

Abstract
Background
Objective measures of Parkinson's disease (PD) are needed for purposes of diagnosis and prognostication, as well as identification of those at risk of PD. In this qualitative review, we provide an overview of the current state of the field of PD biomarker development, delineate challenges, and discuss how the field is evolving.
Methods
A search of PubMed was conducted for articles pertaining to objective biomarkers for PD. Articles were selected based on relevance and methodology; where available, meta‐analyses, systematic reviews, and comprehensive qualitative review articles were preferentially referenced.
Results
There are several potential sources of objective PD biomarkers including biofluids, peripheral tissue, imaging, genetics, and technology based objective motor testing. Approaches to biomarker identification include the candidate biomarker approach and unbiased discovery methods, each of which has advantages and disadvantages. Several emerging techniques hold promise in each of these areas. Advances in technology and bioinformatics, and the increasing availability of biobanks, are expected to facilitate future PD biomarker development.
Conclusions
The field of objective biomarkers for PD has made great progress but much remains to be done in translating putative biomarkers into tools useful in the clinic and for research. Multimodal biomarker platforms have the potential to capitalize on the utility and strengths of individual biomarkers. Rigorous methodology and standards for replication of findings will be key to meaningful progress in the field.
Keywords: biomarkers, Parkinson's disease
Parkinson's disease (PD) is one of the most common neurodegenerative disorders and its prevalence is increasing.1 It is currently diagnosed and monitored primarily based on clinical features. There is, however, widespread recognition that to better monitor the disease and advance our therapeutic approaches, PD biomarkers are necessary for early detection,. Broadly speaking, a biological marker (or biomarker) is “a characteristic that is objectively measured and evaluated as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention.”2 Conceptually, biomarkers can be further categorized into trait, state, and rate markers,3 though there may be overlap into which category a given biomarker falls. In the context of PD, trait markers indicate susceptibility to the disease, as in genotypes that confer a quantifiable risk of future PD. On the other hand, state markers include those that are diagnostic of disease, an indication of disease subtype, and/or disease severity. Rate markers are surrogates of disease progression. The need for a state biomarker in PD is obvious: to serve as a diagnostic marker for motor PD. The clinical diagnosis of PD is relatively accurate in the hands of movement disorders specialists, but diagnostic inaccuracy continues to be a problem4 particularly soon after diagnosis and in older age groups.5, 6 Diagnostic inaccuracy is obviously harmful to patients, and is also detrimental from a clinical trial standpoint. The main utility of a trait PD biomarker would be in identification of individuals at‐risk for PD. This is of particular importance given the increasing recognition that in order to meaningfully modify disease course, earlier detection of PD is needed,5 possibly even prior to the emergence of motor manifestations currently required as minimal diagnostic criteria. Rate biomarkers are needed on clinical grounds to help inform patients on prognosis. From a clinical trial standpoint, biomarkers are needed as surrogates of clinically meaningful endpoints, to help reduce the required duration of trials as well as the sample size required to detect meaningful differences. Biomarkers to objectively measure therapeutic response in PD could be useful both clinically and in clinical trials, where placebo response plays a large role in PD. Finally, there is increasing recognition that there are subtypes of PD7 and biomarkers of these different subtypes will similarly inform prognosis, clinical trial inclusion criteria, and other trial design considerations.
This qualitative review begins with a summary of the approach to PD biomarker development and how the field is evolving. A broad overview of the different sources of objective PD biomarkers is then provided, including biofluids, imaging, peripheral tissue, imaging, genetics, and technology‐based objective motor testing. Each main topic is divided into two sections, one that covers progress and challenges to date, and another that delineates considerations and exciting prospects for future PD biomarker development (Fig. 1). Where available, systematic reviews and meta‐analyses are referenced. Otherwise, recent qualitative review articles are cited, as are key original articles, though the reference list in relation to the latter is by no means comprehensive.
Figure 1.
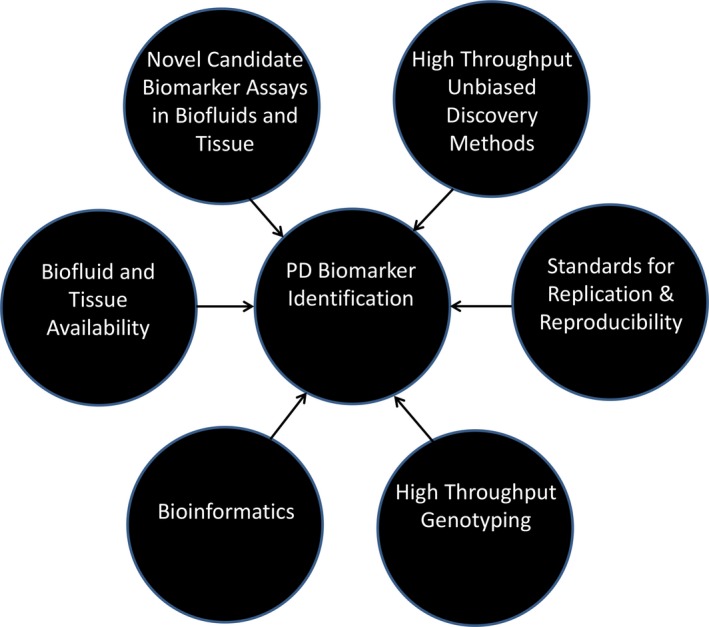
Schematic of key components for successful biomarker development.
As mentioned, this review will focus on objective biomarkers of PD. Symptom‐based markers used for diagnosis and prognosis in PD will not be considered. However, given that much work remains to be done for objective biomarker development, the importance of the latter cannot be over‐emphasized. For example, in one study, disorders of sleep and wakefulness and autonomic dysfunction, clinically diagnosed or ascertained via questionnaire, were superior to candidate CSF biomarkers for predicting progression in early PD.8
Approach to Biomarker Development
Where Are We with the General Approach to Biomarker Development in PD?
