

Abstract
Background
Spinocerebellar ataxias (SCAs) are dominantly inherited, progressive ataxia disorders. Disease progression could be preceded by weight loss.
Objectives
We aimed to study the course of weight loss in patients who had the most common SCAs (SCA1, SCA2 SCA3, and SCA6). Additional objectives were to identify subgroups of weight evolution, to determine the factors influencing these evolutions, and to assess the impact of these evolutions on disease progression.
Methods
In total, 384 patients from the EUROSCA prospective cohort study were analyzed who had SCA1, SCA2, SCA3, or SCA6 and at least 3 measurements of weight. Age was used as a time scale. Clinical outcomes were body mass index (BMI) and the Scale for the Assessment and Rating Ataxia (SARA), with scores ranging from 0 to 40. We used a linear mixed model to analyze the course of BMI and a latent class mixed model to identify subgroup BMI evolution.
Results
Overall, BMI declined over time (−0.11 ± 0.03 kg/m2 per decade; P = 0.0009). Three subgroups of BMI evolution were identified: “decreasing BMI” (n = 88; 23%), “increasing BMI” (n = 70; 18%) and “stable BMI” (n = 226; 59%). Patients in the decreasing BMI group were more severely affected at baseline with higher SARA scores and a higher frequency of non‐ataxia signs (especially motor symptoms) compared with those in the other groups. Weight loss was associated with faster disease progression (5.7 ± 0.7 SARA points per decade; P = 0.036).
Conclusions
The current data have substantial implications for the design of future interventional studies in SCA, as they provide a basis for patient stratification and emphasize the usefulness of BMI as a biomarker for monitoring disease progression.
Keywords: body mass index, latent class mixed model, longitudinal data, spinocerebellar ataxia
Spinocerebellar ataxias (SCAs) are a clinically and genetically heterogeneous group of autosomal‐dominant, inherited, progressive ataxia disorders. Several genetically different SCAs have been defined, and the most common are SCA1, SCA2, SCA3, and SCA6.1 These ataxias are caused by translated CAG repeat expansions, which code for elongated polyglutamine tracts within the different proteins associated with each type. Data from a large prospective study of these disorders yielded important insights into their natural history. Progressive ataxia is the leading symptom in these disorders and increases linearly over time, with different rates in each genotype. Progression is faster in SCA1, intermediate in SCA2 and SCA3, and slower in SCA6. Factors associated with faster disease progression have been identified in all SCAs except SCA3. These factors include older age at baseline and longer repeat expansions in SCA1, younger age at onset in SCA2, and lower baseline Scale for the Assessment and Rating Ataxia (SARA) scores in both SCA2 and SCA6.2
The occurrence of gait ataxia is preceded by a preclinical stage, resulting in dysfunction, degeneration, and metabolic changes in several nervous system compartments.3, 4 Degenerative processes affecting brain structures other than the cerebellum may explain extracerebellar signs in SCAs,5 such as dysphagia and weight loss.6, 7
A significant decline in body weight directly linked with increased CAG repeat length has been observed in Huntington's disease (HD).8 Moreover, several studies that included data from patients with SCA9, 10 and transgenic mouse models of SCA11, 12, 13, 14 have reported the presence of weight loss in SCAs. Dysphagia10 and an increased catabolic state15 have been reported as mechanisms underlying weight loss in SCAs.
The previous studies were either cross‐sectional or retrospective, had case‐control designs, and included small patient numbers. Here, we explore data from a longitudinal EUROSCA cohort with a long‐term follow‐up on body weight and height in relation to ataxia progression.
The objectives of the current analysis were to: (1) specify the course of weight loss, (2) identify different subgroups of weight evolution, (3) determine the factors associated with weight loss, and (4) quantify the association between disease progression and weight loss.
Patients and Methods
Standard Protocol Approvals, Registration, and Patients Consent
The ethics committees of the participating centers approved the study (clinicaltrials.gov identifier NCT02440763). At enrolment, a written consent form was obtained from all study participants.
Study Population
Of 525 patients with SCA who were enrolled between July 1, 2005, and Aug 31, 2006, in the multicenter (17 European centers) EUROSCA prospective cohort study,16 a subsample of 384 patients who had at least 3 measures of weight over a maximum of 6 years of follow‐up were analyzed Fig. S1. These patients were diagnosed with progressive, otherwise unexplained ataxia and had a positive molecular genetic test for SCA1 (n = 80), SCA2 (n = 125), SCA3 (n = 105), or SAC6 (n = 74).
Clinical and Genetics Evaluations
Demographic data included age, sex, age at onset, and disease duration. The following variables were measured at baseline: weight and height; disease stage; symptoms other the ataxia according to Inventory of Non‐ataxia Signs (INAS) scores (range, 0–16) signs, including the extended INAS score, such as dysphagia and double vision17; anxiety or depression status measured by Patient Health Questionnaire‐9 (PHQ‐9) self‐rated items18; the use of any psychiatric medication among antipsychotics, anxiolytics, hypnotics and sedatives, antidepressants, psychostimulants, agents used for attention deficit hyperactivity disorder, and nootropics; and the SARA score. Weight and SARA score were also measured at subsequent visits at 1, 2, and 3 years (±3 months). After the initial 3‐year observation period, the study participants entered an extension phase in which study assessments were done in connection with routine visits, resulting in irregular intervals between the visits. Body mass index (BMI) was calculated with the formula (weight/(height)2). Repeat lengths of the expanded and normal alleles were determined at the Institute of Medical Genetics and Applied Genomics of the University of Tubingen (Tubingen, Germany).
