

Abstract
Background
Huntington's disease (HD) is a rare and fatal inherited genetic disorder characterized by progressive motor, cognitive, and behavioral impairment. It leads to premature death, but data regarding advanced‐stage disease are scarce. We sought to determine HD‐associated survival, mortality, and causes and places of death.
Methods
Data from the European HD Network prospective study (REGISTRY) collected from 2001 through 2013 were used, including the Unified Huntington's Disease Rating Scale and death report forms. Group comparisons were performed using the t test or the χ2 test. Survival analyses were computed through Kaplan‐Meier estimates of median survival. All tests were 2‐sided with a significance level of P = 0.05.
Results
In total, 5164 participants were analyzed. The mean age at diagnosis was 49 years, and the mean age at death was 58 years. At the end of the study period, there were 533 deaths (10.3% of patients). Median survival was 24 years from diagnosis and 35 years from symptom onset. The most frequent causes of death were pneumonia (19.5%), other infections (6.9%), and suicide (6.6%). The most frequent places of death were the hospital (29.8%), the home (23.9%), and nursing houses (19.8%).
Conclusions
Patients with HD tend to die from the same conditions as patients with other neurodegenerative diseases. However, compared with nonhereditary Parkinson's disease and Alzheimer's disease, the median time from onset to death is longer, and the places of death are distinctive.
Keywords: cause of death, Huntington's disease, mortality, survival
Huntington's disease (HD) is a rare and fatal inherited genetic disorder caused by a CAG repeat expansion at the IT15 gene on chromosome 4.1, 2 It is characterized by progressive motor, cognitive, and behavioral impairment, invariably leading to premature disability, with no disease‐modifying treatments currently available.3 Despite this, the advanced stage of this condition has not been extensively studied, and knowledge about this phase of the disease would be essential to plan care and supportive services for individuals with HD and their families4 and to define patient outcomes in clinical studies.
Prognostication,5 advance‐directive decision making, anticipating end‐stage care needs6 and life‐prolonging procedures, and comfort‐maximizing and quality‐of‐life–maximizing approaches are all essential matters for patients with HD and their carers. Because the current literature on this topic is limited and dated, we sought to determine the survival, mortality, and causes and places of death of in patients with HD from the European Huntington's Disease Network's (EHDN) prospective cohort (the REGISTRY study7) and to help contribute to the improvement of care and clinical management and lessen the burden of the disease and improve the quality of life of patients and their families at this late stage in the disease.8
Materials and Methods
Ethical Approval
This study was performed in accordance with the declaration of Helsinki and was approved by the local ethics committees for each study site contributing to the REGISTRY study. All participants gave informed written consent. Participants who lacked the capacity to consent and minors had consent given on their behalf as requested by country‐specific ethical standards.
Participants
All participants were part of the EHDN multicenter, European, prospective observational study–REGISTRY.7 Participants were recruited from 20 countries, although only 17 countries contributed data to our database (Austria, Belgium, Czech Republic, Denmark, Finland, France, Germany, Italy, Netherlands, Norway, Poland, Portugal, Russian Federation, Spain, Sweden, Switzerland, and the United Kingdom). Although REGISTRY recruited healthy controls, individuals at risk of HD, those with premanifest HD, and those with manifest HD, only the latter were included in our study. Participants were recruited from local HD clinics, and data collection followed a standard protocol, including the Unified Huntington's Disease Rating Scale (UHDRS),9 using electronic‐based report forms. After consent, participants were evaluated according to baseline and annual follow‐up visits until study termination or death. EHDN monitors checked data regularly for completeness and plausibility. For the purpose of this analysis, patients with manifest HD were defined as those with a CAG‐expanded allele of the HD gene and with a diagnostic confidence level (DCL) of 4.10 In addition, although included participants had been recruited at the beginning of the study, mortality data were truncated from 2001 through 2013.