There are two main approaches to biomarker development that can be applied across most potential biomarker sources: candidate biomarker development and unbiased biomarker discovery. In candidate biomarker development, a specific target is identified based on biologic plausibility. Unbiased biomarker discovery involves measurement of several variables at once without a priori assumptions.9 Such screens can sometimes yield many potential signals and based on the strength of the detected signal and biologic plausibility, a few key candidates are selected for further testing. Several techniques for unbiased biomarker discovery have been utilized, each with varying degrees of yield, pitfalls, and prospects.9 Some of these techniques can also be applied in a targeted fashion to study specific pathways (metabolic or genetic), but to date, these have been minimally applied in PD and will not be considered here. Two commonly used unbiased methods are proteomics and metabolomics. Proteomics is broadly defined as the study of the structure and function of proteins. Several proteomic methods exist including various immunoassays, 2‐dimensional gel electrophoresis, and liquid chromatography based high‐resolution tandem mass spectrometry.9 Untargeted metabolomics (or metabolomic profiling) is defined as the study of chemical metabolic processes via the measurement of small‐molecule metabolites, including lipids, amino acids, sugars, biogenic amines, and organic acids. Technologies applied in metabolomics include nuclear magnetic resonance spectroscopy, gas chromatography mass spectrometry, and liquid chromatography mass spectroscopy.9 The outcome of metabolomic methods could be single molecules or patterns of co‐occurring molecules that reflect certain metabolic processes. While metabolomics techniques have been applied in various biofluids for biomarker identification in PD, no clearly promising biomarkers have emerged. This is likely due to the many challenges and limitations that come with metabolomics techniques.9 Studies applying metabolomics methods to various biofluids acquired from individuals with PD have largely been limited by small sample sizes and lack of accounting for key influences on the metabolome, such as sex and diet. Proteomics shares some of these challenges,9 but has nevertheless yielded promising biomarkers as discussed below. A major confounder with “‐omics” techniques is batch effect, in which measures vary by the batch tested, resulting at least in part from susceptibility to even minor changes in testing conditions.10 Finally, genotyping and gene expression profiling is another route for unbiased biomarker discovery. For example, untargeted transcriptomics (analysis of mRNA patterns) can help identify differential patterns of gene expression that could be specific to the PD population, as described further below.11
Where Do We Go Next with the General Approach to Biomarker Development in PD?
The large and ever‐increasing number of techniques that can be used to identify biomarkers in PD is exciting and holds great promise. However, the path of a biomarker, from preliminary identification to widespread use (whether in clinical trials or for clinical purposes), is long and arduous and few PD biomarkers have made it through. Among other things, this enforces the necessity for key steps in biomarker development: replication and reproducibility.
Replication and reproducibility within the context of biomarker development involves re‐demonstration of the performance of a putative biomarker in different cohorts and by different investigators. Assay standardization is essential to minimize the impact of technical differences in biomarker measurement on reported results.10 Application of appropriate statistical analyses, public sharing of datasets, and transparent reporting of methods will be key to replication and reproducibility. Higher standards for publication of first‐reported biomarkers are needed, especially in the case of biomarkers identified through an unbiased approach.9, 10
Other important considerations are ease of use, accessibility, and requirements for dissemination of a given biomarker. If a biomarker performs well yet is costly, requires unwieldy equipment, and/or necessitates extensive personnel training, these impracticalities may prevent widespread adoption.
Finally, it is likely that the future of PD biomarker development will see the integration of several biomarkers into multimodal platforms that include biomarkers identified from various sources. These models may combine biomarkers of varying degrees of sensitivity and specificity, capitalizing on test characteristics of the individual biomarkers. This is exemplified in the area of prodromal PD biomarker identification by the Parkinson Associated Risk Study (PARS). In PARS, the combination of hyposmia (a high sensitivity, low specificity marker) and reduced dopamine transporter binding on single photon emission computed tomography (SPECT, a modality discussed further below) accurately predicted future risk of PD.12 Accounting for additional features suggestive of increased PD risk, including constipation and cognitive dysfunction further improved predictive accuracy.13, 14 As more data become available, statistical manipulation of multimodal models will allow for weighting of the different biomarkers (according to their test characteristics) to yield “risk scores.” This approach has been applied in multimodal risk scores proposed for identification of prodromal PD.5 Their application to date has been informative.15, 16 However, it is also clear that additional biomarkers will need to be incorporated to optimize application of these models, particularly outside of the research setting.16, 17 Machine learning will likely assist in the building of these models.18 Great care will be needed in selecting the assumptions taken during the model building process. In order for the findings of models based on machine learning to be replicated and disseminated, clarity and transparency in reporting of these assumptions and details of the statistical approach will be essential, as exemplified in the publication by Dinov et al.19
An important consideration in PD biomarker development pertains to what the “gold standard” for the clinical correlate of a given biomarker is. For example, in searching for a biomarker of dementia risk in PD, PD dementia needs to be clearly defined. Concerns have been raised about anchoring of unbiased biomarker development on clinical criteria.20 It has even been suggested that biomarkers be used to define clinical phenotype, particularly in relation to identifying targets for “personalized” disease‐modifying therapies.20 Whether this biomarker‐to‐clinical‐phenotype model will be of utility and what the place is for the clinical phenotype‐to‐biomarker approach remains to be seen, and will likely be an area of intense investigation and debate in years to come.
Biofluid Biomarker Development
Where Are We with Biofluid Biomarker Development in PD?
Candidate Biomarkers
The potential source of PD biomarkers is large and ever expanding. Several biofluids have the potential to serve as sources of PD biomarkers, including, but not limited to, cerebrospinal fluid (CSF), blood (including whole blood and all blood compartments), saliva, and urine. Of course, identifying biomarkers in biofluids that are easily accessible with low risk procedures (such as saliva, blood, or urine) would be most desirable. However, the peripheral nature of the latter fluids contributes to significant “noise” in measures of various biomarkers. CSF is thus seen as a key source of PD biomarkers as it is thought to best reflect what is going on in the central nervous system (CNS).
