

Abstract
Background
The precise clinical diagnosis of Parkinson's disease (PD) can be difficult in the early stages. Diagnostic criteria include the response of key motor features to levodopa as a supportive prospective criterion. Data are sparse on the diagnostic value of the acute levodopa challenge test (LDCT) in patients with de novo PD. The objective of this study was to validate the LDCT as a tool in the early clinical diagnosis of PD.
Methods
We performed the standardized LDCT with 250 mg levodopa in the prospective longitudinal cohort study “DeNoPa,” comprising 159 patients with de novo PD, and carried out longitudinal clinical follow‐up for 24 months. Motor assessments at baseline using the motor part (part III) of the Unified Parkinson's Disease Rating Scale before and 1 hr after drug administration were documented. The optimal cutoff score on the LDCT was calculated using the Youden index.
Results
Clinical reassessment of 144 patients who returned for follow‐up confirmed the diagnosis of PD in 120 patients (83%). In 24 patients (17%), the initial diagnoses were revised and classified as other neurologic disorders. The optimal cutoff at 33% improvement of motor symptoms on the part 3 of the Unified Parkinson's Disease Rating Scale during the LDCT reached a sensitivity of 70% a specificity of 71%. The positive and negative predictive values were 92% and 32%, respectively. Sensitivity (91%), specificity (79%), and positive/negative (96%/63%) predictive values improved with the addition of further clinical information (urinary incontinence, fainting, asymmetric tremor, and amount of further drug‐intake).
Conclusions
The LDCT is a reliable tool in the early diagnosis of PD. The accuracy of this test can be further improved by additional, easy‐to‐acquire clinical information provided by patients. © 2017 International Parkinson and Movement Disorder Society.
Keywords: DeNoPa cohort, de novo Parkinson's disease, levodopa challenge test, sensitivity, specificity
The diagnosis of idiopathic Parkinson's disease (PD) is currently based on United Kingdom Parkinson's Disease Society Brain Bank (UKPDSBB) criteria as the presence of motor signs as well as their responsiveness to levodopa (l‐dopa).1 These criteria have been fully validated2 and are internationally accepted; however, they produce a high rate of misdiagnosis,3 especially during the first 5 yr of the disease, when symptoms are few and mild and when there is no dyskinesia. Differential diagnosis of PD from other neurologic disorders (ONDs) (e.g., essential tremor) or other neurodegenerative diseases (e.g., multiple system atrophy) can be difficult in the early stages. The accuracy of early diagnosis, however, becomes important4, 5 for clinical trials of upcoming, putative neuroprotective and disease‐modulating substances as well as for patients’ treatment and prognosis. Therefore, easily applicable tests or marker candidates that support the clinical diagnosis are needed.
The responsiveness of motor symptoms to l‐dopa is a key supportive prospective criterion of the UKPDSBB criteria.6 The acute l‐dopa challenge test (LDCT), which involves rating motor symptoms according to the motor part (part 3) of the Unified Parkinson's Disease Rating Scale (UPDRS III) both before and after a “single‐shot” administration of an above‐threshold dose of l‐dopa (or apomorphine), enables this criterion to be tested in a standardized way.7 For a long time, there has been controversial debate regarding the precise cutoff values for the diagnosis of PD as well as the sensitivity and specificity of this test. In a 2001 consensus statement, movement disorder experts called for further studies on the role of acute dopaminergic challenge in PD8; because, until then, only two studies9, 10 had reported on use of the LDCT to evaluate l‐dopa response in patients with de novo PD, but those studies included incomparable dosages and short‐term follow‐up. Furthermore, both studies used different motor tasks: the Webster scale11 (a severity scale of disease symptoms and clinical impairment), tremor, tapping, and a 12‐meter walk in 45 patients9; and tapping and a 3‐meter walk in 22 patients.10 After the consensus statement, Merello et al.12 published results from a prospective study of 82 patients who were first seen at a movement disorders clinic and had a parkinsonian syndrome without specific diagnosis. Each patient underwent an acute LDCT and was followed for 24 months to obtain a definitive clinical diagnosis of idiopathic PD (according to UKPDSBB criteria). The authors calculated the sensitivity (70.9%), specificity (81.4%), and positive (88.6%) and negative (57.9%) predictive ratios of the test to predict a clinical diagnosis of PD. Despite these excellent data, recent European guidelines for the diagnosis of PD2 have not been able to make recommendations on drug challenge tests due to “insufficient evidence to support their role in the differential diagnosis between PD and other parkinsonian syndromes,” mainly because of relatively high rates of false‐negative and false‐positive results. A need for further studies was postulated once again.2
The motor component of the UPDRS13 is frequently used to evaluate and document responses to l‐dopa in daily clinical practice. Cutoff values have been set at a 20% improvement for response to apomorphine (which demonstrated a specificity of 90% and a sensitivity of 88%),14 but these values have mainly been evaluated in patients with established PD and rarely for l‐dopa in those with de novo PD (e.g., see Zappia et al.15). A 30% minimum motor improvement compared with baseline has been considered clinically significant16 and thus was recommended in guidelines.8 For the newer, Movement Disorder Society‐sponsored revision of the UPDRS (MDS‐UPDRS),17 an equivalent 24% motor improvement was proposed for predicting sustained, long‐term l‐dopa response, with a high correlation between the 2 scales.18
The purpose of this study was to provide further evidence of the utility of the LDCT in the differential diagnosis of PD in a prospective cohort study of patients who had de novo PD patients with a long clinical follow‐up and to enhance diagnostic accuracy by providing additional clinical information.
