

Abstract
Background
Clinical diagnostic criteria for PD rely on rest tremor, bradykinesia, and rigidity. These features are non‐specific and neuropathological confirmation remains the gold standard for diagnosis. This study presents data on clinical certainty ratings in autopsy‐proven PD.
Methods
Subjects were assessed annually by a movement disorders specialist and assigned to a clinical certainty group for PD based on multiple clinical features before autopsy. The three groups considered for analysis are as follows: Group I 0–49% certainty, Group II 50‐89% certainty, and Group III 90–100% certainty. All subjects were autopsied and had a standardized neuropathological assessment.
Results
275 subjects were assigned a PD certainty at their last visit before death. Group I had 80 subjects, Group II 56 subjects, and Group III 139 subjects. The clinical features recorded in Group I, II, and III, were as follows: rest tremor, bradykinesia, rigidity, postural instability, asymmetric onset, persistent asymmetry, current response to dopaminergic treatment, motor fluctuations, and dyskinesia. Rigidity, postural instability, asymmetric onset, current response to dopaminergic treatment, motor fluctuation, and dyskinesia were more likely to be present in the group which was rated with higher certainty. The final diagnosis of PD was confirmed by neuropathological assessment in 85% of the patients in Group III as compared to 30% in Group II and 5% in Group I.
Conclusions
High certainty (90–100%) had strong positive predictive value (85%) for autopsy‐proven PD as compared to either lower certainty groups (0–49% and 50–89%) which had lower predictive value (5% and 30% respectively).
Keywords: Parkinson's disease, diagnosis certainty, neuropathology
Introduction
Currently, the lack of a definitive biomarker for a diagnosis of Parkinson's disease (PD) means a diagnosis still requires neuropathological confirmation as the gold standard. The clinical diagnosis of PD relies on the motor symptoms, including bradykinesia, rigidity, and rest tremor.1 The accuracy of diagnosis can be improved if the disease is medication‐responsive and longer than five years.1 Other features, such as dyskinesia, motor fluctuations, and hyposmia also help in improving the accuracy.2 The inaccuracy of diagnosis has implications for therapeutic, genetic, biomarker, and epidemiologic studies. This is particularly relevant for research studies involving early PD patients, especially those never yet treated and where motor fluctuations and dyskinesia are not yet evident.
We performed a retrospective study to evaluate whether assigning a clinical certainty level is helpful in predicting a final neuropathological diagnosis of PD. We also analyzed which clinical features prompted the physicians to assign a higher level of certainty of PD diagnosis. The clinical data obtained in this study were compared with the neuropathological diagnosis.
Methods
Data were obtained from the Arizona Study of Aging and Neurodegenerative Disorders (AZSAND).3 All individuals had signed written informed consent approved by the Banner Sun Health Research Institute (BSHRI) Institutional Review Board. Subjects were examined annually by a movement disorders neurologist and given a clinical diagnosis. Those diagnosed with PD were assigned to one of four clinical certainty groups, and analysis was based on the clinical certainty assigned at the last examination before autopsy (0–9%, 10–49%, 50–89%, and 90–100% certainty). Certainty was based on clinical examination and clinical history obtained by the movement disorders neurologist. There were only five subjects in the 0–9% certainty group so that group was combined with the 10–49% group, resulting in Group I: 0–49%, Group II: 50–89%, and Group III: 90–100%.
A lifetime symptom score was calculated for the certainty groups and is the sum score of the following nine symptoms: bradykinesia, rest tremor, rigidity, postural instability, asymmetric onset, motor fluctuations, and dyskinesia present in one of their clinical visits (cumulative presence in their lifetime) and if they had responded to treatment or persistent asymmetry at their last clinical visit before autopsy (one‐time presence at their last visit).
The final clinical‐neuropathologic diagnosis was made by a single neuropathologist (T.G.B.) based on clinical information, and neuropathologic criteria described previously.4 Parkinsonism NOS was pathologically defined as a subject found to have no clear pathology causing the clinical findings of parkinsonism.
Statistical Analysis
Mean levels were compared among groups by using one‐way analysis of variance and proportions were compared by using the Pearson chi‐square test. The Fisher exact test was used instead of the Pearson chi‐square test if the minimum expected cell count was less than five. The certainty data was also compared between the first and last visit and the change in certainty rating over time was analyzed. Chi‐square test was used to examine the association between the lifetime symptom score and the PD certainty group.