An example of a candidate biomarker receiving great attention in PD is α‐synuclein. This stems from its key role in PD pathophysiology and the potential for α‐synuclein to be a therapeutic target. Various antibody‐based assays that measure different α‐synuclein species (such as total, phosphorylated, oligomeric, and fibrillar α‐synuclein) have been developed and applied to CSF and plasma. Contradictory results have been found in plasma.21 A complicating factor in measuring α‐synuclein in components of blood is the abundance of α‐synuclein within red blood cells. In the CSF, contradictory results have also been reported. The mixed reports likely result, at least in part, from confounding factors, including contamination of the sample with blood and differences in sample acquisition and laboratory procedures used to measure it. Cumulatively, the literature shows lower total CSF α‐synuclein in individuals with PD compared to controls, but with significant overlap, resulting in low specificity.22 While there is also some suggestion that total CSF α‐synuclein may have some utility in sub‐typing PD and as a marker of disease progression,23 data are limited and too contradictory to draw any firm conclusions in this regard. More specific assays to measure both total α‐synuclein as well as different species are therefore needed. How levels of CSF α‐synuclein vary over the course of the disease also needs to be clarified.24
Biofluid biomarkers relevant to other pathologies such as Alzheimer's disease have also been studied in PD. In non‐demented individuals with PD, low baseline CSF amyloid‐β1‐42 predicts development of cognitive impairment.25 While some studies suggest CSF tau and phospho‐tau are higher in PD compared to non‐PD comparators,23 these are not specific enough to be diagnostic markers, and they have not been consistently associated with cognitive function or decline in PD.25 Neurofilament light chain is another example of a candidate biomarker being pursued in PD. It is a marker of axonal injury. It may have some utility in distinguishing PD from other neurodegenerative, atypical parkinsonian syndromes when measured in CSF26, 27 and blood.28 However, it is also not specific and is elevated in several other non‐parkinsonian neurologic disorders.
There are a few other candidate PD biomarkers being pursued in other biofluids. Uric acid is a candidate biomarker based on its anti‐oxidant properties combined with the low levels of uric acid found in the substantia nigra of PD patients.29 Several longitudinal cohort studies have demonstrated a lower risk of PD in individuals with higher uric acid levels at baseline.30, 31, 32 A meta‐analysis demonstrated that the pooled rate ratio of PD associated with 1 standard deviation increase in urate (1.32 mg/dL) is 0.80.31 There are some data to suggest the utility of uric acid as a PD state and rate marker33 as well. Given how non‐specific uric acid levels are, it is unlikely to serve as a biomarker in isolation but rather may be combined in multimodal models.
There is evidence that neuroinflammation contributes to PD pathogenesis34 and markers of inflammation have been studied as potential PD biomarkers.35 Increases in several inflammatory markers including β2‐microglobulin, interleukin‐1β and interleukin‐8 have been reported in PD compared to non‐PD controls. However, studies investigating markers of inflammation as PD biomarkers have had small sample sizes35 and none of the possible biomarkers identified have yet to be validated or replicated.
Biomarkers Identified Through An Unbiased Approach
A few notable blood‐based biomarkers that hold promise in PD have been identified through proteomic analysis. Epidermal Growth Factor (EGF) was first identified as a possible blood‐based biomarker of cognitive impairment in PD via an unbiased approach, utilizing a large‐scale multiplex immunoassay platform36 and replicated by another group.37 However, as is the case with many potential PD biomarkers, the variability of levels of EGF over the course of the disease challenges its utility.38 Similarly, ApoA1 has been identified as a potential biomarker of PD risk and disease severity, and findings have been replicated in independent cohorts.39, 40 ApoA1 is a component of HDL and its biological plausibility as a PD biomarker lies in the role of cholesterol dysmetabolism in PD pathogenesis and prognosis.
Where Do We Go Next in Regards to Biofluid Biomarker Development in PD?
Candidate Biomarkers
Despite the challenges faced, there are many reasons for optimism that an accurate and useful measure of α‐synuclein in biofluids will emerge as a PD biomarker. There are ongoing efforts to develop and standardize immunoassays that measure strains of α‐synuclein specific to PD in various biofluids and yield consistent results across labs and patient populations. Rigorous procedures applied during assay development and validation, including replication at multiple sites (as exemplified by Kruse et al.41) as well as in sample acquisition and processing42 should allow for greater uniformity in future results. Another exciting prospect in development of an α‐synuclein biofluid biomarker is the application of protein misfolded cyclic amplification (PMCA) to detect abnormal α‐synuclein in CSF and other biofluids.43, 44 PMCA assays (or “seeding” assays) call upon the prion‐like conformational properties of pathological α‐synculein to recruit normal α‐synuclein to misfold. One such assay44 was built upon the real‐time quaking‐induced conversion (RT‐QuIC) techniques used to develop a diagnostic CSF test for sporadic Creutzfeldt–Jakob disease.
Biomarkers Identified Through An Unbiased Approach
Advances in computational abilities and bioinformatics resources will likely aid in future unbiased biomarker identification. For example, in recent years several advances in the field of metabolomics have been seen, including the integration of key analytical chemistry techniques with comprehensive database resources and advanced computational methods.45 The future may also see “‐omics” applied more broadly to characterize the “exposome,”46 defined functionally as the “biologically‐active chemicals in a person's blood—from both exogenous and endogenous processes.”47 This approach, while seeming of most utility in examining gene‐environment interactions, also has the potential to aid in discovery of new PD biomarkers.46
Genetics
Where Are We with Genetic Biomarker Development in PD?
Understanding genetic contributions to PD is key to determining PD pathophysiology, but genetics also provide a source of PD biomarkers. One example is the PD genetic risk score, calculated by summing and weighting risk allele counts for 28 loci that have been identified through genome‐wide association studies as being associated with PD risk. This genetic risk score, combined with olfactory function, family history of PD, age, and sex was able to distinguish PD from control with high specificity.48
Untargeted transcriptomics (analysis of RNA transcription patterns) have also shown some promise in identifying PD biomarkers. For example, a study identified a unique expression pattern of 8 genes in PD and this was replicated in a separate cohort.11 Others have also reported on the possible utility of transcriptomics for PD biomarker development.9 In addition, preliminary work suggests that expression patterns of micro‐RNA panels may differ in PD compared to non‐PD controls.49 However, studies to date have had relatively small sample sizes and the findings require replication.
