

Abstract
Background & Aims
Liver fibrosis, hepatocellular necrosis, inflammation and proliferation of liver progenitor cells are features of chronic liver injury. Mouse models have been used to study the end-stage pathophysiology of chronic liver injury. However, little is known about differences in the mechanisms of liver injury among different mouse models, due to our inability to visualize the progression of liver injury in vivo in mice. We developed a method to visualize bile transport and blood-bile barrier (BBlB) integrity in live mice.
Methods
C57BL/6 mice were fed a choline-deficient ethionine supplemented (CDE) diet or a diet containing 0.1% 3, 5-diethoxycarbonyl-1, 4-dihydrocollidine (DDC) for up to 4 weeks to induce chronic liver injury. We used quantitative liver intravital microscopy (qLIM) for real-time assessment of bile transport and blood-bile barrier (BBlB) integrity in the intact livers of the live mice fed the CDE, DDC, or chow (control) diets. Liver tissues were collected from mice and analyzed by histology, immunohistochemistry, real-time PCR, and immunoblots.
Results
Mice with liver injury induced by a CDE or a DDC diet had breaches in the BBlB and impaired bile secretion, observed by qLIM compared to control mice. Impaired bile secretion was associated with reduced expression of several tight-junction proteins (claudins 3, 5, and 7) and bile transporters (NTCP, OATP1, BSEP, ABCG5 and ABCG8). A prolonged (two weeks) CDE but not DDC diet, led to re-expression of tight-junction proteins as well as bile transporters, concomitant to the reestablishment of BBlB integrity and bile secretion.
Conclusions
We used qLIM to study chronic liver injury, induced by a choline-deficient or DDC diet, in mice. Progression of chronic liver injury was accompanied by loss of bile transporters and tight-junction proteins.
Keywords: diet induced liver injury, Blood bile barrier, Hepatocyte tight junction, claudins
Introduction
Chronic liver disease (CLD) is the 12th leading cause of death in the United States1,2. Current treatments for CLD are limited and liver transplant is the only treatment available to patients diagnosed with liver failure. The onset of CLD is primarily attributed to excessive alcohol consumption, viral hepatitis, non-alcoholic fatty liver disease, autoimmune hepatitis, primary biliary cholangitis, primary sclerosing cholangitis, and genetic conditions such as hereditary hemochromatosis2–7. Although chronic liver injury is the hallmark of all types of CLDs, the cellular, molecular and biophysical mechanisms that contribute to progression of chronic liver injury are incompletely understood. Identifying the common molecular pathways that promote liver injury in diverse experimental models of chronic liver injury would enable the development of improved therapies to prevent or halt the progression of CLDs.
Chronic liver injury is characterized by iterative cycles of insult to the liver parenchyma leading to inflammation, matrix deposition, hyper-bilirubinemia, angiogenesis and progressive fibrosis3. Recently8, we have identified that chronic liver injury is associated with loss of Blood-Bile-Barrier (BBlB). BBlB is a physical barrier formed by liver epithelial cells or hepatocytes, which separates bile from sinusoidal blood. The BBlB function is enabled by the junctional adhesion complexes known as tight junctions (TJs), which are maintained by the homotypic interaction of adhesion molecules such as claudins (1–31), occludin, coxsackievirus and adenovirus receptor, and zonula occludens (ZO-1, -2, and -3)9. In addition to TJs, BBlB function is also regulated by highly polarized basolateral and apical transporters present on hepatocytes that modulate the collection and release of bile acids from the blood and into the bile-canaliculi, respectively10–12.
Here, we have utilized two widely used experimental models of chronic liver injury in mice fed choline-deficient ethionine-supplemented (CDE)13–15 or 3, 5-diethoxycarbonyl 1, 4-dihydrocollidine (DDC)16–19 diet, respectively. Using our recently developed quantitative Liver Intravital Microscopy (qLIM) approach, we reveal that chronic liver injury involves physical breach of BBlB, mixing of blood with bile and loss of bile transport across hepatocytes. Our data suggest that loss of TJ adhesion-molecules such as claudins and basolateral or canalicular bile transporters from hepatocytes is the central pathophysiology in chronic liver injury irrespective of the experimental model. Finally, we demonstrate that the recovery from liver injury is dictated by the ability of BBlB to regain physical integrity through reappearance of TJ adhesion-molecules and bile transporters on hepatocytes.
Methods
Mice
C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and Taconic Biosciences (Hudson, NY). Mice used in this study were ages 4–6 weeks and weighed around 18kgs. All animal experiments and procedures were performed under the guidelines of the NIH and under an animal protocol approved by the Institutional Animal Use and Care Committees at the University of Pittsburgh.
Diet
DDC-diet
Mice were fed a special diet containing 0.1% 3, 5-diethoxycarbonyl-1, 4-dihydrocollidine (DDC, Bioserve,) for up to four weeks.
CDE-diet
Mice were given unlimited access to Choline Deficient Diet (Envigo, Frederick, MD) and drinking water supplemented with 0.15% DL-Ethionine (MP Biomedicals, Santa Ana, CA) for up to four weeks.
Surgical preparation and qLIM imaging
Mice were anesthetized with an intraperitoneal (i.p.) injection of 100 mg kg−1 of body weight ketamine HCl (100 mg ml−1; Henry Shein Animal Health; Dublin, OH) and 20 mg kg−1 of body weight xylazine (20 mg ml−1; LLOYD Laboratories; Shenandoah, IA). When anesthetized, mice were given a 1 ml i.p. injection of warmed saline and placed on a heated stage in the supine position. A tracheotomy was performed and a short length of PE90 tubing was inserted into the incision site and tied to the trachea using a silk suture (Fig S1). Next, the right carotid artery was cannulated with heparinized PE10 tubing. Mice were repositioned in the right lateral decubitus position. The right lobe of the liver was exposed through removal of the overlying skin and fat. To gently immobilize the liver, we used a micro-machined thoracic liver window inspired by 20, which provided a light suction through the use of a vacuum pump (Roscoe Medical Inc; Strongsville, OH). The modified thoracic liver device had a larger viewing window (diameter ~ 5 mm) that allows more stable imaging over extended durations. A round coverslip (diameter 12 mm) was placed on top of the window and held in place using vacuum grease. Once the vacuum was applied, the liver window was gently lowered to immobilize a small region of the lower half of the right liver against the coverslip. Next, intravascular fluorescent dyes were injected through the carotid artery catheter (Fig S1). Intravascular fluorescent dyes included 200 μg of TXR dextran, or 100 μg of Carboxyflurescein (CF). TXR dextran (MW 70,000) was used to visualize the blood flow through the liver sinusoids whereas CF (Molecular weight 377) 21 was used to visualize uptake of the dye from blood to hepatocytes 3m post-injection and then from the hepatocyte to the bile-canaliculi within 5–8m. Microscopy was performed using a Nikon MPE multi-photon excitation microscope. This study was done in collaboration with CBI, University of Pittsburgh.