Clinical Information
Age at the onset of motor, cognitive, and behavioral symptoms; sex; and ethnicity from all manifest REGISTRY participants were extracted from study forms. For individuals who died, data were also extracted from the REGISTRY death report form, including age, place, and cause of death. These forms were completed by trained raters, and data were often gathered from patients' families. Although it was optional to include autopsy information, most participants who died did not undergo this procedure.
Statistical Analysis
Statistical analysis was performed with SPSS software, version 18 (SPSS, Inc., Chicago, IL) and R software (R Foundation for Statistical Computing, Vienna, Austria). Descriptive statistics are presented as mean (±standard deviation [SD], minimum–maximum) for continuous variables and proportions for categorical variables. Group comparisons were performed using the t test or the χ2 test, respectively. Survival analyses were computed through Kaplan‐Meier estimates of median survival and 95% confidence intervals (CIs). Sex, the number of CAG repeats, age of onset, first manifestation, and country were planned to be tested as covariates. Unfortunately, this was not possible due to lack of statistical power. All tests were 2‐sided with a significance level of P = 0.05.
Results
Participants
In total, 5164 participants with manifest HD were analyzed (men, 49.0%, Caucasians, 98.0%; mean ± SD age at diagnosis, 47 ± 12 years [range, 5–86 years]). For detailed descriptions of the REGISTRY protocol and participants, please refer to publications by the EHDN7 and Orth et al.,11 respectively. At the end of the study period, from 2001 to 2013, there were 533 deaths (10.3% of patients). When considering only those who had died (men, 57.4%; Caucasians, 98.0%), the mean ± SD age at diagnosis was 49 ± 14 years (range, 9–85 years), and the mean ± SD age at death was 58 ± 13 years (range, 16–93 years). Differences between sex (P < 0.001) and the mean age at diagnosis (P = 0.03) were observed between those who had died and those who remained alive at the end of the study period. Most patients were from the United Kingdom (23.3%), followed by Germany (17%), Spain (16.3%), Italy (9.4%), and France (8.7%).
Survival
Median survival was defined as the time after which only 50% of the participants were still alive. The median survival from symptom onset to death was 35 years (95% CI, 29.2–40.8 years) (Fig. 1, left). Survival estimates for sex or other covariables could not be computed due to lack of statistical power. The median survival from motor diagnosis to death was 24 years (95% CI, 20.8–27.2 years) (Fig. 1, right). Again, survival estimates for sex or other covariables could not be computed.
Figure 1.
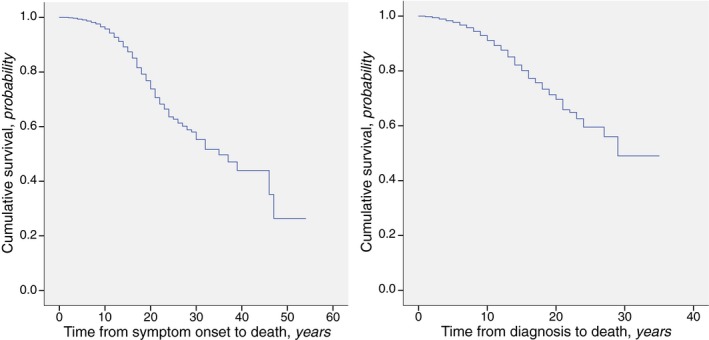
The survival of patients with manifest Huntington's disease included in the REGISTRY study, (Left) after symptom onset and (Right) after the motor diagnosis.
Causes of Death
The most frequent cause of death was pneumonia (19.5%), followed by other infections (6.9%), suicide (6.6%), cancer (3.4%), stroke (2.6%), and trauma (0.9%). Other causes were identified in 36.4% of participants, and the cause of death was undetermined in 23.6% of deaths (Table 1). No significant difference was observed in the distribution of causes of death between countries (P = 0.372) (Table S1).
Table 1.