Where Do We Go Next in Regards to Genetic Biomarker Development in PD?
Advances in powerful high throughput sequencing technologies such as next‐generation sequencing (NGS) enable the assessment of large numbers of genetic loci at once. This will allow for the identification of genomic signatures specific to the PD population. As mentioned, progress has already been made in identifying genomic features within coded DNA sequences that may serve as PD biomarkers. Another area that is actively being explored and holds promise for the future is epigenetics. The field of epigenetics focuses not within the DNA or RNA sequence itself but within the molecules and structures associated with DNA and RNA, such as the methylation pattern of DNA or post‐translational modification of histones. This is exemplified in preliminary work showing that there are DNA methylation patterns that may be unique to PD.50
It is also expected that untargeted transcriptomics will be increasingly applied to identify differential gene expression patterns in PD.9, 11, 49 Availability of various genetic samples on well‐characterized PD patients is expected to facilitate this, as exemplified by the unprecedented resources made available by studies such as the Parkinson Progression Markers Initiative (PPMI)51 and Biofind.52
Peripheral Tissue Biomarker Development
Where Are We with Peripheral Tissue Biomarker Development in PD?
There is a growing body of evidence that α‐synuclein pathology occurs outside of the CNS in PD and it may even precede CNS involvement. Thus, there is great interest in peripheral tissue biomarkers in PD and prodromal PD. Several tissues have been examined as a possible source of PD biomarkers including skin, salivary glands, and other regions of the GI tract. Abnormal α‐synuclein in the submandibular gland of individuals with PD but not healthy controls has been the most consistently reported potential tissue PD biomarker (in regards to sensitivity and specificity).21, 53 Importantly, aggregated α‐synuclein has also been reported in individuals with the prodromal disorder REM sleep behavior disorder (RBD).54, 55 Earlier studies examining skin α‐synuclein as a PD biomarker were contradictory,21, 56 but more recent studies suggest promise for this as a PD biomarker as well.57 Small studies have also shown α‐synuclein in skin biopsies from various body regions in RBD but not in healthy controls.58, 59 These findings require further replication in larger studies.
Aggregated α‐synuclein has been reported in biopsies taken from various regions of the GI tract (besides the salivary glands), including stomach, appendix, ascending and descending colon, sigmoid colon, and rectum. However, the specificity of extra‐salivary GI α‐synuclein for PD has been questioned, as α‐synuclein has been reported in biopsies taken from these regions in Multiple System Atrophy and healthy controls as well.60, 61, 62 It is likely that non‐specific staining of non‐neuronal α‐synuclein in the enteric nervous system confounds these results, largely reflecting suboptimal staining techniques.62, 63 Other potential reasons for the difficulty in replicating reports of abnormal α‐synuclein in peripheral tissue in PD include differences in preanalytical tissue acquisition (including both technique and biopsy site) and processing, differences in the immunoassays used, and the subjective and operator‐dependent nature of neuropathological characterization.64 Thus, assays and interpreters that distinguish between neuronal aggregated α‐synuclein that represents a pathologic state from non‐specific staining are needed. Safety is also an important consideration in peripheral tissue PD biomarker development. For example, adverse events are not uncommon in studies reporting on GI tract tissue as a PD biomarker, though fortunately the adverse events have been largely mild.64
Where Do We Go Next in Regards to Peripheral Tissue Biomarker Development in PD?
There is good reason to believe that the future will bring a robust measure of tissue α‐synuclein that will be a useful biomarker for PD and prodromal PD. The ongoing Systemic Synuclein Sampling Study (S4) is a multisite study in which three tissues (submandibular gland, skin, and colon) and three fluids (CSF, saliva, and blood) are being acquired in the same subject in a standardized manner.65 Total α‐synuclein will be measured in the biolfuids and neuropathologists, blinded to diagnosis and uniformly trained, will make semi‐quantitative determinations of α‐synuclein immunohistochemical staining in the tissues. S4 is unique in its design and implementation, including standardized tissue acquisition and processing across multiple sites, rigorous methodology used to develop the biofluid and tissue assays used, and expertise and training of the neuropathologists.65 S4 will yield not only valuable data to inform future development of tissue α‐synuclein biomarkers in PD, but serves to help establish a biobank of various tissue which will be available to the PD research community, thus facilitating the challenging logistical aspect of tissue biomarker development. In addition, several assays are under development that may have advantages over conventional immunohistochemical staining techniques in identifying pathologic forms of α‐synuclein specific to the PD state.66 For example, PMCA assays such as RT‐QuIC (discussed in the biofluilds section) may be applied using tissue specimens as the “seeding” source.
Imaging
Where Are We with Imaging Biomarker Development in PD?
Many imaging modalities have been utilized in the search for PD biomarkers.67, 68 Conventional MRI studies have shown reduced volumes in various cortical and subcortical regions in PD compared to healthy and disease controls. MR‐based high‐resolution morphometric and volumetric analysis of substantia nigra has yielded several promising findings that may serve as PD biomarkers.67, 68 Techniques applied include diffusion tensor imaging (DTI) which measures tissue microstructure and structural connectivity, proton magnetic resonance spectroscopy, and quantitative susceptibility mapping which measures iron deposition. Signal changes thought to result from neuromelanin deposition have also been identified. However, there has been little reproducibility across studies67 and some contradictory results reported. Thus, the preliminarily promise seen in MR‐based PD biomarkers awaits validation and replication.67, 68
Radioligand‐based imaging (utilizing both SPECT and positron emission tomography (PET)) has also been widely applied in the search for PD biomarkers.67 Neurotransmitter systems have been investigated using several radioligands reflective of the dopaminergic, serotonergic, and cholinergic systems. Reduced binding of presynaptic dopamine transporter (DAT) ligand is well established as a surrogate marker for nigrostriatal degeneration,67 and dissemination of radioligand measuring DAT binding into the clinical space has been successful. However, DAT binding deficit is not specific to PD69 and utility as a rate marker is not clear. Emerging data suggest DAT SPECT is also a trait marker for risk of PD in individuals with other prodromal features,12, 70 but in order to maximize its specificity and predictive value it is unlikely that it can be used in isolation. Other targets of radioligand imaging have included markers of neuroinflammation,67 but sample sizes have been small and findings are yet to be replicated. Radioligand biomarkers of underlying protein deposition are actively being pursued, with the most progress made for cerebral β‐amyloid. Finally, cardiac imaging as a measure of autonomic denervation may be useful but requires further validation and dissemination as well.67
Functional imaging with MRI, PET, and SPECT has also been applied to identify specific patterns of cortical and/or subcortical blood flow or metabolism. Fluorodeoxyglucose PET imaging in the resting state has revealed a PD‐related metabolic pattern (PDRP) using a technique called special covariance mapping.71 This pattern was also found in individuals with RBD, a prodromal PD state.72 While these and other imaging findings have demonstrated high sensitivity and specificity for PD,67, 68 most functional imaging studies are small in sample size and conclusions are hard to draw. While in many cases the results have been reproducible in collaboration with the group that reported on them,71, 73 their replication by other groups has been limited, also raising concerns for the disseminability of such imaging biomarkers.
