

Abstract
Determining the impact of lipid electrophile-mediated protein damage that occurs during oxidative stress requires a comprehensive analysis of electrophile targets adducted under pathophysiological conditions. Incorporation of ω-alkynyl linoleic acid into the phospholipids of macrophages prior to activation by Kdo2-lipid A, followed by protein extraction, click chemistry, and streptavidin affinity capture, enabled a systems-level survey of proteins adducted by lipid electrophiles generated endogenously during the inflammatory response. Results revealed a dramatic enrichment for membrane and mitochondrial proteins as targets for adduction. A marked decrease in adduction in the presence of MitoTEMPO demonstrated a primary role for mitochondrial superoxide in electrophile generation and indicated an important role for mitochondria as both a source and target of lipid electrophiles, a finding that has not been revealed by prior studies using exogenously provided electrophiles.
Graphical Abstract
Oxidative stress is associated with numerous pathologies, including cardiovascular disease, neurodegenerative diseases, and cancer.1 Reactive oxygen species (ROS) generated during oxidative stress react with polyunsaturated fatty acids (PUFAs), leading to increased production of electrophilic lipid species that can form adducts on nucleophilic groups of cellular macromolecules, including proteins.2 Increased levels of lipid electrophile-adducted proteins have been observed in many disease states,3–5 and adduction can impact protein function, altering cellular signaling.2,6,7 However, the role of lipid electrophiles in human pathology is not fully understood, partially due to the inability to globally identify and quantify protein targets of adduction.
Currently, there are three general approaches to study protein adduction by lipid electrophiles. Antibodies8 and probes9 have been used to identify gross changes in adduction and to capture and identify proteins targeted by a specific lipid electrophile. Alternatively, proteomics approaches have been employed to identify and quantify proteins adducted by known electrophiles in vivo10 and in cells.11 Both of these methods can be applied to physiologically relevant settings but are constrained to studying electrophile adducts of known structure. Other analyses have relied on bolus dosing of exogenously formed electrophiles to cultured cells.6,12,13 This method enables the accurate identification of proteins that are susceptible to adduction by exogenous electrophiles and the assignment of relative reactivities to nucleophilic amino acid residues. However, electrophile generation in vivo does not occur at discrete concentrations, at distinct time points, or extracellularly, so these experiments miss potential cellular microenvironmental factors that modulate electrophile formation and reactivity. Collectively, each method that has been used to study lipid electrophile adduction of proteins fails to address at least one aspect of electrophile formation, including physiologically relevant time scales, cellular location, concentration, or the diverse array of electrophiles generated.
Recently, we expanded the study of lipid electrophile adduction of proteins by introducing alkyne tags remote from the electrophilic group. Placement of the alkyne distant from the electrophilic groups results in no differences in adduction chemistry when compared to the native electrophiles.14,15 The alkynyl group of these electrophiles enables the selective recovery of adducted proteins by attaching an affinity tag via click chemistry.12,15,16 In these experiments, the tagged electrophiles were added exogenously at a fixed concentration and time point. Thus, the previously mentioned criticisms of the approach still apply. Consequently, to study lipid electrophiles in a physiological setting, we have developed a series of ω-alkynyl PUFAs that can be incorporated into cells as sources of endogenously generated lipid electrophiles.
We have reported that the susceptibility to and products of in vitro autoxidation of ω-alkynyl linoleic acid (aLA) and ω- alkynyl arachidonic acid (aAA) are similar to those of native linoleic acid (LA) and arachidonic acid (AA), respectively.17 Therefore, we expect equivalent electrophilic products of native and alkynyl fatty acids to be generated when the latter are oxidized in physiological settings. Here, we demonstrate that aLA is transformed into aAA and that both fatty acids are broadly distributed into the phospholipids of RAW264.7 macrophages. Oxidation of the incorporated alkynyl fatty acids upon activation of the cells with the lipopolysaccharide- mimetic Kdo2-lipid A (KLA)18 leads to increased electrophile formation and protein adduction. We combined stable isotope labeling with amino acids in cell culture (SILAC),19 click chemistry, and previously described affinity enrichment techniques12,15,16 to explore both protein expression changes and adduction targets of lipid electrophiles endogenously generated during macrophage activation. These studies identified over 1000 protein adduction targets, including enzymes involved in energy generation and oxidant defense. Furthermore, this comprehensive and biologically relevant assessment of lipid electrophile-mediated protein damage demonstrates a striking difference between protein targets of endogenously generated and those of exogenously provided lipid electrophiles.