Causes and places of death among patients with manifest Huntington's disease included in the REGISTRY study
| Variable | No. of patients (%) | Age at death: Mean ± SD, y | Sex, % women | Disease duration: Mean ± SD, y | Symptom duration: Mean ± SD, y |
|---|---|---|---|---|---|
| Causes of death | |||||
| Pneumonia | 104 (19.5) | 57.27 ± 1.46 | 34.6 | 10.33 ± 0.52 | 15.96 ± 0.71 |
| Other infection | 37 (6.9) | 54.32 ± 2.53 | 51.4 | 9.11 ± 0.88 | 14.24 ± 0.96 |
| Suicide | 35 (6.6) | 49.05 ± 1.76 | 28.6 | 5.12 ± 0.76 | 8.68 ± 0.93 |
| Cancer | 18 (3.4) | 65.06 ± 2.13 | 27.8 | 7.53 ± 1.14 | 14.18 ± 1.44 |
| Stroke | 14 (2.6) | 62.73 ± 3.55 | 35.7 | 7.50 ± 1.32 | 14.29 ± 1.72 |
| Trauma | 5 (0.9) | 54.00 ± 5.39 | 20.0 | 5.00 ± 1.84 | 10.20 ± 3.15 |
| Other | 194 (36.4) | 58.60 ± 0.93 | 43.8 | 9.02 ± 0.33 | 13.11 ± 0.44 |
| Unknown | 126 (23.6) | 57.83 ± 1.16 | 52.4 | 9.48 ± 0.39 | 13.42 ± 0.50 |
| Places of death | |||||
| Hospital | 152 (29.8) | 57.83 ± 1.19 | 32.2 | 8.48 ± 0.37 | 13.08 ± 0.52 |
| Home | 122 (23.9) | 56.32 ± 1.21 | 49.2 | 8.23 ± 0.48 | 12.72 ± 0.58 |
| Nursing home | 101 (19.8) | 59.70 ± 1.52 | 47.5 | 11.01 ± 0.63 | 15.95 ± 0.86 |
| Hospice care | 17 (3.3) | 50.88 ± 3.97 | 35.3 | 9.13 ± 1.18 | 13.06 ± 1.38 |
| Unknown | 118 (23.1) | 57.42 ± 1.84 | 44.9 | 8.08 ± 0.66 | 13.75 ± 0.91 |
SD, standard deviation.
Places of Death
The most frequent place of death was the hospital (29.8%), followed by home (23.9%), nursing houses (19.8%), and hospice care facilities (3.3%). The place of death was undetermined in 23.1% of deaths (Table 1). No significant difference was observed in the distribution of places of death between countries (P = 0.419) (Table S2).
Discussion
To the best of our knowledge, this is the first report of survival, mortality, causes and places of death in patients with manifest HD from an international, multicenter prospective study. In Europe, individuals with HD die a median of 35 years after symptoms onset, and 24 years after formal diagnosis, at a mean age of 58 years (see Fig. 1). In addition, death occurs most frequently from pneumonia and other infections, and the most frequent places of death are the hospital and home (see Table 1).
Apart from the biological characteristics of HD, such as the low kinetics of brain degeneration,12 other independent factors could contribute to our results and justify their variability. Ideally, individual characteristics, such as the number of CAG repeats, age of onset, sex, and severity of motor, cognitive, and behavioral phenotype, should have been included in our analysis. Unfortunately, we did not have enough statistical power to study these characteristics, but we expect this to be possible with Enroll‐HD,13 a prospective, observational, multinational platform that will include North American, Australian, Latin American, Asian, and European centers.
