

Abstract
Yeast HSF is constitutively trimeric and DNA bound. Heat shock is thought to activate HSF by inducing a conformational change. We have developed an assay in which we can follow a conformational change of HSF that correlates with activity and thus appears to be the active conformation. This conformational change requires two HSF trimers bound cooperatively to DNA. The conformational change can be induced in whole cell extracts, and is thus amenable to biochemical analysis. We have purified a factor that triggers the conformational change. The factor is sensitive to dialysis, insensitive to NEM, and is not extractable by phenol. It is small, and apparently not a peptide. Mass spectroscopy identifies a novel guanine nucleotide that tracks with activity on columns. This novel nucleotide, purchased from Sigma, induces the conformational change (although this does not prove the identity of the activating factor unambiguously, because Sigma's preparation is contaminated with other compounds). What is the source of this nucleotide in cells? Activity can be generated by treating extracts with ribonuclease; this implicates RNA degradation as a source of HSF-activating activity. The heat shock response is primarily responsible for monitoring the levels of protein chaperones; how can RNA degradation be involved? Synthetic lethal interactions link HSF activity to ribosome biogenesis, suggesting a possible model. Ribosomal proteins are produced in large quantities, and in excess of rRNA; unassembled r-proteins are rapidly degraded (t 1/2 ≈ 3 min). Unassembled r-proteins aggregate readily. It is likely that unassembled r-proteins represent a major target of chaperones in vivo, and for proteasome-dependent degradation. Interference with rRNA processing (e.g., by heat shock) requires hsp70s to handle the aggregation-prone r-proteins, and proteasome proteins to help degrade the unassembled r-proteins before they aggregate. A nucleotide signal could be generated from the degradation products of the rRNA itself.
Keywords: Heat shock, Conformational change, HSF, Saccharomyces cerevisiae, DNA binding protein, Transcription factor
IT is believed that, during stresses such as heat shock, proteins become destabilized and unfold, at least partially. Other stresses interfere with protein synthesis, resulting in the premature release of nascent peptides that cannot fold into a stable conformation. The production of aberrant proteins triggers the heat shock response, which elevates the level of expression of the protein chaperones such as hsp70. These bind to the aberrant proteins, and minimize their likelihood of aggregation, and facilitate their refolding.
Not all aberrant proteins produced during stress can be refolded. These are degraded through the ubiquitin-dependent proteasome system, some components of which are also produced more abundantly during stress. Thus, the heat shock system represents a mechanism by which cells can monitor the level of aberrant proteins, and refold or degrade them.
In eukaryotes the “aberrant protein signal” is communicated to the heat shock transcription factor, HSF, which is the transcription factor primarily responsible for the stress-dependent regulation of the heat shock genes. In the species examined thus far, HSF (HSF1 in species with multiple HSF-related genes) is expressed constitutively, and is “activated” when cells are stressed. HSF activation appears to be a two-step process. The first step is the assembly of HSF monomers into trimers that are capable of binding DNA. The second step of HSF activation is a conformational change that exposes the otherwise masked transcriptional activation domains of the protein. In human cells, the monomer-to-trimer transition can be induced by treatment with salicylate (14), or by overexpression of HSF1 (24). In budding yeast such as Saccharomyces cerevisiae, trimerization and DNA binding occur constitutively (13,29). The second step of HSF activation requires a stress, such as heat shock.
Most attention has been given to the monomer-to-trimer transition. It is clear that this involves both hsp70 (2,3,20,27) and hsp90 (32), which interact with the HSF1 monomer. Presumably, these chaperones maintain the monomeric conformation of HSF, preventing it from assembling into trimers. If the concentration of aberrant proteins increases, however, these may titrate the available chaperones, leaving HSF free to change conformation, and assemble into trimers.
Less is known about the second step of HSF activation, the “unmasking” of the transcriptional activation domains. In S. cerevisiae, it is known that the N-terminal 165 amino acids of HSF participate in masking the transcriptional activators, which reside at the N-terminus and in the C-terminal quarter of the protein (6,8,21,28). It is also known that the central portion of the protein, comprising the DNA binding and trimerization domains, plays a role in heat shock inducibility (6,21). Indeed, a single missense mutation in a highly conserved residue in the DNA binding domain renders HSF at least 40-fold more active under nonstressed conditions (6), indicating that this mutation interferes with the mechanism whereby the transcriptional activation domains are masked. Here, we use this mutation to provide an entry into the analysis of the second step of HSF activation.
MATERIALS AND METHODS
β-Galactosidase Assays
Cultures of YJB378 (a/α trp1-289::TRP1-HSE-lacZ/trpl-289::TRPl-HSE-lacZ ura3-52/ura3-52 leu2-3,112;leu2-3,112 his3Δl/his3ΔHindIII met2/MET2 his4-519/HIS4 adel-100/ADEl LYS2/LYS2::HSE-HIS3 hsflΔ/hsflΔ [YEpHSF·LEU2]) or YJB368 (isogenic with YJB378, but carrying YEpHSFM232V·LEU2) were grown in YPD at 25°C, until cell density reached A600 = 0.7–1.0. Cells were heat shocked at 37°C in a shaking water bath. After the appropriate time, cells were harvested and assayed as described previously (6).