Statistical analysis
Descriptive statistics (frequencies and percentages or means ±standard deviations) were used to describe demographics and clinical characteristics of the participants at baseline. BMI at baseline was compared between genotypes using an analysis of variance with Tukey's Honestly Significant Difference test to account for multiple comparisons. To describe changes in BMI over time, the standard linear mixed (LM) model19 was used with the cubic function of age as a time scale and without any adjustment for covariates. The cubic function was the best of the tested models (linear, quadratic, and cubic models). Correlated, autoregressive, random intercept, linear and quadratic slope accounted for inter‐patient variability. The age variable was centered at 18 years (the minimum age in the cohort) and is indicated in 10‐year increments (i.e., ages 18, 28, 38, 48 years, etc.). We tested the effect of genotype on BMI evolution by interaction between the given factor and the time variable.
To identify subgroups of participants who exhibited different trajectories of BMI, a latent class mixed (LCM) model that accounted for individual and latent group structure variability through random effects was used. The same fixed and random‐effects that were included in the LM model were used in the LCM model.19 We selected the best‐fitting model with the optimal number of latent classes using a compromise between the Bayesian Information Criterion, a mean posterior probability >0.70 for each latent class, and a number of patients in each class >5.20 Distributions of the baseline variables across these classes were compared a posteriori using a Chi‐squared (X2) test for the categorical variables and an analysis of variance for continuous variables with the Honestly Significant Difference test to account for multiple comparisons. To assess whether the rate of change in the SARA score was directly related to these classes, a posteriori LM model was used for the SARA score, and the interaction between class variable and time was tested.
Furthermore, a previous report on long‐term disease progression revealed a substantial dropout rate, mainly for disease‐related reasons during the open extension period, which followed the initial 3‐year period with annual visits. Patients who had a maximum follow‐up of 3 years had a high dropout rate and faster disease progression.2 In these patients, the dropout rate is informative, and standard estimation methods like LM models and LCM models are biased. Informative dropout leads to a “missing not at random” (MNAR) assumption that the observation probability depends on unobserved outcomes.19 To account for an informative dropout process and ensure that estimates from “missing at random” assumptions are not biased, a sensitivity analysis under an MNAR assumption was performed using joint models for longitudinal data and the time to dropout.20 The estimates from the joint model under MNAR were then compared with those obtained from the LM model under a missing at random assumption. All data analyses were performed using R version 3.2.0 (R Foundation for Statistical Computing Vienna, Austria) and SAS (version 9.4; SAS Institute, Cary, NC).
Results
Baseline Characteristics
The 384 patients included in the current study had a total of 1795 visits, with a median of 3 visits (interquartile range, 3–4 visits) for each patient. Baseline characteristics of the patients are provided in Table 1. BMI at baseline differed between SCA types (P < 0.05), with a significantly lower BMI in patients who had SCA3 compared with all others.
Table 1.
Population characteristics at baseline
| Characteristic | Frequency/No. (%) or Mean ± SD | |||
|---|---|---|---|---|
| SCA1, n = 80 | SCA2, n = 125 | SCA3, n = 105 | SCA6, n = 74 | |
| Men | 51 (64) | 59 (47) | 55 (52) | 40 (54) |
| Age, y | 46 ± 12 | 46 ± 14 | 49 ± 12 | 64 ± 11 |
| Age at onset, y | 38 ± 11 | 35 ± 13 | 38 ± 11 | 54 ± 10 |
| Disease duration, y | 8 ± 5 | 11 ± 6 | 11 ± 6 | 10 ± 6 |
| CAGa | 47 ± 5 | 39 ± 3 | 67 ± 4 | 22 ± 1 |
| Diabetes | 3 (4) | 5 (4) | 2 (2) | 5 (7) |
| Cancer | 0 (0) | 0 (0) | 2 (4) | 2 (6) |
| Gastrointestinal diseases | 7 (9) | 7 (6) | 8 (8) | 10 (10) |
| BMI, kg/m2 | 24.9 ± 4 | 25.1 ± 4 | 23.2 ± 4 | 25.4 ± 4 |
| Underweight | 13 (17) | 13 (11) | 13 (14) | 13 (14) |
| Normal weight | 34 (46) | 62 (51) | 57 (62) | 34 (44) |
| Overweight | 18 (24) | 34 (28) | 16 (17) | 18 (32) |
| Obese | 10 (13) | 12 (10) | 6 (7) | 10 (11) |
| SARA score | 12.9 (7) | 14.9 (7) | 13.5 (7) | 14.6 (6) |
| Disease stage | ||||
| 1 | 56 (70.9) | 79 (69.3) | 49 (48.0) | 34 (47.9) |
| 2 | 19 (24.1) | 24 (21.1) | 37 (36.3) | 33 (46.5) |
| 3 | 4 (5.1) | 11 (9.6) | 16 (15.7) | 4 (5.6) |
| Anxiety or depression, yes | 37 (46.3) | 59 (47.2) | 49 (46.7) | 35 (47.3) |