Patients and Methods
Recently diagnosed, drug‐naive patients with PD were recruited between September 2008 and January 2012 at the Paracelsus‐Elena Klinik in Kassel, Germany. As previously reported.19 Patients were screened independently by 2 neurologists (C.T./F.S.‐D. and B.M./J.E.) who specialize in movement disorders to determine whether they met the inclusion/exclusion criteria (including UKPDSBB criteria6). Patients had to be between ages 40 and 85 yr and newly diagnosed with PD. To be considered as de novo, patients’ l‐dopa exposure was allowed to be no greater than 2 weeks and must not have been within 4 weeks before study entry. Patients who had known, severe vascular encephalopathy or normal‐pressure hydrocephalus on magnetic resonance imaging and/or signs or symptoms of multiple system atrophy (MSA), progressive supranuclear palsy, or dementia with Lewy bodies according to consensus criteria16, 20, 21 were excluded.
Baseline Investigations
Upon study entry, patients were examined by a neurologist and a movement disorder specialist to determine their UPDRS III (motor component) score and also underwent a general neurologic examination. The medical history, past medical conditions, comorbidities, and current medications were assessed by standardized interview. Patients also answered several questionnaires (the Scale for Outcomes in Parkinson's Disease for Autonomic Symptoms [SCOPA‐AUT],22 the Non‐motor Symptoms Scale,23 and the remaining parts of the UPDRS).13 Because malabsorption can hamper the test, serum glucose and vitamin B12 levels were also evaluated from a blood sample collected by venous puncture in the morning (between 7 and 8 am) under fasting conditions. For further details on baseline investigations, see also Mollenhauer et al., 2013.19 After the baseline assessments, all patients started dopaminergic therapy according to accepted guidelines.
The Acute LDCT
Patients were pretreated with domperidone (20 mg) 12 hr before and again 1 hr before the administration of l‐dopa to minimize side effects. The UPDRS part III (motor component) score13 was determined by a movement disorder specialist in the morning between 8 and 9 am. The, 250 mg l‐dopa (soluble preparation; Madopar LT, Roche Pharma AG, Basel, Switzerland) was then administered under fasting conditions. One hour after administration, the UPDRS part III (motor component) was performed once again by the same investigator. The procedure was documented by video recording. Patients knew about the purpose of the DeNoPa study to evaluate and characterize biomarkers in de novo PD. They were further informed about the LDCT as a standardized test for the responsiveness of disease symptoms to l‐dopa. They were not informed about the test results or about the prognosis for the course of the disease in case of a negative or positive result.
Clinical Follow‐up
To improve diagnostic accuracy, patients were followed clinically after a mean follow‐up of 24 months after initial diagnosis. The clinical diagnosis was reassessed by 2 teams of independent neurologists (C.T./F.S.‐D. and B.M./J.E.) applying UKPDSBB criteria for PD and international consensus criteria for the various ONDs.16, 20, 21 Investigators were blinded to results from the acute LDCT at baseline, but they took subjective responses to anti‐Parkinson drugs (by history) into account in re‐evaluating the diagnosis (e.g., if the patient experienced worsening of symptoms when they forgot their l‐dopa medication).
Statistical Analysis
Normally distributed data were described as mean ± standard deviation values; non‐normal and ordinal variables were described as the median with minimum and maximum values; and, in case of dichotomous parameters, the absolute and relative frequencies were stated. Furthermore, all parameters were compared between patients with and without PD using the t test for continuous, normally distributed variables; the Wilcoxon‐Mann‐Whitney U test in for ordinal or non‐normally distributed variables; and the Fisher exact test for dichotomous variables. The multiple testing was regarded as a screening process to identify variables that might be suitable for predicting PD; therefore, P values were not adjusted.