Gel Mobility Shift DNA Binding Assays
Extracts were prepared as previously described (5) from YJB371 (a prcl-407 prbl-1122 pep4-3 leu2trpl ura3-52 hsflΔ [YEpHSF·LEU2]) or isogenic cultures in which YEpHSF·LEU2 was replaced with different HSF plasmids, as indicated in the text. For each binding assay, extract (35 μg of protein) was suspended in 30 (μl of binding reaction [20 mM N-2-hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES; pH 7.9), 1 mM EDTA, 60 mM KC1, 12% glycerol, 1 mM dithiothreitol, 1 μg/1 bovine serum albumin (BSA), 0.1 (iμg/ml sonicated E. coli DNA]. The binding reaction was initiated by addition of 1.3 μg of oligonucleotide probe HSE4T (5′-ACAGGGATCCTGAAGCTTCTAGAAGCTTCCTAGAGT CGACCTGCAG-3′) or HSE6T (5′-ACAGGGATCC TGAAGCTTCTAGAAGCTTCTAGAAGCTTCCT AGAGTCGACCTGCAG-3′) labeled with [oc-32P] dCTP by fill-in synthesis of a second strand after priming with oligo Bsb(5′-CTGCAGGTCGACTC-TAG-3′). ATP (1 mM) was included in the reactions. Reactions were incubated at room temperature for 30 min. To assay column fractions of complex Ill-inducing activity, incubations were supplemented, after 30 min at 25°C, with 5 μl of test fraction dissolved in FCBM (50 mM Tris, 7.4, 50 mM KC1, 10% glycerol, 3 mM MgCl2), and incubated an additional 30 min. After incubation, reactions were chilled and loaded at 4°C onto a 4–10% polyacrylamide gel (prepared and run in 22.5 mM Tris-base, 22.5 mM boric acid, 0.63 mM EDTA, or, for the experiment of Fig. 3, 50 mM Tris-base, 380 mM glycine, 0.63 mM EDTA). Gels were run at 4°C for 4800 volt-hours.
FIG. 3.
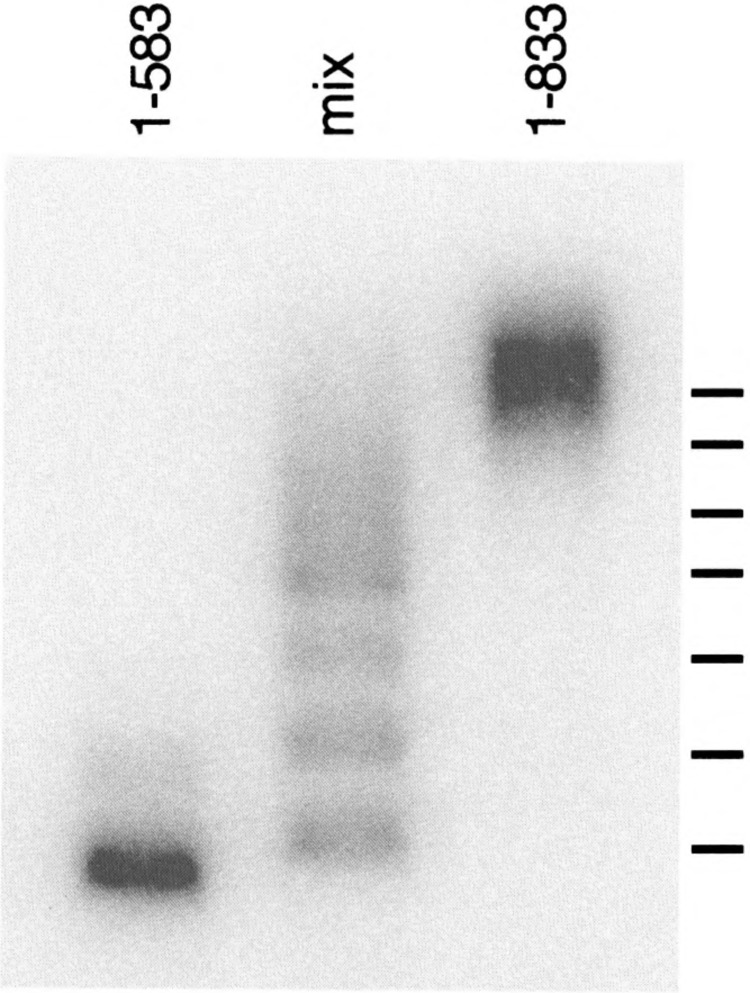
Heteromultiraer analysis of complex III. Extracts were prepared from heat-shocked cells carrying either truncated HSF (HSF1–583) or full-length HSF (HSF1–833), each with the M232V mutation. The extracts were used alone, or mixed, in a gel mobility shift DNA binding assay using Tris-glycine gel buffer. The positions of predicted mobilities for heteromultimers formed from six HSF monomers are indicated. The HSF1–583 expression vector was prepared by inserting a 9-base oligonucleotide carrying an in-frame stop codon into a Stul restriction site. The stop codon was later discovered to be subject to translational readthrough [see (15)]; cells carrying this plasmid express a small amount of full-length readthrough product (detectable by Western blot, not shown), which accounts for the appearance of heteromultimers in the “1-583 only” lane.