Results
A total of 275 subjects were included in the study. The demographics and baseline characteristics are listed in Table 1. Individuals rated with the highest certainty (90–100%) had an earlier age at last visit (mean 79.4 years), earlier age at death (mean 80.6 years), earlier age at onset (mean 66.1 years), and longer disease duration (mean 14.5 years). Group III also had more severe clinical findings with higher UPDRS III score (mean 40.8) and higher Hoehn & Yahr stage (mean 3.31).
Table 1.
Demographics of Subjects Included in the Study
|
Group I (0–49% certainty) |
Group II (50–89% certainty) |
Group III (90–100% certainty) |
p | |
|---|---|---|---|---|
| N | 80 | 56 | 139 | ‐‐ |
| Female, n (%) | 44 (55%) | 22 (39%) | 45 (32%) | 0.004 |
| Age at last visit, years, mean (SD) | 84.6 (6.2) | 83.4 (6.3) | 79.4 (7.0) | <0.001 |
| Age at death, years, mean (SD) | 89.1 (6.2) | 86.4 (6.6) | 80.6 (6.9) | <0.001 |
| Age at onset, years, mean (SD) [number of subjects with available information] | 84.0 (7.8) [18] | 77.6 (9.0) [42] | 66.1 (10.4) [139] | <0.001 |
| Disease duration, years, mean (SD) [number of subjects with available information] | 5.6 (3.6) [18] | 8.2 (5.2) [42] | 14.5 (7.2) [139] | <0.001 |
| Hoehn & Yahr stage, mean (SD) [number of subjects with available information] | 1.12 (1.26) [79] | 2.78 (1.18) [55] | 3.31 (0.90) [138] | <0.001 |
| Total UPDRS III (OFF state), mean (SD) [number of subjects with available information] | 11.2 (7.4) [79] | 24.5 (19.2) [54] | 40.8 (16.6) [94] | <0.001 |
The clinical features for Groups I, II, and III are shown in Table 2. Rigidity, postural instability, asymmetric onset, current response to treatment, motor fluctuations, and dyskinesia were more likely to be present in Group III. As shown in Fig. 1, the final diagnosis of PD was confirmed by neuropathological assessment in 85% of the subjects in group III, 30% in group II, and 5% in group I.
Table 2.
Clinical Features
|
Group I (0–49% certainty) |
Group II (50–89% certainty) |
Group III (90–100% certainty) |
p | |
|---|---|---|---|---|
| Rest tremor | 55% | 69% | 64% | .22 |
| Bradykinesia | 66% | 98% | 100% | <.001 |
| Rigidity | 15% | 67% | 89% | <.001 |
| Postural instability | 47% | 67% | 93% | <.001 |
| Asymmetric onset | 52% | 71% | 84% | <.001 |
| Persistent asymmetry | 58% | 67% | 49% | .13 |
| Never treated | 80 (100%) | 32/55 (58%) | 0/138 (0%) | <.001 |
| Current response to treatment | NA | 11/23 (48%) | 129/138 (93%) | <.001 |
| Motor fluctuations | 0% | 18% | 55% | .001 |
| Dyskinesia | 0% | 18% | 37% | .12 |
Figure 1.
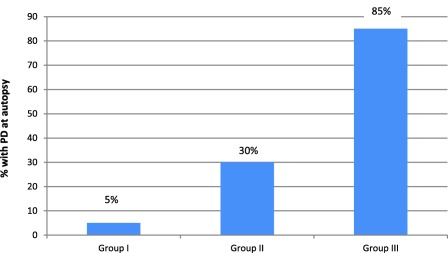
Final diagnosis of PD confirmed by autopsy.
Among the 275 subjects, 106 had only one clinical visit with PD certainty data before their autopsy; 169 subjects had two or more clinical visits before their autopsy. For these 169 subjects, PD certainty data were compared from first to the last visit. The mean duration between the first and last visit was 4.4 years.
As shown in Table 3, PD certainty increased in 24 (14.2%) subjects, stayed the same in 139 (82.2%) subjects, and decreased in 6 (3.6%) subjects. Of the 24 subjects with increased PD certainty, 2 of 7 subjects who had PD certainty increased from 0%–49% to 50%–89% had PD confirmed by autopsy, while 14 of the 17 subjects who had PD certainty increase from 0%–49% or 50%–89% to 90%–100% had PD confirmed by autopsy.
Table 3.