| Any psychotropic use, yes | 15 (18.8) | 18 (14.4) | 20 (19.1) | 7 (9.5) |
| INAS score, no. of signs | 4.4 ± 2 | 4.1 ± 2 | 4.8 ± 2 | 2.0 ± 1 |
| Hyperreflexia, yes | 56 (70.0) | 18 (14.5) | 39 (37.9) | 15 (20.3) |
| Areflexia, yes | 13 (16.2) | 78 (62.9) | 59 (56.2) | 16 (21.6) |
| Extensor plantor, yes | 34 (46.6) | 38 (33.3) | 41 (39.4) | 2 (2.8) |
| Spasticity, yes | 49 (61.3) | 13 (10.7) | 43 (41.3) | 9 (12.5) |
| Paresie, yes | 15 (18.8) | 18 (14.5) | 23 (22.1) | 5 (6.8) |
| Muscle atrophy, yes | 17 (21.2) | 25 (20.3) | 38 (36.9) | 9 (12.3) |
| Fasciculations, yes | 28 (34.5) | 51 (41.1) | 33 (31.4) | 1 (1.4) |
| Myoclonus, yes | 1 (1.2) | 18 (14.5) | 4 (3.8) | 0 (0.0) |
| Rigidity, yes | 1 (1.2) | 7 (5.6) | 11 (10.7) | 6 (8.1) |
| Chorea/dyskinesia, yes | 3 (3.8) | 11 (8.9) | 7 (6.7) | 2 (2.7) |
| Dystonia, yes | 5 (6.2) | 18 (14.4) | 20 (19.0) | 3 (4.1) |
| Resting tremor, yes | 6 (7.5) | 17 (13.8) | 4 (3.8) | 1 (1.4) |
| Sensory symptoms, yes | 45 (60.0) | 88 (72.1) | 65 (66.3) | 36 (50.0) |
| Urinary dysfunction, yes | 25 (31.2) | 50 (40.3) | 49 (47.1) | 26 (35.1) |
| Cognitive impairment, yes | 12 (15.0) | 32 (25.8) | 19 (18.8) | 7 (9.5) |
| Brainstem oculomotor signs, yes | 29 (36.2) | 44 (35.5) | 51 (49.5) | 11 (14.9) |
| Dysphagia, yes | 42 (53.2) | 62 (54.4) | 59 (57.8) | 39 (54.9) |
| Double vision, yes | 13 (16.2) | 26 (20.8) | 58 (55.2) | 36 (48.6) |
SD, standard deviation; SCA, spinocerebellar ataxia; BMI, body mass index; SARA, Scale for the Assessment and Rating Ataxia; INAS, Inventory of Non‐ataxia Signs.
Values indicate the length of the expanded allele (repeat units).
Rate of BMI Change
Based on an LM model, BMI declined cubically over time (−0.11 ± 0.03 kg/m2 per decade; P = 0.0009) (Fig. 1A). The rate of decline did not differ significantly between the 4 SCA genotypes (P = 0.4400).
Figure 1.
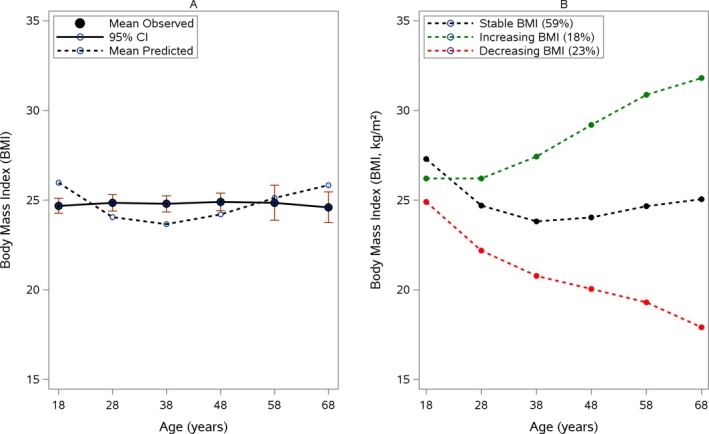
(A) Mean decline in body mass index (BMI) and (B) and BMI group trajectories. (A) The mean observed BMI (in kg/m2; solid line) with 95% confidence intervals (CIs) is compared with the predicted BMI (dashed line) over time according a linear mixed model equation: BMI = 25.9–3.1 * age + 1.1 * age2 − 0.11 * age,3 where age was measured in decades starting from age 18 years. (B) Weighted, subject‐specific predictions of BMI trajectories are illustrated according to a latent class mixed model equation: stable BMI (black line) = 27.3–3.7 * age + 1.2 * age2 − 0.11 * age3; increasing BMI (green line) = 26.2–0.83 * age + 0.94 *age2 − 0.11 * age3, and decreasing BMI (red line) = 24.9–3.6 * age + 0.99 * age2 − 0.11 * age3.
Trajectories of BMI and their Relationship to Ataxia Severity and Non‐ataxia Signs
To identify different trajectories of BMI, the LCM model with several latent classes, ranging from 1 to 5, were estimated. The model with the optimal number of classes selected by the compromise criterion included 3 classes (Tables S1 and S2). This model identified 3 different BMI evolutions (Fig. 1B). Class 1 (n = 226; 59%) was characterized by stable BMI evolution over time; Class 2 (n = 70; 18%) corresponded to patients who had an increase in BMI over time; and Class 3, which included 23% of patients (n = 88), was characterized by a decrease in BMI over time. According to the posterior classification provided in Table 2, the 3 classes differed for the most part on the baseline characteristics tested. Pairwise comparisons demonstrated that patients in the decreasing BMI group, compared with those in the increasing and stable BMI groups, were more severely affected at baseline and had higher SARA scores (16 ± 8 points), were more often permanently dependent on a wheelchair (19%), were younger at the time of disease onset (28 1± 9 years), had double vision (50%), and had more CAG repeats in the SCA1 (51 ± 6 CAG repeats) and SCA2 (43 ± 3 repeat units) cohorts. These patients had also a greater frequency of number of non‐ataxia signs (INAS score, 5 ± 2 signs), especially pyramidal and peripheral motor symptoms: spasticity (44%), hyperreflexia (47%), extensor plantar (41%), muscle atrophy (39%), fasciculation (43%), and brainstem oculomotor signs (59%). Paresis, rigidity, resting tremor, sensory symptoms, urinary dysfunction, cognitive impairment, dysphagia, anxiety or depression, the use of any psychiatric medication, cancer, and gastrointestinal and endocrine diseases like diabetes all were similar between groups. Moreover, we failed to identify any factors that were associated with a BMI increase. Although we did not find a correlation with dysphagia, the decreasing BMI group included 64% of patients who had dysphagia, whereas the increasing and stable BMI groups included 54% and 48% of patients, respectively.