Image Analysis
Movies were processed using Nikon’s NIS Elements (Nikon Elements 3.10). A median filter with a kernel size of 3 was applied over each video frame to improve signal-to-noise ratio. Signal contrast in each channel of a multicolor image was further enhanced by adjusting the maxima and minima of the intensity histogram of that channel. Spectral unmixing was performed using NIS Elements to separate out tissue autofluorescence from the FITC fluorescence and to reduce bleed through among different channels of bicolour images.
RESULTS
CDE and DDC-diet promote physical breach of BBlB and impaired bile transport
Recently, we developed qLIM that uses multi-photon-excitation fluorescence microscopy to enable real-time assessment of BBlB integrity and kinetics of bile transport in the intact liver of live mice8. Here, we have used qLIM to determine whether BBlB integrity and bile transport are effected in CDE and DDC-diet induced models of chronic liver injury. Mice fed regular-chow, CDE or DDC-diet for 4–6 days (d) were intravenously (IV) administered with TXR-dextran (red fluorescence) and carboxy-fluorescein-di-acetate (CFDA; green fluorescence)22 to visualize the blood flow through liver sinusoids and bile flow through the bile-canaliculi, respectively. Blood cells flowing in sinusoids were visible as dark cells moving over a red background (Fig. 1A, S2A–A″). Previously8, we have shown that CFDA in the blood can be internalized by hepatocytes, where it is hydrolyzed by esterase22 into the fluorescent green metabolite carboxy-fluorescein (CF-green fluorescence). Also, the translocation of CFDA across the hepatocytes has been used previously as a surrogate for assessment of bile transport across the BBlB22,23. As shown by representative images in Fig. 1A and Movie-S1, CFDA was internalized by hepatocytes and hydrolyzed to CF (green fluorescence in hepatocytes marked with white dotted trace) within 1–3 min (m) following IV administration in mice fed regular chow diet. CF (green fluorescence) was released by hepatocytes into the bile-canaliculi (green fluorescence) at 4 to 6m (Fig. 1B, S2B–B″, Movie-S2) and started clearing away from the bile-canaliculi by 20–22m post-CFDA administration (Fig. 1C, S2C–C″, Movie-S3) (complete absence of green fluorescence was seen at 30m post-administration; data not shown). At all-time points, the vascular dye was restricted to the liver sinusoids and CF was absent from liver sinusoids suggesting that the BBlB was physically intact in mice fed regular chow-diet. The physical integrity of BBlB in mice fed regular-chow was quantitatively confirmed (Fig. S2D) using Pearson’s colocalization coefficient analysis, which revealed complete absence of colocalization between blood (red) and CF (green). The intensity analysis (Fig S2E–G) also revealed the absence of blood (red) and CF (green) from intrahepatic space and sinusoids, respectively suggesting a complete separation of blood and bile flow in mice fed chow-diet.
Figure 1. qLIM enables real-time assessment of BBlB in the intact liver of live mice.

Time series images of (A) 3m, (B) 10m and (C) 20m of mouse liver fed on normal-chow shows localization of CF (green) and TXR-dextran (red) in bile-canaliculi and sinusoids, respectively. (D) qLIM schematic diagram of mouse liver section fed with normal-chow. Time series images of (E) 3m, (F) 10m and (G) 20m of (n=1) mouse liver fed on CDE-diet for 4d show complete localization of CF (green) and TXR-dextran (red) in liver sinusoids. (H) qLIM schematic diagram of mouse liver section fed with CDE-diet for 4d. Time series images of (I) 3m, (J) 10m and (K) 20m of (n=1) mouse liver fed on DDC-diet for 6d show localization of CF (green) and TXR-dextran (red) in hepatocytes and sinusoids, respectively. (L) qLIM schematic diagram of mouse liver section fed with DDC-diet for 6d. Arrow denotes presence of this conduits through which blood and bile are leaking and dotted lines represents hepatocytes.
Unlike mice fed chow-diet, mice fed CDE-diet for 4d showed signs of physical breach in BBlB, vascular leakage and defect in bile transport. CF was retained in the liver sinusoids evident by the complete overlap (yellow fluorescence caused by the overlap of red and green fluorescence) of vascular dye (red) and CF (green) at 3m post CFDA administration (Fig. 1E, S3A–A″, movie-S4). This observation was confirmed by quantitative analysis showing significantly high colocalization of TXR-dextran and CF in liver sinusoids (Fig. S3D) indicating a defect in blood-bile separation (Fig. S3D). We did not observe any uptake of CF into the hepatocytes (absence of green stain in hepatocytes) or secretion into the bile-canalicular network (absence of green fluorescence in bile-canaliculi) even at later time points (Fig. 1E–G, S3A–C movie-S5, 6). This was further confirmed by the significantly high intensity of CF in liver sinusoids compared to hepatocytes or bile-canaliculi (Fig S3H–J) suggesting a defect in the uptake of bile by hepatocytes. TXR-dextran positive bright spots and a few CF positive bright spots were observed outside the sinusoidal region (arrow, Fig. 1E–G, S3A–C), which increased over time (T=10 min in Fig. 1F vs T=20 min Fig. 1G) suggestive of vascular leakage into the intrahepatic space. Indeed, quantitative fluorescent intensity-analysis revealed that TXR-dextran was not only confined to liver sinusoids but also accumulated in extra-sinusoidal space (Fig. S3H–H). Interestingly, we also observed thin conduits (connecting sinusoids) through which a mixture of CF and TXR-dextran was flowing between the hepatocytes (marked with arrow Fig. 1E–G, arrow, S3A–C). We posit that such conduits represent widening of space between adjacent hepatocytes leading to a physical breach of BBlB.
In mice fed DDC-diet for 6d, qLIM revealed presence of TXR-dextran (red) and CF (green) in sinusoids and hepatocyte, respectively with no overlap at 5m post-CFDA administration (Fig. 1I, S4A–A″ movie-S7). Quantitative analysis also confirmed that DDC fed mice showed almost no colocalization (Fig. S4D) of TXR-dextran and CF at 5m and all later time points (Fig. S4D). Similarly, quantitative fluorescent intensity analysis also revealed that TXR-dextran and CF exclusively localized to liver sinusoids and extra sinusoidal space (hepatocytes), respectively with minimal overlap (Fig. S4G–I). This phenotype was identical to that observed in mice fed regular chow but contrary to mice fed CDE-diet, and suggested that the DDC-diet did not affect CF uptake by hepatocytes. However, unlike the canalicular release of CF observed in chow-fed mice, CF was absent from bile-canaliculi but confined in hepatocytes at 5m post CFDA administration (Fig. 1I, S4A–A″, movie-S7). QLIM revealed that CF was not released in bile-canaliculi even after 10m (Fig. 1J, S4B–B″, movie-S8) and continued to be retained by hepatocytes at 20m post-administration (Fig. 1K, S4C–C″ movie-S9) suggesting a loss of canalicular bile secretion in mice fed DDC-diet.
