

Abstract
Molecular responses to genotoxic stress are complex and are mediated by a variety of regulatory pathways. One key element in cellular response is the stress gene transcription factor p53, which can regulate nearly 100 genes that have already been identified. Although p53 plays a central role in the cellular response to DNA-damaging agents such as ionizing radiation (IR), other pathways can also have important roles. One example is the transcriptional responses associated with IR-induced apoptosis, where induction of some genes is limited to p53 wild-type (wt) cells that also have the ability to undergo rapid apoptosis after irradiation. In contrast, other genes are triggered after IR in lines undergoing rapid apoptosis regardless of p53 status. From this and other examples, it is apparent that the pattern of stress gene expression is cell type specific in both primary and transformed lines. The premise will be developed that such differences in stress gene responsiveness can be employed as molecular markers using a combination of informatics and functional genomics approaches. An example is given using the panel of lines of the NCI anticancer drug screen where both the p53 status and sensitivity to a large collection of cytotoxic agents have been determined. The utility of cDNA microarray hybridization to measure IR-stress gene responses has recently been demonstrated and a large number of additional IR-stress genes have been identified. The responses of some of these genes to IR and other DNA-damaging agents varied widely in cell lines from different tissues of origin and different genetic backgrounds, highlighting the importance of cellular context to genotoxic stress responses; this also highlights the need for informatics approaches to discover and prioritize hypotheses regarding the importance of particular cellular factors. The aim of this review is to demonstrate the utility of combining an informatics approach with functional genomics in the study of stress responses.
Keywords: Ionizing radiation, p53, Microarray, Genotoxic stress
REMARKABLE progress has been made over the last decade in our understanding of the molecular responses to ionizing radiation (IR) and other types of genotoxic stress. Before then (3,20), the prevailing opinion was that mammalian cells lacked inducible responses to DNA damage, whereas in simpler eu-karyotes, early studies in yeast (45) indicated complex responses to genotoxic stress, involving up to 1% or more of the yeast genome. More recently, an increasing number of genes have been found to be induced in mammalian cells by DNA-damaging agents including IR, and the list of such genes now numbers in the hundreds (7,19,21,27,64). As shown in Table 1, mammalian genotoxic stress genes have been associated with a variety of cellular processes. In some cases, such as for genes with roles in DNA repair and virus activation, one could argue that the responses are specific for genotoxic stress. Many stress-induced genes, however, appear to be more general responses to cell and tissue injury. In addition, many stress-induced genes, such as growth factors, cytokines, and many oncogenes, overlap with genes involved in other physiologic signaling processes. For example, the “UV response” (26) in volves induction of the immediate early genes c-fos and c-jun, genes involved in a variety of physiologic processes.
TABLE 1.
GENERAL CATEGORIES OF GENOTOXIC-RESPONSIVE GENES
| Growth control |
| Apoptosis associated |
| “UV response” |
| Immediate-early genes—transcription factors and oncogenes |
| Inflammation and tissue injury |
| Oxidant stress responses |
| DNA repair |
| Hypoxia inducible |
| Viral genes |
| [Ah] receptor gene battery |
| Heat shock protein related |
Although a variety of important signaling mechanisms can be activated by genotoxic stress, p53 has been described as the universal sensor of genotoxic stress (32). It can be activated by a wide spectrum of DNA-damaging agents as well as other stresses, including hypoxia and nutrient starvation (2,29). As outlined briefly in Fig. 1, the activation of p53 is primarily posttranslational and probably involves specific phosphorylation (48,49), acetylation (24), and perhaps other modifications. The signaling events leading to these changes are the focus of intense ongoing investigation, but the ATM protein is clearly involved in the case of IR. A variety of other kinases, including members of the MAPK pathway (28,29), are likely to have roles in stress signal transduction to p53.
FIG. 1.
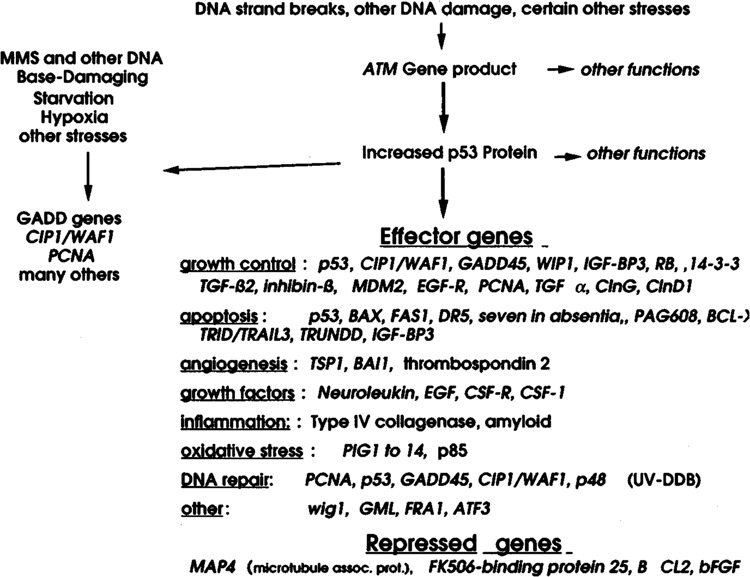
The p53 transcriptional pathway. A simplified scheme is shown for genes regulated by p53. In some cases, such as for GADD45 (28), CIP1/W1P1 (17), and MDM2 (5,38), p53 binding sites have been identified in regulatory regions of these genes; the inclusion of others, such as PIG] to PIG14 (43), is based on induction by p53 expression vectors.
Following its activation, p53 has been shown to function as a sequence-specific transcription factor. The total number of p53 binding sites in the human genome has been predicted to be between 200 and 300 (53), and the number of known p53-regulated genes is approximately 100, many with roles in a variety of important cellular processes (Fig. 1). In many cases, these genes appear to have opposing roles. Presumably, such competing signals play a role in limiting the duration of the particular response (e.g., Mdm2 blocks the action of p53 itself) (38). It should also be noted that many stress genes, like CIPU WAF1 and GADD45 (22,72), can be induced by p53-independent mechanisms after exposure to stresses such as UV radiation and other base-damaging agents, whereas their responsiveness to IR is primarily p53 dependent (72). p53 may also contribute to the activation of other stress pathways by upregulating certain components of these pathways. In the case of cell surface receptors such as DR5 (65), Fas (39), or EGFR (13), increased expression by p53 could conceivably lead to activation in the absence of li-gand with resultant triggering of apoptotic signaling or the MAPK pathway. More recently, evidence has been presented that p53 can also contribute to the increased expression of the stress genes GADD45 (71) and WIP1 (Appella, personal communication), by indirect binding via protein-protein interactions rather than direct DNA binding. An important implication of these recent observations is that p53 may contribute to the induction of an even larger number of genes than originally predicted (53).
MOLECULAR RESPONSES TO IONIZING RADIATION
Unlike many other types of DNA-damaging agents where the lesion frequency is much greater, IR causes little S phase delay at biologically relevant doses, and with the exception of interphase death in apoptosis-susceptible cells, cell death from IR appears to be due primarily to errors in mitosis caused by relatively infrequent double strand breaks (dsb) (15). In most cell types, cytogenetic studies have revealed a variety of chromosomal aberrations following irradiation, and often a correlation between the frequency of chromosomal aberrations and cell killing (25). It was originally believed that aberrations leading to cell death were mostly those associated with the loss of essential gene function. It has recently been shown in yeast, however, that a single persistent dsb in an artificial chromosome, which contained no essential genes, resulted in cell lethality (6). These results suggest that a chromosome break, presumably caused by an unrepaired or misrepaired dsb, can lead to cell death by an as yet undefined signaling pathway, or perhaps simply by disruption of the mitotic machinery. Considering the size of the mammalian genome and the low frequency of radiation-induced dsb, these issues have been difficult to resolve. For the majority of cells, most of the radiation-induced cell lethality appears to be caused by mitosis-linked loss of replicative ability. This mitosis-linked loss of cell viability can culminate in frank necrosis, apoptosis, or sometimes a senescent appearance (15,59). This form of cell death can be distinguished from the more rapid onset of apoptosis in noncycling lymphocytes, thymocytes, and hemato-poietic cells, which have been shown to undergo an interphase mode of apoptotic cell death without progressing through mitosis (2).