The parameter “l‐dopa‐change” (i.e, the percentage of improvement in the UPDRS III motor score after the LDCT) was analyzed with respect to PD by a receiver operating characteristic (ROC) curve. Different cutoff points, as well as sensitivity, specificity, and predictive values, were reported; and the optimal cutoff point was determined using the Youden criterion.24 The point on the ROC curve at which the Youden index is maximal relates to the optimal cutoff, which accounts for the trade‐off between sensitivity and specificity. Moreover, each parameter was described for the patients who had false‐negative LDCT results.
Multiple imputations were performed for the parameters that differed significantly between patients with and without PD in order to fill in the missing values. For each of the 100 imputed data sets, all parameters were combined by a logistic regression model, and a backward variable‐selection algorithm was applied to the full model. The variables that remained in the final model over all data sets more than 95 times were included in the final logistic regression for multiple imputed data sets. For each patient, the mean over all fitted probabilities was calculated, and these means were regarded as a combined classifier, which was evaluated by the ROC curve analysis.
Furthermore, the correlation between “l‐dopa‐change” and each continuous and ordinal variable was determined using Pearson's r or Kendall's tau coefficient, respectively. Because of the multiple testing situations, raw P values were adjusted using the Bonferroni‐Holm method.
The significance level was set to α = 5% for all statistical tests. All analyses were performed with the statistic software R (version 3.0.2; R Foundation for Statistical Computing, Austria, Vienna; http://www.r-project.org) using the R‐package “pROC”25 for the ROC analyses and the package “mice”26 for multiple imputations.
Standard Protocol Approvals, Registrations, and Patient Consents
We conformed to the Declaration of Helsinki, and the study was approved by the Ethics Committee of the Physician's Board Hesse, Germany (approval no. FF89/2008). The study was registered at the German Register for Clinical Trials (DRKS00000540) according to the World Health Organization Trial Registration Data Set. All patients provided informed written consent.
Results
In total, 147 patients completed 24 months of follow‐up. Three patients were excluded from analysis because of severe nausea caused by l‐dopa and, thus, invalid testing. The remaining 144 patients were included in the statistical analysis (for details of patients’ study flow, see Fig. 1). Basic demographic data are shown in Table 1. In total, 118 patients (82%) fulfilled at least 1 of 2 applicable supportive UKPDSBB criteria (strictly unilateral rest tremor and/or rigor, rest tremor present) upon baseline investigation (ONDs, 12 [50%]; PD, 106 [88%]). The reassessment of clinical diagnosis revealed ONDs in 24 patients (17%) after 24 months of follow‐up: 4 were classified with progressive supranuclear palsy, 4 had MSA parkinsonian subtype (MSA‐P), 1 had dementia with Lewy bodies, 2 had vascular parkinsonism, 1 had corticobasal degeneration, and, in 3 patients, the predominant movement abnormality was classified as essential tremor (n = 2) or cerebellar tremor (n = 1). In 8 patients, the final diagnosis remained unclear. One patient with MSA‐P died after the 24‐month follow‐up visit, and an autopsy confirmed the diagnosis of MSA‐P.
Figure 1.
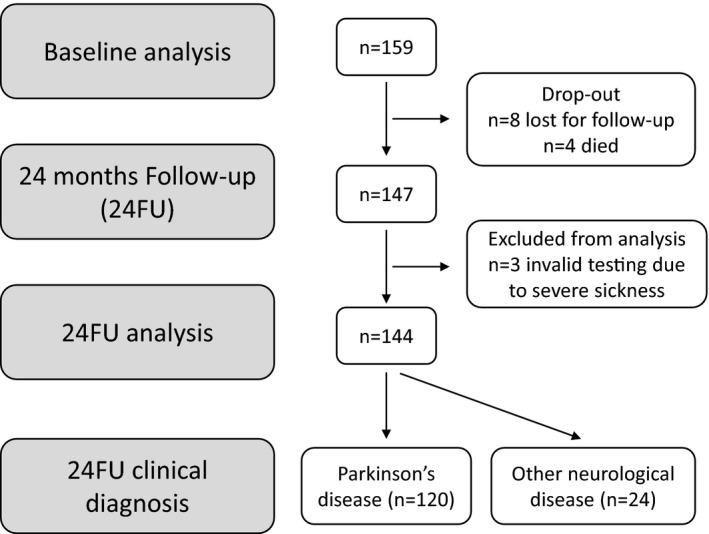
Patient flow chart.