Table 2.
Description of the 3 posterior latent classes according to the patient's baseline characteristics
| Characteristic | No. (%) or Mean ± SDa | P valueb | ||
|---|---|---|---|---|
| Decreasing BMI, n = 88 | Increasing BMI, n = 70 | Stable BMI, n = 226 | ||
| Genotype | <0.0001c | |||
| SCA1 | 19 (21.6) | 20 (28.6) | 41 (18.1) | |
| SCA2 | 30 (34.1) | 27 (38.6) | 68 (30.1) | |
| SCA3 | 38 (43.2) | 12 (17.1) | 55 (24.3) | |
| SCA6 | 1 (1.1) | 11 (15.7) | 62 (27.4) | |
| Women | 47 (53.4) | 41 (58.6) | 91 (40.3) | 0.0095c |
| Age, y | 38.4 ± 11.8a | 50.1 ± 12.0b | 55.8 ± 12.1c | < 0.0001c |
| Age at onset, y | 27.7 ± 9.2a | 40.6 ± 11.5b | 45.5 ± 11.5c | < 0.0001c |
| Disease duration, y | 10.7 ± 6.3 | 9.5 ± 5.8 | 10.2 ± 5.3 | 0.4470 |
| SARA score | 15.9 ± 7.5a | 12.8 ± 7.4b | 13.6 ± 6.6b | 0.0108c |
| Disease stage | 0.0241c | |||
| 1 | 47 (55.3) | 40 (59.7) | 131 (61.2) | |
| 2 | 22 (25.9) | 22 (32.8) | 69 (32.2) | |
| 3 | 16 (18.8) | 5 (7.5) | 14 (6.5) | |
| Diabetes | 0 (0.0) | 5 (7.5) | 10 (4.8) | 0.0569 |
| Cancer | 1 (4.0) | 0 (0.) | 3 (3.8) | 0.6202 |
| Gastrointestinal diseases | 7 (8.2) | 9 (13.4) | 16 (7.6) | 0.3361 |
| CAGd | ||||
| SCA1 | 51.3 ± 6.3a | 45.4 ± 3.8b | 45.7 ± 3.7b | <0.0001c |
| SCA2 | 42.5 ± 3.4a | 39.2 ± 2.9b | 37.7 ± 2.4b | < 0.0001c |
| SCA3 | 70.9 ± 3.1a | 68.6 ± 3.8a,b | 67.1 ± 4.2b | < 0.0001c |
| SCA6 | 22.0 | 22.3 ± 0.5 | 22.5 ± 1.2 | 0.4904 |
| Anxiety or depression, yes | 44 (50.0) | 36 (51.4) | 100 (44.2) | 0.4597 |
| Any psychotropic use, yes | 14 (15.9) | 14 (20.0) | 32 (14.2) | 0.4991 |
| INAS score | 5.2 ± 2.3a | 3.8 ± 2.2b | 3.5 ± 2.1b | < 0.0001c |
| Hyperreflexia, yes | 41 (47.1) | 19 (27.1) | 69 (30.4) | 0.0087c |
| Areflexia, yes | 35 (39.8) | 29 (41.4) | 102 (45.3) | 0.6300 |
| Extensor plantar, yes | 34 (41.0) | 26 (38.8) | 55 (25.9) | 0.0175c |
| Spasticity, yes | 38 (43.7) | 17 (24.6) | 59 (26.6) | 0.0071c |
| Paresis, yes | 19 (21.6) | 12 (17.1) | 30 (13.5) | 0.2030 |
| Muscle atrophy, yes | 34 (39.1) | 12 (17.1) | 43 (19.4) | <0.0001c |
| Fasciculations, yes | 37 (42.5) | 24 (34.3) | 52 (23.1) | 0.0022c |
| Myoclonus, yes | 10 (11.4) | 4 (5.7) | 9 (4.0) | 0.0488c |
| Rigidity, yes | 7 (8.0) | 3 (4.3) | 15 (6.7) | 0.6342 |
| Chorea/dyskinesia, yes | 11 (12.5) | 2 (2.9) | 10 (4.4) | 0.0124c |
| Dystonia, yes | 20 (22.7) | 4 (5.7) | 22 (9.7) | 0.0013c |
| Resting tremor, yes | 11 (12.8) | 5 (7.1) | 12 (5.4) | 0.0806 |
| Sensory symptoms, yes | 50 (61.7) | 49 (72.1) | 135 (61.9) | 0.2881 |
| Urinary dysfunction, yes | 29 (33.3) | 33 (47.1) | 88 (39.1) | 0.2115 |
| Cognitive impairment, yes | 18 (20.7) | 14 (20.0) | 38 (17.0) | 0.6937 |
| Brainstem oculomotor signs, yes | 51 (58.6) | 19 (27.1) | 65 (29.0) | < 0.0001c |
| Dysphagia, yes | 54 (63.5) | 32 (47.8) | 116 (54.2) | 0.1374 |
| Double vision, yes | 44 (50.0) | 18 (25.7) | 71 (31.4) | 0.0018c |
SD, standard deviation; SCA, spinocerebellar ataxia; BMI, body mass index; SARA, Scale for the Assessment and Rating Ataxia; INAS, Inventory of Non‐ataxia Signs.