Perhaps due to the unique properties of DNA strand breaks and the relatively low lesion frequency compared to other DNA-damaging agents (19,27,50), molecular responses to IR can differ from those to base-damaging agents. For example, activation of MAPK-related signaling mechanisms by agents like UV radiation probably contributes much of the response to these agents (22,32), whereas activation of these pathways by physiologic doses of IR is often much less (see below). In addition, CIP1/WAF1 and GADD45 show a universal responsiveness to base-damaging agents in mammalian cells, whereas in most cases their IR responsiveness is strictly p53 dependent. As shown in Fig. 2, appreciable induction of CIP1/WAF1 and GADD45 by even a high dose of IR was primarily seen only in human tumor lines with wt p53 (37). It should be noted that tumor lines have a myriad of genetic abnormalities compared to untransformed cells; nonetheless, the correlation with p53 status is striking (62).
FIG. 2.
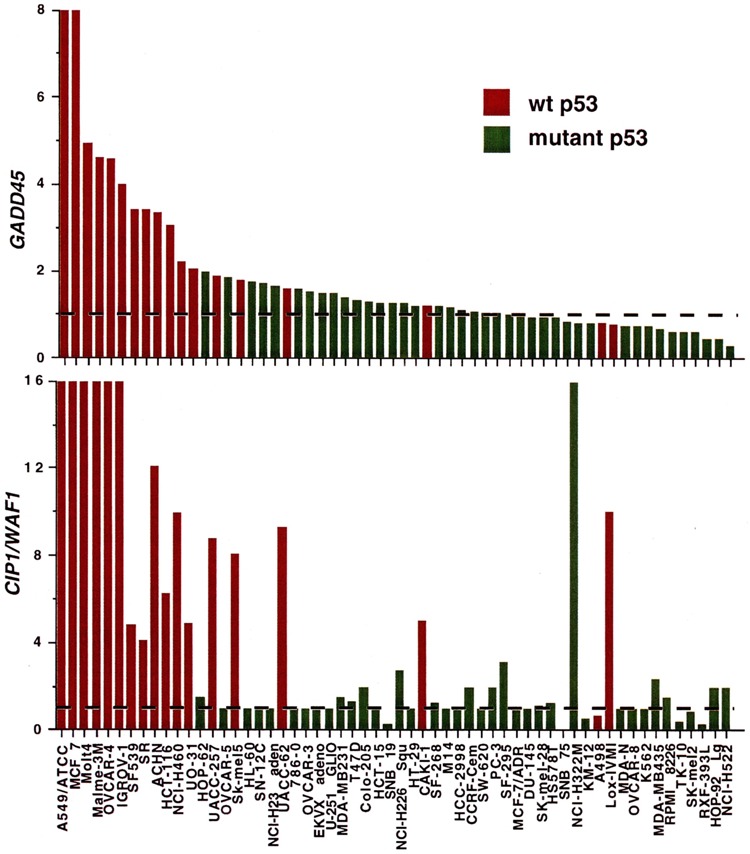
Degree of γ-ray responsiveness of the GADD45 and CIP1/WAF1 genes in tumor lines from the NCI screen. The relative increase in mRNA levels compared to that in untreated cells was determined in the lines of the NCI anticancer drug screen following a 4-h incubation after 20 Gy. Data are taken from O’Connor et al. (36) and only lines with known p53 status have been included. Lines with wt p53 genotype are designated in red, and lines containing mutant alleles in green. A value of 1 (designated by the dotted lines) indicates no change (no induction) from that of untreated controls.
APOPTOSIS AND RADIATION STRESS RESPONSES
Whereas cellular context is often a factor in determining the signaling responses triggered by IR, rapid apoptotic responses occur in only a limited number of cell types, such as those of the myeloid and lymphoid lineages. A variety of signaling pathways, including p53, the sphingomyelin pathway, and the MAPK pathway (2,27,58), are probably involved in IR apoptosis. IR also triggers the rapid activation of a variety of tyrosine kinases (54) with possible roles in apoptotic signaling. For example, Bruton’s tyrosine kinase has been shown to mediate radiation apoptosis in the chicken DT-40 lymphoma line (55).
One characteristic outcome of the apoptotic signaling pathways is chromatin cleavage in internucleosomal linker sites that produces a typical DNA laddering pattern as seen on standard agarose gels. This hallmark of apoptosis can typically be observed 24–48 h after irradiation of apoptosis-susceptible cells (73). At earlier times, however, DNA is cleaved into fragments of 50 kbp and larger, which can be detected by pulse field gel electrophoresis (36). A convenient and quantitative approach to detect these larger molecular weight cleavage products is with a simple adaptation of the alkaline/neutral elution approach; cells are deposited on an inert filter and then lysed in detergent at neutral pH. DNA fragments of approximately 100 kbp and less flow through the filter in the lysis solution, whereas the higher molecular weight DNA is retained. Within several hours of irradiation of myeloid or lymphoid cells, substantial cleavage could be detected using this approach (73). For example, more than 75% of the cellular DNA showed cleavage within 4 h of irradiation of HL-60 myeloid cells, clearly demonstrating that IR-induced apoptosis can occur rapidly and in interphase cells.
The ability to undergo rapid apoptosis also correlated with gene induction in a variety of human cell lines, as shown in Table 2. The p53-regulated genes BAX and BCL-X were induced only in cell lines that underwent rapid apoptosis, whereas induction of GADD45 and CIP1/WAF1 occurred in all the p53 wt lines. Other immediate-early genes, such as MCL1, GADD153, GADD34, and c-JUN were induced regardless of p53 status but only in those cells where rapid apoptosis was also triggered. These results may indicate an “apoptotic proficiency” factor that channels the stress response signal toward apoptosis in specific cell types. c-JUN induction reflects activation of the SAPK portion of the MAPK pathway, and there are numerous other associations between MAPK activation and apoptosis in irradiated cells [discussed in (7,15,63,66)]. For example, IR triggered activation of SAPK in thymocytes, whereas dexamethasone did not, despite the fact that both cause rapid apoptosis (52). Thus, the SAPK pathway may contribute to an IR-specific signal transduction pathway in some cells. The nature of the “apoptosis proficiency” factor(s) shaping stress responses is uncertain, but recent results suggest that early event(s) in the caspase cascade itself could be involved, because overexpression of Bcl2 in a lymphoid line was shown to blunt p53 transcriptional responses after IR (74).
TABLE 2.
GENE RESPONSES TO IONIZING RADIATION
| GADD45 | CIP1 | BAX | BCL-X | MCL1 | GADD153 | GADD34 | c-JUN | |
|---|---|---|---|---|---|---|---|---|
| Cell lines with normal p53 function | ||||||||
| Apoptosis | + | + | + | + | + | + | + | + |
| No apoptosis | + | + | - | - | - | - | - | - |
| Cell lines with defective p53 | ||||||||
| Apoptosis* | - | - | - | - | + | + | + | + |
| No apoptosis | - | - | - | - | - | - | - | - |
Induction, as measured by increased mRNA levels 4 h after IR, was determined in various human lines with wild-type or deficient p53 status and whether ionizing radiation induced a rapid apoptotic response as determined by filter elution (67-69,72,73), which detects high mw (approximately 50 kbp) DNA cleavage. The results were then separated into the four groups shown above. + indicates appreciable induction whereas — does not.