Failure of BBlB and bile transport is associated with liver injury
Next, we determined whether the breach of BBlB and loss of bile transport manifested at 4d and 6d in mice fed CDE and DDC-diet, respectively is also associated with development of liver injury. Identical to previous reports13–15,18,19,24, mice fed either CDE or DDC-diet for 4d and 6d, respectively had significantly high levels of serum alanine-aminotransferase (ALT), alkaline-phosphatase (ALP) and both total and direct bilirubin compared to mice fed normal-chow diet (Fig 2A).
Figure 2. Characterization of liver injury progression in CDE and DDC fed mice based of qLIM.
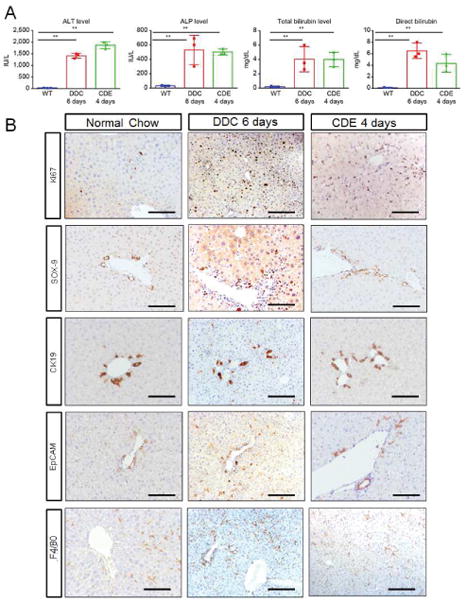
(A) Serum-analysis from CDE and DDC-diet fed mice indicate significant increase in alkaline-phosphatase (ALP), alanine-aminotransferase (ALT), total and direct bilirubin on 4 and 6d respectively. (B) IHC of mice on normal-chow, CDE-diet fed mice at 4d and DDC-diet fed mice at 6d shows increased number of cells (especially ductular cells) by ki67 staining; a prominent increase in Sox-9 staining in CDE and a modest increase in DDC liver sections; increased ductular-reaction by CK19 positive staining. Both CDE and DDC fed liver shows highly enriched EpCAM signal. F480 staining indicates increased macrophage activation in CDE-diet at 4d and DDC-diet at 6d compared to WT-liver.
Compared to chow-fed mice, mice fed CDE or DDC-diet had disorganized hepatic architecture (Fig. S5A–A″) in the liver suggestive of hepatic injury. Sirius-red (Fig. S5B–B″), a marker for fibrosis was mildly increased but TUNEL, which labels dying cells was increased in the livers of CDE and DDC fed as compared to chow fed mice (Fig. S5C–C″). Increase in CD45 (Fig. S5D–D″) as well as F4/80 staining (Fig. 2B) indicated increased inflammation, and more α-SMA (Fig. S5E–E″) suggests hepatic stellate cell activation in CDE and DDC compared to chow-fed mice. Both CDE and DDC-diet also promoted cell proliferation in the liver, which was evident by the increase in Ki67 staining compared to chow fed mice (Fig. 2B). In addition to increase in bilirubinemia, inflammation and fibrosis, increased staining for Sox-9 (Fig. 2B), CK19 (Fig. 2B) and epithelial cell adhesion molecule (EpCAM) (Fig. 2B) suggested that both CDE and DDC-diet also led to an increase in ductular-reaction and appearance of transiently amplifying hepatic progenitors. Altogether, these findings suggest that breach of BBlB and loss of bile transport at 4d and 6d in mice fed CDE and DDC-diet, respectively is associated with development of hepatic injury, inflammation and bilirubinemia and may be setting the stage for hepatic fibrosis.
Loss of claudins promotes BBlB breach and failure of bile secretion
Recently, we have established a role for TJ claudins and occludin in the maintenance of BBlB integrity8. Claudins can be classified into plaque-forming claudins (1, 3, 5, 7 and 9) responsible for maintenance of trans-epithelial resistance, and pore-forming claudins (2, 10 and 17) known to decrease trans-epithelial resistance25,26. We found that knocking down claudin-3 in vitro in Hep-3B cells caused significant increase in profibrotic cytokines/factors including TGFβ, procollagen, collagen 1 and 2 (Fig. S13A) and aberrant expression of claudin-1 and ZO-2 proteins (Fig. S13B) that are associated with cholestasis27–31. Based on this in vitro observation and our in vivo qLIM data revealing mixing of blood with bile and bile secretion defect in mice fed CDE or DDC-diet for 4d and 6d (Fig. 1E–L), respectively, we determined whether the failure of BBlB is a result of the loss of claudins from hepatocytes. Remarkably, we found that the mRNA levels of claudins-3, 5 and 7 were significantly reduced, while the occludin mRNA levels were upregulated in the liver of mice fed CDE-diet for 4d (Fig. 3A–B). This finding was further supported by immunofluorescence as well as western-blot analyses, which revealed significant reduction in claudin-3 (Fig. 3C), 5 (Fig. 3D, F), and 7 (Fig. 3E, F), and increase in occludin (Fig. 3C,) in hepatocytes of mice fed CDE-diet for 4d. Similarly, mice fed DDC-diet for 6d also exhibited significant reduction in claudins-3, 5 and 7, and significant increase in occludin at both mRNA (Fig. 4A–B) and protein level (Fig. 4C–D). Taken together, these results suggest that the progression of liver injury and breach of BBlB in mice fed CDE or DDC-diet for 4 or 6d, respectively is linked to loss of claudins-3, 5 & 7 from hepatocyte TJs. We also assessed the mRNA and protein expression of TJ proteins in mice fed CDE or DDC-diet for 14d. Surprisingly, the mRNA levels of claudins-3, 5, and 7 rebound while mRNA levels of occludin were further upregulated in hepatocytes of mice fed CDE-diet for 14d (Fig. 3A–B). Immunofluorescence analysis validated reappearance of claudins-3 (Fig. 3C), 5 (Fig. 3D, F), and 7 (Fig. 3E, F) and further increase in localization of occludin after 14d of CDE-diet (Fig. 3C, F). In contrast, mRNA (Fig. 4A–B) levels as well as membrane localization of claudins-3, 5 and 7 (Fig. 4C–D) was found to be further reduced in hepatocytes of mice fed DDC-diet for 14d. Similar to CDE-diet, occludin mRNA and expression continued to increase through 14d of DDC-diet (Fig. 4A, C).