Means with the same letter did not differ significantly from each other according to a Tukey's Honestly Significant Difference multiple comparison test (P > 0.05).
The chi‐square test was used to analyze qualitative covariates, and an analysis of variance was used for quantitative covariates.
P < 0.05 (statistically significant).
Values indicate the length of the expanded allele (repeat units).
Trajectories of BMI and Disease Progression
The association between the BMI groups and disease progression, as determined with the SARA score, was analyzed (Fig. 2). Disease progression was significantly faster for the decreasing BMI group (SARA score, 5.7 ± 0.73 points every decade), intermediate for the stable BMI group (SARA score, 4.2 ± 0.47 points every decade), and slower for the group increasing BMI group (SARA score 3.3 ± 0.78 points every decade).
Figure 2.
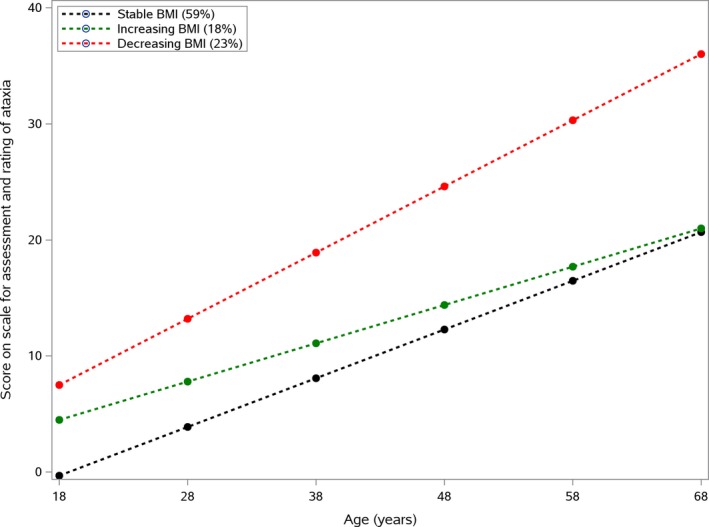
Progression of scores on the Scale for the Assessment and Rating Ataxia according to body mass index (BMI) evolution. Stable BMI (in kg/m2; black curve) = −0.32 + 4.2 * age; increasing BMI (green curve) = 4.5 + 3.3 * age; and decreasing BMI (red curve) = 7.5 + 5.7 * age.
Discussion
In this first long‐term follow‐up study of body weight changes in a large cohort of patients with SCA, a decline in BMI over time was observed. Furthermore, three trajectories of BMI evolution were identified: decreasing, increasing, and stable. There was a significant association between weight loss and disease progression, but no significant correlation of BMI change with the presence of dysphagia was observed.
At baseline, patients with SCA3 had a lower BMI than the other patients. These results are consistent with findings from the Brazilian cohort at the Hospital de Clinicas de Porto Alegre (HCPA), who demonstrated that that BMI was lower in patients who had SCA3 compared with controls and inversely was correlated with expanded CAG repeat lengths.9 In addition, these results are in line with observations in several transgenic SCA models, in which mice had progressive body weight loss over the course of the disease.11, 12, 13, 14 We observed that BMI declined over time. Similar results were reported in longitudinal clinical studies of Huntington's disease over 3 years, in which BMI declined with time, and patients with higher CAG repeat numbers had faster rates of weight loss.8
To our knowledge, this is the first study to allow the identification of subgroups with respect to the evolution of body weight among patients with polyglutamine disorders. Unlike the LM model, which shows an average decrease BMI over time, our subgroup analysis showed that only 23% of patients lost weight during follow‐up. Three groups were objectified with decreasing, increasing, and stable BMI over time. The most interesting finding was that the decreasing BMI group had faster disease progression, suggesting that weight loss may serve as a biomarker to monitor disease progression. In addition, longer CAG expansions were associated with BMI declines in patients with SCA1, SCA2, and SCA3. This was not replicated for those with SCA6, because there was only 1 patient in the decreasing BMI group. These results reinforced the assumption of a correlation between genetic findings, disease progression, and weight loss for patients with SCA1, SCA2, and SCA3, but not for those with SCA6.
Although ataxia is the major symptom of all SCAs, various non‐ataxia signs may accompany ataxia. We observed a greater frequency of non‐ataxia signs (especially motor symptoms) in patients who had declining BMI over time. On average, each patient in the group with decreasing BMI had 5 non‐ataxia symptoms. From our results, muscle atrophy is very likely to contribute to total weight loss, because 39% of patients who had decreasing BMI over time had muscle atrophy. This frequency increases to 75% in patients with stage 3 disease. In line with that finding, a significant correlation was observed between disease stage and BMI: 45% of patients who were permanently wheelchair dependent had a declining BMI over time. By contrast, no such association was reported in the Brazilian cohort study.9
The rates of progression in our study were slower than the values reported in a previous analysis of the same cohort.2 This discrepancy is explained by the different time scales used in the analyses. Here, we analyzed data on an age scale, as recommended for chronic diseases, in which age is the main disease evolution factor.20 In addition, unlike the previous report, all SCA groups were pooled for the analysis, because the sample size for each SCA group separately did not allow the fit of LCM models with more than 2 subgroups.