Table 1.
Baseline demographic data and motor assessments of patients diagnosed with and wwithout parkinson's disease at follow‐up
| Variable | PD, n = 120 | OND, n = 24 | P value |
|---|---|---|---|
| Age, y | 63.86 ± 9.34 | 69.71 ± 7.95 | 0.0029 |
| No. of men/women (%) | 79/41 (65.83/34.17) | 16/8 (66.67/33.33) | 1.0000 |
| No. of comorbidities [min, max] | 2 [0, 8] | 2.5 [1, 8] | 0.1965 |
| No. of co‐medications [min, max] | 2 [0, 10] | 5 [1, 12] | <0.001 |
| Duration of first motor symptom [min, max], mo | 13.5 [2, 240] | 12 [2.5, 147] | 0.9121 |
| UPDRS motor score | |||
| Before l‐dopa | 17.98 ± 9.94 | 22.33 ± 10.19 | 0.0641 |
| After l‐dopa | 10.4 ± 7.01 | 16 ± 7.32 | 0.0016 |
| Relative change (%) | 42.51 ± 19.42 | 25.51 ± 17.81 | <0.001 |
| Hoehn & Yahr stage [min, max] | 1.5 [1, 3] | 2 [1, 3] | 0.0473 |
PD, Parkinson's disease; OND, other neurologic disorders; min, minimum; max, maximum; UPDRS, Unified Parkinson's Disease Rating Scale; l‐dopa, levodopa.
All values are reported as mean ± standard deviation, or median [minimum, maximum], or absolute frequency (relative frequency in %).
Comparison of PD and OND
All parameters of the investigations were analyzed with respect to differences between PD and ONDs. Demographics demonstrated that the OND group had more severe motor phenotype according to Hoehn and Yahr stage (P < 0.05) (see Table 1). Additional significantly different values between patients with and without PD are provided in Table S1 (see online supporting information). Results from the final logistic regression of these parameters, excluding the variable “l‐dopa‐change,” along with the consecutive ROC analyses also are provided in the online supporting information (see Tables S2 and S3).
Results of the Acute LDCT
Patients with PD improved with a mean ± standard deviation score of 42.5% ± 19.4% on the UPDRS III (motor component) after drug intake, whereas patients from the OND group improved only with a mean ± standard deviation of 25.5% ± 17.81%. This difference was statistically significant (P < 0.001; t test). Different cutoff points and the corresponding sensitivity, specificity, and predictive values are listed in Table 2. The optimal cutoff point (according to the Youden criterion) was 32.85%. The parameter yields an area under the curve (AUC) of 74.3%, which can be regarded as an estimate for the correct classification rate (see Fig. 2). Although patients in the OND group were significantly older, statistical analysis did not reveal a significant correlation between “age” and “l‐dopa‐change” (percentage improvement in UPDRS III motor score after the LDCT). Furthermore, no significant correlation between “l‐dopa‐change” and “body weight” or tremor‐reflecting items from the UPDRS III motor score could be revealed.
Table 2.
Different cutoff points for levodopa change with responding sensitivity, specificity, positive and negative predictive values, and Youden indexa
| T | Sens | Spec | PPV | NPV | Youden index |
|---|---|---|---|---|---|
| 20 | 85.0 | 37.5 | 87.2 | 33.3 | 22.5 |
| 25 | 82.5 | 45.8 | 88.4 | 34.4 | 28.3 |
| 30 | 76.7 | 50.0 | 88.5 | 30.0 | 26.7 |
| 32 | 70.8 | 58.3 | 89.5 | 28.6 | 29.2 |
| 32.85b | 70.0 | 70.8 | 92.3 | 32.1 | 40.8 |
| 33 | 70.0 | 70.8 | 92.3 | 32.1 | 40.8 |
| 35 | 64.2 | 75.0 | 92.8 | 29.5 | 39.2 |
| 46 | 45.0 | 90.9 | 96.4 | 23.3 | 35.9 |
T = threshold (relative change in Unified Parkinson's Disease Rating Scale motor scores after drug intake); Sens, sensitivity; Spec, specificity; PPV, positive predictive value; NPV, negative predictive value.
All data shown are percentages.
This is the optimal cutoff point for levodopa change according to the Youden index.
Figure 2.
