

Short abstract
http://onlinelibrary.wiley.com/journal/10.1002/(ISSN)2330-1619/homepage/mdc312582-sup-v001.htm
Keywords: progressive supranuclear palsy, corticobasal degeneration, 4r‐tauopathies
In 1964, Steele et al described a rapidly progressive neurodegenerative disease “involving the brain stem, basal ganglia, and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia, and dementia.”1 They coined the term progressive supranuclear palsy (PSP),1 which is currently used for the neuropathologically defined disease entity, whereas the initially described clinical manifestation is referred to as Richardson's syndrome (RS).2
In 1968, Reibeiz et al reported another neurodegenerative condition with “corticodentatonigral degeneration with neuronal achromasia.”3 Today, the corresponding neuropathological entity is now termed corticobasal degeneration (CBD)3 and the initially described clinical manifestation is called corticobasal syndrome (CBS).4, 5
Clinical diagnoses of RS and CBS have a reported prevalence of 2.3 to 10.6 and 4.9 to 7.3 cases per 100,000 individuals in the general population, respectively.6, 7 However, in a community‐based autopsy series of individuals aged between 77 and 87, neuropathological diagnoses of PSP and CBD were present more frequently in 3% and 0.4%, respectively.8
Although described as separate entities initially, it might be advantageous to classify CBD and PSP as the same disorder. This is due to overlapping genetic, biochemical, pathological, clinical, and therapeutic aspects discussed in detail in the following paragraphs.
Pathogenesis
PSP and CBD share a risk genetic variant in MOBP (myelin‐associated oligodendrocyte basic protein),9, 10 but homozygosity for the H1 haplotype, comprising MAPT (microtubule‐associated protein tau) is clearly the major risk factor for both PSP (odds ratio 5.5, P = 1.5 × 10−116) and CBD (odds ratio 3.7, P = 1.4 × 10−12; Fig. 1).9, 10 The H1c sub‐haplotype is particularly relevant for this predisposition.9, 10
Figure 1.
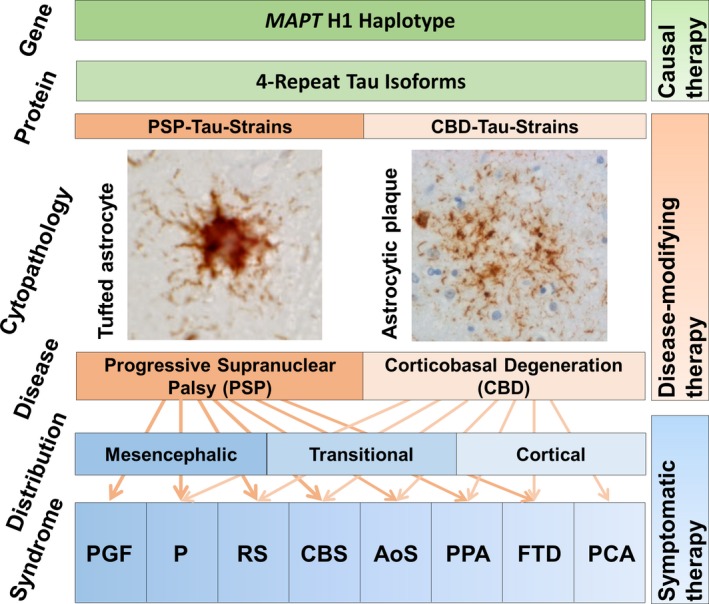
Joint pathophysiological concept of progressive supranuclear palsy (PSP) and corticobasal degeneration (CBD). Homozygosity for the MAPT H1 haplotype predisposes for increased expression of aggregation‐prone 4‐repeat tau isoforms. Postulated tau‐strains appear to propagate tau‐pathology from cell to cell in a prion‐like manner and to imprint disease‐specific ultrastructural and cytopathological aggregation patterns. Tufted astrocytes with tau aggregates (AT8‐tau immunoreactive) in proximal more than distal processes are the defining astrocytic lesion in PSP. Astrogytic plaques, i.e. annular clusters of short tau aggregates in distal segments of astrocytic processes, with little aggregation in the perinuclear cytoplasm, are the defining astrocytic lesion in CBD (AT8‐tau immunostaining; courtesy Dr. Sigrun Roeber, Munich). While PSP pathology is most frequently observed with mesencephalic predominance, also cortical or transitional patterns do occur. Inversely, CBD pathology most frequently occurs in cortical and transitional patterns. Following the anatomical topical lesion pattern, PSP and CBD present a broad spectrum of clinical syndromes, including progressive gait freezing (PGF), predominant parkinsonism (P), Richardson syndrome (RS), corticobasal syndrome (CBS), apraxia of speech (AoS), non‐fluent agrammatical variant primary progressive aphasia (PPA), behavioral variant frontotemporal dementia (FTD), and posterior cortical atrophy syndrome (PCA). Symptomatic therapy is targeting the clinical syndromes by physiotherapy, speech therapy, and pharmacological modulation of neurotransmitter systems in the affected brain regions. Disease‐modifying therapy aiming to slow down disease progression might be achieved by interfering in mechanisms involved in spreading of the pathology. Causal therapy aiming to prevent or cure the disease might be possible by interfering at the primary, i.e. most upstream disease mechanisms.