Figure 3. Chronic liver injury in CDE-diet fed mice is associated with loss of TJ adhesion-molecules.

(A) RT-PCR analysis shows significant decrease in mRNA expression of claudins-3, 5, 7 and increase in occludin in CDE-diet for 4d as compared to mice on normal-diet. Rescue was seen in claudins-3, 5, 7 level whereas further increase in occludin at 14d on CDE-diet. (B) Gel showing PCR product for claudins-3, 5, 7 and occludin. (C) In WT liver, claudin-3 and occludin localizes to the membrane in a punctate manner. Localization of claudin-3 to the membrane along with its colocalization with occludin is reduced in liver of mice fed on CDE-diet for 4d. Occludin levels are more at this stage. At 14d, claudin-3 level reappears to the membrane and colocalizes with occludin which continues to show an increase. (D) Membrane localization of claudin-5 and phalloidin staining marking the hepatocyte cytoskeleton in WT-liver. Membrane localization of claudin-5 is lost in CDE-diet fed mice (4d) and phalloidin staining indicates increases steatosis. At day 14 on CDE, claudin-5 level reappears whereas phalloidin staining indicates improved hepatic architecture. (E) In WT-liver, claudin-7 localizes to the membrane in a punctate manner. Localization of claudin-7 to the membrane in liver of mice fed on CDE-diet for 4d shows reduced expression. At 14d on CDE-diet, claudin-7 level reappears to the membrane. (F) Representative WB of junctional proteins shows reduction in claudins-3, 5, 7 at 4d post CDE-diet administration which is rescued at 14d. Occludin levels are increased at both 4d and 14d. GAPDH shows protein loading. * denotes p=0.05
Figure 4. Misexpression of TJ proteins is associated with DDC-diet induced chronic liver injury.
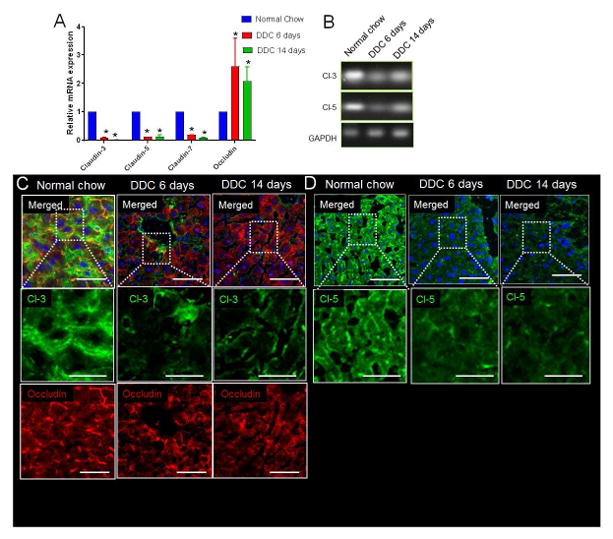
(A) RT-PCR analysis shows significant decrease in mRNA expression of claudins-3, 5, 7 and increase in mRNA expression of occludin in DDC-diet for 6das compared to WT mice on normal-diet. Further decrease was seen in claudins-3, 5, 7 level whereas occludin were increased at 14d on DDC-diet. (B) Gel showing PCR product for claudins-3, 5 and GAPDH. (C) In WT liver, claudin-3 and occludin localizes to the membrane in a punctate manner. Localization of claudin-3 to the membrane is reduced in liver of mice fed on DDC-diet for 6d. Occludin levels are more at this stage. At 14d on DDC-diet, claudin-3 continues to decrease whereas occludin levels are still upregulated. (D) Membrane localization of claudin-5 in WT-liver. Membrane localization of claudin-5 is lost in at 6d and 14d on DDC-diet fed mice. * denotes p=0.05
Loss of bile transporters contribute to impaired bile flow
Basolateral and canalicular bile transporters expressed on hepatocytes promote the uptake of bile acids from the sinusoidal blood and release into the bile-canaliculi, respectively12,32–34. Based on our qLIM data identifying loss of CF uptake by hepatocytes in mice fed CDE-diet for 4d (Fig. 1E–H) and impaired CF secretion into bile-canaliculi in mice fed DDC-diet for 6d (Fig. 1I–L), we assessed the expression and function of bile transporters in the liver of mice fed either CDE or DDC-diet. We found a significant reduction in the mRNA levels of basolateral transporters NTCP and OATP-1 in the liver of mice fed CDE-diet for 4d (Fig. 5A). Moreover, we found that the mRNA level of canalicular transporter BSEP was also reduced in the liver of mice CDE-diet for 4d (Fig. 5A). Similar to mRNA levels, immunofluorescence analysis of frozen liver section also revealed a significant reduction in expression of OATP-1 (Fig. 5B), NTCP (Fig. 5C) and BSEP (Fig. 5D) in the hepatocytes of mice fed CDE-diet. We also found that 6d of DDC-diet led to significant reduction in mRNA levels and expression of major canalicular transporters BSEP, ABCG5 and ABCG8 (Fig. 6A, B) as well as the basolateral transporter NTCP, OATP-1 in the hepatocytes (Fig. 6A, B). Based on the observed rescue (Fig. 3) or further reduction (Fig. 4) in the expression of claudins following 14d of CDE or DDC-diet, respectively, we also assessed the expression of basolateral and canalicular transporters following 14d of CDE or DDC-diet. Indeed, the mRNA (Fig. 5A) as well as expression (Fig. 5b) of NTCP, OATP-1 and BSEP was found to recover following 14d of CDE-diet. However, the mRNA levels (Fig. 6A, B) or the expression of OATP-1 (Fig. 6C), ABCG8 (Fig. 6D) and BSEP (Fig. 6E) did not recover but further deteriorated after 14d of DDC-diet.
Figure 5. Altered expression of basolateral and apical bile transporters in CDE mediated chronic liver injury.