Finally, ultrasound based imaging, and specifically transcranial ultrasonography (TCS), has shown an area of substantia nigra hyperechogenicity that has strong potential as a diagnostic marker for PD as well as a biomarker of risk in prodromal individuals.74 However, challenges of TCS include difficulty obtaining ultrasound windows into the skull (especially in older adults and in certain ethnicities), significant operator‐dependence of result interpretation, and specific equipment needs (operator training and technical requirements for the more widely used application of transcranial sonography, namely transcranial Doppler sonography of cerebral blood vessels, are different from those used to measure substantia nigra hyperechogenicity).
Where Do We Go Next with Imaging Biomarker Development in PD?
Advances in MR technology, such as ultra‐high field imaging with 7 tesla MRI, is allowing for increased spatial resolution and sensitivity to susceptibility. This will allow for significantly improved visualization of structures relevant to PD such as the substantia nigra, but also smaller nuclei that cannot be well visualized with lower strength MRIs.67 Many research‐based MR techniques have not yet made it to the conventional scanners used in the clinic, so the logistics and feasibility of dissemination will be important to consider.
Another potential imaging biomarker source is “connectomics” which studies patterns of co‐activation as a measure of connectivity between different brain regions. Altered connectivity is thought to underlie the pathophysiology of many neuropsychiatric disorders, and certain brain activation patterns could serve as state markers as well.75 Changes in connectivity patterns, following therapeutic intervention, suggest a potential use as a surrogate outcome in clinical trials. However, replication of findings has been limited and will be key to validation and dissemination of connectivity‐based imaging biomarkers. The improved resolution available with ultra‐high field imaging will also be useful in functional imaging, including resting state MRI, which has so far been somewhat limited in resolution of subcortical structures (basal ganglia and brainstem). It is likely that PD imaging biomarkers of the future will be multimodal: fusion of images acquired via different modalities has been proposed as a means of improving discrimination between studied groups compared to single markers alone.68
It will be incumbent upon the imaging biomarker field to set high standards for reproducibility. Calls for standardization have been made for functional, structural, and molecular imaging. Many aspects of imaging studies are amenable to standardization. For example, there could be minimum sample size requirements, guidelines for correction for multiple comparisons, suggested corrections for effects of age, motion, partial volume effects, and artifact removal, and standardization of analysis pipelines for different imaging modalities.76 Detailed reporting of methods (including imaging acquisition, processing, and statistical analysis) and data sharing will also contribute to the success of this standardization and to the ultimate goal of replication and reproducibility.
Finally, there is great interest in imaging α‐synuclein in vivo, and intensive efforts are underway to develop appropriate synuclein imaging ligands.77 Partnerships between industry and academia will likely be essential to the development and ultimate dissemination of such agents.
Objective Motor Testing
Where Are We with Objective Motor Biomarker Development in PD?
Technology‐based objective measures (TOMs) using various technologies are an attractive source of PD biomarkers for various reasons.78, 79 First, TOMs may allow for detection of subtle motor abnormalities prior to the threshold required for clinical detection, thus serving as both a state marker (diagnostic of early PD) as well a trait marker (to help identify individuals at risk for PD). Second, objective measures of motor function may be useful as rate markers, since in longitudinal studies baseline motor abnormalities are strongly predictive of rate of change and future outcome.80 There is a wide and ever increasing array of technologies available for objective motor testing in PD.78 Some allow for cross‐sectional assessments via administration of specific tasks, whereas others are comprised of sensors (including “wearables”) that allow continuous monitoring. With the exciting advances seen in sensor‐based data come several challenges. The sheer amount of data collected by wearable sensors poses a bioinformatics challenge. Much work is needed to translate digital signals into measures of clinically meaningful manifestations. The proprietary nature of some of the algorithms applied in data collection makes interpretation and, more importantly, replication challenging.
Where Do We Go Next with Objective Motor Biomarker Development in PD?
The field of TOMs is in its infancy and the future is likely to bring several advances that will hopefully translate into a useful PD biomarker. Involvement of formal task forces will help facilitate definition and standardization of measures and maximize complementary efforts by different groups while minimizing duplication.78 Development of computational methods that can integrate massive amounts of data into practical, easily interpretable, but not over‐simplified measures will be key. Synthesis of the collected data into composite scores may be of utility.79 Efforts to relate objective motor measures to biomarkers from other sources, as they emerge, will be essential.
Conclusion
Since the description of PD over two centuries ago, much progress has been made in characterizing it clinically. With this progress has come recognition that objective markers are needed to diagnose PD, measure its severity and progression, define subtypes, and predict who is at risk of PD. The need for biomarkers is clinical and research‐based, both for PD clinical trials and for exploring the application and implications of precision medicine in PD. The source of PD biomarkers is large and ever increasing, including, but not limited to, biofluifds, peripheral tissue, imaging, genetics, and technology‐based objective measures of motor and other functions. Multimodal biomarker platforms, incorporating combinations of biomarkers from these different sources, can be used to capitalize on the strengths of individual biomarkers. While there are many promising and exciting prospects for PD biomarker development, there is also the need to be thoughtful and methodical in our quest to find those markers that are consistent and reproducible. Adoption of modality‐specific standards will be key to this.