In brains of PSP and CBD patients, the protein tau is forming insoluble fibrillary aggregates. This feature is shared with numerous other neurodegenerative diseases, jointly termed tauopathies.11 Since there are no major aggregates of other proteins, PSP and CBD are considered as primary tauopathies, unlike Alzheimer's disease, where tau aggregation is believed to occur as consequence of amyloid‐beta pathology.11
The aggregates of PSP and CBD consist of tau isoforms with four repeats in the microtubule‐binding domain (4R‐tau isoforms), but not of isoforms with three repeats (3R‐tau isoforms) (Fig. 1).11, 12 This peculiarity is shared with argyrophilic grain disease and globular glial tauopathy and defines the group of 4R‐tauopathies.11 In contrast, tau aggregates in Pick's disease contain mainly 3R‐tau isoforms (3R‐tauopathy) and in Alzheimer's disease both 3R‐ and 4R‐tau isoforms (mixed 3R/4R‐tauopathy).13
Interestingly, the MAPT H1c risk haplotype increases transcription of tau, particularly of 4R‐isoforms, providing a rational pathophysiological basis for this genetic predisposition in PSP and CBD.14
Despite these communalities, some molecular distinctions have been reported between PSP and CBD (e.g., different carboxy‐terminal tau cleavage fragments have been observed in insoluble brain extracts [PSP: 33 kDa, CBD 37 kDa]).15 Ultrastructurally, intraneuronal tau filaments have been reported as 13 to 14 nm straight tubules in PSP, but a mix of 15 nm straight tubules and 20 nm twisted filaments in CBD.16
Apart from intracellular tau aggregation causing neuronal dysfunction and degeneration, transcellular propagation of tau also appears to be relevant. Brain homogenates from PSP and CBD patients do actually induce tau aggregates in nerve cell bodies after injection into the brain tissue of tau‐transgenic mice.17 This prion‐like spreading of tau seems to mediate the successive affection of widespread brain regions, which goes along with progressively increasing severity and broadening in the spectrum of clinical symptoms.18
While the molecular identity of the spreading tau specimen has not been identified, it has been convincingly demonstrated that tau immunoprecipitated from brains of PSP and CBD patients induces tau aggregates in cultured cells.19 Although there was remarkable heterogeneity in the aggregation patters induced with tau extracted from individual PSP and CBD patients, these patterns were clearly different from those observed with Alzheimer's disease patients.19 Thus, it was hypothesized that different tau strains may cause different diseases. It remains to be shown whether such hypothetical heterogeneity in tau strains does actually exist between PSP and CBD, and is it distinctive enough to define two separate disease entities (Fig. 1).