Quantitation was performed after phosphorimager analysis by integrating under the peaks of traces of individual lanes, in order to avoid complications due to background (and thus the baseline above which the signal is detected) or “smiling” of the bands. Guanosine 5′-phosphate 2′,3′-cyclic phosphate (5G2^3) was purchased from Sigma Chemical Company and dissolved in water. The relative concentration of 5G2^3 was determined by mass spectroscopy; concentrations given in Fig. 6 are based on this determination.
FIG. 6.
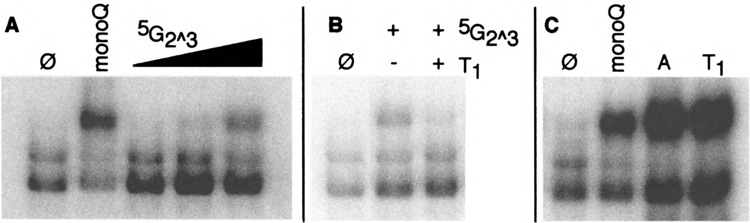
Induction of complex ffl by 5G2^3 and ribonuclease. (A) Commercially prepared 5G2^3 was tested at 0.625, 1.45, and 2.5 mM. Untreated extract (Ø) and extract treated with the monoQ fraction from Fig. 5 were run in parallel. (B) 5G2^3, was treated with RNAse T1 then analyzed. (C) Extracts were treated with RNAse A or RNAse T1 during the DNA binding reaction and assayed by gel mobility shift, with untreated extract (Ø) and extract treated with the monoQ fraction as controls
Preparation of Complex Ill-Inducing Activity
Extracts were prepared from heat-shocked cells, as for gel mobility shift DNA binding assays, then boiled for 10 min and clarified by centrifugation at 13,000 rpm at 4°C. The supernatant was then passed over affigel blue and heparin agarose, and the unbound fraction passed over DEAE sephadex. The bound material was eluted at 500 mM NaCl, and precipitated with 6 volumes of 95% ethanol. The precipitate was resuspended in FCBM, and subjected to HPLC fraction on monoQ as described in the text. The active fraction was again precipitated and resuspended in FCBM for assay of activity, or in H20 for mass spectroscopy.
Mass Spectroscopy
Analyses of the samples by mass spectrometry were performed using a delayed-extraction linear time-of-flight matrix-assisted laser desorption/ioniza-tion (MALDI) mass spectrometer in negative ion mode. A matrix of 0.2 μl of 3-hydroxypicolinic acid (saturated) and 0.2 μl of trihydroxyammonium citrate (1 molar) was combined with 1 μ1 of a sample aliquot. The dried solution of matrix and sample was laser desorbed using a frequency-tripled neodymium/ yttrium-aluminum garnet (Nd:YAG) laser at 355 run. Masses were calibrated using a range of nucleotide bases and nucleotides of known mass and purity.
RESULTS
Replacement of methionine232 with valine (M232V) drastically elevates the activity of yeast HSF (Fig. 1A), such that the expression of a lacZ reporter gene is elevated 40-fold under nonstressed conditions. Upon heat shock, HSFM232V mutant cells nonetheless display a modest increase in lacZ expression, indicating that HSFM232V is highly, but not completely, deregulated. Interestingly, HSFM232V displays at least as much activity in nonshocked cells as does wild-type HSF in heat-shocked cells, and even greater activity after heat shock. This suggests that wild-type HSF does not completely unmask its transcriptional activation domains upon heat shock, or that heat shock induces only a fraction of the HSF molecules in the cell to adopt the high-activity conformation.
FIG. 1.
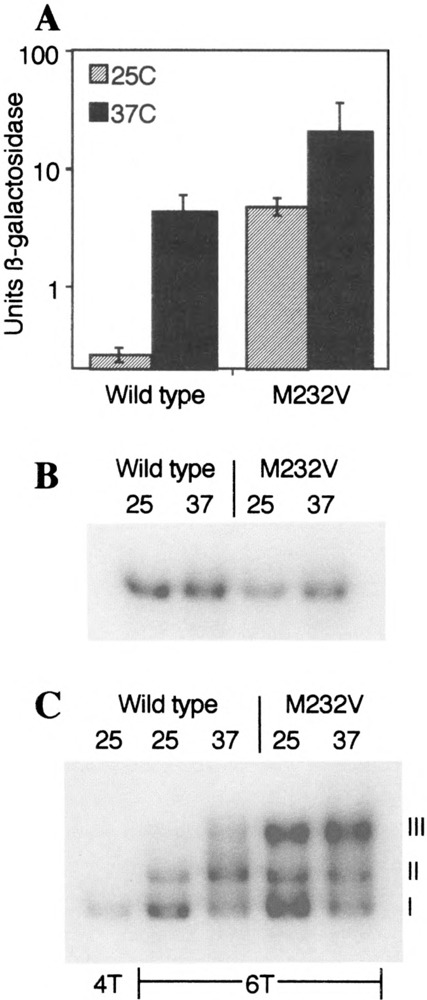
The effect of the M232V mutation. (A) Yeast carrying a HSE-lacZ reporter gene, the hsfJ-Δ disruption, and an episomal plasmid expressing either wild-type HSF or HSFM232V were grown at 25°C, then assayed for β-galactosidase activity either directly or after 1 h at 37°C. (B) Protease-deficient yeast expressing either wild-type HSF or HSFM232V were used to prepare whole cell extracts after growth at 25°C or at 25°C followed by a 30-min heat shock at 37°C. Extracts were then used for gel mobility shift DNA binding assay using a 32P-labeled DNA probe (HSE4T) to which a single HSF trimer can bind. (C) The extracts from (B) were reanalyzed using a DNA probe (HSE6T) to which two trimers can bind simultaneously. The 25°C sample of wild-type was also assayed with DNA probe 4T, in order to identify the position of the DNA-bound trimer.