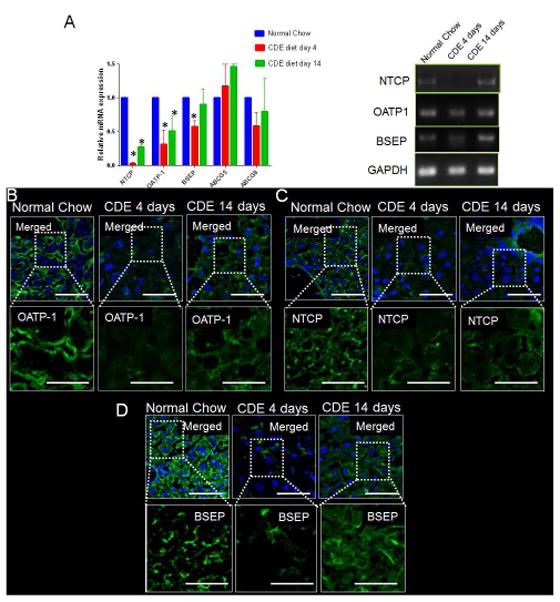
(A) RT-PCR analysis shows significant decrease in mRNA expression of basolateral transporters NTCP, OATP-1 and apical transporter BSEP on CDE-diet for 4d as compared to mice on normal-diet. Rescue was seen in BSEP, NTCP and OATP-1 level at 14d on CDE-diet. Gel showing PCR product for BSEP, NTCP, OATP-1 and GAPDH. (B) Localization of OATP-1 to the basolateral membrane of hepatocytes in a WT-liver. Membrane localization of OATP-1 is reduced in liver of mice fed on CDE-diet for 4d. At 14d on CDE diet, OATP-1 level reappears to the basolateral membrane. (C) Basolateral localization of transporter NTCP in a WT liver. Mice on CDE-diet for 4d shows reduced localization of NTCP in basolateral membrane. Mild increase in ABCG8 level in seen at 14d on CDE-diet. (D) BSEP localization is seen in the basolateral membrane of hepatocytes in a control liver. BSEP levels are strongly reduced in CDE-diet fed mice at 4d. At 14d on CDE-diet, BSEP levels reappears to the basolateral side of the membrane. * denotes p=0.05
Figure 6. Misexpression of basolateral and apical bile-transporters in DDC mediated chronic liver injury.
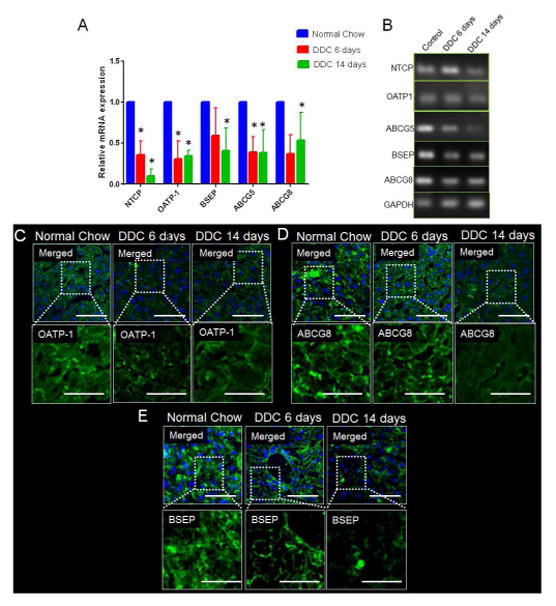
(A) RT-PCR analysis shows significant decrease in mRNA expression of apical transporters ABCG5, ABCG8, BSEP and basolateral transporters NTCP and OATP-1, on DDC-diet for 6d as compared to mice on normal diet. Continued reduction is seen in NTCP, OATP-1, ABCG5, ABCG8 and BSEP at 14d on DDC-diet. (B) Gel showing PCR product for NTCP, OATP-1, ABCG-5, ABCG-8, BSEP and GAPDH. (C) Localization of OATP-1 to the basolateral membrane of hepatocytes in a WT-liver. OATP-1 is reduced in liver of mice fed on DDC-diet for 6d. At 14d on DDC-diet, OATP-1 remains absent from the basolateral membrane. (D) Basolateral localization of ABCG8 in a WT-liver. Mice on DDC-diet for 6d and 14d shows reduced localization of ABCG8 in basolateral membrane. (E) BSEP localization is seen in the basolateral membrane of hepatocytes in a control liver. BSEP levels are strongly reduced in DDC-diet fed mice at 6d and 14d. * denotes p=0.05
Reappearance of claudins and bile transporters rescues BBlB integrity
Based on the recovery of claudins and bile transporters in mice fed CDE-diet for 14d, and continued deterioration and almost complete ablation of claudins and transporters in mice fed DDC-diet for 14d, we next determined the status of liver injury and BBlB integrity at 14d in both models. We found that the reappearance of claudins and bile transporters at 14d of CDE-diet was associated with significant amelioration of liver injury and hyperbilirubinemia compared to 4d of CDE-diet. Blood ALP, ALT, total and direct bilirubin levels in mice fed CDE-diet for 14d were comparable to that in mice fed chow-diet for 14d (Fig. S8A). The reduction in hyperbilirubinemia in CDE-diet at 14d also led to recovery of body-weight and liver-weight (Fig. S8B). Like mice fed CDE-diet for 4d, mice fed CDE-diet for 14d continued to lack activation of hepatic stellate cells (α-SMA staining in Fig. S9A–A′) and additionally showed less accumulation of macrophages (F4/80 staining in Fig. S9B–B′), reduced injury, and reduced inflammation (Fig. S9C–C′ and H&E staining in Fig S9D–D′). Remarkably, qLIM revealed that the attenuation of liver injury and bilirubinemia in mice fed CDE-diet for 14d was associated with complete rescue of BBlB physical integrity and bile transport across hepatocytes (Fig. 7E–H, S6A–C compared to Fig. 7A–D). Similar to mice fed chow-diet, CF localization was evident inside hepatocytes including a few bile-canaliculi within 5m of IV administration of CFDA. At 5m time point, TXR-dextran was confined exclusively to liver sinusoids (Fig. 7E, S6A–A″, movie-S10). Identical to our observations in mice fed chow-diet, we found that TXR-dextran and CF continued to mark the sinusoids and hepatocytes including bile ducts, respectively at 10 (Fig. 7F, S6B–B″, movie-S11) and 20m (Fig. 7G, S6C–C″, movie- S12,) post IV administration of CFDA. These observations were verified using colocalization and intensity analyses, which revealed exclusive localization of TXR-dextran and CF in liver sinusoids and bile-canaliculi, respectively (Fig. S6D; Fig. S6E–G). Interestingly, the number of bile-canaliculi seen at 14d in CDE-diet was still less (Fig. 7E–G) compared to mice fed chow-diet (Fig. 7A–C). Although we did not find a marked difference in the structure of canaliculi, fewer canaliculi does suggest notable but incomplete resolution of liver injury at 14d of CDE-diet (Fig. 7E–G, S6A–C).
Figure 7. Imaging liver injury progression in CDE and DDC regimen using qLIM.