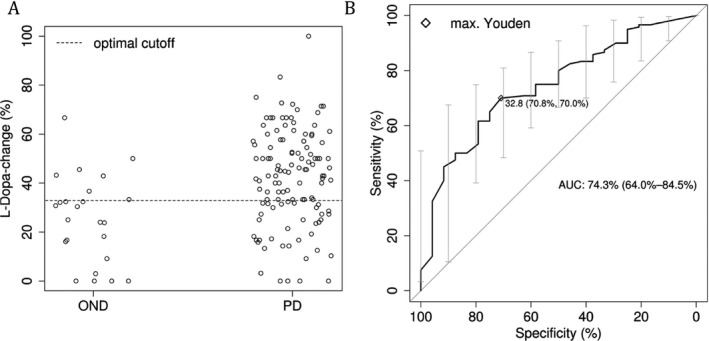
(A) Distribution of levodopa change between patients with and without Parkinson's disease (PD) and the optimal cutoff point according to the Youden criterion. (B) The receiver operating characteristic (ROC) curve and the point of the maximal Youden value. OND = other neurologic disease; max. = maximal; AUC = area under the ROC curve.
Improving Diagnostic Accuracy of the Acute LDCT
To further improve diagnostic accuracy of the acute LDCT, we included significantly different variables between PD and ONDs (see Table S1), indicating either atypical Parkinson syndromes or intestinal malabsorption in further statistical analysis. The final logistic regression included the parameter “L‐dopa‐change,” “number of co‐medications,” “SCOPA‐AUT_9” (involuntary loss of urine), “SCOPA‐AUT_16” (fainted in the past 6 months), and “UPDRS III_20b” (asymmetrical tremor at rest of the upper extremity). In the subsequent ROC curve analysis, this combined classifier yielded an AUC of 90.3% (Fig. 3), which was better than the single parameter “l‐dopa‐change.” Subsequently, sensitivity (90.83%), specificity (79.17%), and positive (95.61%) and negative (63.33%) predictive values also improved. However, the confidence intervals of the AUCs for the combined classifier and “l‐dopa‐change” overlapped (Figs. 2 and 3), so the gain of the combined classifier was not significant.
Figure 3.
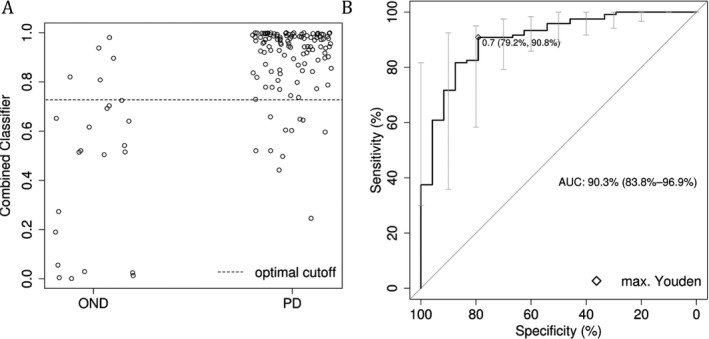
(A) Distribution of probabilities from the combined classifier between patients with and without Parkinson's disease (PD) and the optimal cutoff point according to the Youden criterion. (B) The receiver operating characteristic (ROC) curve and the point of the maximal (max.) Youden value. OND = other neurologic disease; AUC = area under the ROC curve.
False‐negative Acute LDCT
An additional look at serum glucose and vitamin B12 levels (as possible indicators of malabsorption) in patients with PD who had negative LDCT results (defined as a change <33% in the UPDRS part III motor score) compared with those who had PD with positive LDCT results revealed no significant differences.
Discussion
In the quest to improve diagnostic accuracy in early PD (which is reportedly very poor3), we systematically investigated the standardized acute LDCT in 144 patients with de novo PD in a prospective, single‐center study who had their diagnoses clinically reconfirmed after a 2‐year follow‐up visit according to UK Brain Bank criteria. We found that the LDCT was highly accurate in differentiating patients who had PD from those who were assessed with a different disease after 2‐year follow‐up, with a sensitivity of 70% and a specificity of 71% according to international diagnostic criteria. Our analysis demonstrated that the suggested cutoff value of a 33% improvement in the UPDRS III motor score has high sensitivity and specificity. We were able to confirm previous data from Merello et al.12 in an independent cohort, thereby strengthening evidence for the utility of the acute LDCT as an additional tool for the clinical diagnosis of early PD in drug‐naive patients. With the upcoming clinical trials for disease modification in PD, there is a tremendous need to improve the diagnostic accuracy in early stages of the disease. The proposed LDCT can reduce the enrolment numbers of those with misdiagnoses.