The M232V mutation is in the DNA-contact α-helix of HSF (11,30). To determine whether the mutation affects DNA binding, we examined the ability of the mutant protein to bind to an oligonucleotide that accommodates only a single HSF trimer, using an electrophoretic mobility shift assay. We were unable to detect any differences in binding between the wild-type protein and the M232V mutant (Fig. 1B), even when the assays were performed kinetically to examine association and dissociation rates (not shown).
Although HSFM232V showed no difference from wild-type with respect to the binding of a single HSF trimer to DNA, the mutation had a remarkable effect on the binding of two trimers cooperatively. Figure 1C shows that HSF from nonshocked wild-type cells exhibits primarily two bands in this assay, the lower of which (complex I) corresponds to the trimer, the upper of which (complex II) corresponds to two trimers (5). HSF from heat-shocked cells displays, in addition, a third band (complex III) that is of slower mobility than either complex I or complex II. HSFM232V forms much more complex III; indeed, the majority of the probe is in complex III after heat shock.
The appearance of complex III in heat-shocked wild-type extracts, and in HSFM232V extracts, suggested that complex III might be the high-activity conformation of HSF. To assess how well complex III correlates with activity, we examined both β-galactosidase activity and complex III formation during the course of a heat shock and subsequent recovery. There is a very good correlation between HSF activity, as measured by the lacZ reporter gene (Fig. 2A), and the formation of complex III (Fig. 2B). We also asked whether other stress agents, such as ethanol or hydrogen peroxide, might also induce the formation of complex III; they did (Fig. 2C). These correlations suggest that complex III does, indeed, represent the “active” form of HSF.
FIG. 2.
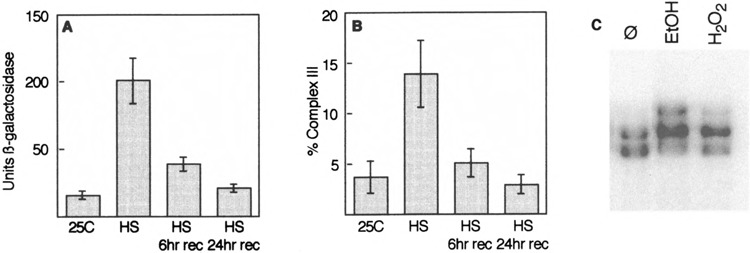
Complex III correlates with HSF activity. (A) Cells carrying a HSE-lacZ reporter gene were grown at 25°C, then heat shocked at 37°C for 1 h, then returned to 25°C for recovery. Samples were taken at the indicated times for assay of (β-galactosidase activity. (B) Cells were grown as for (A), then used to prepare extracts for gel mobility shift DNA binding assay. The distribution of label in complexes I, II, and III was determined by phosphorimager, and the percentage of complex III calculated. (C) Cells were exposed to 6% ethanol or to 125 pM H202 for 30 min, then used to prepare extracts for gel mobility shift DNA binding assay. An extract from untreated cells (Ø) was run on the same gel for comparison.
We have previously shown that complex II represents two trimers of HSF bound to DNA (5); complex III exhibits a slower mobility. Is complex III composed of more protein subunits, or does it have the same composition as complex II, but in a different conformation? Our gel mobility shift assays were performed with the addition of ATP, which dissociates hsp70 from HSF (23). We were unable to detect hsp70 or any other protein associated with complex III by EGS cross-linking (not shown). If non-HSF proteins exist in complex III, they have thus far escaped detection by our techniques.
To determine how many HSF subunits are in complex III, we took advantage of the fact that C-terminally truncated HSF molecules also form complex III both upon heat shock and when they carry the M232V mutation. Using HSF1–583 as a short form, and full-length HSF1–833 as a long form, both carrying M232V, we performed a mixed-extract DNA binding experiment, in which the different forms of HSF can associate as heteromultimers. To ensure that we examined only complex III, we used heat-shocked cells, and ran the gel mobility shift assays in a buffer system (Tris-glycine) in which only complex III is detected. The mixed extract (Fig. 3) exhibits a large number of bands, the mobilities of which are nearly identical to those predicted for the heteromulitimers of a complex containing six HSF monomers. These observations lead us to conclude that complex III represents two trimers of HSF, but in a different conformation from those that comprise complex II.
It is possible to induce the formation of complex III by an in vitro heat shock (Fig. 4A). We took advantage of this finding to ask whether complex III forms de novo, or from preexisting complexes I and II. To do so, we incubated a nonshocked extract with labeled probe, then added a 1000-fold excess of unlabeled competitor, and shifted the extract to 37°C and loaded samples onto a gel at various times thereafter. Although the total amount of label in complexes I, II, and III decreased as expected after adding competitor (Fig. 4B, C), the amount of label in complex III actually increased at 37°C (Fig. 4C), whereas the amounts of complexes I and II decreased much more rapidly than they did at 25°C (Fig. 4B). Because the competitor blocks new binding of HSF to the labeled probe (not shown), the increase in complex III can be explained only by conversion of complexes I and/or II into complex III.