GADD45 and CIP1/WAF1 sometimes showed a weak response in a p53-deficient myeloid line (HL-60) undergoing apoptosis.
USE OF RADIATION STRESS RESPONSES AS MOLECULAR MARKERS
Considering the heterogeneous responses of different cell lines to genotoxic stress, and the cross-talk between the many molecules we currently know to be involved, transcriptional stress responses can be considered a type of molecular marker that may be used to characterize different cell types and lines. In the case of cancer treatment, such molecular marker “fingerprints” will probably prove to have clinical utility in designing customized therapy for individual tumors (62). Both from standard molecular biologic techniques and newer functional genomic approaches (see below), an increasing number of such molecular markers have been described. Sophisticated pattern analysis and database mining methods, coupled with informatics systems, are also being developed to digest the results of these database-sized experiments. One example of such a database-intensive approach is the National Cancer Institute’s antineoplastic drug screen panel (23,34,35,37,40,41,44,51,56). This is a collection of 60 human tumor cell lines that have been characterized with respect to over 300 molecular markers measured as individual gene mutations, mRNA expression patterns, protein expression patterns, or activity. For instance, there was a striking correlation between p53 status and IR induction of CIP1/WAF1, GADD45, and MDM2 (36). As shown in Fig. 2, strong induction of CIP1/WAF1 correlated closely with wt p53 expression, with only one exception. In the case of this one exception, the basal expression was substantially lower than that of the p53 wt lines, meaning that the induction in absolute terms was low compared to induction in the p53 wt lines. (The ordinate in Fig. 2 represents expression in irradiated cells relative to that of untreated controls, not absolute levels.) More information can be gained from this study by comparing the basal expression of CIP1AVAF1 mRNA in unirradiated lines; again, expression was substantially higher in the p53 wt lines compared to the p53 mutant lines (Amundson et al., manuscript in preparation).
Similar results were obtained for GADD45 (Fig. 2) when again only the p53 wt lines showed substantial induction. However, in contrast to the finding for CIP1/WAF1, a substantial number of p53 wt lines did not fall within the strongly induced category and showed responses similar to those of the p53 mutant lines. This subset of GADD45 response-deficient lines, which were primarily melanomas, showed a substantial perturbation in p53 responsiveness, including an apparent compensatory prolonged induction of CIP1/WAF1 (4). To consider the possibility that this attenuated expression of GADD45 after stress may affect’ chemosensitivity, the p53 wt lines with normal GADD45 responsiveness were compared to the subset with deficient responsiveness (Fig. 3A). An initial analysis of the National Cancer Institute Anticancer Drug Screen (61), using a set of 141 agents with well-defined mechanisms of action, indicated that the sensitivity to topoisomerase inhibitors was significantly less in this subset compared to the other lines having a wt p53 genotype (8). The initial study was then expanded to encompass the approximately 43,000 compounds that had been tested in the NCI screen at the time of this analysis. The compounds were divided into three groups for comparison: one group composed of 421 Top2 (topoisomerase II) inhibitors [identified using the COMPARE computer program (40,41)], another of 167 Topi (topoisomerase I) inhibitors (identified as camptothecin analogues), and the other of a very diverse remaining group of agents. Because these lines are not isogenic and many variables affect the actual sensitivity, Pearson correlation coefficients were calculated using the levels of GADD45 induction as one variable and cytotoxicity as the other. A Wilcoxon rank sum test showed that sensitivity to compounds in both of the topoisomerase inhibitor groups was greater in the subset of p53 wt cells that showed normal GADD45 induction than for the case with the bulk of other compounds tested in the screen (p < 0.0001). This indicates that higher GADD45 expression after stress correlated with enhanced sensitivity to Top1 and Top2 inhibitors. To more clearly show the difference in sensitivity for this subset of p53 wt lines, the Wilcoxon one-tailed p-values are shown in Fig. 3 where the sensitivity to specific agents was compared to whether or not the cell line was a member of this subset. For 400 Top2 inhibitor-type compounds the p-values clustered near 1 whereas an essentially random distribution of p-values was seen for 43,000 other compounds. As presented in Carrier et al. (8), this finding was an important lead to the elucidation of a role of Gadd45 in chromatin maintenance and accessiblity. It is presented here to illustrate the power of an informatics approach to gain basic mechanistic insight as well as more applied information of potential clinical usefulness.
FIG. 3.
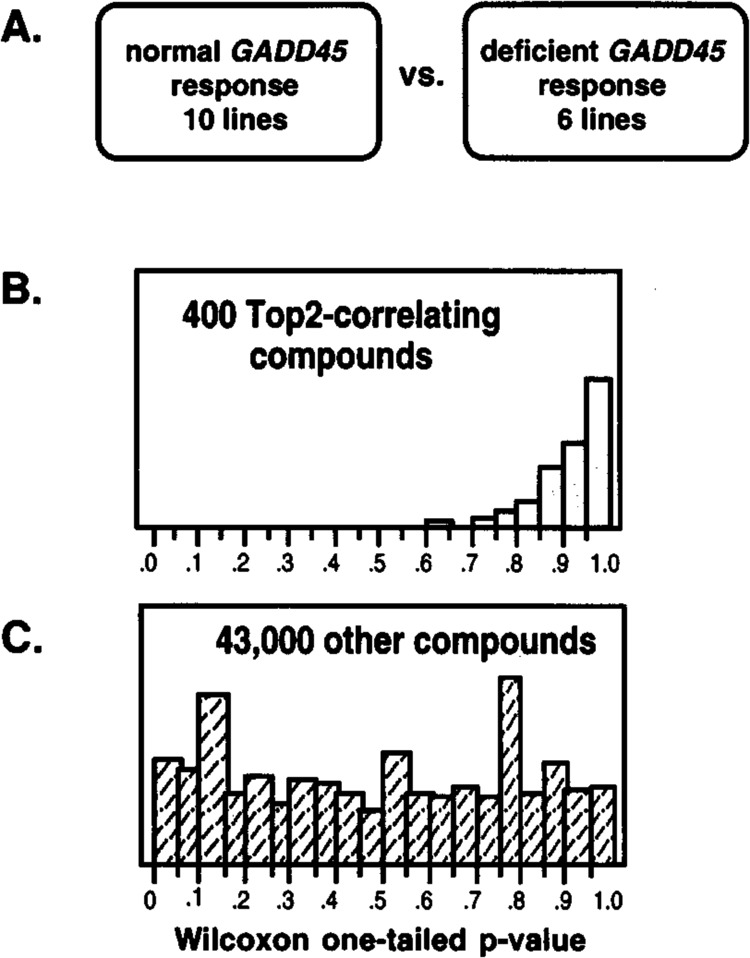
A clue to Gadd45 function by use of an informatics approach. (A) Six of 16 lines with functional p53 were found to show deficient γ-ray induction of GADD45 in the NCI screen. (B) Wilcoxon rank sum test was used to determine whether individual compounds tested were more active in cell lines with competent GADD45 induction (p-value = 1) or more active in cell lines with deficient GADD45 induction (p-value = 0); results are shown for a selection of COMPARE-identified topoisomerase II inhibitors (8). (C) Results for the remainder of the database of tested compounds.