Pathology
In brains of PSP and CBD patients, tau aggregates can be found in astrocytes, oligodendrocytes, and neurons.11, 20
The defining astrocytic pathology, almost exclusively seen in PSP, is presence of tufted astrocytes (i.e., tau aggregates in proximal more than distal astrocytic processes; Fig. 1).11, 20 In contrast, the defining astrocytic lesions in CBD are astrocytic plaques (i.e., annular clusters of short tau aggregates in distal segments of astrocytic processes, with little aggregation in the perinuclear cytoplasm; Fig. 1).11, 20 Astrocytic plaques and tufts usually do not coexist and appear mutually exclusive in CBD and PSP.21
Comma‐shaped tau aggregates in oligodendroglia (coiled bodies) are present and non‐pathognomonic in PSP, CBD, and other tauopathies.11, 20
Neuropil threads (i.e., short, irregular neuronal and oligodendroglial processes) are sparsely seen in PSP, but are numerous in gray and white matter in cortical and subcortical regions in CBD.11, 20
The most characteristic neuronal lesions in PSP are globose neurofibrillary tangles and in CBD diffuse or granular pretangles without apparent formation of fibrillary structures.11, 20
Ballooned neurons, which are achromatic in HE staining, immunoreactive for alpha‐B‐crystallin, and inconsistently immunoreactive for tau are considered a hallmark of CBD without being entirely specific. In PSP, they are only seen in association with concurrent argyrophilic grain disease.11, 20
Overall, these histopathological stigmata predominate in hindbrains in PSP and forebrains in CBD (Fig. 1). Macroscopically, PSP brains typically show marked atrophy of the mesencephalon, subthalamic nucleus, superior cerebellar peduncle, mild atrophy of the frontal cortex, and e vacuo dilatation of the third ventricle. Asymmetric focal frontoparietal or paracentral cortical atrophy is the most characteristic macroscopic finding in CBD. Unusual lesion and atrophy distributions, however, do exist in patients with atypical clinical presentations.11, 20
Clinical Manifestations
PSP and CBD are sporadic diseases with typical onset in the fifth to seventh decade and average disease durations of 7.9 and 6.8 years.22 PSP and CBD present a largely overlapping spectrum of clinical syndromes, reflecting the variable topography of the cerebral lesions (Fig. 1). The initially described and most frequent clinical manifestation of PSP is RS, a combination of progressive postural instability, slowing of saccades, and vertical supranuclear gaze palsy.1, 2, 22 Although RS it is still believed to be highly specific for PSP,2, 22 it is acknowledged that patients with CBD can also present a clinical RS manifestation.5, 23 CBS is a combination of at least one cortical symptom (apraxia, cortical sensory loss, alien limb phenomenon) and at least one extrapyramidal symptom (akinesia, rigidity, dystonia, myoclonus).3, 4, 5 CBS was initially considered the predominant manifestation of CBD.3, 4, 5 Meanwhile, it is known that CBD patients can also present clinical syndromes other than CBS5, 22, 23 and that CBS patients can have other underlying neuropathologies, including PSP.2, 22, 23 Progressive parkinsonism (i.e., bradykinesia and rigidity with or without tremor), may also predominate the early clinical picture in both PSP and CBD.2, 4, 22, 23 The non‐fluent, “agrammatical” variant of primary progressive aphasia and apraxia of speech have more recently been identified as clinical manifestations of PSP and CBD.2, 5, 22, 24 Predominant frontal presentations of PSP and CBD may evoke behavioral and cognitive symptoms of frontotemporal dementia.2, 5, 22, 23 A posterior cortical atrophy syndrome has been reported with diverse neuropathologies, including CBD.23 Progressive gait freezing as initial manifestation has so far been reported to be highly specific for PSP.2, 22 In summary, while PSP and CBD have preferential clinical manifestations each, the width of their possible clinical manifestations encompasses the same spectrum.
Therapy
The therapies currently recommended for PSP and CBD are symptomatic in nature, relying on physiotherapy, speech therapy, and pharmacological modulation of neurotransmitter systems in the affected brain regions (Fig. 1). They include dopaminergic stimulation for akinetic‐rigid symptoms, clonazepam for myoclonus, and botulinum toxin A for focal dystonia.25 The choice of therapies is based on the presence of alike symptoms in both PSP and CBD.