Time series images of (A) 3m, (B) 10m and (C) 20m of mouse liver fed on normal-chow shows localization of CF (green) and TXR-dextran (red) in bile-canaliculi and sinusoids, respectively. (D) qLIM schematic diagram of mouse liver section fed with normal-chow. Time series images of (E) 3m, (F) 10m and (G) 20m of mice liver fed on CDE-diet for 14d shows localization of CF (green) and TXR-dextran (red) in bile-canaliculi and sinusoids, respectively. (H) qLIM schematic diagram of mouse liver section fed with CDE-diet for 14d. Time series images of (I) 3m, (J) 10m and (K) 20m of mouse liver fed on DDC-diet for 14d shows complete localization of CF (green) and TXR-dextran (red) in liver sinusoids. (L) qLIM schematic diagram of mouse liver section fed with DDC-diet for 14d. Arrow denotes presence of this conduits through which blood and bile are leaking and dotted-lines represents hepatocytes.
In contrast to CDE-diet, mice fed DDC-diet for 14d did not show any improvement in liver injury and bilirubinemia compared to mice fed DDC-diet for 6d. Compared to mice fed DDC-diet for 6d, serum levels of ALT, ALP, total and direct bilirubin were significantly elevated at 14d suggesting worsening of liver injury and bilirubinemia (Fig. S8C). Unlike CDE-diet, both body and liver weight continued to reduce at 14d compared to 6d of DDC-diet (Fig. S8D). The worsening rather than amelioration of liver injury in mice fed DDC-diet for 14d was further supported by increased inflammation (CK19 in Fig. S10C–C′, EpCAM in Fig. S10D–D′ and SOX-9 in Fig. S10-E–E′), characteristic onion skin fibrosis (H&E in Fig. S10B–B′), proliferation and ductular reaction (Sirius red in Fig. S10A–A′). These findings suggest that 14d of DDC-diet, which is associated with significant ablation of claudins (3, 5 and 7) and bile transporters from hepatocytes, also contributes to worsening of liver injury. In support of this finding, qLIM revealed that unlike the recovery of BBlB and bile transport observed at 14d of CDE-diet, 14d of DDC-diet led to breach of the BBlB and complete impairment of bile transport measured in the form of CF uptake or release by hepatocytes (Fig. 7I–L, S7, A–C, movies-S13–15). The failure in bile transport and breach of BBlB appeared to be more severe at 14d compared to 6d of DDC-diet (Fig. 1I–K compared Fig. 7I–K). Whereas CF was uptaken by hepatocytes but not released into canaliculi at 6d, colocalization analysis revealed complete overlap of CF (green) with TXR-dextran (red) in the liver sinusoids at all time points (Fig. 7D) suggesting that both uptake and release of CF by hepatocytes was impaired at 14d of DDC-diet (Fig. 7I–K, S7A–C). Finally, the physical breach of BBlB was confirmed in mice fed DDC-diet for 14d by the presence of thin conduits through which a mixture (yellow) of TXR- dextran (red) and CF (green) was flowing into the extra-sinusoidal space between hepatocytes (arrow; Fig. 7I–K, S7A–C, movies-S13–15), and intensity analysis showing accumulation of TXR-dextran (red) into the extra sinusoidal space between hepatocytes and bile-canaliculi (Fig. S7E–G). Finally, to elucidate the long-term effect of these diets on liver injury, we analyzed the livers of mice fed CDE and DDC diet for 30d. Remarkably, DDC diet led to further aggravation of injury and hyperbilirubinemia based on blood serum analysis (Fig. S8C, D). IHC analysis revealed increased injury, inflammation, fibrosis, ductular reaction and trans-differentiation (Fig. S10A–O), whereas qLIM imaging confirmed persistent loss of BBlB (Fig S12A–I) post 30d of DDC diet. In contrast, mice fed CDE diet for 30d manifested almost complete reversal of the liver injury based on blood serum analyses (Fig. S8A, B), immunohistochemistry (Fig. S9) and qLIM imaging (Fig. S11A–I). Furthermore, RT-PCR analysis revealed a continuous loss of key TJ proteins and bile acid transporters post 30 day of DDC diet (Fig. S12J, K), which was absent in mice fed CDE diet for 30 days (Fig. S11J, K). Taken together, our data supports the role of BBlB loss in chronic liver injury progression and maintenance.
Discussion
CLD results in over 70,000 deaths every year in the US35 and the current treatment is limited to supportive therapy36–40. Although liver transplantation is offered to patients diagnosed with liver failure, the dearth of qualified liver donors underscores that better therapies designed to halt the progression of CLD are needed. Chronic liver injury that involves inflammation of the liver parenchyma, hyperbilirubinemia, ductular reaction, liver fibrosis and cirrhosis, is the predominant pathophysiology underlying CLD. CDE and DDC-diet induced models of chronic liver injury in mice have been instrumental in studying the progression of liver injury. However, the cellular and molecular basis that underlies the histopathological manifestation of liver injury have remained elusive due to our inability to visualize the liver pathophysiology in mice in real-time in vivo. Here, we have used qLIM microscopy (qLIM) to visualize the BBlB interface and the kinetics of bile transport in the intact liver of live mice fed chow, CDE or DDC-diet.
We found that mice fed CDE or DDC-diet developed hyperbilirubinemia and liver injury, which was concomitant to physical breach of BBlB and failure of bile transport across hepatocytes. Interestingly, we found differences in the dynamics of induction, progression and resolution of liver injury by the two diets. Although the two diets led to comparable liver injury and hyperbilirubinemia, the injury was evident at 4d on CDE but 6d on DDC-diet suggesting that the progression of liver injury was faster in CDE than DDC fed mice. In mice fed CDE-diet for 4d, the uptake of bile by hepatocytes was absent and inter-hepatocyte TJs were widened leading to para-hepatocyte leakage of sinusoidal blood and mixing of blood with bile. In contrast, the blood and bile did not mix and the uptake of bile by hepatocytes was unaffected, but the release of bile by hepatocytes into the bile-canaliculi was absent post 6d of DDC-diet.
We found that the physical breach of BBlB was associated with the reduction in mRNA as well as protein expression of TJ adhesion molecules (claudins-3, 5 and 7) at 4d of CDE or 6d of DDC-diet. Previously, the expression of TJ adhesion-molecule occludin was shown to increase following blood-brain-barrier injury41 to compensate for the loss of other TJ adhesion-molecules including claudins, however, a similar role for occludin in BBlB breach has never been reported. We found that the loss of claudins-3, 5 and 7 from hepatocytes in CDE or DDC-fed mice was indeed associated with a compensatory increase in the expression of occludin. We also found that the failure of bile uptake by hepatocytes post 4d of CDE and failure of bile release by hepatocytes into bile-canaliculi post 6d of DDC was associated with loss of basolateral bile transporters (NTCP and OATP1) and both basolateral (OATP1 and NTCP) as well as canalicular (BSEP, ABCG5 and ABCG8) bile transporters from hepatocytes, respectively. This finding further suggested that the failure of bile release in canaliculi post 6d of DDC-diet is probably associated with a collective failure of bile uptake from sinusoidal blood as well as release into canaliculi by hepatocytes. This observation is also supported by previous investigations on the role of bile transporters in DDC-diet induced chronic liver injury18, 42.