The mechanism underlying weight loss in patients with SCA is not clear. It has been suggested that the occurrence of dysphagia may be an important cause of weight loss.10 However, the available results are contradictory. The current findings agree with those from the Brazilian study, in which no relation of BMI with dysphagia was reported.9 In contrast, a systematic post‐mortem study on thick serial tissue sections from 12 dysphasic patients with SCA2, SCA3, SCA6, and SCA7 reported the presence of widespread neurodegeneration of the brainstem nuclei involved in the ingestive process that may cause nutritional deficiencies, weight loss, and dehydration.10
In addition, only 12 patients (3%) from the EUROSCA cohort developed severe dysphagia during follow‐up, requiring the use a tube feeding. In theory, this type of diet should increase the patient's weight. However, among those patients, there were 4 (42%) in the increasing BMI group, 5 (42%) in the decreasing BMI group, and 7 (58%) in the stable BMI group. These results suggest that severe dysphagia has no impact on increases in BMI, and it is also evident that weight loss is a primary consequence of disease progression.
The increased catabolic state in patients with SCA1 was reported as a possible mechanism involved in weight loss.15 In that study, the authors demonstrated that patients with SCA1 had a 22% increase in energy expenditure per kilogram of fat‐free mass and a 28% increase in fat oxidation compared with controls. This increasing catabolic state might relate to the presence of autonomic dysfunction, as has been suggested in HD.21 Several studies have reported the presence of autonomic dysfunction in patients with symptomatic SCA1, SCA2, and SCA3 who may be able to reduce parasympathetic activity.22, 23 In addition, the lower motor neuron degeneration, which is described in SCA1,SCA2, and SCA3, but not in SCA6, may have an influence on BMI decline.24 In amyotrophic lateral sclerosis, lower BMI might be caused by hypermetabolism.25 It is likely that the same mechanisms are present in patients with SCA1, SCA2, and SCA3. The greater prevalence of hypermotoric signs like dystonia or dyskinesia in the group with decreasing BMI point in the same direction, because such signs have been associated with metabolic changes and weight loss in other disorders like Parkinson's disease.26
Alterations in the insulin/insulin‐like growth factor‐1 system have been involved as another mechanism for weight loss in transgenic animal models and in data from patients with SCA3.27, 28 In the Brazilian cohort study, investigators observed that patients with SCA3 had reduced insulin levels and that the insulin levels were directly associated with BMI.9, 28 Similar results were reported in HD8 but were not replicated in all studies in which, despite high caloric intake, the weight loss took place. This allowed the use of a plasma biomarker to identify early stages of the disease.29 It also allowed the development of a spectroscopic cerebral marker of brain energy metabolism, which could be improved by using triheptanoin to provide substrates to the Krebs cycle.30
Our study has several strengths. First, it had a multicenter, prospective design with long observational periods. Second, to account of informative dropout, we applied a sensitivity analysis using a joint model for longitudinal data and the time to dropout.20 The results obtained from this joint model were similar to estimations using standard methods, suggesting an absence of bias in parameter estimations (Table S3). Third, we used a compromise criterion to select the best model instead of using only the Bayesian Information Criterion. The average probability of a model with 3 classes was relatively high, ranging from 0.74 to 0.83, suggesting an unambiguous classification. However, the data have limitations: Information about caloric intake, lipid profile, daily exercise, physiotherapy, food habits, gait training, occupational therapy, and olfactory impairment, all of which may influence BMI, was not assessed in the EUROSCA study. In addition, other anthropometric measurements, such as skin‐fold thickness or waist circumference, which could possibly serve as biomarkers to monitor disease progression, were missing; and the small sample size per genotype negated the possibility of performing a trajectory analysis for each SCA type.
The current data allowed us to identify 3 different groups of BMI evolution and provided evidence of an association between weight loss and disease progression. These findings suggest monitoring of body weight as a potentially useful biomarker of disease progression, because patients who had the greatest number of CAG repeats were more likely to lose weight. In addition, the results provide more information to help with patient selection and stratification for the design of interventional clinical trials.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
A.Diallo: 2A, 2B, 2C, 3A, 3B
H.J.: 1A, 1B, 1C, 2C, 3B
T.S.H.: 1B, 1C, 2C, 3B
A.C.: 1B, 1C, 2C, 3B
R.L.: 1B, 1C, 2C, 3B
A.Durr: 1B, 1C, 2C, 3B
A.B.: 1B, 1C, 2C, 3B
P.C.: 1B, 1C, 2C, 3B
C.Marelli: 1B, 1C, 2C, 3B
C.Mariotti: 1B, 1C, 2C, 3B
L.N.: 1B, 1C, 2C, 3B
M.Panzeri: 1B, 1C, 2C, 3B
M.R.: 1B, 1C, 2C, 3B
A.Sobanska: 1B, 1C, 2C, 3B
A.Sulek: 1B, 1C, 2C, 3B
L.S.: 1B, 1C, 2C, 3B
H.H.: 1B, 1C, 2C, 3B
B.M.: 1B, 1C, 2C, 3B
A.F.: 1B, 1C, 2C, 3B
A.A.: 1B, 1C, 2C, 3B
J.I.: 1B, 1C, 2C, 3B
J.B.: 1B, 1C, 2C, 3B
B.P.v.d.W.: 1B, 1C, 2C, 3B
D.T.: 1B, 1C, 2C, 3B
S.B.: 1B, 1C, 2C, 3B
M.Pandolfo: 1B, 1C, 2C, 3B
J.B.S.: 1B, 1C, 2C, 3B
P.B.: 1B, 1C, 2C, 3B
P.G.: 1B, 1C, 2C, 3B
L.B.: 1B, 1C, 2C, 3B
M.H.P.: 1B, 1C, 2C, 3B
J.S.K.: 1B, 1C, 2C, 3B
T.K.: 1A, 1B, 1C, 2A, 2C 3B
S.T.d.M.: 1A, 1B, 1C, 2A, 2C, 3A 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: The sponsors of this study had no role in design, data collection, data analysis, data interpretation, or writing the report. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication. The EU FP6 (EUROSCA), the German Ministry of Education and Research (GeneMove), the Polish Ministry of Science, and the EU FP7 (Neuromics) supported the study. This study was funded within the framework of EU FP6 (EUROSCA). Additional funds came from the German Ministry of Education and Research (BMBF; GeneMove), the Polish Ministry of Scientific Research and Information Technology, and the European Community's Seventh Framework Programme (FP7/2007–2013) under grant agreement 2012‐305121, “Integrated European‐Omics Research Project for Diagnosis and Therapy in Rare Neuromuscular and neurodegenerative diseases (NEUROMICS)” and Funding by Neuromics (EU FP7).