When considering REGISTRY, as far as age of onset and survival are concerned, our results indicate that HD has a distinctive natural history among neurodegeneration: in contrast to patients with nonmonogenetic Alzheimer's disease and Parkinson's disease, patients with HD tend to be diagnosed sooner in life–between the fourth and fifth decades of life–and to live longer after motor diagnosis14, 15, 16, 17 and after symptoms onset, although reports on other conditions indicate a large variation possibly explained by age of onset. A similar analogy may be applicable to other forms of dementia, such as vascular dementia and mixed dementia.14, 15 These results were unexpectedly higher than the median survival times previously reported.18, 19, 20 We believe this difference could be explained at least in part by a hypothetical better standard of care of patients who are followed at centers belonging to the EHDN. Some evidence suggests that collaboration with clinical and research networks may positively influence outcomes.21 For the same reason, a drawback from our results is that they may not be extrapolated to patients who are followed outside these networks.
It is noteworthy that the mean ages of diagnosis (49 years) and death (58 years) were calculated assuming a normal distribution and that the median survival times (35 and 24 years after symptom onset and the formal diagnosis, respectively) were calculated using a nonparametric distribution. As such, these results cannot be directly compared.
The patients in this study died most frequently from the same conditions described in previous studies of HD22, 23, 24 and other neurodegenerative diseases, such as Alzheimer's disease, vascular dementia,14, 25 and Parkinson's disease.26 Those studies demonstrated that pneumonia leads to death 4.7 times more often in patients who have HD compared with the general population,27 representing the primary cause of death.22, 23, 24, 28, 29 According to the same studies, other frequent causes of death were other infectious processes,22, 23 suicide,22, 29, 30 cardiovascular diseases,22, 23, 24, 28, 29 and nutritional unbalances, such as cachexia.22, 23, 29 Strikingly, these 4 conditions seemed to be as prevalent in patients with HD as they were in the general population.27
Two seminal studies from the 1980s based on death certificates in the United States reported on 2 major discrepancies compared with our results. Haines et al. described a higher frequency of cardiovascular diseases and of HD as a cause of death,24 and Lanska and colleagues supported the former results on cardiovascular diseases.22, 27 There are reasons to believe that the overall improvement in cardiovascular primary and secondary prevention strategies may justify these differences. We are also convinced that the diagnosis of HD as a cause of death is an issue of the past, which vanished with the introduction of more modern death certificate registries.
A great deal of research has been undertaken on suicide and associated behaviors in HD. Overall, in the general population, less than 1% of all deaths are caused by suicide31; however, in the REGISTRY data set, almost 7% of patients with HD committed suicide, a figure significantly higher than that in previous reports.22, 23 A previous analysis of this database reported that a high proportion (9.9%) of gene carriers present suicidal ideation32; whereas, in other data sets, the rate was even higher after isolating patients who had manifest HD, affecting one‐fifth of the population with early HD.33, 34 As in the general population35 and in patients with other neurodegenerative diseases like Parkinson's disease,36 the frequency of completed suicide in patients with manifest HD may be less than one‐half of the frequency of suicidal ideation, which is in line with these results.
Surprisingly, the place of death differed greatly from that of other patients with dementia (i.e., nursing homes)37, 38 and was comparable to the place of death of cancer patients (at hospital and at home).37, 38 We believe this may reflect the burden of the disease and the lack of competence and training in nursing homes to deal with the serious behavioral problems associated with HD. The possible willingness of HD families to care for their relatives also cannot be excluded. On the other hand, the high percentage of hospital deaths should ease tissue contributions to biobanks—a much‐needed offer for advancing research into better understanding the disease.