Furthermore, we demonstrated that additional parameters taken from medical history and UPDRS III motor scores could improve accuracy of the LDCT even further—up to a sensitivity of 91% and a specificity of 78%—by adding parameters concerning involuntary loss of urine in the past 3 months, fainting in the past 6 months, apparent asymmetric tremor, and the number of co‐medications. Nevertheless, even by adding these parameters, approximately 10% of patients judged to have PD (and over one‐quarter of those without these parameters) had an LDCT result that was considered false‐negative for the diagnosis of idiopathic PD. Therefore, a negative result should not prevent the attending physician from performing a full trial of anti‐Parkinson drugs in clinical practice.
The OND group included patients who had other neurodegenerative syndromes that occasionally might be responsive to medication with l‐dopa. For instance, a core feature for clinical diagnosis of dementia with Lewy bodies (according to international diagnostic criteria21) is parkinsonism, and these patients partly exhibit l‐dopa responsiveness, although to a lesser extent.27, 28 A further look at our data revealed that exclusion of the 2 patients who had “probable” dementia with Lewy bodies at the 24‐month follow‐up visit did not alter sensitivity (70%) or specificity (72%) at the cutoff value of a 33% change in the LDCT score. In this alternative calculation, the optimal cutoff value according to the Youden criterion would be slightly elevated to 37% (sensitivity, 62%; specificity, 82%).
We are aware of certain limitations to our study. The LDCT was conducted in an open, unblinded fashion with no placebo control. We intentionally rejected using a placebo control, because the objective of the study was not to establish the efficacy of l‐dopa but to determine the relevance of the test (including the drug's placebo effect) as a biomarker for diagnostic criteria. To reduce bias, investigators for the re‐assessment of diagnosis after follow‐up were blinded to the outcome of the LDCT at baseline. Furthermore, all motor investigations were videotaped and re‐evaluated by a second investigator (in case of disagreement, a decision was made upon agreement). The UPDRS part III (motor score) remains a partly subjective, rater‐dependent test; all of our patients were assessed by experts in movement disorders who were trained to use the UPDRS. To ensure a strict, standardized protocol for the acute LDCT, our patients were tested 60 min after drug intake. The peak efficacy of oral l‐dopa occurs at 45 to 90 min after intake and can last for several hr.8 Considering that the 60‐min time point might not have been the most optimal ON‐medication state for every single patient, it is possible that there were patients who had a formal negative result on the LDCT that would have been positive upon later testing. In clinical practice, this should be kept in mind, and some patients with a low response after 1 hr should be tested again approximately 30 to 60 min later.
Clinical diagnosis (even with longstanding experts in PD and with strict adherence to international criteria) has its uncertainty, which is a general problem for all studies in PD.6 A final diagnosis can only be pathologically rendered. DeNoPa is a longitudinal study with biannual follow‐up visits and clinical reevaluation with each visit to increase the diagnostic accuracy (along with a brain donation program, which is also established). In that study, as of December 2016, all diagnoses of PD were confirmed, no further ONDs were diagnosed, and the ONDs remained in this group separate from the PD group. In addition, to date, 2 cases have been neuropathologically confirmed post‐mortem.
Another limitation is the rather small (and heterogeneous) group of patients who had ONDs (n = 24) compared with those who had PD (n = 120). Interestingly, the distribution of about 16% primarily misdiagnosed patients replicates results from earlier neuropathologic/empirical studies.6 Furthermore, three patients (all from the PD group) had to be excluded from statistical analysis of the LDCT due to severe nausea and consequently had inadequate testing. These patients might have been nonresponders but also may have been excellent responders with a lower dose of l‐dopa and less side effects. In such patients, a chronic dopaminergic trial with slowly increasing doses of l‐dopa should be considered in clinical practice.
Finally, our cohort is a single‐center study with a homogenous ethnic group from a rather small catchment area; thus, an ethnic and/or genetic bias cannot be excluded. Our findings need to be validated in an independent cohort that includes different recruitment centers and larger numbers.