Financial Disclosures for previous 12 months: Thomas Klockgether reports grants from EU FP6 during the conduct of the study and personal fees from Biohaven, ICON Clinical Research, and Atheneum outside the submitted work. Anna Sobanska takes part in the clinical trial “Clinical TeleNeuroforma‐clinically verified home rehabilitation system for people suffering from selected neurological conditions” sponsored by The National Center for Research and Development. Sophie Tezenas du Montcel reports grants from EU FP6 during the conduct of the study. Bart P. van de Warrenburg reports grants from ZonMW, BBMRI‐NL, the Gossweiler Foundation, Hersenstichting, Radboud University Medical Center, the European Union, and Bioblast Pharma and other support from the Movement Disorder Society outside the submitted work. Jörg B. Schulz reports service on the scientific advisory boards for Lundbeck, TEVA, Novartis, and Lilly; funding for travel and personal fees from GlaxoSmithKline, Merz Pharmaceuticals, Medical Tribune, Lundbeck, Pfizer, Boehringer, and Bayer; and research support from the German Ministry of Education and Research and the European Union. Peter Bauer reports a European Commission research grant (EUROSCA), during the conduct of the study and personal fees from Actelion Pharmaceutis and Aschwil CH and personal fees from Centogene AG and Rostock GER outside the submitted work. The remaining authors report no sources of funding and no conflicts of interest.
Supporting information
Table S1. Summary of the estimated latent class mixed model on the EUROSCA sample (n=384).
Table S2. Posterior classification.
Table S3. Sensitivity analysis.
Figure S1. Flow‐chart.
Acknowledgments
We thank the Fondation pour la Recherche Médicale for the financial support of Alhassane Diallo. We also thank Fatoumata Diallo from East Carolina University for critical reading of the article; and Thomas Klopstock (Department of Neurology, University of Munich, Munich, Germany), Jens Petersen (Department of Neurology, University of Munich, Munich, Germany, Department of Neurology, University of Zurich, Zurich, Switzerland), Catherine Delnooz (Department of Neurology, Radboud University Nijmegen Medical Center, Nijmegen, Netherlands), and Rafal Rola (First Department of Neurology, Institute of Psychiatry and Neurology, Warsaw, Poland) for their contribution of patients and help with patient assessments.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Durr A. Autosomal dominant cerebellar ataxias: polyglutamine expansions and beyond. Lancet Neurol 2010;9:885–894. [DOI] [PubMed] [Google Scholar]
- 2. Jacobi H, du Montcel ST, Bauer P, et al. Long‐term disease progression in spinocerebellar ataxia types 1, 2, 3, and 6: a longitudinal cohort study. Lancet Neurol 2015;14:1101–1108. [DOI] [PubMed] [Google Scholar]
- 3. Maas RP, van Gaalen J, Klockgether T, van de Warrenburg BP. The preclinical stage of spinocerebellar ataxias. Neurology 2015;85:96–103. [DOI] [PubMed] [Google Scholar]
- 4. Jacobi H, Reetz K, du Montcel ST, et al. Biological and clinical characteristics of individuals at risk for spinocerebellar ataxia types 1, 2, 3, and 6 in the longitudinal RISCA study: analysis of baseline data. Lancet Neurol 2013;12:650–658. [DOI] [PubMed] [Google Scholar]
- 5. Seidel K, Siswanto S, Brunt ER, den Dunnen W, Korf HW, Rub U. Brain pathology of spinocerebellar ataxias. Acta Neuropathol (Berl) 2012;124:1–21. [DOI] [PubMed] [Google Scholar]
- 6. Pedroso JL, Braga‐Neto P, Escorcio‐Bezerra ML, et al. Non‐motor and extracerebellar features in spinocerebellar ataxia type 2. Cerebellum 2017;16:34–39. [DOI] [PubMed] [Google Scholar]
- 7. Pedroso JL, Franca MC Jr, Braga‐Neto P, et al. Nonmotor and extracerebellar features in Machado‐Joseph disease: a review. Mov Disord 2013;28:1200–1208. [DOI] [PubMed] [Google Scholar]
- 8. Aziz NA, van der Burg JM, Landwehrmeyer GB, Brundin P, Stijnen T, EHDI Study Group ; Roos RA. Weight loss in Huntington disease increases with higher CAG repeat number. Neurology 2008;71:1506–1513. [DOI] [PubMed] [Google Scholar]
- 9. Saute JA, Silva AC, Souza GN, et al. Body mass index is inversely correlated with the expanded CAG repeat length in SCA3/MJD patients. Cerebellum 2012;11:771–774. [DOI] [PubMed] [Google Scholar]
- 10. Rub U, Brunt ER, Petrasch‐Parwez E, et al. Degeneration of ingestion‐related brainstem nuclei in spinocerebellar ataxia type 2, 3, 6 and 7. Neuropathol Appl Neurobiol 2006;32:635–649. [DOI] [PubMed] [Google Scholar]