FIG. 4.
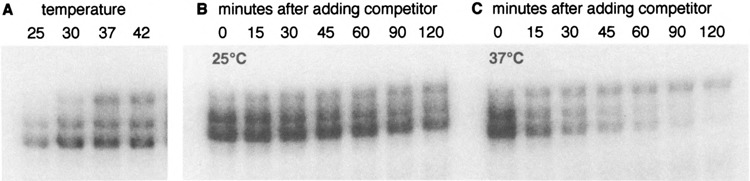
In vitro heat shock induces complex III. (A) Extracts from nonshocked cells were used in gel mobility shift DNA binding assays, in which the DNA binding reactions were performed at various temperatures. (B) A nonshocked extract was incubated in a 25°C DNA binding reaction for 30 min, then a 1000-fold excess of unlabeled HSE6T probe was added; incubation was continued at 25°C. At various times thereafter, samples were withdrawn and applied to a (running) gel for analysis. (C) Analogous to (B), except that the reaction was shifted to 37°C after addition of unlabeled competitor.
The ability to generate complex III by in vitro heat shock of an extract from nonshocked cells suggested to us that the biochemistry of complex III formation might be accessible using this system. Thus, we sought to determine whether it might be possible to identify an activity in extracts from heat-shocked cells that, when added to a DNA binding reaction using extract from nonshocked cells, would induce complex III formation in the recipient extract. We initially expected to identify a kinase or other protein factor that might act to modify HSF, thus triggering the conformational change. We were therefore surprised to discover that, while a complex Ill-inducing activity can be purified, it is dialyzable, resistant to extraction with phenol, unaffected by N-ethylmaleimide, and stable to boiling. Figure 5 shows its fractionation on monoQ by HPLC. The active fraction has a UV absorbance profile indicative of nucleotides, not amino acids.
FIG. 5.
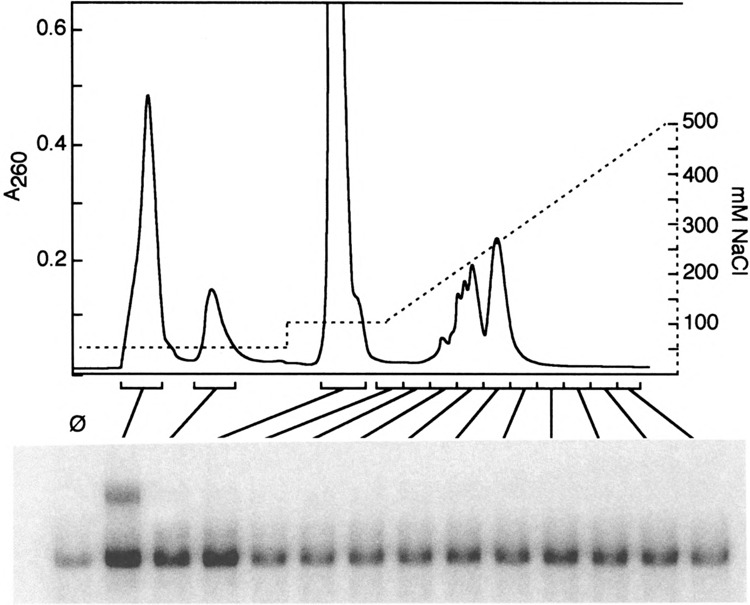
HPLC fractionation of complex Ill-inducing activity. Partially purified complex Ill-inducing activity was loaded onto monoQ at 50 mM NaCl, washed with 100 mM NaCl, then eluted with a 100–500 mM NaCl gradient. Fractions were assayed for their ability to induce formation of complex III. Ø indicates the 25°C extract with no additions.
Mass spectroscopy of the active fractions showed multiple peaks, only one of which (422.5 mass units) correlated with activity. This is the mass of guanosine 5′-phosphate 2′,3′-cyclic phosphate, which we will refer to as 5G2^3. Addition of a commercial preparation of 5G2^3 also showed activity (Fig. 6A). Unfortunately, this does not confirm the identity of the inducing factor as 5G2^3, for the simple reason that the commercial preparation was quite impure, as determined by mass spectroscopy. Activity could potentially be due to a minor component of our partially purified factor, which exists as a contaminant of the 5G2^3 preparation.
2′,3′-Cyclic nucleotides are intermediates in RNA degradation by a variety of ribonucleases. If 5G2^3 were able to change HSF conformation, then it seemed likely that treatment of 5G2^3 with ribonuclease Ti should eliminate activity; it did (Fig. 6B). It furthermore seemed likely that addition of ribonuclease to a whole cell extract should induce the formation of complex III. This prediction was borne out only in part (Fig. 6C). Both ribonuclease A (which cleaves after G residues) and ribonuclease A (which cleaves after pyrimidines) induced complex III, even though the latter cannot produce oligo- or mononucleotides with terminal guanosine 2′,3′-cyclic phosphates. This suggests either that nucleotides besides 5G2^3 can signal HSF (e.g., 5U2^3) or that “aberrent” fragmented RNAs (such as might be generated by ribonuclease treatment in our extracts) may be ultimately degraded by a “scavenger” ribnonuclease that generates the true HSF signaling molecule. Although the impurity of the preparations available to us at present precludes a definitive identification of the complex Ill-inducing activity, these observations suggest that it derives from an aspect of RNA metabolism, rather than protein denaturation.