Author Roles:
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
L.M.C.: 1A, 1B, 1C, 3A
M.B.S: 1A, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the Journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: No specific funding was received for this work. The authors declare that there are no conflicts of interest relevant to this work.
Financial Disclosures for the previous 12 months: Lana M. Chahine receives support from the NIH (P50 NS053488), receives support from The Michael J Fox Foundation, and receives royalties from Wolters Kluwel (for book authorship). Matthew B Stern is a consultant for Impax, Neuroderm, Adamas, Acorda, and Teva and holds stock options in Adamas.
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Dorsey ER, Constantinescu R, Thompson JP, et al. Projected number of people with Parkinson's disease in the most populous nations, 2005 through 2030. Neurology 2007;68(5):384–386. [DOI] [PubMed] [Google Scholar]
- 2. Biomarkers Definitions Working Group . Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther 2001;69(3):89–95. [DOI] [PubMed] [Google Scholar]
- 3. Fox N, Growdon JH. Biomarkers and surrogates. NeuroRx 2004;1(2):181. [Google Scholar]
- 4. Rizzo G, Copetti M, Arcuti S, Martino D, Fontana A, Logroscino G. Accuracy of clinical diagnosis of Parkinson's disease: a systematic review and meta‐analysis. Neurology 2016;86(6):566–576. [DOI] [PubMed] [Google Scholar]
- 5. Berg D, Postuma RB, Adler CH, et al. MDS research criteria for prodromal Parkinson's disease. Mov Disord 2015;30(12):1600–1611. [DOI] [PubMed] [Google Scholar]
- 6. Postuma RB, Berg D, Stern M, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord 2015;30(12):1591–1601. [DOI] [PubMed] [Google Scholar]
- 7. Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson's disease: Biomarkers and longitudinal progression. Brain 2017;140(7):1959–1976. [DOI] [PubMed] [Google Scholar]
- 8. Mollenhauer B, Zimmermann J, Sixel‐Doring F, et al. Monitoring of 30 marker candidates in early Parkinson's disease as progression markers. Neurology 2016;87(2):168–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chen‐Plotkin AS. Unbiased approaches to biomarker discovery in neurodegenerative diseases. Neuron 2014;84(3):594–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McShane LM. In pursuit of greater reproducibility and credibility of early clinical biomarker research. Clin Transl Sci 2017;10(2):58–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Scherzer CR, Eklund AC, Morse LJ, et al. Molecular markers of early Parkinson's disease based on gene expression in blood. Proc Natl Acad Sci USA 2007;104(3):955–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jennings D, Siderowf A, Stern M, Seibyl J, Eberly S, Oakes D, et al. Conversion to Parkinson's disease in the PARS hyposmic and dopamine transporter‐deficit prodromal cohort. JAMA Neurol 2017;74(8):933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jennings D, Siderowf A, Stern M, et al. Imaging prodromal Parkinson's disease: the Parkinson's associated risk syndrome study. Neurology 2014;83:1739–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chahine LM, Weintraub D, Hawkins KA, et al. Cognition in individuals at risk for Parkinson's: Parkinson associated risk syndrome (PARS) study findings. Mov Disord 2016;31(1):86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fereshtehnejad SM, Montplaisir JY, Pelletier A, Gagnon JF, Berg D, Postuma RB. Validation of the MDS research criteria for prodromal Parkinson's disease: longitudinal assessment in a REM sleep behavior disorder (RBD) cohort. Mov Disord 2017;32(6):865–873. [DOI] [PubMed] [Google Scholar]
- 16. Pilotto A, Heinzel S, Suenkel U, et al. Application of the movement disorder society prodromal Parkinson's disease research criteria in 2 independent prospective cohorts. Mov Disord 2017;32(7):1025–1034. [DOI] [PubMed] [Google Scholar]
- 17. Teipel SJ, Sabri O, Grothe M, et al. Perspectives for multimodal neurochemical and imaging biomarkers in alzheimer's disease. J Alzheimers Dis 2013;33(Suppl 1):S329–S347. [DOI] [PubMed] [Google Scholar]
- 18. Kubota KJ, Chen JA, Little MA. Machine learning for large‐scale wearable sensor data in Parkinson's disease: Concepts, promises, pitfalls, and futures. Mov Disord 2016;31(9):1314–1326. [DOI] [PubMed] [Google Scholar]
- 19. Dinov ID, Heavner B, Tang M, et al. Predictive big data analytics: a study of Parkinson's disease using large, complex, heterogeneous, incongruent, multi‐source and incomplete observations. PLoS ONE 2016;11(8):e0157077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Espay AJ, Schwarzschild MA, Tanner CM, et al. Biomarker‐driven phenotyping in Parkinson's disease: a translational missing link in disease‐modifying clinical trials. Mov Disord 2017;32(3):319–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Malek N, Swallow D, Grosset KA, Anichtchik O, Spillantini M, Grosset DG. Alpha‐synuclein in peripheral tissues and body fluids as a biomarker for Parkinson's disease ‐ a systematic review. Acta Neurol Scand 2014;130(2):59–72. [DOI] [PubMed] [Google Scholar]
