, 10614–10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulusma CC, Folmer DE, Ho-Mok KS, de Waart DR, Hilarius PM, Verhoeven AJ, and Oude Elferink RP (2008). ATP8B1 requires an accessory protein for endoplasmic reticulum exit and plasma membrane lipid flippase activity. Hepatology 47, 268–278. [DOI] [PubMed] [Google Scholar]
- Poe AR, Tang L, Wang B, Li Y, Sapar ML, and Han C (2017). Dendritic space-filling requires a neuronal type-specific extracellular permissive signal in Drosophila. Proc. Natl. Acad. Sci. USA 114, E8062–E8071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen JP, Sack GS, Martin SM, and Sagasti A (2015). Vertebrate epidermal cells are broad-specificity phagocytes that clear sensory axon debris. J. Neurosci 35, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran KS (2010). Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J. Exp. Med 207, 1807–1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiero L, Connor MP, Chen J, Langen R, and Finnemann SC (2012). Diurnal, localized exposure of phosphatidylserine by rod outer segment tips in wild-type but not Itgb5−/−or Mfge8−/−mouse retina. Proc. Natl. Acad. Sci. USA 109, 8145–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, and Tanaka K (2004). Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol. Biol. Cell 15, 3418–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salter MW, and Stevens B (2017). Microglia emerge as central players in brain disease. Nat. Med 23, 1018–1027. [DOI] [PubMed] [Google Scholar]
- Scheffer LL, Sreetama SC, Sharma N, Medikayala S, Brown KJ, Defour A, and Jaiswal JK (2014). Mechanism of Ca2+-triggered ESCRT assembly and regulation of cell membrane repair. Nat. Commun 5, 5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenmann Z, Assa-Kunik E, Tiomny S, Minis A, Haklai-Topper L, Arama E, and Yaron A (2010). Axonal degeneration is regulated by the apoptotic machinery or a NAD+-sensitive pathway in insects and mammals. J. Neurosci 30, 6375–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner O, and Yaron A (2015). Mechanisms of developmental neurite pruning. Cell. Mol. Life Sci 72, 101–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K, and Nagata S (2015). An apoptotic ‘eat me’ signal: phosphatidylserine exposure. Trends Cell Biol 25, 639–650. [DOI] [PubMed] [Google Scholar]
- Segawa K, Suzuki J, and Nagata S (2011). Constitutive exposure of phosphatidylserine on viable cells. Proc. Natl. Acad. Sci. USA 108, 19246–19251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Weimer RM, McLaughlin T, Kallop D, Stanger K, Yang J, O’Leary DD, Hannoush RN, and Tessier-Lavigne M (2012). A caspase cascade regulating developmental axon degeneration. J. Neurosci 32, 17540–17553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Umeda M, Sims PJ, and Nagata S (2010). Calcium-dependent phospholipid scrambling by TMEM16F. Nature 468, 834–838. [DOI] [PubMed] [Google Scholar]
- Suzuki J, Denning DP, Imanishi E, Horvitz HR, and Nagata S (2013). Xkrelated protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science 341, 403–406. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Fujimura-Kamada K, and Yamamoto T (2011). Functions of phospholipid flippases. J. Biochem 149, 131–143. [DOI] [PubMed] [Google Scholar]
- Tang X, Halleck MS, Schlegel RA, and Williamson P (1996). A subfamily of P-type ATPases with aminophospholipid transporting activity. Science 272, 1495–1497. [DOI] [PubMed] [Google Scholar]
- Tao J, and Rolls MM (2011). Dendrites have a rapid program of injuryinduced degeneration that is molecularly distinct from developmental pruning. J. Neurosci 31, 5398–5405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tufail Y, Cook D, Fourgeaud L, Powers CJ, Merten K, Clark CL, Hoffman E, Ngo A, Sekiguchi KJ, O’Shea CC, et al. (2017). Phosphatidylserine exposure controls viral innate immune responses by microglia. Neuron 93, 574–586.e578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki S, and Araki T (2017). Specific phospholipid scramblases are involved in exposure of phosphatidylserine, an ‘‘eat-me’’ signal for phagocytes, on degenerating axons. Commun. Integr. Biol 10, e1296615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki S, Tokunaga S, Shibata M, and Araki T (2017). GSK3B-mediated phosphorylation of MCL1 regulates axonal autophagy to promote Wallerian degeneration. J. Cell Biol 216, 477–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DW, Kondo S, Krzyzanowska A, Hiromi Y, and Truman JW (2006). Local caspase activity directs engulfment of dendrites during pruning. Nat. Neurosci 9, 1234–1236. [DOI] [PubMed] [Google Scholar]
- Zhu X, Libby RT, de Vries WN, Smith RS, Wright DL, Bronson RT, Seburn KL, and John SW (2012). Mutations in a P-type ATPase gene cause axonal degeneration. PLoS Genet 8, e1002853. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.