Change in the Level of Certainty Between First and Last Visit
| First visit | Last visit before autopsy | ||
|---|---|---|---|
| 0%–49% | 50%–89% | 90%–100% | |
| 0%–49%, (n = 41) | 32 | 7 | 2 |
| 50%–89%, (n = 35) | 2 | 18 | 15 |
| 90%–100%, (n = 93) | 0 | 4 | 89 |
| Total | 34 | 29 | 106 |
Of the six subjects who had decreased PD certainty, two went from 50%–89% at first visit to 0%–49% at last visit and neither had PD at autopsy. Four subjects had PD certainty decrease from 90%–100% at first visit to 50%–89% at last visit yet all had PD pathologically.
Table 4 shows the correlation between the lifetime symptom score and the PD certainty group. This correlation was significant by Chi‐square test (p < 0.0001).
Table 4.
Lifetime Symptom Score
| Lifetime symptoms pooled together (lifetime symptom score) | ||||
|---|---|---|---|---|
| Frequency | Group I | Group II | Group III | Total |
| 1 | 13 (16.3%) | 0 (0%) | 0 (0%) | 13 |
| 2 | 20 (25.0%) | 1 (1.8%) | 0 (0%) | 21 |
| 3 | 26 (32.5%) | 13 (23.2%) | 0 (0%) | 39 |
| 4 | 11 (13.8%) | 18 (32.1%) | 4 (2.9%) | 33 |
| 5 | 10 (12.5%) | 5 (8.9%) | 16 (11.5%) | 31 |
| 6 | 0 (0%) | 13 (23.2%) | 30 (21.6%) | 43 |
| 7 | 0 (0%) | 3 (5.4%) | 39 (28.1%) | 42 |
| 8 | 0 (0%) | 3 (5.4%) | 26 (18.7%) | 29 |
| 9 | 0 (0%) | 0 (0%) | 24 (17.3%) | 24 |
| Total | 80 | 56 | 139 | 275 |
The pathologic findings in Group I did not reveal one particular disease by pathology. Some subjects were found to have dementia with Lewy bodies (DLB; 4%, all with AD), incidental Lewy body disease (ILBD; 14%), progressive supranuclear palsy (PSP; 4%), Alzheimer's disease (25%), vascular dementia (10%), and aging‐related tau astrogliopathy (ARTAG; 15%). In Group III subjects with the highest certainty for PD, multiple system atrophy (2 cases), PSP (9 cases), parkinsonism NOS (9 cases), and DLB (1 case) were the final diagnoses in those who did not have PD by autopsy criteria.
Discussion
Giving a clinical certainty rating for Parkinson's disease may have value depending on the clinical or research question being asked. This study showed that subjects rated with a higher certainty (90–100%) for a clinical diagnosis of PD had strong positive predictive value (85%) for autopsy‐proven PD compared to those with a lower certainty rating (0–49% and 50–89%) having lower predictive value (5% and 30%, respectively). Assigning a level of certainty for the diagnosis of PD may be a useful adjunct to enrolment criteria in a research trial or stratification for genetic, epidemiologic or biomarker studies.
The clinicians assigned a higher level of certainty for subjects with advanced disease (mean disease duration 14.5 years) as the mean total UPDRS scores and Hoehn & Yahr stage were higher in the highest certainty group. The subjects assigned with a low level of certainty had shorter disease duration, reconfirming that positive predictive value of PD diagnosis is poor in short‐duration subjects.1 With longitudinal assessment, few subjects had a decrease in certainty while 24 subjects had an increase in certainty on follow‐up. Certain features, such as dyskinesia and motor fluctuations, appear later in the disease course and can prompt clinicians to assign a higher level of certainty.1 The overall lifetime symptom score was highest in Group III as the patients with PD eventually develop other signs that support the diagnosis. The higher lifetime symptom score also correlated with a higher level of certainty. Despite being in the highest certainty group, the positive predictive value for pathologically proven PD is only 85% and reinforces the fact that post‐mortem pathological diagnosis remains the gold standard. There are overlapping clinical features such as initial response to dopaminergic treatment, dyskinesia, and motor fluctuation in tauopathies and synucleinopathies, making an accurate clinical diagnosis difficult.5
The most frequent neuropathologic finding in subjects with the highest certainty that did not have PD was PSP and parkinsonism NOS; this, despite clear findings that suggested typical PD even after long duration follow‐up.6 This data are similar to the diagnostic accuracy of PD, which was found to be 76% in a study performed by Hughes et al. The percentage increased to 82% after applying the recommended criteria.7 Applying the criteria proposed by Hughes et al. to the current three groups (0% of Group I, 32% of Group II, and 100% of Group III) met their diagnostic criteria for PD. This is because the subjects in Group I had never been treated for PD. Our finding is in line with the previous studies where the accuracy of a clinical diagnosis of PD ranged between 70 to 94% when pathology was considered as a gold standard for diagnosis.8