Our study has 3 major implications. First, we can easily recommend the further use of the acute LDCT as a timesaving, easy, and fast additional tool for improving the accuracy of clinical differential diagnosis. It offers a quick read‐out and limits bias due to the investigators’ long‐term memory attenuated by a high patient turn‐over or long time intervals between visits compared with a chronic dopaminergic trial. In addition, the test is safe and is more widely applicable than nuclear imaging, especially in less developed regions. The proof of responsiveness to l‐dopa also helps the physician with further therapeutic decisions. Second, the LDCT is feasible in patients with de novo PD, which is important especially for clinical studies that rely on an early diagnosis. Third, when using the UPDRS III motor scale,13 we propose an improvement of one‐third (33%) of the total score as the best cutoff point for a positive versus negative test result, thereby confirming previous data reported by Merello et al.12 According to a previous correlation study,18 this would be equivalent to a 24% change in the MDS‐UPDRS motor score.17 Sensitivity and specificity can be further improved if there is no involuntary loss of urine and no fainting in the recent past, if an asymmetric tremor is apparent, and if there is low drug intake. This may be important for selecting patients with de novo PD for future clinical trials of neuromodulatory drugs. However, because the LDCT does not separate diseases by neuropathologic entities (e.g., synuclein aggregation disorders), further specific diagnostic tests are needed (e.g., for α‐synuclein–targeting strategies).
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
S.S.: 2C, 3A, 3B.
F.S.‐D.: 1A, 1B, 1C, 3B.
J.E.: 1C, 3B.
X.S.: 2A, 2B, 3B.
C.T.: 1C, 3B.
B.M.: 1A, 1B, 1C, 2C, 3A, 3B.
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This study was supported by unrestricted research grants from the Paracelsus‐Elena‐Klinik (Kassel, Germany) and the Michael J. Fox Foundation for Parkinson's Research. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the previous 12 months: Jens Ebentheuer reports personal fees from Desitin and nonfinancial support from AbbVie. Friederike Sixel‐Döring reports lecture fees from AbbVie, Medtronic, UCB Pharma, Zambon and Licher MT as well as congress participation fees from Licher MT. Claudia Trenkwalder reports honoraria from and serves on scientific advisory boards for UCB Pharma, GlaxoSmithKline, Mundipharma, Desitin, Britannia, Vifor, and Grünenthal. Brit Mollenhauer reports research support from TEVA Pharma, Desitin, Boehringer Ingelheim, GE Healthcare, the Michael J. Fox Foundation for Parkinson's Research, the German Federal Ministry of Education and Research (BMBF), and the European Union and personal fees from Bayer Schering Pharma AG, Roche, AbbVie, TEVA Pharma, Biogen, Orion Pharma, and GlaxoSmithKline. Sebastian Schade and Xenia Schultz report no sources of funding and no conflicts of interest.
Supporting information
Table S1. Further significantly different parameters of patients with and without Parkinson's disease.
Table S2. Results of the final logistic regression for multiple imputed data sets excluding the variable “l‐dopa‐change”.
Table S3. Optimal cutoff point for the combined classifier (resulting from the model in Table S2) according to the Youden criterion as well as sensitivity, specificity, and positive and negative predictive values.
Acknowledgments
We thank all participants in the study and their families as well as referring neurologists. We also thank Valentina Krenz, Friederike Panzer, Maria Rudolph, Miriam Schanze, Carolin Sengstock and Olivia Steuer for support with sample collection and data entry; Dr. Klaus Jung for statistical advice; as well as Anne‐Marie Williams for revision of the article.
Supporting information may be found in the online version of this article
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Lees AJ, Hardy J, Revesz T. Parkinson's disease. Lancet 2009;373:2055–2066. [DOI] [PubMed] [Google Scholar]
- 2. Berardelli A, Wenning GK, Antonini A, et al. EFNS/MDS‐ES/ENS [corrected] recommendations for the diagnosis of Parkinson's disease. Eur J Neurol 2013;20:16–34. [DOI] [PubMed] [Google Scholar]
- 3. Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson disease: clinicopathologic study. Neurology 2014;83:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord 2015;30:1600–1611. [DOI] [PubMed] [Google Scholar]
- 5. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 6. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Clarke CE, Davies P. Systematic review of acute levodopa and apomorphine challenge tests in the diagnosis of idiopathic Parkinson's disease. J Neurol Neurosurg Psychiatry 2000;69:590–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Albanese A, Bonuccelli U, Brefel C, et al. Consensus statement on the role of acute dopaminergic challenge in Parkinson's disease. Mov Disord 2001;16:197–201. [DOI] [PubMed] [Google Scholar]