Surprisingly, the loss of claudins- and bile transporters was partially rescued by 14d and completely rescued by 30d on CDE-diet. In contrast, both claudins and bile transporters were further reduced post 14d on DDC-diet and continued to reduce at 30d of DDC-diet. Remarkably, we found that the presence or lack of rescue of claudins and bile transporters at later time points of CDE or DDC-diet, respectively was associated with amelioration or worsening of BBlB physical integrity, active bile transport, liver injury, hyperbilirubinemia and fibrosis. Taken together, our findings suggest that chronic liver injury involves loss of TJ adhesion-molecules claudins and bile transporters from hepatocytes, which is associated with physical breach of BBlB and failure of bile transport. The reappearance of TJ proteins and bile transporters on hepatocytes is accompanied by the recovery of BBlB integrity and resolution of liver injury. This finding was further supported by the biochemical analyses (data shown in Fig. S13C) of liver biopsies showing aberrant expression of claudins and bile transporters in subset of primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC) patients.
The interpretation of our data also leads to several new questions that need to be addressed in future studies. First, the DDC-diet induced loss of canalicular bile transporters (BSEP, ABCG5 and ABCG8) could promote the intra-hepatocyte accumulation of bile acids. Bile accumulation within hepatocytes may lead to reduced expression of basolateral (OATP1 and NTCP) and increased expression of efflux (MRP3 and MRP4) bile transporters to reduce the uptake and increase the efflux of bile acids to sinusoidal blood, respectively. Indeed, loss of both basolateral and canalicular transporters as well as hyperbilirubinemia was evident in mice fed DDC-diet. Interestingly, CFDA emits green fluorescence only following hydrolysis to CF in hepatocytes. Although the presence of fluorescent CF within the sinusoids of mice fed DDC or CDE-diet suggest a possible contribution of bile efflux from hepatocytes, the synergistic role of canalicular, basolateral and efflux bile transporters remains unknown. Second key question that warrants future investigations is the hierarchy of molecular and biophysical events identified by the current study. Although the current study is the first to establish that loss or rescue of claudins and bile transporters is associated with failure or recovery, respectively of BBlB integrity and bile transport, whether there is a cause and effect relationship between these events or they all are secondary to another unidentified event remains to be elucidated.
The primary objective of our study was to identify cellular, molecular and biophysical events that are common to the progression of injury in diverse experimental models of chronic liver injury in mice. Irrespective of the chosen model (CDE or DDC-diet), we have identified that loss of TJ adhesion-molecules and bile transporters is linked to physical disruption of BBlB, mixing of blood with bile, and failure of bile uptake or release by hepatocytes. Our findings suggest that preventing the loss or stabilizing the expression of claudins or bile transporters could be a useful approach to prevent or delay liver failure in CLD and the feasibility of such a therapy should be tested in future studies using mice models of chronic liver injury.
Supplementary Material
Acknowledgments
Grant Support: This study was supported in part by NIH grants 1R01DK62277, 1R01DK100287, R01CA204586 and an Endowed Chair for Experimental Pathology to SPM. This study was also supported in part by NIH-NHLBI 1R01HL128297-01 to PS. This study was also supported by training grant number 5T32DK639-22 (13), NIH, NIDDK to TP-S.
Footnotes
The authors have no conflict of interest to declare.
Study concept and design: TP-S, SPM and PS; acquisition of data: TP-S, RV, SS, AM; analysis and interpretation of data: TP-S, SPM, PS, drafting of the manuscript: TP-S, SPM, PS critical revision of the manuscript for important intellectual content: TP-S, SPM, PS; statistical analysis:: TP-S, obtained funding: TP-S, SPM, PS; technical, or material support: JR, LM, MP, KN-B, SS, PC, SK, SW, study supervision: SPM, PS
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu J, Murphy SL, Kochanek KD, Bastian BA. Deaths3: Final Data for 2013. National VItal Statistics Reports. 2016;64 [PubMed] [Google Scholar]
- 2.Younossi ZM, et al. Changes in the Prevalence of the Most Common Causes of Chronic Liver Diseases in the United States From 1988 to 2008. Clin Gastroenterol Hepatol. 2011;9 doi: 10.1016/j.cgh.2011.03.020. [DOI] [PubMed] [Google Scholar]
- 3.Pellicoro A, Ramachandran P, Iredale JP, Fallowfield JA. Liver fibrosis and repair: immune regulation of wound healing in a solid organ. Nat Rev Immunol. 2014;14:181–194. doi: 10.1038/nri3623. [DOI] [PubMed] [Google Scholar]
- 4.Blachier M, Leleu H, Peck-Radosavljevic M, Valla DC, Roudot-Thoraval F. The burden of liver disease in Europe: a review of available epidemiological data. J Hepatol. 2013;58:593–608. doi: 10.1016/j.jhep.2012.12.005. [DOI] [PubMed] [Google Scholar]
- 5.Bataller R, Brenner D. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology. 2014;147:765–783.e4. doi: 10.1053/j.gastro.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schuppan D, Schattenberg JM. Non-alcoholic steatohepatitis: Pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol. 2013;28:68–76. doi: 10.1111/jgh.12212. [DOI] [PubMed] [Google Scholar]
- 8.Pradhan-Sundd T, et al. Dual catenin loss in murine liver causes tight junctional deregulation and progressive intrahepatic cholestasis. Hepatology. 2017 doi: 10.1002/hep.29585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matter K, Balda MS. Signalling to and from tight junctions. Nat Rev Mol Cell Biol. 2003;4:225–36. doi: 10.1038/nrm1055. [DOI] [PubMed] [Google Scholar]
- 10.Rao RK, Samak G. Bile duct epithelial tight junctions and barrier function. Tissue barriers. 2013;1:e25718. doi: 10.4161/tisb.25718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kojima T, et al. Regulation of the blood-biliary barrier: Interaction between gap and tight junctions in hepatocytes. Medical Electron Microscopy. 2003;36:157–164. doi: 10.1007/s00795-003-0220-5. [DOI] [PubMed] [Google Scholar]