FUNCTIONAL GENOMICS APPROACH TO STRESS GENE ANALYSIS
Genotoxic stress responses are too complex to be analyzed solely by the classic reductionist approach to molecular biology, whereby genes are analyzed individually. Modern integrative “omic” (60) approaches are necessary if we are to clarify our understanding of the many factors interacting in the p53 and other stress response pathways. Techniques for obtaining expression data simultaneously for thousands of genes, potentially even for the entire genome, are becoming more readily accessible. Combined with the informatics methods discussed earlier, these approaches may allow us to unravel the pathways of stress signal transduction, and may help identify the determinants of cell fate following DNA damage in different cell types. New techniques, including serial analysis of gene expression (SAGE), oligonucleotide microarrays, and cDNA microarrays, are being developed to obtain expression profiling information for thousands of molecular targets in a single experiment. SAGE allows comparison of relative mRNA abundance in any two samples (57). For example, SAGE has been used to study p53 as a tran-scriptional activator, identifying around 30 transcripts apparently induced by p53 expression after very high transient expression of p53 via an adenoviral vector (43). SAGE analysis of rat embryo fibroblasts expressing temperature-sensitive p53 yielded 14 transcripts induced by wt p53 and 3 downregulated by p53 (33). An advantage of the SAGE technique is that it does not require previous identification of genes or representation of sequence in any database. This could be crucial if some genes of interest are not expressed under nonstressed conditions, as EST collections are derived primarily from libraries made from untreated cells. A disadvantage is that a thorough analysis requires sequencing of several hundred thousand sequence tags, making routine analysis of multiple samples prohibitively expensive.
Another functional genomics approach that has been applied to the study of p53 and stress response is cDNA microarray hybridization (16,46,47). This method requires some preexisting sequence information, usually in the form of EST clones. It has been successfully applied to characterization of gene expression patterns in human cancers (14), and studies are under way to use this technique to characterize expression patterns of the cell lines in the NCI-Anti-cancer Drug Screen for up to 10,000 genes (P. O. Brown, personal communication). The methodology employed in this approach is shown schematically in Fig. 4. Briefly, each target or spot on the microarray contains DNA from a single cDNA clone; it is hybridized with fluorescent-labeled cDNA probes pre-pared from the RNA of control or treated (irradiated) cells. Because many stress genes encode low abundance transcripts that are frequently induced only several fold (19), its application requires sensitivity to measure both weak signals (low abundance transcript) and small changes in gene expression. Substantial progress has made in this regard recently [discussed in (16)]. For example, the DeArray program (12) has been developed to identify targets by image segmentation, to calibrate relative ratios, and to develop confidence intervals for testing the significance of the ratios obtained even for small differences in relative expression.
FIG. 4.
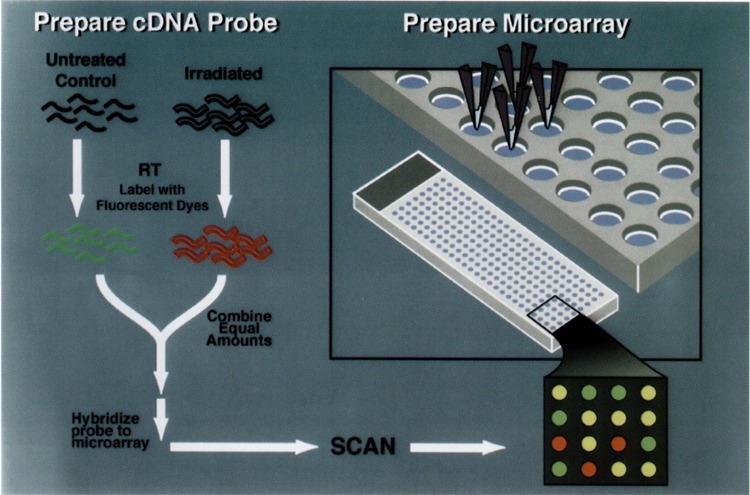
Schema for analysis of ionizing radiation-inducible genes by cDNA microarray hybridization. The black wavy lines represent mRNA from untreated control and irradiated ML-1 cells. Labeled cDNA is synthesized from this mRNA by a single round of reverse transcription to produce the complex probe for hybridization. Targets for the array are prepared by PCR amplification of inserts from EST clones. These are printed onto glass slides using a robotic print head, and the control (green) and irradiated (red) cDNA probes are mixed and hybridized to the microarray. The slide is then scanned and analyzed with a fluorescent reader (1,14,18,46).
In our laboratory, we are applying cDNA hybridization technology to the study of molecular responses to IR and other DNA-damaging agents. For our initial studies, we chose the human ML-1 myeloid line (30) based on our previous experience in many different cell lines. Unlike some of the other functional genomics studies discussed earlier, activation of signaling pathways, such as for p53, employed the cells’ normal response mechanisms to IR, and no recombinant vectors were utilized (1). This p53 wt line undergoes rapid apoptosis after high-dose IR and has shown induction of a broad range of genes (see Table 2). As shown in Fig. 5, induced genes (red) such as CIP1/WAF1 and JUN-B as well as repressed genes (green) such as c-MYC could be detected. Relative induction or repression ratios, as calculated with the DeArray program (12), are shown in Fig. 6 for results with a 1238 member array (1.2K chip) consisting of a selected collection of EST sequences including many implicated in cancer biology. Several conclusions can be drawn from these initial studies (1). First, IR responses in this line are quite complex, involving many genes related to a variety of cellular processes such as growth control (e.g., CIP1/WAF1, GADD45, MDM2, c-MYC), apoptosis (e.g., FAS, IAP-1, BCL-X), cell signaling (e.g., ATF3, RELB, FRAl, c-FOS, JUN-B), and DNA metabolism (e.g., TOP2 and DNA ligase III). Second, even low abundance transcripts could be detected (e.g., we estimated that the expression of some of these genes in untreated cells was 10−5). Third, there were surprisingly few false positives; of the 19 transcripts measured by quantitiative single probe hybridization, only one did not show a change in expression of twofold or more. Finally, the estimate of relative gene expression after IR compared to unirradiated cells employing the DeArray program agreed in the majority of cases with results from quantitiative single probe hybridization (see http://rex.nci.nih.gov/RESEARCH/basic/lbc/fornace.htm for more details). In particular, 68% of the transcripts examined showed less than a twofold difference in relative expression as determined by the two approaches. In other cases a difference in results greater than twofold was observed due to signal compression for the values determined by microarray hybridization; the cause for this is currently under investigation but appears to be sequence related.
FIG. 5.

Example of cDNA microarray hybridization. Results are shown employing RNA from untreated control ML-1 cells (green fluorochrome) and ML-1 cells 4 h after treatment with 137Cs γ-rays (red fluorochrome) (1). Targets appearing as yellow spots have equal representation of both fluorochromes and indicate no change in expression by the IR treatment. Red spots, such as CIP1/WAF1 and JUN-B, are targets increased by the treatment, and green targets, such as MYC, are decreased.
FIG. 6.