- 9. Hughes AJ, Lees AJ, Stern GM. Challenge tests to predict the dopaminergic response in untreated Parkinson's disease. Neurology 1991;41:1723–1725. [DOI] [PubMed] [Google Scholar]
- 10. D'Costa DF, Sheehan LJ, Phillips PA, Moore‐Smith B. The levodopa test in Parkinson's disease. Age Ageing 1995;24:210–212. [DOI] [PubMed] [Google Scholar]
- 11. Webster DD. Critical analysis of the disability in Parkinson's disease. Mod Treat 1968;5:257–282. [PubMed] [Google Scholar]
- 12. Merello M, Nouzeilles MI, Arce GP, Leiguarda R. Accuracy of acute levodopa challenge for clinical prediction of sustained long‐term levodopa response as a major criterion for idiopathic Parkinson's disease diagnosis. Mov Disord 2002;17:795–798. [DOI] [PubMed] [Google Scholar]
- 13. Fahn S, Elton R. UPDRS Development Committee. The Unified Parkinson's Disease Rating Scale In: Fahn S, Marsden C, Calne D, Goldstein M, editors. Recent Developments in Parkinson's Disease. 2nd ed. Florham Park, NJ: Macmillan Healthcare Information; 1987:153–163, 293–304. [Google Scholar]
- 14. Gasser T, Schwarz J, Arnold G, Trenkwalder C, Oertel WH. Apomorphine test for dopaminergic responsiveness in patients with previously untreated Parkinson's disease. Arch Neurol 1992;49:1131–1134. [DOI] [PubMed] [Google Scholar]
- 15. Zappia M, Montesanti R, Colao R, et al. Short‐term levodopa test assessed by movement time accurately predicts dopaminergic responsiveness in Parkinson's disease. Mov Disord 1997;12:103–106. [DOI] [PubMed] [Google Scholar]
- 16. Gilman S, Low P, Quinn N, et al. Consensus statement on the diagnosis of multiple system atrophy. American Autonomic Society and American Academy of Neurology. Clin Auton Res 1998;8:359–362. [DOI] [PubMed] [Google Scholar]
- 17. Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129–2170. [DOI] [PubMed] [Google Scholar]
- 18. Merello M, Gerschcovich ER, Ballesteros D, Cerquetti D. Correlation between the Movement Disorders Society Unified Parkinson's Disease Rating Scale (MDS‐UPDRS) and the Unified Parkinson's Disease Rating Scale (UPDRS) during L‐dopa acute challenge. Parkinsonism Relat Disord 2011;17:705–707. [DOI] [PubMed] [Google Scholar]
- 19. Mollenhauer B, Trautmann E, Sixel‐Doring F, et al. Nonmotor and diagnostic findings in subjects with de novo Parkinson disease of the DeNoPa cohort. Neurology 2013;81:1226–1234. [DOI] [PubMed] [Google Scholar]
- 20. Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele‐Richardson‐Olszewski syndrome): report of the NINDS‐SPSP international workshop. Neurology 1996;47:1–9. [DOI] [PubMed] [Google Scholar]
- 21. McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology 2005;65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 22. Visser M, Marinus J, Stiggelbout AM, Van Hilten JJ. Assessment of autonomic dysfunction in Parkinson's disease: the SCOPA‐AUT. Mov Disord 2004;19:1306–1312. [DOI] [PubMed] [Google Scholar]
- 23. Chaudhuri KR, Martinez‐Martin P, Brown RG, et al. The metric properties of a novel non‐motor symptoms scale for Parkinson's disease: results from an international pilot study. Mov Disord 2007;22:1901–1911. [DOI] [PubMed] [Google Scholar]
- 24. Youden WJ. Index for rating diagnostic tests. Cancer 1950;3:32–35. [DOI] [PubMed] [Google Scholar]
- 25. Pepe MS, Cai T, Longton G. Combining predictors for classification using the area under the receiver operating characteristic curve. Biometrics 2006;62:221–229. [DOI] [PubMed] [Google Scholar]
- 26. van der Heijden GJ, Donders AR, Stijnen T, Moons KG. Imputation of missing values is superior to complete case analysis and the missing‐indicator method in multivariable diagnostic research: a clinical example. J Clin Epidemiol 2006;59:1102–1109. [DOI] [PubMed] [Google Scholar]
- 27. Molloy S, McKeith IG, O'Brien JT, Burn DJ. The role of levodopa in the management of dementia with Lewy bodies. J Neurol Neurosurg Psychiatry 2005;76:1200–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lucetti C, Logi C, Del Dotto P, et al. Levodopa response in dementia with Lewy bodies: a 1‐year follow‐up study. Parkinsonism Relat Disord 2010;16:522–526. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Further significantly different parameters of patients with and without Parkinson's disease.
Table S2. Results of the final logistic regression for multiple imputed data sets excluding the variable “l‐dopa‐change”.
Table S3. Optimal cutoff point for the combined classifier (resulting from the model in Table S2) according to the Youden criterion as well as sensitivity, specificity, and positive and negative predictive values.