- 12.Arrese M, Ananthananarayanan M, Suchy FJ. Hepatobiliary transport: molecular mechanisms of development and cholestasis. Pediatr Res. 1998;44:141–147. doi: 10.1203/00006450-199808000-00001. [DOI] [PubMed] [Google Scholar]
- 13.Akhurst B, et al. A modified choline-deficient, ethionine-supplemented diet protocol effectively induces oval cells in mouse liver. Hepatology. 2001;34:519–522. doi: 10.1053/jhep.2001.26751. [DOI] [PubMed] [Google Scholar]
- 14.Croager EJ, Smith PGJ, Yeoh GCT. Ethanol interactions with a choline-deficient, ethionine-supplemented feeding regime potentiate pre-neoplastic cellular alterations in rat liver. Carcinogenesis. 2002;23:1685–93. doi: 10.1093/carcin/23.10.1685. [DOI] [PubMed] [Google Scholar]
- 15.Ueberham E, Böttger J, Ueberham U, Grosche J, Gebhardt R. Response of sinusoidal mouse liver cells to choline-deficient ethionine-supplemented diet. Comp Hepatol. 2010;9:8. doi: 10.1186/1476-5926-9-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Preisegger KH, et al. Atypical ductular proliferation and its inhibition by transforming growth factor beta1 in the 3,5-diethoxycarbonyl-1,4-dihydrocollidine mouse model for chronic alcoholic liver disease. Lab Invest. 1999;79:103–109. [PubMed] [Google Scholar]
- 17.Fickert P, et al. Bile acid-induced Mallory body formation in drug-primed mouse liver. Am J Pathol. 2002;161:2019–26. doi: 10.1016/S0002-9440(10)64480-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fickert P, et al. A New Xenobiotic-Induced Mouse Model of Sclerosing Cholangitis and Biliary Fibrosis. Am J Pathol. 2007;171:525–536. doi: 10.2353/ajpath.2007.061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thompson MD, et al. Spontaneous repopulation of β-catenin null livers with β-catenin-positive hepatocytes after chronic murine liver injury. Hepatology. 2011;54:1333–1343. doi: 10.1002/hep.24506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Looney MR, et al. Stabilized imaging of immune surveillance in the mouse lung. Nat Methods. 2011;8:91–96. doi: 10.1038/nmeth.1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Massou S, Albigot R, Prats M. Carboxyfluorescein fluorescence experiments. Biochem Educ. 2000;28:171–173. [PubMed] [Google Scholar]
- 22.Liu Y, et al. Visualization of hepatobiliary excretory function by intravital multiphoton microscopy. J Biomed Opt. 2011;12:14014. doi: 10.1117/1.2710237. [DOI] [PubMed] [Google Scholar]
- 23.Li FC, et al. In vivo dynamic metabolic imaging of obstructive cholestasis in mice. Am J Physiol Gastrointest Liver Physiol. 2009;296:G1091–7. doi: 10.1152/ajpgi.90681.2008. [DOI] [PubMed] [Google Scholar]
- 24.Fickert P, et al. A New Xenobiotic-Induced Mouse Model of Sclerosing Cholangitis and Biliary Fibrosis. Am J Pathol. 2007;171:525–536. doi: 10.2353/ajpath.2007.061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Angelow S, Ahlstrom R, Yu ASL. Biology of claudins. Am J Physiol Renal Physiol. 2008;295:F867–F876. doi: 10.1152/ajprenal.90264.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koval M. Claudins—Key Pieces in the Tight Junction Puzzle. Cell Commun Adhes. 2006;13:127–138. doi: 10.1080/15419060600726209. [DOI] [PubMed] [Google Scholar]
- 27.Sambrotta M, et al. Mutations in TJP2 cause progressive cholestatic liver disease. Nat Genet. 2014;46:326–328. doi: 10.1038/ng.2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sambrotta M, Thompson RJ. Mutations in TJP2, encoding zona occludens 2, and liver disease. Tissue barriers. 2015;3:e1026537. doi: 10.1080/21688370.2015.1026537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlton VEH, et al. Complex inheritance of familial hypercholanemia with associated mutations in TJP2 and BAAT. Nat Genet. 2003;34:91–96. doi: 10.1038/ng1147. [DOI] [PubMed] [Google Scholar]
- 30.Maly IP, Landmann L. Bile duct ligation in the rat causes upregulation of ZO-2 and decreased colocalization of claudins with ZO-1 and occludin. Histochem Cell Biol. 2008;129:289–299. doi: 10.1007/s00418-007-0374-7. [DOI] [PubMed] [Google Scholar]
- 31.Huang S, et al. Altered expression levels of occludin, claudin-1 and myosin light chain kinase in the common bile duct of pediatric patients with pancreaticobiliary maljunction. BMC Gastroenterol. 2016;16:7. doi: 10.1186/s12876-016-0416-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boyer JL. Bile formation and secretion. Compr Physiol. 2013;3:1035–1078. doi: 10.1002/cphy.c120027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meier PJ, Stieger B. Molecular Mechanisms in Bile Formation. News Physiol Sci. 2000;15:89–93. doi: 10.1152/physiologyonline.2000.15.2.89. [DOI] [PubMed] [Google Scholar]
- 34.Wagner M, et al. Role of farnesoid X receptor in determining hepatic ABC transporter expression and liver injury in bile duct-ligated mice. Gastroenterology. 2003;125:825–838. doi: 10.1016/s0016-5085(03)01068-0. [DOI] [PubMed] [Google Scholar]
- 35.Vong S, Bell BP. Chronic liver disease mortality in the United States, 1990–1998. Hepatology. 2004;39:476–83. doi: 10.1002/hep.20049. [DOI] [PubMed] [Google Scholar]
- 36.Takami T, Terai S, Sakaida I. Stem cell therapy in chronic liver disease. Curr Opin Gastroenterol. 2012;28:203–208. doi: 10.1097/MOG.0b013e3283521d6a. [DOI] [PubMed] [Google Scholar]
- 37.Nicolas CT, Wang Y, Nyberg SL. Cell therapy in chronic liver disease. Curr Opin Gastroenterol. 2016;1 doi: 10.1097/MOG.0000000000000262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Arora G, Keeffe EB. Management of chronic liver failure until liver transplantation. Med Clin North Am. 2008;92:839–60. ix. doi: 10.1016/j.mcna.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 39.Fox RK. When to consider liver transplant during the management of chronic liver disease. Medical Clinics of North America. 2014;98:153–168. doi: 10.1016/j.mcna.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 40.Solà E, Ginès P. Chronic kidney disease: A major concern in liver transplantation in the XXI century. Journal of Hepatology. 2014;61:196–197. doi: 10.1016/j.jhep.2014.05.011. [DOI] [PubMed] [Google Scholar]
- 41.Pan R, et al. Blood Occludin Level as a Potential Biomarker for Early Blood Brain Barrier Damage Following Ischemic Stroke. Sci Rep. 2017;7:40331. doi: 10.1038/srep40331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamazaki Y, Moore R, Negishi M. Nuclear receptor CAR (NR1I3) is essential for DDC-induced liver injury and oval cell proliferation in mouse liver. Lab Investig. 2011;91:1624–1633. doi: 10.1038/labinvest.2011.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.