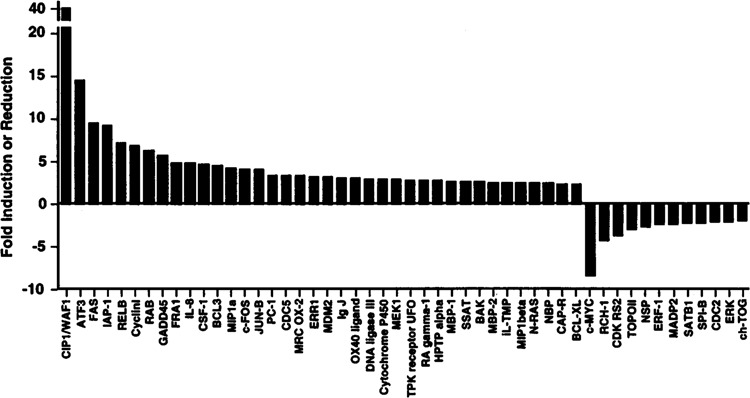
Ionizing radiation-responsive genes as determined by cDNA microarray hybridization. Relative changes in gene expression for irradiated ML-1 cells were estimated using the DeArray program (12) for results with a 1,238 member cDNA array (1).
Our expectation is that cDNA microarray hybridization will be a valuable high-throughput approach to the analysis of stress gene responses. In addition to the results shown in Fig. 6, preliminary results for several other stress gene studies are also summarized in Table 3. Although not a DNA-damaging agent, hypoxia does trigger p53 induction and is an important variable in radiotherapy. As shown in this table, a very large fraction of the genes tested showed altered expression with this treatment. Our initial IR studies with the 1.2K chip used a high dose of IR to search for rapidly induced genes. Important issues are whether such responses occur at more biologically relevant doses of IR, such as those used in cancer treatment, and whether there are more prolonged or late IR responses, which could potentially provide biomarkers for environmental exposure. To address these issues, a larger 5K chip was used with a lower dose of IR. As shown in the last two rows of Table 3, a substantial number of genes responded both acutely (3 h) and at a later time (24 h) after IR. More than 10 years ago it was estimated that 1% or more of the yeast genome may be responsive to genotoxic stress (45); our results with IR indicate that a comparable fraction of the human genome may also be responsive to genotoxic stress.
TABLE 3.
SUMMARY OF RESULTS FOR MICROARRAY ANALYSIS OF STRESS RESPONSES
| Array* | Targets | Treatments | Induced† | Reduced‡ | 99% Conf.§ |
|---|---|---|---|---|---|
| 1.2Kchip | 1238 | 20Gy + 4h | 38 | 11 | 0.63-2.37 |
| 1238 | 6 h hypoxia | 135 | 19 | 0.51-2.47 | |
| 1238 | 24 h hypoxia | 202 | 8 | 0.55-2.38 | |
| 5K chip# | 5408 | 2 Gy + 3 h | 67 | 21 | 0.68-2.32 |
| 5408 | 2Gy + 24h | 161 | 26 | 0.69-1.89 |
Results are summarized for microarray hybridizations conducted in ML-1 cells. Many of the results for rows 1 and 3 have been verified by quantitative single-probe hybridization.
Two different microarrays (chips) used contained only limited overlap in the genes represented.
The number induced refers to cDNA clones showing significant induction (> 99% confidence).
For cDNA clones showing a significant reduction, values are shown for those having at least a twofold reduction in expression compared to untreated cells; all these values exceeded 99% confidence.
Values represent range of relative expression of irradiated sample compared to untreated control (e.g., only targets showing an increase in the relative mRNA level of 2.37-fold or more were scored as induced in the first row).
Preliminary results with larger microarray are shown, but have not been verified by single-probe hybridization.
THE IMPORTANCE OF CELLULAR CONTEXT IN GENOTOXIC STRESS RESPONSES
One advantage of the cDNA microarray approach is that it has allowed us to identify a large number of human genes that were not previously known to be IR responsive or, in many cases, genotoxic stress responsive. To further investigate the importance of cellular context, nine of these newly identified IR-responsive genes along with three known IR-responsive genes were studied in a panel of human lines following exposure to IR or to the base-damaging agents methylmethane sulfonate (MMS) or UV radiation (1). The cell lines used in this comparison included six lines of myeloid-lymphoid lineage [ML-1 (myeloid), Molt4 (lymphoid), SR (lymphoid), CCRF-CEM (lymphoid), HL60 (myeloid), and K562 (myeloid)], two lung cancer lines (A549 and HI299), two breast carcinoma lines (MCF7 and T47D), and the colon cancer line RKO along with its derivative transfected with E6 (RKO/E6), which blocks p53 function (70). Quantitative single probe hybridization analysis was carried out in independent experiments on the cell lines indicated in Fig. 7. In the case of ML-1 cells, all showed IR responsiveness, indicating the reproducibility of the earlier experiments. Many genes were not responsive to MMS in ML-1 cells, however, suggesting that the regulation of these genes may have some radiation-specific features. Striking differences were found between ML-1 and the other cell lines (Fig. 7). For example, many of the p53 mutant lines showed little induction by IR, and K562 showed no appreciable change in expression for any of the genes after IR. Consistent with its ability to trigger multiple stress pathways (2,19,27), MMS was able to induce, or in the case of RCH1 repress, a variety of genes in different cell lines. The most wide-ranging response was seen for activating transcription factor 3 (ATF3) (31), which was induced by both MMS and UV radiation in all cell lines tested. ATF3 has previously been shown to be induced by serum stimulation, to 12-0-tetradecanoylphorbol-13-acetate (9), and by physiological stresses such as wounding, CC14 and alcohol intoxication, ischemia/reperfusion, and brain seizure (10). With such a strong and pervasive response, ATF3 is likely to play an important role in generalized genotoxic stress responses.
FIG. 7.
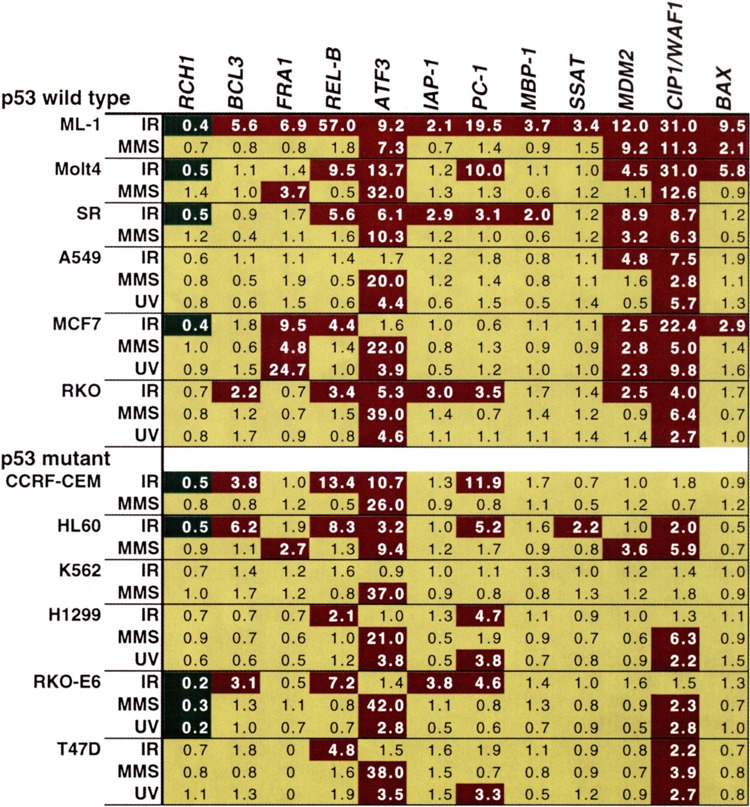
Responsiveness of transcripts identified by cDNA microarray hybridization in a panel of human tumor cell lines. Numbers shown are the relative induction or repression for each gene over levels in untreated controls 4 h after treatment with ionizing radiation (IR), methyl methanesulfonate (MMS), or ultraviolet (UV) radiation as measured by quantitative single-probe hybridization; data are taken from Amundson et al. (1). The results are color coded: red for > twofold induction, green for > twofold reduction, and yellow for < twofold change from untreated control. A zero indicates no detectable expression in either control or treated cells.