- 22. Gao L, Tang H, Nie K, et al. Cerebrospinal fluid alpha‐synuclein as a biomarker for Parkinson's disease diagnosis: a systematic review and meta‐analysis. Int J Neurosci 2015;125(9):645–654. [DOI] [PubMed] [Google Scholar]
- 23. Kang JH, Mollenhauer B, Coffey CS, et al. CSF biomarkers associated with disease heterogeneity in early Parkinson's disease: the Parkinson's progression markers initiative study. Acta Neuropathol 2016;131(6):935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Majbour NK, Vaikath NN, Eusebi P, et al. Longitudinal changes in CSF alpha‐synuclein species reflect Parkinson's disease progression. Mov Disord 2016;31(10):1535–1542. [DOI] [PubMed] [Google Scholar]
- 25. Leaver K, Poston KL. Do CSF biomarkers predict progression to cognitive impairment in Parkinson's disease patients? A systematic review Neuropsychol Rev 2015;25(4):411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sako W, Murakami N, Izumi Y, Kaji R. Neurofilament light chain level in cerebrospinal fluid can differentiate Parkinson's disease from atypical parkinsonism: evidence from a meta‐analysis. J Neurol Sci 2015;352(1–2):84–87. [DOI] [PubMed] [Google Scholar]
- 27. Gong Y, Xiong KP, Mao CJ, et al. Clinical manifestations of Parkinson's disease and the onset of rapid eye movement sleep behavior disorder. Sleep Med 2014;15(6):647–653. [DOI] [PubMed] [Google Scholar]
- 28. Hansson O, Janelidze S, Hall S, et al. Blood‐based NfL: a biomarker for differential diagnosis of parkinsonian disorder. Neurology 2017;88(10):930–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Church WH, Ward VL. Uric acid is reduced in the substantia nigra in Parkinson's disease: Effect on dopamine oxidation. Brain Res Bull 1994;33(4):419–425. [DOI] [PubMed] [Google Scholar]
- 30. Davis JW, Grandinetti A, Waslien CI, Ross GW, White LR, Morens DM. Observations on serum uric acid levels and the risk of idiopathic Parkinson's disease. Am J Epidemiol 1996;144(5):480–484. [DOI] [PubMed] [Google Scholar]
- 31. Weisskopf MG, O'Reilly E, Chen H, Schwarzschild MA, Ascherio A. Plasma urate and risk of Parkinson's disease. Am J Epidemiol 2007;166(5):561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. de Lau LM, Koudstaal PJ, Hofman A, Breteler MM. Serum uric acid levels and the risk of Parkinson's disease. Ann Neurol 2005;58(5):797–800. [DOI] [PubMed] [Google Scholar]
- 33. Schwarzschild MA, Schwid SR, Marek K, et al. Serum urate as a predictor of clinical and radiographic progression in Parkinson's disease. Arch Neurol 2008;65(6):716–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McGeer PL, McGeer EG. Inflammation and neurodegeneration in Parkinson's disease. Parkinsonism Relat Disord 2004;10(Suppl 1):S3–S7. [DOI] [PubMed] [Google Scholar]
- 35. Andersen AD, Binzer M, Stenager E, Gramsbergen JB. Cerebrospinal fluid biomarkers for Parkinson's disease ‐ a systematic review. Acta Neurol Scand 2017;135(1):34–56. [DOI] [PubMed] [Google Scholar]
- 36. Chen‐Plotkin AS, Hu WT, Siderowf A, et al. Plasma epidermal growth factor levels predict cognitive decline in Parkinson's disease. Ann Neurol 2011;69(4):655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pellecchia MT, Santangelo G, Picillo M, et al. Serum epidermal growth factor predicts cognitive functions in early, drug‐naive Parkinson's disease patients. J Neurol 2013;260(2):438–444. [DOI] [PubMed] [Google Scholar]
- 38. Jiang QW, Wang C, Zhou Y, et al. Plasma epidermal growth factor decreased in the early stage of Parkinson's disease. Aging Dis 2015;6(3):168–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Qiang JK, Wong YC, Siderowf A, et al. Plasma apolipoprotein A1 as a biomarker for Parkinson's disease. Ann Neurol 2013;74(1):119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Swanson CR, Berlyand Y, Xie SX, Alcalay RN, Chahine LM, Chen‐Plotkin AS. Plasma apolipoprotein A1 associates with age at onset and motor severity in early Parkinson's disease patients. Mov Disord 2015;30(12):1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kruse N, Persson S, Alcolea D, et al. Validation of a quantitative cerebrospinal fluid alpha‐synuclein assay in a european‐wide interlaboratory study. Neurobiol Aging 2015;36(9):2587–2596. [DOI] [PubMed] [Google Scholar]
- 42. Andreasson U, Perret‐Liaudet A, Van Waalwijk van Doorn LJ, et al. A practical guide to immunoassay method validation. Front Neurol 2015;6:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson's disease by detection of alpha‐synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 2017;74(2):163–172. [DOI] [PubMed] [Google Scholar]
- 44. Fairfoul G, McGuire LI, Pal S, et al. Alpha‐synuclein RT‐QuIC in the CSF of patients with alpha‐synucleinopathies. Ann Clin Transl Neurol 2016;3(10):812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. GenomeNet . transparent. http://www.genome.jp/. Accessed March 1, 2017.