Returning, briefly, to the HSFM232V mutant, we have asked whether cells carrying this mutation have a greater concentration of the complex Ill-inducing factor than do wild-type cells. They do not (data not shown). This indicates that the M232V mutation renders HSF particularly sensitive to the conformational change that is caused by this factor.
DISCUSSION
Before offering interpretations of our findings, it is necessary to point out the caveats. First, our evidence that complex III represents the high-activity form of HSF is correlative, not direct. Nonetheless, complex III does appear and disappear in parallel with HSF activity during the course of a heat shock and recovery, it does appear when cells are stressed in several different ways, and it appears very efficiently with a mutant form of HSF (M232V) that has exceptionally high activity. Together, these findings are strongly suggestive of complex III being an active form of HSF.
Second, our evidence that complex III consists solely of two HSF trimers bound to DNA is based, in part, on negative results. We have been unable to identify any other, non-HSF component of this complex, but cannot rule out the possibility that one may exist that is undetectable by our methods.
Third, we remain uncertain as to the true identity of the activating factor. 5G2^3 appears to be an excellent candidate, but until such time as we have purified the activating factor to homogeneity, and/or such time as we can test a pure preparation of 5G2^3, its identity must remain in doubt.
Lastly, we have thus far used whole cell extracts for our assays. We have shown that these extracts can respond to in vitro heat shock and to the addition of ribonuclease, and can thus generate complex Ill-inducing activity. It is therefore possible that 5G2^3 represents a precursor to the active molecule. The use of whole cell extracts also leaves one other question unanswered: whether the activating factor acts directly on HSF as an allosteric regulator, or whether it triggers the activity of an HSF-modifying enzyme. We believe that our assay system should have identified the latter, should it exist, and therefore favor the concept of allosteric regulation.
With these caveats in mind, it is intriguing to consider our most striking findings. First, it is surprising that the conformational change that gives rise to complex III is detectable only when HSF is bound to DNA as two trimers, side-by-side. A single DNA-bound trimer shows no change in gel mobility under otherwise identical conditions. This could indicate that, for single trimers, the conformational change does not lead to a sufficient change in cross-sectional area to affect gel mobility. Alternatively, it is possible that the conformational change requires the cooperative interaction of two trimers. This latter possibility is intriguing in view of several observations. First, we have previously shown that a heat shock element that favors cooperative interactions is more effective in yeast cells than one that does not (5). Second, Cohen and Meselson have shown that the cooperative interaction is necessary for heat shock-induced transcription of the Drosophila hsp70 gene (9). Third, natural heat shock elements contain sufficient nGAAn motifs for the cooperative interaction of two or more trimers of HSF (26). These latter findings may indicate that, in vivo as well as in our in vitro assays, the conformational change of HSF is facilitated by interactions between DNA-bound trimers.
A second surprising observation is the apparent involvement of a small molecule—perhaps a nucleotide—in the conformational change of HSF. These data suggest that an allosteric effector may trigger HSF activation. The possibility of allosteric regulation, although popular at one time, has fallen into disfavor. This has been due partly to the lack of discovery of such a molecule, which is not surprising given the dependence of the conformational change, in our assays, on two DNA-bound trimers. It has also been due to the findings that heat shock proteins such as hsp70 and hsp90 can influence HSF activity (2,3,20,27,32), thus providing a satisfying explanation for HSF regulation. However, much of the information on the interaction of these chaperones with HSF has focused on the monomer-to-trimer transition of metazoan HSF, rather than on conformational changes that occur to HSF once it has bound DNA. Our data do not speak to the involvement of these chaperones in the regulation of yeast HSF, which is structurally distinct from metazoan HSF, and for which the monomer-to-trimer transition is constitutive. Rather, our findings suggest an additional layer of regulation beyond the role of the chaperones themselves.
Our most unexpected finding is that ribonuclease can induce the formation of complex III. This suggests that RNA degradation may play a role in the activation of HSF. This is entirely unexpected, inasmuch as the heat shock system is well known to be involved in the regulation of expression of protein chaperones, and has not heretofore been strongly linked to RNA metabolism.
In searching for models to link RNA metabolism and the heat shock response, we suggest two general possibilities. First, it is conceivable that RNA degradation in our extracts causes a variety of RNA binding proteins to lose their stable folding patterns, and trigger an unfolded protein response. This mechanism may play a role in our experiments, and cannot be ruled out. This is the simplest explanation that retains its focus on unfolded proteins—but it does not offer an origin for the small molecule that induces the conformational change. A second possibility, however, is suggested by synthetic lethal interactions between HSF, RNA binding proteins, and proteasome sub-units (J. J. Bonner, unpublished; to be described elsewhere). These interactions have led us to consider ribosomal RNA processing, and ribosome biogenesis as a major metabolic system for which the heat shock response may be expected to be important.