As shown in Table 1, subjects in Group I were older, had an older age at the onset of symptoms, shorter disease duration, and less severe disease (low UPDRS scores and H & Y stage). It is possible that this group represents the mild extrapyramidal signs (EPS) seen in elderly individuals.9 Mild parkinsonian signs seen in the elderly population is associated with evidence of cognitive decline, with one study suggesting that this could represent a prodrome of PD.10 In our study, 9 of 41 subjects in Group I had an increase in the level of certainty on follow‐up visit and four of them had pathologically confirmed PD. Thus, there may be a subgroup of elderly individuals with mild EPS who go on to develop PD. However, care is needed, as 30% of the subjects in Group I did not have any neurodegenerative pathology in the brain. As previously shown, mild EPS can be seen in AD and cognition declines more rapidly than others.11 In our study, 25% of the subjects in Group I had a final diagnosis of AD, which was a probable cause of mild EPS. The presence of small vessel disease can also cause mild EPS, which can lead to diagnostic confusion.12 Ten percent of the subjects in Group I had a final neuropathological diagnosis of vascular dementia, therefore, this etiology of parkinsonism should also be considered in elderly patients with mild EPS. ARTAG has been shown to be present in elderly brains without any cognitive complaints or neurodegenerative disease.13 It is possible that this pathology in the brain can lead to mild EPS as it was present in 15% of the subjects in Group I, but further studies are needed to confirm this finding. The diagnosis of parkinsonism is challenging in elderly subjects because of a mixed clinical picture due to the presence of concomitant pathologies in the brain found at the time of autopsy.8 Since studies focusing on the accuracy of diagnosis in late‐onset PD are lacking, assigning a level of certainty can be useful in late‐onset disease as elderly individuals in a higher certainty group are more likely to have PD.8 The importance of the predictive value for PD with a certainty rating of 1–49% may be useful for research purposes in an elderly population, especially since these subjects had not been treated when evaluated.
The current data show that the presence of tremor is not significantly associated with the level of certainty for PD. A possible explanation for this finding could be the presence of tremor associated with dystonia, drug‐induced tremor, or essential tremor (ET) in the elderly.14 Some patients enrolled in PD studies, with PD, have been found to have normal presynaptic dopaminergic imaging (DaT scan). Many of these patients had a predominant tremor and are labeled SWEDDs (Scans Without Evidence of Dopaminergic Deficit). A long‐term follow‐up study of these patients showed that there was minimal progression and these patients were unlikely to have PD.15 The tremor in these patients is usually due to an alternative cause, such as dystonia, vascular, iatrogenic, etc.16 Rest tremor can also be seen in as many as 50% of the patients with ET.17 Similarly, the presence of bradykinesia did not distinguish among higher certainties. It is possible that the presence of confounding factors in the elderly (e.g., arthritis, weakness) interfere with the interpretation of bradykinesia.2 Slower movements can also be observed in some patients with ET, leading to a diagnostic confusion due to the presence of a concomitant tremor.18 Rigidity is a useful sign when assigning a level of certainty. The MDS clinical diagnostic criteria for PD published in 2015 advocate testing for lead‐pipe rigidity as isolated cogwheeling without rigidity (Froment's sign) can be seen in ET which can be misleading.2, 19
No asymmetry on physical examination is considered as a red flag for the diagnosis of PD,2 but persistent asymmetry had no relationship with the level of certainty. The current response to dopaminergic therapy, documented by improvement in the symptoms with increasing doses, motor fluctuations (on/off), and dyskinesia were very helpful in assigning a higher level of certainty.
Although recent papers have discussed that DLB, Parkinson's disease dementia (PDD), and PD should be considered as Lewy body disorders with different phenotypic presentations.20 In our study, we have considered DLB as a misdiagnosis because the recruitment in AZSAND is directed at Alzheimer's disease (AD) and PD patients and not patients with DLB.3 Also, there is a subtype of DLB patients who do not have parkinsonism and their clinical presentation can be confused with AD.20
One limitation of this study is the recruitment of subjects from the same geographical location, racial background, level of education, and non‐availability of some data for all the subjects.3 The current response to treatment, motor fluctuations, and dyskinesia can only be assessed with dopaminergic therapy, therefore, using certainty rating may not be helpful in drug naïve subjects (i.e., Group I).