- 46. Miller DB, O'Callaghan JP. Biomarkers of Parkinson's disease: present and future. Metabolism 2015;64(3 Suppl 1):S40–S46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rappaport SM. Biomarkers intersect with the exposome. Biomarkers 2012;17(6):483–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nalls MA, McLean CY, Rick J, et al. Diagnosis of Parkinson's disease on the basis of clinical and genetic classification: a population‐based modelling study. Lancet Neurol 2015;14(10):1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Batistela MS, Josviak ND, Sulzbach CD, de Souza RL. An overview of circulating cell‐free microRNAs as putative biomarkers in alzheimer's and Parkinson's diseases. Int J Neurosci 2017;127(6):547–558. [DOI] [PubMed] [Google Scholar]
- 50. Jakubowski JL, Labrie V. Epigenetic biomarkers for Parkinson's disease: from diagnostics to therapeutics. J Parkinsons Dis 2017;7(1):1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Initiative Parkinson Progression Marker. The Parkinson's progression marker initiative (PPMI). Prog Neurobiol 2011;95(4):629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kang UJ, Goldman JG, Alcalay RN, et al. The BioFIND study: characteristics of a clinically typical Parkinson's disease biomarker cohort. Mov Disord 2016;31(6):924–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Adler CH, Dugger BN, Hentz JG, et al. Peripheral synucleinopathy in early Parkinson's disease: submandibular gland needle biopsy findings. Mov Disord 2016;31(2):250–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mahlknecht P, Iranzo A, Hogl B, et al. Olfactory dysfunction predicts early transition to a lewy body disease in idiopathic RBD. Neurology 2015;84(7):654–658. [DOI] [PubMed] [Google Scholar]
- 55. Sprenger FS, Stefanova N, Gelpi E, et al. Enteric nervous system alpha‐synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 2015;85(20):1761–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Michell AW, Luheshi LM, Barker RA. Skin and platelet alpha‐synuclein as peripheral biomarkers of Parkinson's disease. Neurosci Lett 2005;381(3):294–298. [DOI] [PubMed] [Google Scholar]
- 57. Gibbons CH, Garcia J, Wang N, Shih LC, Freeman R. The diagnostic discrimination of cutaneous alpha‐synuclein deposition in Parkinson's disease. Neurology 2016;87(5):505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Doppler K, Volkmann J, Sommer C. Skin biopsies in the differential diagnosis of Parkinsonism: are we ready for simplified protocols? Brain 2016;139(Pt 1):e5. [DOI] [PubMed] [Google Scholar]
- 59. Antelmi E, Donadio V, Incensi A, Plazzi G, Liguori R. Skin nerve phosphorylated alpha‐synuclein deposits in idiopathic REM sleep behavior disorder. Neurology 2017;88(22):2128–2131. [DOI] [PubMed] [Google Scholar]
- 60. Visanji NP, Marras C, Hazrati LN, Liu LW, Lang AE. Alimentary, my dear watson? the challenges of enteric alpha‐synuclein as a Parkinson's disease biomarker. Mov Disord 2014;29(4):444–450. [DOI] [PubMed] [Google Scholar]
- 61. Chung SJ, Kim J, Lee HJ, et al. Alpha‐synuclein in gastric and colonic mucosa in Parkinson's disease: Limited role as a biomarker. Mov Disord 2016;31(2):241–249. [DOI] [PubMed] [Google Scholar]
- 62. Visanji NP, Marras C, Kern DS, et al. Colonic mucosal a‐synuclein lacks specificity as a biomarker for Parkinson's disease. Neurology 2015;84(6):609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Corbille AG, Letournel F, Kordower JH, Lee J, Shanes E, Neunlist M, et al. Evaluation of alpha‐synuclein immunohistochemical methods for the detection of lewy‐type synucleinopathy in gastrointestinal biopsies. Acta Neuropathol Commun 2016;4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lee JM, Derkinderen P, Kordower JH, et al. The search for a peripheral biopsy indicator of alpha‐synuclein pathology for Parkinson's disease. J Neuropathol Exp Neurol 2017;76(1):2–15. [DOI] [PubMed] [Google Scholar]
- 65. Visanji NP, Mollenhauer B, Beach TG, et al. The systemic synuclein sampling study: toward a biomarker for Parkinson's disease. Biomark Med 2017;11(4):359–368. [DOI] [PubMed] [Google Scholar]
- 66. Ruffmann C, Parkkinen L. Gut feelings about alpha‐synuclein in gastrointestinal biopsies: Biomarker in the making? Mov Disord 2016;31(2):193–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Saeed U, Compagnone J, Aviv RI, et al. Imaging biomarkers in Parkinson's disease and parkinsonian syndromes: current and emerging concepts. Transl. Neurodegener. 2017;6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Lehericy S, Vaillancourt DE, Seppi K, et al. The role of high‐field magnetic resonance imaging in parkinsonian disorders: Pushing the boundaries forward. Mov Disord 2017;32(4):510–525. [DOI] [PubMed] [Google Scholar]
- 69. Strafella AP, Bohnen NI, Perlmutter JS, Eidelberg D, Pavese N, Van Eimeren T, et al. Molecular imaging to track Parkinson's disease and atypical parkinsonisms: new imaging frontiers. Mov Disord 2017;32(2):181–192. [DOI] [PubMed] [Google Scholar]
- 70. Iranzo A, Valldeoriola F, Lomena F, et al. Serial dopamine transporter imaging of nigrostriatal function in patients with idiopathic rapid‐eye‐movement sleep behaviour disorder: a prospective study. Lancet Neurol 2011;10(9):797–805. [DOI] [PubMed] [Google Scholar]
- 71. Poston KL, Eidelberg D. Network biomarkers for the diagnosis and treatment of movement disorders. Neurobiol Dis 2009;35(2):141–147. [DOI] [PubMed] [Google Scholar]
- 72. Holtbernd F, Gagnon JF, Postuma RB, et al. Abnormal metabolic network activity in REM sleep behavior disorder. Neurology 2014;82(7):620–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ma Y, Tang C, Spetsieris PG, Dhawan V, Eidelberg D. Abnormal metabolic network activity in Parkinson's disease: test‐retest reproducibility. J Cereb Blood Flow Metab 2007;27(3):597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Berg D. Transcranial ultrasound as a risk marker for Parkinson's disease. Mov Disord 2009;24(Suppl 2):S677–S683. [DOI] [PubMed] [Google Scholar]
- 75. Deco G, Kringelbach ML. Great expectations: using whole‐brain computational connectomics for understanding neuropsychiatric disorders. Neuron 2014;84(5):892–905. [DOI] [PubMed] [Google Scholar]
- 76. Griffanti L, Rolinski M, Szewczyk‐Krolikowski K, et al. Challenges in the reproducibility of clinical studies with resting state fMRI: an example in early Parkinson's disease. NeuroImage 2016;124(Pt A):704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Eberling JL, Dave KD, Frasier MA. Alpha‐synuclein imaging: a critical need for Parkinson's disease research. J Parkinsons Dis 2013;3(4):565–567. [DOI] [PubMed] [Google Scholar]
- 78. Espay AJ, Bonato P, Nahab FB, et al. Technology in Parkinson's disease: challenges and opportunities. Mov Disord 2016;31(9):1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Horak FB, Mancini M. Objective biomarkers of balance and gait for Parkinson's disease using body‐worn sensors. Mov Disord 2013;28(11):1544–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Marras C, Rochon P, Lang AE. Predicting motor decline and disability in Parkinson's disease: a systematic review. Arch Neurol 2002;59(11):1724–1728. [DOI] [PubMed] [Google Scholar]