Cells expend a tremendous amount of energy to make ribosomes. The majority of cellular RNA is rRNA; in dividing cells, the major class of mRNAs represents genes for ribosomal proteins. Every cell division, the vast store of ribosomes must be duplicated. With 80 or more ribosomal proteins, and the several rRNAs, it is no easy matter to coordinate the synthesis of all these individual components. In yeast, there is some degree of transcriptional regulation of ribosomal proteins (12,16,19), but the most striking regulation is at the level of protein stability. Unassembled ribosomal proteins have remarkably short half-lives, on the order of a few minutes; due to this rapid degradation, overexpression of ribosomal protein genes leads to virtually no increase in the abundance of the proteins; the excess protein is degraded (1,18,31).
We can imagine the evolutionary advantage of such exquisite regulation. Ribosomal proteins are highly prone to aggregation. It is thus advantageous to keep the pool of unassembled ribosomal proteins from increasing to a level sufficient to build toxic intracellular aggregates. Rapid degradation of excess ribosomal proteins ensures that this is unlikely to occur.
Unasssembled ribosomal proteins thus represent a major class of short-lived proteins. Short-lived proteins are typically degraded by the proteasome system, some components of which are heat shock inducible. Furthermore, unassembled ribosomal proteins are quite likely not in their mature conformations, and may thus represent a major class of chaperone substrates in vivo. Thus, for two reasons—chaperoning, and the degradation of excess protein—a case can be made for a strong link between ribosome biogenesis and the activity of HSF.
Upon heat shock, ribosomal RNA processing is blocked (4,7,10,22,25). With ribosome biogenesis thus blocked, the pool of unassembled ribosomal proteins would seem likely to increase markedly—with the potential for formation of protein aggregates. This, in turn, would be expected to increase demand for proteolysis as well as chaperone activity. It is a reasonable inference that this increased demand is met through the activation of HSF.
Although the unassembled ribosomal proteins themselves should serve as a trigger for HSF activation by means of the chaperone-dependent regulatory mechanisms that have been previously described (2,3,20,27,32), our data suggest that an additional mechanism works as well, via a potential allosteric effector of HSF. How is this factor generated in vivo? One clear possibility is that it, like the alarmones described by Ames and colleagues (17), is synthesized de novo by one or more enzymes involved in protein synthesis. An alternative that is particularly attractive in view of our finding that ribonuclease can stimulate the HSF conformational change is that pre-rRNA, having been blocked from normal processing, is itself degraded by a scavenger ribonuclease that generates the signaling nucleotide as part of its catalytic mechanism.
It remains to be seen whether a model such as this is at all realistic. Even if the details prove somewhat different from what we have suggested, it will have been significant in itself that these findings have alerted us to the ribosomal proteins as serious candidates for critical targets of the heat shock system, ones whose assembly and stability must be carefully monitored both under normal conditions, and during stress.
ACKNOWLEDGMENT
This work was supported by grant GM51853 from the National Institutes of Health.
REFERENCES
- 1. Abovich N.; Gritz L.; Tung L.; Rosbash M. Effect of RP51 gene dosage alterations on ribosome synthesis in Saccharomyces cerevisiae . Mol. Cell. Biol. 5:3429–3435; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Abravaya K.; Myers M. P.; Murphy S. P.; Morimoto R. I. The human heat shock protein hsp70 interacts with HSF, the transcription factor that regulates heat shock gene expression. Genes Dev. 6:1153–1164; 1992. [DOI] [PubMed] [Google Scholar]
- 3. Baler R.; Welch W. J.; Voellmy R. Heat shock gene regulation by nascent polypeptides and denatured proteins: hsp70 as a potential autoregulatory factor. J. Cell Biol. 117:1151–1159; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bell J.; Neilson L.; Pellegrini M. Effect of heat shock on ribosome synthesis in Drosophila melanogaster . Mol. Cell. Biol. 8:91–95; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bonner J. J.; Ballou C.; Fackenthal D. L. Interactions between DNA-bound trimers of the yeast heat shock factor. Mol. Cell. Biol. 14:501–508; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bonner J. J.; Heyward S.; Fackenthal D. L. Temperature-dependent regulation of a heterologous transcriptional activation domain fused to yeast HSF. Mol. Cell. Biol. 12:1021–1030; 1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bouche G.; Raynal F.; Amalric F.; Zalta J. P. Unusual processing of nucleolar RNA synthesized during a heat shock in CHO cells. Mol. Biol. Rep. 7:253–258; 1981. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y.; Bariev N. A.; Westergaard O.; Jakobsen B.K. Identification of the C-terminal activator domain in yeast heat shock factor: Independent control of transient and sustained transcriptional activity. EMBO J. 12:5007–5018; 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cohen R. S.; Meselson M. Perrodic interactions of heat shock transcriptional elements. Nature 332:856–858; 1988. [DOI] [PubMed] [Google Scholar]