In conclusion, further development and incorporation of PD clinical certainty ratings may be a useful step for clinical practice and research. In this paper, we have pointed out certain clinical factors which increase the diagnostic certainty and others, such as tremor, which may not improve diagnostic accuracy.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
H.G.: 1C, 2C, 3A, 3B
S.M.: 1C, 2C, 3B
N.Z.: 2B, 3B
J.H.: 2A, 2B, 3B
H.S.: 1A, 1B, 1C, 2C, 3B
E.D.‐D.: 1B, 1C, 2C, 3B
M.S.: 1A, 1B, 1C, 2C, 3B
C.B.: 1A, 1B, 1C, 2C, 3B
B.D.: 1C, 2C, 3B
T.B.: 1A, 1B, 1C, 2C, 3B
G.S.: 1C, 2C, 3B
L.S.: 1B, 1C, 3B
K.D.: 1C, 3B
C.A.: 1A, 1B, 1C, 2C, 3A, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: The National Institute of Neurological Disorders and Stroke, grant number: U24 NS072026 (National Brain and Tissue Resource for Parkinson's Disease and Related Disorders); the National Institute on Aging, grant number: P30 AG19610 (Arizona Alzheimer's Disease Core Center); the Arizona Department of Health Services, contract number: 211002 (Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission, contract numbers: 4001, 0011, 05‐901, and 1001 (Arizona Parkinson's Disease Consortium); the Michael J. Fox Foundation for Parkinson's Research; and Mayo Clinic Foundation. The authors report no conflicts of interest.
Financial Disclosures for the previous 12 months: H.V. Gupta reports no disclosures. S.H. Mehta has consulted with Allergan Inc, US World Meds, Merz Pharma, and Cynapsus Therapeutics. J.G. Hentz received grant funding from the National Institute of Neurological Disorders and Stroke. N. Zhang reports no disclosures. H.A. Shill has served as a consultant with Cynapsus/Sunovion, Abbvie, and Lundbeck; Dr. Shill has received research support from Cynapsus/Sunovion, Axovant, Impax, US World Meds, Michael J. Fox Foundation, and the NIH. E. Driver‐Dunckley reports no disclosures. M.N. Sabbagh received grant/research support from AstraZeneca, Avid Pharmaceuticals, Axovant, Genentech, Inc., Lilly Pharmaceuticals, Merek & Co, Pfizer, Roche Diagnostics Corporation, vTv Therapeutics, Piramal Imaging; consultant for Axovant, Biogen, Grifols, Humana, Lilly Pharmaceuticals, Sanofi, vtv Therapeutics; and a stock shareholder of Brain Health, Muses Labs, and Versanum. C.M. Belden receives research support from AstraZeneca, Avid Pharmaceuticals, Axovant, Genentech, Inc., Lilly Pharmaceuticals, Merek & Co, Pfizer, Roche Diagnostics Corporation, Intracellular, ABBvie, Janssen, Avanir, Novartis, Biogen, Takeda, Functional Neuromodulation, Neuronix, Navidea, Transtech Pharma, Suven; supported by grants from the National Institutes of Health, Miche J Fox Foundation, and state of Arizona. B.N. Dugger is supported by grants AG002132 (Core C; PI: Dugger) from the National Institutes of Health, the Alzheimer's Association (PI:Dugger), as well as the CurePSP foundation (PI: Dugger), the Henry M. Jackson Foundation (HU0001‐15‐2‐0020, PI: Prusiner), and Daiichi Sankyo Co., Ltd. (PI: Prusiner). T.G. Beach is a consultant for Avid Radiopharmaceuticals, GE Healthcare, Genentech, and Roche; he does contracted research for Avid Radiopharmaceuticals and Navidea Biopharmaceuticals; and is supported by grants from the National Institutes of Health, the Michael J Fox Foundation, and the state of Arizona. G.E. Serrano reports no disclosures. L.I. Sue reports no disclosures. K. Davis reports no disclosures. C.H. Adler is a consultant for Acadia, Acorda, Adamas, Cynapsus, Jazz, Lundbeck, Merz, Minerva, Neurocrine, and Sunovion.
Acknowledgments
The authors are grateful to the subjects who have volunteered to participate in the Banner Sun Health Research Institute Brain and Body Donation Program. The authors thank Mr. Bruce Peterson for developing and maintaining the database, and other members of the Arizona Parkinson's Disease Consortium for helpful discussions, including Kathy Jo Davis.