- 11. Iizuka A, Nakamura K, Hirai H. Long‐term oral administration of the NMDA receptor antagonist memantine extends life span in spinocerebellar ataxia type 1 knock‐in mice. Neurosci Lett 2015;592:37–41. [DOI] [PubMed] [Google Scholar]
- 12. Jafar‐Nejad P, Ward CS, Richman R, Orr HT, Zoghbi HY. Regional rescue of spinocerebellar ataxia type 1 phenotypes by 14‐3‐3epsilon haploinsufficiency in mice underscores complex pathogenicity in neurodegeneration. Proc Natl Acad Sci USA 2011;108:2142–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hubener J, Casadei N, Teismann P, et al. Automated behavioral phenotyping reveals presymptomatic alterations in a SCA3 genetrap mouse model. J Genet Genomics 2012;39:287–299. [DOI] [PubMed] [Google Scholar]
- 14. Boy J, Schmidt T, Wolburg H, et al. Reversibility of symptoms in a conditional mouse model of spinocerebellar ataxia type 3. Hum Mol Genet 2009;18:4282–4295. [DOI] [PubMed] [Google Scholar]
- 15. Mahler A, Steiniger J, Endres M, Paul F, Boschmann M, Doss S. Increased catabolic state in spinocerebellar ataxia type 1 patients. Cerebellum 2014;13:440–446. [DOI] [PubMed] [Google Scholar]
- 16. Schmitz‐Hubsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 2006;66:1717–1720. [DOI] [PubMed] [Google Scholar]
- 17. Schmitz‐Hubsch T, Coudert M, Bauer P, et al. Spinocerebellar ataxia types 1, 2, 3, and 6: disease severity and nonataxia symptoms. Neurology 2008;71:982–989. [DOI] [PubMed] [Google Scholar]
- 18. Spitzer RL, Kroenke K, Williams JB. Validation and utility of a self‐report version of PRIME‐MD: the PHQ primary care study. Primary Care Evaluation of Mental Disorders. Patient Health Questionnaire. JAMA 1999;282:1737–1744. [DOI] [PubMed] [Google Scholar]
- 19. Verbeke G, Molenberghs G. Linear Mixed Models for Longitudinal Data. New York: Springer Science + Business Media; 2009. [Google Scholar]
- 20. Commenges D, Jacqmin‐Gadda H. Dynamical Biostatistical Models. Boca Raton, FL: CRC Press; 2015. [Google Scholar]
- 21. Aziz NA, Pijl H, Frolich M, Snel M, Streefland TC, Roelfsema F, Roos RA. Systemic energy homeostasis in Huntington's disease patients. J Neurol Neurosurg Psychiatry 2010;81:1233–1237. [DOI] [PubMed] [Google Scholar]
- 22. Netravathi M, Sathyaprabha TN, Jayalaxmi K, Datta P, Nirmala M, Pal PK. A comparative study of cardiac dysautonomia in autosomal dominant spinocerebellar ataxias and idiopathic sporadic ataxias. Acta Neurol Scand 2009;120:204–209. [DOI] [PubMed] [Google Scholar]
- 23. Montes‐Brown J, Sanchez‐Cruz G, Garcia AM, Baez ME, Velazquez‐Perez L. Heart rate variability in type 2 spinocerebellar ataxia. Acta Neurol Scand 2010;122:329–335. [DOI] [PubMed] [Google Scholar]
- 24. Liang L, Chen T, Wu Y. The electrophysiology of spinocerebellar ataxias. Neurophysiol Clin 2016;46:27–34. [DOI] [PubMed] [Google Scholar]
- 25. Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM. Body mass index, not dyslipidemia, is an independent predictor of survival in amyotrophic lateral sclerosis. Muscle Nerve 2011;44:20–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kashihara K. Weight loss in Parkinson's disease. J Neurol 2006;253(suppl 7):VII38–VII41. [DOI] [PubMed] [Google Scholar]
- 27. Duarte‐Silva S, Neves‐Carvalho A, Soares‐Cunha C, Teixeira‐Castro A, Oliveira P, Silva‐Fernandes A, Maciel P. Lithium chloride therapy fails to improve motor function in a transgenic mouse model of Machado‐Joseph disease. Cerebellum 2014;13:713–727. [DOI] [PubMed] [Google Scholar]
- 28. Saute JA, da Silva AC, Muller AP, et al. Serum insulin‐like system alterations in patients with spinocerebellar ataxia type 3. Mov Disord 2011;26:731–735. [DOI] [PubMed] [Google Scholar]
- 29. Mochel F, Charles P, Seguin F, et al. Early energy deficit in Huntington disease: identification of a plasma biomarker traceable during disease progression. PLoS ONE 2007;2:e647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Adanyeguh IM, Rinaldi D, Henry PG, et al. Triheptanoin improves brain energy metabolism in patients with Huntington disease. Neurology 2015;84:490–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Summary of the estimated latent class mixed model on the EUROSCA sample (n=384).
Table S2. Posterior classification.
Table S3. Sensitivity analysis.
Figure S1. Flow‐chart.