Interventions in the molecular mechanisms involved in spreading of the pathology will hopefully become available as disease‐modifying therapies to slow down disease progression (Fig. 1). Ideally, interventions at the primary, most upstream disease mechanisms might even allow development of causal therapies to prevent or to cure the diseases (Fig. 1). Currently, such approaches are being developed to target expression or degradation of the tau protein (specifically the 4R‐tau isoforms), disease‐associated post‐translational modifications, or the trans‐cellular tau spread. None of these targets appear to be specific to either PSP or CBD.26
Conclusion
In summary, PSP and CBD are sporadic, adult‐onset, primary 4R‐tauopathies, affecting neurons, oligodendrocytes, and astrocytes alike, with shared genetic risk factors in MAPT and MOBP. The overlapping clinical spectrum in absence of distinctive imaging or fluid biomarkers still requires neuropathological examination for a definite differential diagnosis.
Therefore, joint classification of overlapping PSP and CBD syndromes as clinically probable 4R‐tauopathy has now been operationalized for the first time by the Movement Disorders Society Criteria.2 This approach avoids diagnostic ambitions to differentiate PSP from CBD in clinical situations, where these are unattainable with currently available methods. Moreover, this concept links clinical syndrome to a molecular diagnosis, defined by presence of a molecular target for therapeutic interventions, and thus selects patients for 4R‐tau‐targeting therapies.
At the latest, when the therapeutic efficacy of future 4R‐tau‐targeting therapies will not be compromised by the subtle histological and molecular differences between PSP and CBD, it will be time to seriously reconsider the need to classify PSP and CBD as different disorders, but to adopt the diagnostic concept of 4R‐tauopathies more broadly.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
G.U.H.: 1A, 1B, 1C, 2A, 2B, 3A
Disclosures
Ethical Compliance Statement: The author confirms that the approval of an institutional review board was not required for this work. I confirm that I have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources & Conflicts of Interest: This work was supported by the European Union Innovative Medicines Initiative 2 Joint Undertaking (IMI‐JU, grant number: 116060‐IMPRIND) and the German Federal Ministry for Education and Research Program for Individualized Medicine (BMBF, grant HIT‐Tau). The author reports no conflicts of interest.
Financial Disclosures for previous 12 months: Günter U. Höglinger has served on the advisory boards for AbbVie, Alzprotect, Asceneuron, Bristol‐Myers Squibb, Novartis, Roche, Sellas Life Sciences Group, UCB; has received honoraria for scientific presentations from Abbvie, Roche, Teva, UCB, has received research support from CurePSP, the German Academic Exchange Service (DAAD), German Parkinson's Disease Foundation (DPG), German PSP Association (PSP Gesellschaft), German Research Foundation (DFG) and the German Ministry of Education and Research (BMBF), International Parkinson's Fonds (IPF), Neuropore; has received institutional support from the German Center for Neurodegenerative Diseases (DZNE).
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Steele JC, Richardson JC, Olszewski J. Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 1964;10:333–359. [DOI] [PubMed] [Google Scholar]
- 2. Höglinger GU, Respondek G, Stamelou M, et al.; Movement Disorder Society‐endorsed PSP Study Group . Clinical diagnosis of progressive supranuclear palsy: the Movement Disorder Society criteria. Mov Disord 2017;32:853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rebeiz JJ, Kolodny EH, Richardson EP Jr. Corticodentatonigral degeneration with neuronal achromasia. Arch Neurol 1968;18:20–33. [DOI] [PubMed] [Google Scholar]
- 4. Boeve BF, Lang AE, Litvan I. Corticobasal degeneration and its relationship to progressive supranuclear palsy and frontotemporal dementia. Ann Neurol 2003;54(Suppl 5):S15–S19. [DOI] [PubMed] [Google Scholar]