Financial information: The National Institute of Neurological Disorders and Stroke, grant number: U24 NS072026 (National Brain and Tissue Resource for Parkinson's Disease and Related Disorders); the National Institute on Aging, grant number: P30 AG19610 (Arizona Alzheimer's Disease Core Center); the Arizona Department of Health Services, contract number: 211002 (Arizona Alzheimer's Research Center), the Arizona Biomedical Research Commission, contract numbers: 4001, 0011, 05‐901, and 1001 (Arizona Parkinson's Disease Consortium); the Michael J. Fox Foundation for Parkinson's Research; and Mayo Clinic Foundation.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Adler CH, Beach TG, Hentz JG, et al. Low clinical diagnostic accuracy of early vs advanced Parkinson's disease: clinicopathologic study. Neurology 2014;83:406–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30:1591–1601. [DOI] [PubMed] [Google Scholar]
- 3. Beach TG, Adler CH, Sue LI, et al. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology 2015;35354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 2009;8:1150–1157. [DOI] [PubMed] [Google Scholar]
- 5. Litvan, I. Progressive supranuclear palsy In Litvan I. (Ed.), Atypical parkinsonian disorders. 2005; Clinical and research aspects (pp. 287–308). Totowa, NJ: Humana Press. [Google Scholar]
- 6. Respondek G, Stamelou M, Kurz C, et al. The phenotypic spectrum of progressive supranuclear palsy: a retrospective multicenter study of 100 definite cases. Mov Disord 2014;29:1758–1766. [DOI] [PubMed] [Google Scholar]
- 7. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rizzo G, Copetti M, Arcuti S, et al. Accuracy of clinical diagnosis of Parkinson's disease: A systematic review and meta‐analysis. Neurology 2016. Feb;86:566–576. [DOI] [PubMed] [Google Scholar]
- 9. Richards M, Touchon J, Ledesert B, Ritchie K. Mild extrapyramidal signs and functional impairment in ageing. Int J Geriatr Psychiatry 2002;17:150–153. [DOI] [PubMed] [Google Scholar]
- 10. Lerche S, Hobert M, Brockmann K, et al. Mild parkinsonian signs in the elderly—is there an association with PD? Crossectional findings in 992 individuals. PLoS One 2014;9:e92878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Portet F, Scarmeas N, Cosentino S, Helzner EP, Stern Y. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol 2009;66:1120–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Staekenborg SS, van der Flier WM, van Straaten EC, et al. Neurological signs in relation to type of cerebrovascular disease in vascular dementia. Stroke 2008;39:317–322. [DOI] [PubMed] [Google Scholar]
- 13. Liu AK, Goldfinger MH, Questari HE, Pearce RK, Gentleman SM. ARTAG in the basal forebrain: widening the constellation of astrocytic tau pathology. Acta Neuropathol Commun 2016;4:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen W, Hopfner F, Becktepe JS, Deuschl G. Rest tremor revisited: Parkinson's disease and other disorders. Transl Neurodegener 2017;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marek K, Seibyl J, Eberly S, et al. Longitudinal follow‐up of SWEDD subjects in the PRECEPT Study. Neurology 2014;82:1791–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erro R, Schneider SA, Stamelou M, Quinn NP, Bhatia KP. What do patients with scans without evidence of dopaminergic deficit (SWEDD) have? New evidence and continuing controversies. J Neurol Neurosurg Psychiatry 2016;87:319–323. [DOI] [PubMed] [Google Scholar]
- 17. Louis ED, Hernandez N, Michalec M. Prevalence and correlates of rest tremor in essential tremor: cross‐sectional survey of 831 patients across four distinct cohorts. Eur J Neurol 2015;22:927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Thenganatt MA, Louis ED. Distinguishing essential tremor from Parkinson's disease: bedside tests and laboratory evaluations. Expert Rev Neurother 2012;12:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deuschl G, Bain P, Brin M. Consensus statement of the Movement Disorder Society on Tremor. Ad Hoc Scientific Committee. Mov Disord 1998;13:2–23. [DOI] [PubMed] [Google Scholar]
- 20. Boeve BF, Dickson DW, Duda JE, et al. Arguing against the proposed definition changes of PD. Mov Disord 2016;31:1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brooks DJ. Parkinson's disease: diagnosis. Parkinsonism Relat Disord 2012;18:S31–33. [DOI] [PubMed] [Google Scholar]