- 10. Ghoshal K.; Jacob S. T. Heat shock inhibits pre-rRNA processing at the primary site in vitro and alters the activity of some rRNA binding proteins. J. Cell. Biochem. 62:506–515; 1996. [DOI] [PubMed] [Google Scholar]
- 11. Harrison C. J.; Bohm A. A.; Nelson H. C. M. Crystal structure of the DNA binding domain of the heat shock transcription factor. Science 263:224–227; 1994. [DOI] [PubMed] [Google Scholar]
- 12. Herruer M. H.; Mager W. H.; Woudt L. P.; Nieuwint R. T.; Wassennar G. M.; Groenveld P.; Planta R. J. Transcriptional control of yeast ribosomal protein synthesis during carbon-source upshift. Nucleic Acids Res. 15:10133–10144; 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jakobsen B. K.; Pelham H. R. B. Constitutive binding of yeast heat shock factor to DNA in vivo. Mol. Cell. Biol. 8:5040–5042; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jurivich D. A.; Sistonen L.; Kroes R. A.; Morimoto R. I. Effect of sodium salicylate on the human heat shock response. Science 255:1243–1245; 1992. [DOI] [PubMed] [Google Scholar]
- 15. Kopczynski J. B.; Raff A. C.; Bonner J. J. Transitional readthrough at nonsense mutations in the HSF1 gene of Saccharomyces cerevisiae . Mol. Gen. Genet. 234:369–378; 1992. [DOI] [PubMed] [Google Scholar]
- 16. Kraakman L. S.; Griffioen G.; Zerp S.; Groenveld P.; Thevelein J. M.; Mager W. H.; Planta R. J. Growth-related expression of ribosomal protein genes in Saccharomyces cerevisiae . Mol. Gen. Genet. 239:196–204; 1993. [DOI] [PubMed] [Google Scholar]
- 17. Lee P. C.; Bochner B. R.; Ames B. N. AppppA, heat-shock stress, and cell oxidation. Proc. Natl. Acad. Sci. USA 80:7496–7500; 1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maicas E.; Pluthero F. G.; Friesen J. D. The accumulation of three yeast ribosomal proteins under conditions of excess mRNA is determined primarily by fast protein decay. Mol. Cell. Biol. 8:169–175; 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moehle C. M.; Hinnebusch A. G. Association of RAP1 binding sites with stringent control of ribosomal protein gene transcription in Saccharomyces cerevisiae . Mol. Cell. Biol. 11:2723–2735; 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mosser D. D.; Duchaine J.; Massie B. The DNA-binding activity of the human heat shock transcription factor is regulated in vivo by hsp70. Mol. Cell. Biol. 13:5427–5438. 1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nieto-Sotelo J.; Wiederrecht G.; Okuda A.; Parker C. S. The yeast heat thock transcription factor contains a transcriptional activation domain whose activity is repressed under nonshock conditions. Cell 62:907–917; 1990. [DOI] [PubMed] [Google Scholar]
- 22. Nover L.; Munsche D.; Neumann D.; Ohme K.; Scharf K. D. Control of ribosome biosynthesis in plant cell cultures under heat-shock conditions. Ribosomal RNA. Eur. J. Biochem. 160:297–304; 1986. [DOI] [PubMed] [Google Scholar]
- 23. Palleros D. R.; Reid K. L.; Shi L.; Welch W. J.; Fink A. L. ATP-induced protein-hsp70 complex dissociation requires K+ but not ATP hydrolysis. Nature 365:664–666; 1993. [DOI] [PubMed] [Google Scholar]
- 24. Rabindran S. K.; Wisniewski J.; Li L.; Li G. C.; Wu C. Interaction between heat thock factor and hsp70 is insufficient to suppress induction of DNA-binding activity in vivo. Mol. Cell. Biol. 14:6552–6560; 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sadis S.; Hickey E.; Weber L. A. Effect of heat shock on RNA metabolism in HeLa cells. J. Cell Physiol. 135:377–386; 1988. [DOI] [PubMed] [Google Scholar]
- 26. Scharf K. D.; Materna T.; Treuter E.; Nover L. Heat stress promoters and transcription factors. Results Probl. Cell Differ. 20:125–162; 1994. [DOI] [PubMed] [Google Scholar]
- 27. Shi Y.; Mosser D. D.; Morimoto R. I. Molecular chaperones as HSF1-specific transcriptional repressors. Genes Dev. 12:654–666; 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sorger P. K. Yeast heat shock factor contains separable transient and sustained response transcriptional activators. Cell 62:793–805; 1990. [DOI] [PubMed] [Google Scholar]
- 29. Sorger P. K.; Lewis M. J.; Pelham H. R. B. Heat shock factor is regulated differently in yeast and HeLa cells. Nature 329:81–84; 1987. [DOI] [PubMed] [Google Scholar]
- 30. Torres F. A. G.; Bonner J. J. Genetic identification of the site of DNA contact in the yeast heat shock transcription factor. Mol. Cell. Biol. 15:5063–5070; 1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Warner J. R.; Mitra G.; Schwindinger W. F.; Stu-deny M.; Fried H. M. Saccharomyces cerevisiae coordinates accumulation of yeast ribosomal proteins by modulating mRNA splicing, translational initiation, and protein turnover. Mol. Cell. Biol. 5:1512–1521; 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zou J.; Guo Y.; Guettouche T.; Smith D. F.; Voellmy R. Repression of heat shock transcription factor HSF1 activation by HSP90 (HSP90 complex) that forms a stress-sensitive complex with HSF1. Cell 94:471–80; 1998. [DOI] [PubMed] [Google Scholar]