Several aspects make REGISTRY an ideal database for answering our current questions. Because HD is a rare disease, its phenotype must be studied in large, multicenter, and multinational studies to achieve robust results. The 9221 participants, the 165 study sites, and the 20 countries signed up (although not all contributed data to our results) to the REGISTRY study qualify this data set as methodologically satisfactory. The annual follow‐up and the HD‐specific scales applied to patients enrolled in REGISTRY may help mitigate 2 of the major problems identified when studying advanced‐stage HD: the irregular patient follow‐up and the difficulty in quantifying disease status.39 In addition, the REGISTRY protocol assures that generated data are of high quality by means of frequent monitoring carried out both locally at each site and centrally by trained monitors.11
Unfortunately, even after taking into account its advantages, our analysis included large numbers of patients without a specified cause or place of death. This potentially may lead to an under‐representation of specific causes or places of death. This is likely to have happened, because the follow‐up of patients with advanced‐stage HD rarely takes place in hospitals, where EHDN sites are usually located, but occur in smaller and more personalized, continuous, and palliative care units, making it difficult to ascertain socially complex details about this period of life in patients with HD. Also, it was outside our power and ethical permits to gather this information from families of patients who had died. Nevertheless, local EHDN sites should increase their compliance with this practice in the future. In addition, very few causes of death were confirmed by autopsy, which may further decrease the strength of our conclusions in this regard. Some of these issues would be solved at least in part by the introduction of a codification system for causes of death and with the use of electronic death certificates, a tool currently available in some countries. Importantly, generating uniform results is complex when using multinational studies.
In conclusion, our inferences give robustness to the results derived from previous smaller studies, establishing the overall survival of 24 years after motor diagnosis and 35 years after symptom onset, the most frequent cause of death (pneumonia), and the most important places of death (the hospital and the patient's home).
Finally, it would interesting to attempt to validate our findings with data from the COHORT study,40 which included American and Australian patients, and with the Enroll‐HD data set,13 in which, because of the expected increased statistical power, more analyses should be feasible.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
F.B.R.: 1A, 1B, 2C, 3A
D.A.: 2A, 2B, 3B
J.D.: 3B
N.G.: 2A, 2B, 3B
L.C.G.: 2C, 3B
M.C.: 2C, 3B
J.J.F.: 1A, 2C, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: Filipe Brogueria Rodrigues received a Fundação AstraZeneca grant for this project. REGISTRY data were provided by the European Huntington's Disease Network free of cost. The remaining authors report no sources of funding for this project and no conflicts of interest.
Financial disclosures from previous 12 months: Filipe Brogueria Rodrigues received a CHDI fellowship, grants from the Guarantors of Brain charity and the European Academy of Neurology, and Movement Disorder Society honorarium. Joaquim J. Ferreria received grants from GlaxoSmithKline, Grunenthal, Fundação MSD (Portugal), Teva, MSD, Allergan, and Novartis and consultancy fees from GlaxoSmithKline, Novartis, TEVA, Lundbeck, Solvay, Abbott, BIAL, Merck‐Serono, Merz, Ipsen, Biogen, and Sunovion Pharmaceuticals. The remaining authors report no sources of funding and no conflicts of interest.
Supporting information
Appendix S1: Investigators of the European Huntington's Disease Network
Table S1. Causes of death per country
Table S2. Places of death per country
Acknowledgments
We acknowledged Fundação AstraZeneca and the European Huntington's Disease Network for supporting our work. In addition, we thank all European Huntington's Disease Network REGISTRY Study Group investigators for collecting the data and all participating REGISTRY patients for their time and efforts.
Supporting information may be found in the online version of this article.
The first two authors contributed equally to this work.
Relevant disclosures and conflicts of interest are listed at the end of this article.
Members of the REGISTRY Investigators of the European Huntington's Disease Network are listed in Appendix S1
References
- 1. Gusella JF, Wexler NS, Conneally PM, et al. A polymorphic DNA marker genetically linked to Huntington's disease. Nature 1983;306:234–238. [DOI] [PubMed] [Google Scholar]
- 2. The Huntington's Disease Collaborative Research Group . A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell 1993;72:971–983. [DOI] [PubMed] [Google Scholar]
- 3. Mestre T, Ferreira J, Coelho MM, Rosa M, Sampaio C. Therapeutic interventions for disease progression in Huntington's disease. Cochrane Database Syst Rev 2009;3:CD006455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Travers E, Jones K, Nichol J. Palliative care provision in Huntington's disease. Int J Palliat Nurs 2007;13:125–130. [DOI] [PubMed] [Google Scholar]
- 5. Dawson S, Kristjanson LJ, Toye CM, Flett P. Living with Huntington's disease: need for supportive care. Nurs Health Sci 2004;6:123–130. [DOI] [PubMed] [Google Scholar]
- 6. Klager J, Duckett A, Sandler S, Moskowitz C. Huntington's disease: a caring approach to the end of life. Care Manag J 2008;9:75–81. [DOI] [PubMed] [Google Scholar]
- 7. European Huntington's Disease Network (EHDN) . REGISTRY—an observational study of the European Huntington's Disease Network (EHDN). Available at: https://ClinicalTrials.gov/show/NCT01590589. Accessed on the 20 March 2017.