The response patterns in Fig. 7 may hold clues to the stress regulation of these genes. For instance, the effect of the p53 status of a cell on its ability to induce CIP1/WAF1 (17,72) and MDM2 (11,42) in response to IR has been well documented. This effect is reflected in the weak to absent IR induction of these two transcripts in p53 mutant cell lines in this panel compared to the clear IR induction in all p53 wt lines examined. Although not so widely induced, FRA-1 showed a similar pattern in that it was not induced by IR in any of the p53 mutant lines in this panel. Similarly, the fivefold IR induction of ATF3 in RKO was attenuated in the RKO/E6 cell line, although some IR induction was seen among the other p53 mutant cell lines. These results raised the possibility that the IR induction of FRA-1 and ATF3 may involve a p53 regulatory component. This premise was subsequently confirmed in isogenic systems including in vivo irradiation of p53 wt and p53−/− mice (1). As isogenic models are developed for other stress regulatory pathways, similar approaches may help to decipher the mechanistic basis for the heterogeneity of the responses seen in Fig. 7.
Even the relatively modest window onto the complexity of cellular stress response presented here should make clear the need for high-throughput measurements coupled with sophisticated analysis techniques for the meaningful interpretation of stress response pathways. Mammalian cells appear to have evolved many redundant mechanisms for directing and responding to stress signals. Some pathways may be active only in certain cell types, whereas some mechanisms have likely been disrupted in transformed cells. The application of functional genomics techniques to systems with defined genetic differences will generate data of challenging complexity and great potential value.
REFERENCES
- 1. Amundson S. A.; Bittner M.; Chen Y. D.; Trent J.; Meltzer P.; Fomace A. J. Jr. cDNA microarray hybridization reveals complexity and heterogeneity of cellular genotoxic stress responses. Oncogene (in press). [DOI] [PubMed]
- 2. Amundson S. A.; Myers T. G.; Fornace A. J. Jr. Roles for p53 in growth arrest and apoptosis: Putting on the brakes after genotoxic stress. Oncogene 17:3287–3300; 1999. [DOI] [PubMed] [Google Scholar]
- 3. Angel P.; Poting A.; Mallick U.; Rahmsdorf H. J.; Schorpp M.; Herrlich P. Induction of metallothionein and other mRNA species by carcinogens and tumor promoters in primary human skin fibroblasts. Mol. Cell. Biol. 6:1760–1766; 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bae I.; Smith M. L.; Sheikh M. S.; Zhan Q.; Scudiero D. A.; Friend S. H.; O’Connor P. M.; Fornace A. J. Jr. An abnormality in the p53 pathway following γ-irradiation in many wild-type p53 human melanoma lines. Cancer Res. 56:840–847; 1996. [PubMed] [Google Scholar]
- 5. Barak Y.; Juven T.; Haffner R.; Oren M. mdm2 expression is induced by wild type p53 activity. EMBO J. 12:461–468; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bennett C. B.; Westmoreland T. J.; Snipe J. R.; Resnick M. A. A double-strand break within a yeast artificial chromosome (YAC) containing human DNA can result in YAC loss, deletion or cell lethality. Mol. Cell. Biol. 16:4414–4425; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Boothman D. A.; Burrows H. L.; Yang C. R.; Davis T. W.; Wuerzberger S. M.; Planchon S. M.; Odegaard E.; Lewis J. E.; Pink J.; Meyers M.; Patten C. W.; Sharda N.; Kinsella T. J. Damage-sensing mechanisms in human cells after ionizing radiation. Stem Cells 15(Suppl. 2):27–42; 1997. [DOI] [PubMed] [Google Scholar]
- 8. Carrier F.; Georgel P. T.; Pourquier P.; Blake M.; Kontny H. U.; Antinore M. J.; Gariboldi M.; Myers T. G.; Weinstein J.; Pommier Y.; Fornace A. J. Jr. Gadd45, a p53-responsive stress protein, modifies DNA accessibility on damaged chromatin. Mol. Cell. Biol, (in press). [DOI] [PMC free article] [PubMed]
- 9. Chen B. P.; Liang G.; Whelan J.; Hai T. ATF3 and ATF3 delta Zip. Transcriptional repression versus activation by alternatively spliced isoforms. J. Biol. Chem. 269:15819–15826; 1994. [PubMed] [Google Scholar]
- 10. Chen B. P.; Wolfgang C. D.; Hai T. Analysis of ATF3, a transcription factor induced by physiological stresses and modulated by gaddl53/Chop10. Mol. Cell. Biol. 16:1157–1168; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen C. Y.; Oliner J. D.; Zhan Q.; Fornace A. J. Jr.; Vogelstein B.; Kastan M. B. Interactions between p53 and MDM2 in a mammalian cell cycle checkpoint pathway. Proc. Natl. Acad. Sci. USA 91:2684–2688; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Y.; Dougherty E. R.; Bittner M. L. Ratio-based decisions and the quantitative analysis of cDNA microarray images. J. Biomed. Opt. 2:364–374; 1997. [DOI] [PubMed] [Google Scholar]
- 13. Deb S. P.; Munoz R. M.; Brown D. R.; Subler M. A.; Deb S. Wild-type human p53 activates the human epidermal growth factor receptor promoter. Oncogene 9:1341–1349; 1994. [PubMed] [Google Scholar]
- 14. DeRisi J.; Penland L.; Brown P. O.; Bittner M. L.; Meltzer P. S.; Ray M.; Chen Y.; Su Y. A.; Trent J. M. Use of a cDNA microarray to analyse gene expression patterns in human cancer. Nat. Genet. 14:457–460; 1996. [DOI] [PubMed] [Google Scholar]
- 15. Dewey W. C.; Ling C. C.; Meyn R. E. Radiation-induced apoptosis: Relevance to radiotherapy. Int. J. Radiat. Oncol. Biol. Phys. 33:781–796; 1995. [DOI] [PubMed] [Google Scholar]
- 16. Duggan D. J.; Bittner M.; Chen Y.; Meltzer P.; Trent J. M. Expression profiling using cDNA microarrays. Nat. Genet. 21:10–14; 1999. [DOI] [PubMed] [Google Scholar]
- 17. El-Deiry W. S.; Tokino T.; Velculescu V. E.; Levy D. B.; Parsons R.; Trent J. M.; Lin D.; Mercer W. E.; Kinzler K. W.; Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell 75:817–825; 1993. [DOI] [PubMed] [Google Scholar]
- 18. Ermolaeva O.; Rastogi M.; Pruitt K. D.; Schuler G. D.; Bittner M. L.; Chen Y.; Simon R.; Meltzer P.; Trent J. M.; Boguski M. S. Data management and analysis for gene expression arrays. Nat. Genet. 20:19–23; 1998. [DOI] [PubMed] [Google Scholar]
- 19. Fornace A. J. Jr. Mammalian genes induced by radiation; activation of genes associated with growth control. Annu. Rev. Genet. 26:507–526; 1992. [DOI] [PubMed] [Google Scholar]