- 5. Armstrong MJ, Litvan I, Lang AE, et al. Criteria for the diagnosis of corticobasal degeneration. Neurology 2013;80:496–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schrag A, Ben‐Shlomo Y, Quinn NP. Prevalence of progressive supranuclear palsy and multiple system atrophy: a cross‐sectional study. Lancet 1999;354:1771–1775. [DOI] [PubMed] [Google Scholar]
- 7. Togasaki DM, Tanner CM. Epidemiologic aspects. Adv Neurol 2000;82:53–59. [PubMed] [Google Scholar]
- 8. Kovacs GG, Milenkovic I, Wöhrer A, et al. Non‐Alzheimer neurodegenerative pathologies and their combinations are more frequent than commonly believed in the elderly brain: a community‐based autopsy series. Acta Neuropathol 2013;126:365–384. [DOI] [PubMed] [Google Scholar]
- 9. Höglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet 2011;43:699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kouri N, Ross OA, Dombroski B, et al. Genome‐wide association study identifies microtubule‐associated protein tau (MAPT) and myelin‐associated oligodendrocytic basic protein (MOBP) as shared genetic risk factors for corticobasal degeneration and progressive supranuclear palsy. Nat Commun 2015;6:7247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kovacs GG. Neuropathology of tauopathies: principles and practice. Neuropathol Appl Neurobiol 2015;41:3–23. [DOI] [PubMed] [Google Scholar]
- 12. Sergeant N, Wattez A, Delacourte A. Neurofibrillary degeneration in progressive supranuclear palsy and corticobasal degeneration: tau pathologies with exclusively “exon 10” isoforms. J Neurochem 1999;72:1243–1249. [DOI] [PubMed] [Google Scholar]
- 13. Delacourte A, Sergeant N, Wattez A, Gauvreau D, Robitaille Y. Vulnerable neuronal subsets in Alzheimer's and Pick's disease are distinguished by their tau isoform distribution and phosphorylation. Ann Neurol 1998;43:193–204. [DOI] [PubMed] [Google Scholar]
- 14. Myers AJ, Pittman AM, Zhao AS, et al. The MAPT H1c risk haplotype is associated with increased expression of tau and especially of 4 repeat containing transcripts. Neurobiol Dis 2007;25:561–570. [DOI] [PubMed] [Google Scholar]
- 15. Arai T, Ikeda K, Akiyama H, et al. Identification of amino‐terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann Neurol 2004;55:72–79. [DOI] [PubMed] [Google Scholar]
- 16. Arima K. Ultrastructural characteristics of tau filaments in tauopathies: immuno‐electron microscopic demonstration of tau filaments in tauopathies. Neuropathology 2006;26:475–483. [DOI] [PubMed] [Google Scholar]
- 17. Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci USA 2013;110:9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Williams DR, Holton JL, Strand C, et al. Pathological tau burden and distribution distinguishes progressive supranuclear palsy‐parkinsonism from Richardson's syndrome. Brain 2007;130:1566–1576. [DOI] [PubMed] [Google Scholar]
- 19. Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 2014;82:1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dickson DW. Neuropathologic differentiation of progressive supranuclear palsy and corticobasal degeneration. J Neurol 1999;246(Suppl 2):II6–II15. [DOI] [PubMed] [Google Scholar]
- 21. Komori T, Arai N, Oda M, et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol 1998;96:401–408. [DOI] [PubMed] [Google Scholar]
- 22. Respondek G, Kurz C, Arzberger T, et al.; Movement Disorder Society‐endorsed PSP study group . Which ante mortem clinical features predict progressive supranuclear palsy pathology?. Mov Disord 2017;32:995–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ling H, O'Sullivan SS, Holton JL, et al. Does corticobasal degeneration exist? A clinicopathological re‐evaluation. Brain 2010;133:2045–2057. [DOI] [PubMed] [Google Scholar]
- 24. Josephs KA, Duffy JR. Apraxia of speech and nonfluent aphasia: a new clinical marker for corticobasal degeneration and progressive supranuclear palsy. Curr Opin Neurol 2008;21:688–692. [DOI] [PubMed] [Google Scholar]
- 25. Levin J, Kurz A, Arzberger T, Giese A, Höglinger GU. The differential diagnosis and treatment of atypical parkinsonism. Dtsch Arztebl Int 2016;113:61–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Höglinger GU. Targeting tau therapeutics to PSP: support from new diagnostic criteria and biomarkers. Lancet Neurol 2017;16:552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]