- 8. Kristjanson LJ, Aoun SM, Oldham L. Palliative care and support for people with neurodegenerative conditions and their carers. Int J Palliat Nurs 2006;12:368–377. [DOI] [PubMed] [Google Scholar]
- 9. Huntington's Study Group . Unified Huntington's Disease Rating Scale: reliability and consistency Huntington Study Group. Mov Disord 1996;11:136–142. [DOI] [PubMed] [Google Scholar]
- 10. Reilmann R, Leavitt BR, Ross CA. Diagnostic criteria for Huntington's disease based on natural history. Mov Disord 2014;29:1335–1341. [DOI] [PubMed] [Google Scholar]
- 11. Orth M, Handley OJ, Schwenke C, et al. Observing Huntington's Disease: the European Huntington's Disease Network's REGISTRY. PLoS Curr 2010;2:RRN1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early‐stage Huntington's disease in the TRACK‐HD study: analysis of 36‐month observational data. Lancet Neurol 2013;12:637–649. [DOI] [PubMed] [Google Scholar]
- 13. CHDI Foundation Inc . Enroll‐HD: A Prospective Registry Study in a Global Huntington's Disease Cohort. Available at: https://ClinicalTrials.gov/show/NCT01574053. Accessed on the 20 March 2017.
- 14. Molsa PK, Marttila RJ, Rinne UK. Survival and cause of death in Alzheimer's disease and multi‐infarct dementia. Acta Neurol Scand 1986;74:103–107. [DOI] [PubMed] [Google Scholar]
- 15. Barclay LL, Zemcov A, Blass JP, Sansone J. Survival in Alzheimer's disease and vascular dementias. Neurology 1985;35:834–840. [DOI] [PubMed] [Google Scholar]
- 16. Willis AW, Schootman M, Evanoff BA, Perlmutter JS, Racette BA. Neurologist care in Parkinson disease: a utilization, outcomes, and survival study. Neurology 2011;77:851–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Forsaa EB, Larsen JP, Wentzel‐Larsen T, Alves G. What predicts mortality in Parkinson disease?. A prospective population‐based long‐term study Neurology 2010;75:1270–1276. [DOI] [PubMed] [Google Scholar]
- 18. Roos RA, Hermans J. Vegter‐van der Vlis M, van Ommen GJ, Bruyn GW. Duration of illness in Huntington's disease is not related to age at onset. J Neurol Neurosurg Psychiatry 1993;56:98–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rinaldi C, Salvatore E, Giordano I, et al. Predictors of survival in a Huntington's disease population from southern Italy. Can J Neurol Sci 2012;39:48–51. [DOI] [PubMed] [Google Scholar]
- 20. Foroud T, Gray J, Ivashina J, Conneally PM. Differences in duration of Huntington's disease based on age at onset. J Neurol Neurosurg Psychiatry 1999;66:52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tolson D, McIntosh J, Loftus L, Cormie P. Developing a managed clinical network in palliative care: a realistic evaluation. Int J Nurs Stud 2007;44:183–195. [DOI] [PubMed] [Google Scholar]
- 22. Lanska DJ, Lavine L, Lanska MJ, Schoenberg BS. Huntington's disease mortality in the United States. Neurology 1988;38:769–772. [DOI] [PubMed] [Google Scholar]
- 23. Heemskerk AW, Roos RA. Aspiration pneumonia and death in Huntington's disease. PLoS Curr 2012;4:RRN1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Haines JL, Conneally PM. Causes of death in Huntington disease as reported on death certificates. Genet Epidemiol 1986;3:417–423. [DOI] [PubMed] [Google Scholar]