- 20. Fornace A. J. Jr.; Alamo I. J.; Hollander M. C. DNA damage-inducible transcripts in mammalian cells. Proc. Natl. Acad. Sci. USA 85:8800–8804; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Friedberg E. C.; Walker G. C.; Siede W. DNA repair and mutagenesis. Washington, DC: ASM Press; 1995. [Google Scholar]
- 22. Gorospe M.; Martindale J. L.; Sheikh M. S.; Fornace A. J. Jr.; Holbrook N. J. Regulation of p21CIPI/WAFI expression by cellular stress: p53-dependent and p53-independent mechanisms. Mol. Cell. Differ. 4:47–65; 1996. [Google Scholar]
- 23. Grever M. R.; Schepartz S. A.; Chabner B. A. The National Cancer Institute: Cancer drug discovery and development program. Semin. Oncol. 19:622–638; 1992. [PubMed] [Google Scholar]
- 24. Gu W.; Roeder R. G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 90:595–606; 1997. [DOI] [PubMed] [Google Scholar]
- 25. Hall E. J. Radiobiology for the radiobiologist, 3rd ed Philadelphia:J. P. Lippincott; 1988. [Google Scholar]
- 26. Herrlich P.; Ponta H.; Rahmsdorf H. J. DNA damage induced gene expression: Signal transduction and relation to growth factor signaling. Rev. Physiol. Biochem. Pharmacol. 119:187–223; 1992. [DOI] [PubMed] [Google Scholar]
- 27. Holbrook N. J.; Liu Y.; Fornace A. J. Jr. Signaling events controlling the molecular response to genotoxic stress. In: Feige U.; Morimoto R. I.; Yahara I.; Polla B. S., eds. Stress-inducible cellular responses. Basel: Birkhauser Verlag; 1996:273–288. [DOI] [PubMed] [Google Scholar]
- 28. Kastan M. B.; Zhan Q.; El-Deiry W. S.; Carrier F.; Jacks T.; Walsh W. V.; Plunkett B. S.; Vogelstein B.; Fornace A. J. Jr. A mammalian cell cycle checkpoint utilizing p53 and GADD45 is defective in ataxia telangiectasia. Cell 71:587–597; 1992. [DOI] [PubMed] [Google Scholar]
- 29. Ko L. J.; Prives C. p53: Puzzle and paradigm. Genes Dev. 10:1054–1072; 1996. [DOI] [PubMed] [Google Scholar]
- 30. Kozopas K. M.; Yang T.; Buchan H. L.; Zhou P.; Craig R. W. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc. Natl. Acad. Sci. USA 90:3516–3520; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liang G.; Wolfgang C. D.; Chen B. P.; Chen T. H.; Hai T. ATF3 gene. Genomic organization, promoter, and regulation. J. Biol. Chem. 271:1695–1701; 1996. [DOI] [PubMed] [Google Scholar]
- 32. Liu Z. G.; Baskaran R.; Lea-Chou E. T.; Wood L. D.; Chen Y.; Karin M.; Wang J. Y. Three distinct signalling responses by murine fibroblasts to genotoxic stress. Nature 384:273–276; 1996. [DOI] [PubMed] [Google Scholar]
- 33. Madden S. L.; Galella E. A.; Zhu J.; Bertelsen A. H.; Beaudry G. A. SAGE transcript profiles for p53-dependent growth regulation. Oncogene 15:1079–1085; 1997. [DOI] [PubMed] [Google Scholar]
- 34. Monks A.; Scudiero D.; Skehan P.; Shoemaker R.; Pauli K.; Vistica D.; Hose, C; Langley J.; Cronise P.; Vaigro-Wolff A.; et al. Feasibility of a high-flux anticancer drug screen using a diverse panel of cultured human tumor cell lines. J. Natl. Cancer Inst. 83:757–766; 1991. [DOI] [PubMed] [Google Scholar]
- 35. Myers T. G.; Anderson N. L.; Waltham M.; Li G.; Buolamwini J. K.; Scudiero D. A.; Pauli K. D.; Sausville E. A.; Weinstein J. N. A protein expression database for the molecular pharmacology of cancer. Electrophoresis 18:647–653; 1997. [DOI] [PubMed] [Google Scholar]
- 36. Oberhammer F.; Wilson J. W.; Dive C.; Morris I. D.; Hickman J. A.; Wakeling A. E.; Walker P. R.; Sikorska M. Apoptotic death in epithelial cells: Cleavage of DNA to 300 and/or 50 kb fragments prior to or in the absence of internucleosomal fragmentation. EMBO J. 12:3679–3684; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O’Connor P. M.; Jackman J.; Bae I.; Myers T. G.; Fan S.; Mutoh M.; Scudiero D.; Monks A.; Sausville E. A.; Weinstein J. N.; Friend S.; Fornace A. J. Jr.; Kohn K. W. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 57:4285–1300; 1997. [PubMed] [Google Scholar]
- 38. Oliner J. D.; Pietenpol J. A.; Thiagalingam S.; Gyuris J.; Kinzler K. W.; Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor p53. Nature 362:857–860; 1993. [DOI] [PubMed] [Google Scholar]
- 39. Owen-Schaub L. B.; Zhang W.; Cusack J. C.; Angelo L. S.; Santee S. M.; Fujiwara T.; Roth J. A.; Deisseroth A. B.; Zhang W. W.; Kruzel E. et al. Wild-type human p53 and a temperature-sensitive mutant induce Fas/APO-1 expression. Mol. Cell. Biol. 15:3032–3040; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Paull K. D.; Hamel E.; Malspeis L. Prediction of biochemical mechanism of action from the in vitro antitumor screen of the National Cancer Institute. In: Foye W. O., ed. Cancer chemotherapeutic agents. Washington, DC: American Cancer Society; 1995. [Google Scholar]
- 41. Pauli K. D.; Shoemaker R. H.; Hodes L.; Monks A.; Scudiero D. A.; Rubinstein L.; Plowman J.; Boyd M. R. Display and analysis of patterns of differential activity of drugs against human tumor cell lines: Development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst. 81:1088–1092; 1989. [DOI] [PubMed] [Google Scholar]
- 42. Perry M. E.; Piette J.; Zawadzki J. A.; Harvey D.; Levine A. J. The mdm-2 gene is induced in response to UV light in a p53-dependent manner. Proc. Natl. Acad. Sci. USA 90:11623–11627; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Polyak K.; Xia Y.; Zweler J. L.; Kinzler K. W.; Vogelstein B. A model for p53-induced apoptosis. Nature 389:300–306; 1997. [DOI] [PubMed] [Google Scholar]
- 44. Rubinstein L. V.; Shoemaker R. H.; Pauli K. D.; Simon R. M.; Tosini S.; Skehan P.; Scudiero D. A.; Monks A.; Boyd M. R. Comparison of in vitro anti-cancer-drug-screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J. Natl. Cancer Inst. 82:1113–1118; 1990. [DOI] [PubMed] [Google Scholar]
- 45. Ruby S. W.; Szostak J. W. Specific Saccharomyces cerevisiae genes are expressed in response to DNA-damaging agents. Mol. Cell. Biol. 5:75–84; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schena M.; Shalon D.; Davis R. W.; Brown P. O. Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270:467–170; 1995. [DOI] [PubMed] [Google Scholar]
- 47. Schena M.; Shalon D.; Heller R.; Chai A.; Brown P. O.; Davis R. W. Parallel human genome analysis: Microarray-based expression monitoring of 1000 genes. Proc. Natl. Acad. Sci. USA 93:10614–10619; 1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shieh S. Y.; Dceda M.; Taya Y.; Prives C. DNA damage-induced phosphorylation of p53 alleviates inhibition by MDM2. Cell 91:325–334; 1997. [DOI] [PubMed] [Google Scholar]