- 25. Beard CM, Kokmen E, Sigler C, Smith GE, Petterson T, O'Brien PC. Cause of death in Alzheimer's disease. Ann Epidemiol 1996;6:195–200. [DOI] [PubMed] [Google Scholar]
- 26. Beyer MK, Herlofson K, Arsland D, Larsen JP. Causes of death in a community‐based study of Parkinson's disease. Acta Neurol Scand 2001;103:7–11. [DOI] [PubMed] [Google Scholar]
- 27. Lanska DJ, Lanska MJ, Lavine L, Schoenberg BS. Conditions associated with Huntington's disease at death. A case‐control study. Arch Neurol 1988;45:878–880. [DOI] [PubMed] [Google Scholar]
- 28. Farrer LA. Suicide and attempted suicide in Huntington disease: implications for preclinical testing of persons at risk. Am J Med Genet 1986;24:305–311. [DOI] [PubMed] [Google Scholar]
- 29. Sorensen SA, Fenger K, Olsen JH. Significantly lower incidence of cancer among patients with Huntington disease: an apoptotic effect of an expanded polyglutamine tract? Cancer 1999;86:1342–1346. [PubMed] [Google Scholar]
- 30. Di Maio L, Squitieri F, Napolitano G, Campanella G, Trofatter JA, Conneally PM. Suicide risk in Huntington's disease. J Med Genet 1993;30:293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Turecki G, Brent DA. Suicide and suicidal behaviour. Lancet 2016;387:1227–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hubers AA, van Duijn E, Roos RA, et al. Suicidal ideation in a European Huntington's disease population. J Affect Disord 2013;151:248–258. [DOI] [PubMed] [Google Scholar]
- 33. Paulsen JS, Hoth KF, Nehl C, Stierman L. Critical periods of suicide risk in Huntington's disease. Am J Psychiatry 2005;162:725–731. [DOI] [PubMed] [Google Scholar]
- 34. Wetzel HH, Gehl CR, Dellefave‐Castillo L, Schiffman JF, Shannon KM. Paulsen JS; Huntington Study Group. Suicidal ideation in Huntington disease: the role of comorbidity. Psychiatry Res 2011;188:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Borges G, Nock MK, Haro Abad JM, et al. Twelve‐month prevalence of and risk factors for suicide attempts in the World Health Organization World Mental Health Surveys. J Clin Psychiatry 2010;71:1617–1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kummer A, Cardoso F, Teixeira AL. Suicidal ideation in Parkinson's disease. CNS Spectr 2009;14:431–436. [DOI] [PubMed] [Google Scholar]
- 37. Mitchell SL, Teno JM, Miller SC, Mor V. A national study of the location of death for older persons with dementia. J Am Geriatr Soc 2005;53:299–305. [DOI] [PubMed] [Google Scholar]
- 38. Houttekier D, Cohen J, Bilsen J, Addington‐Hall J, Onwuteaka‐Philipsen BD, Deliens L. Place of death of older persons with dementia. A study in five European countries. J Am Geriatr Soc 2010;58:751–756. [DOI] [PubMed] [Google Scholar]
- 39. Harezlak J, Gao S, Hui SL. An illness‐death stochastic model in the analysis of longitudinal dementia data. Stat Med 2003;22:1465–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Huntington Study Group COHORT Investigators ;Dorsey E. Characterization of a large group of individuals with Huntington disease and their relatives enrolled in the COHORT study. PLoS ONE 2012;7:e29522. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Investigators of the European Huntington's Disease Network
Table S1. Causes of death per country
Table S2. Places of death per country