- 49. Siliciano J. D.; Canman C. E.; Taya Y.; Sakaguchi K.; Appella E.; Kastan M. BB DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 11:3471–3481; 1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Smith M. L.; Fornace A. J. Jr. Mammalian DNA damage-inducible genes associated with growth arrest and apoptosis. Mutat. Res. 340:109–124; 1996. [DOI] [PubMed] [Google Scholar]
- 51. Stinson S. F.; Alley M. C.; Kopp W. C.; Fiebig H. H.; Mullendore L. A.; Pittman A. F.; Kenney S.; Keller J.; Boyd M. R. Morphological and immunocytochemical characteristics of human tumor cell lines for use in a disease-oriented anticancer drug screen. Anti-cancer Res. 12:1035–1053; 1992. [PubMed] [Google Scholar]
- 52. Testolin L.; Carson C.; Wang Y.; Walker P. R.; Armato U.; Sikorska M. Jun and JNK kinase are activated in thymocytes in response to VM26 and radiation but not glucocorticoids. Exp. Cell Res. 230:220–232; 1997. [DOI] [PubMed] [Google Scholar]
- 53. Tokino T.; Thiagalingam S.; el-Deiry W. S.; Waldman T.; Kinzler K. W.; Vogelstein B. p53 tagged sites from human genomic DNA. Hum. Mol. Genet. 3:1537–1542; 1994. [DOI] [PubMed] [Google Scholar]
- 54. Uckun F. M.; Tuel-Ahlgren L.; Song C. W.; Waddick K.; Myers D. E.; Kirihara J.; Ledbetter J. A.; Schieven G. L. Ionizing radiation stimulates unidentified tyrosine-specific protein kinases in human B-lymphocyte precursors, triggering apoptosis and clonogenic cell death. Proc. Natl. Acad. Sci. USA 89:9005–9009; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Uckun F. M.; Waddick K. G.; Mahajan S.; Jun X.; Takata M.; Bolen J.; Kurosaki T. BTK as a mediator of radiation-induced apoptosis in DT-40 lymphoma B cells. Science 273:1096–1100; 1996. [DOI] [PubMed] [Google Scholar]
- 56. van Osdol W. W.; Myers T. G.; Pauli K. D.; Kohn K. W.; Weinstein J. N. Use of the Kohonen self-organizing map to study the mechanisms of action of chemotherapeutic agents. J. Natl. Cancer Inst. 86:1853–1859; 1994. [DOI] [PubMed] [Google Scholar]
- 57. Velculescu V. E.; Zhang L.; Vogelstein B.; Kinzler K. W. Serial lnalysis of gene expressson. Science 270:484–487; 1995. [DOI] [PubMed] [Google Scholar]
- 58. Verheij M.; Bose R.; Lin X. H.; Yao B.; Jarvis W. D.; Grant S.; Birrer M. J.; Szabo E.; Zon L. I.; Kyriakis J. M.; Haimovitz-Friedman A.; Fuks Z.; Kolesnick R. N. Requirement for ceramide-initiated SAPK/ JNK signalling in stress-induced apoptosis. Nature 380:75–79; 1996. [DOI] [PubMed] [Google Scholar]
- 59. Vidair C. A.; Chen C. H.; Ling C. C.; Dewey W. C. Apoptosis induced by X-irradiation of rec-myc cells is postmitotic and not predicted by the time after irradiation or behavior of sister cells. Cancer Res. 56:4116–4118; 1996. [PubMed] [Google Scholar]
- 60. Weinstein J. N. Fishing expeditions. Science 282:628–629; 1998. [DOI] [PubMed] [Google Scholar]
- 61. Weinstein J. N.; Kohn K. W.; Grever M. R.; Viswanadhan V. N.; Rubinstein L. V.; Monks A. P.; Scudiero D. A.; Welch L.; Koutsoukos A. D.; Chiausa A. J.; Pauli K. D. Neural computing in cancer drug development: Predicting mechanism of action. Science 258:447–451; 1992. [DOI] [PubMed] [Google Scholar]
- 62. Weinstein J. N.; Myers T. G.; O’Connor P. M.; Friend S. H.; Fornace A. J. Jr.; Kohn K. W.; Fojo T.; Bates S. E.; Rubinstein L. V.; Anderson N. L.; Buolamwini J. K.; Osdol W. W.; Monks A. P.; Scudiero D. A.; Sausville E. A.; Zaharevitz D. W.; Bunow B.; Johnson G. S.; Wittes R. E.; Pauli K. D. An information intensive approach to the molecular pharmacology of cancer. Science 275:343–349; 1997. [DOI] [PubMed] [Google Scholar]
- 63. White E. Life, death, and the pursuit of apoptosis. Genes Dev. 10:1–15; 1996. [DOI] [PubMed] [Google Scholar]
- 64. Woloschak G. E. Radiation-induced responses in mammalian cells. In: Koval T., ed. Stress-inducible processes in higher eukaryotic cells. New York: Plenum Press; 1997:185–219. [Google Scholar]
- 65. Wu G. S.; Burns T. F.; McDonald E. R. III; Jiang W.; Meng R.; Krantz I. D.; Kao G.; Gan D. D.; Zhou J. Y.; Muschel R.; Hamilton S. R.; Spinner N. B.; Markowitz S.; Wu G.; El-Deiry W. S. KILLER/DR5 is a DNA damage-inducible p53-regulated death receptor gene. Nat. Genet. 17:141–143; 1997. [DOI] [PubMed] [Google Scholar]
- 66. Yang E.; Korsmeyer S. J. Molecular thanatopsis: A discourse on the BCL2 family and cell death. Blood 88:386–401; 1996. [PubMed] [Google Scholar]
- 67. Zhan Q.; Alamo I. Jr.; Yu K.; Boise L. H.; O’Connor P. M.; Fornace A. J. Jr. The apoptosis-associated y-ray response of BCL-XL depends on normal p53 function. Oncogene 13:2287–2293; 1996. [PubMed] [Google Scholar]
- 68. Zhan Q.; Bae I.; Fornace A. J. Jr.; Craig R. W. Induction of Bcl-2 family member Mcl-1 as an early response to DNA damage. Oncogene 14:1031–1039; 1997. [DOI] [PubMed] [Google Scholar]
- 69. Zhan Q.; Bae I.; Kastan M. B.; Fomace A. J. Jr. The p53-dependent y-ray response of GADD45 . Cancer Res. 54:2755–2760; 1994. [PubMed] [Google Scholar]
- 70. Zhan Q.; Carrier F.; Fornace A. J. Jr. Induction of cellular p53 activity by DNA-damaging agents and growth arrest. Mol. Cell. Biol. 13:4242–4250; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Zhan Q.; Chen I. T.; Antinore M. J.; Fornace A. J. Jr. Tumor suppressor p53 can participate in transcriptional induction of the GADD45 promoter in the absence of direct DNA binding. Mol. Cell. Biol. 18:2768–2778; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zhan Q.; El-Deiry W.; Bae I.; Alamo I. Jr.; Kastan M. B.; Vogelstein B.; Fornace A. J. Jr. Similarity of the DNA-damage responsiveness and growth-suppressive properties of WAF1 to GADD45 . Int. J. Oncol. 6:937–946; 1995. [DOI] [PubMed] [Google Scholar]
- 73. Zhan Q.; Fan S.; Bae I.; Guillouf C.; Liebermann D. A.; O’Connor P. M.; Fornace A. J. Jr. Induction of BAX by genotoxic stress in human cells correlates with normal p53 status and apoptosis. Oncogene 9:3743–3751; 1994. [PubMed] [Google Scholar]
- 74. Zhan Q.; Kontny U.; Iglesias M.; Alamo I.; Yu K.; Hollander M. C.; Woodworth C. D.; Fornace A. J. Jr. Inhibitory effect of Bcl-2 on p53-mediated transactivation following genotoxic stress. Oncogene (in press). [DOI] [PubMed]