RESULTS/DISCUSSION
aLA and aAA Are Incorporated into Phospholipids of RAW264.7 Macrophages, and aAA Is Released in Response to an Inflammatory Stimulus.
Robust incorporation of alkynyl PUFAs across all phospholipid pools of RAW264.7 macrophages was observed following incubation with either aLA or aAA. As seen in Figure S1, aLA is incorporated into phospholipids at similar levels to those seen for LA in unincorporated cells. In contrast, the total amount of aAA incorporated far exceeds the amount of AA present in the unincorporated cells (Figure S2). This agrees with a previous study demonstrating that, relative to primary resident peritoneal macrophages, RAW264.7 macrophages are AA- deficient and readily incorporate the fatty acid if it is provided.20
Macrophages that had been preincubated in the presence or absence of aLA or aAA were then treated with KLA or a vehicle control. KLA is a chemically defined lipopolysaccharide that acts on the Toll-like receptor-4 (TLR-4) to stimulate the production of ROS and induce the release of large amounts of AA and its oxygenated metabolites from the phospholipid bilayer.18,21 As indicated in Figure S2, KLA treatment mediated the release of both AA and aAA in similar amounts from similar phospholipid pools. This result is consistent with previous studies demonstrating that aAA-incorporated cells generate enzymatic metabolites of aAA when stimulated.17,22 We did not see alterations in the phospholipid levels of LA or aLA in response to KLA treatment (Figure S1).
Both aLA and aAA Are Incorporated into the Phospholipids of aLA-Treated RAW264.7 Macrophages.
Previously, we noted the presence of aAA in aLA-incorporated RAW264.7 macrophages.23 LA is an essential fatty acid that serves as the precursor for the synthesis of AA and other signaling PUFAs.24–26 Thus, we investigated the conversion of aLA to aAA by RAW264.7 macrophages (Figure 1A). The expected elongation pathway of aLA starts with the desaturation of aLA to ω-alkynyl /-linolenic acid (aGLA) by fatty acid desaturase 2, then the elongation of aGLA to ω- alkynyl dihomo-γ-linolenic acid (aDGLA) by an elongase, and finally the desaturation of aDGLA to aAA by fatty acid desaturase 1. All of the expected intermediates were observed, indicating that the placement of the alkyne on the fatty acid does not prevent enzymatic processing in any step in the pathway. Approximately 80% of the aLA taken up by the macrophages was converted to one of the elongation products, with aAA comprising nearly 60% of the detected alkynyl fatty acids.
Figure 1.
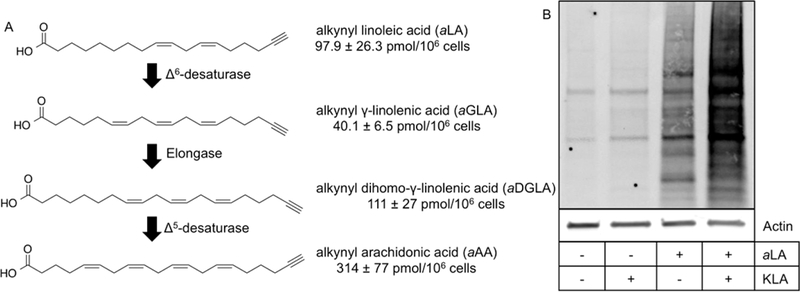
(A) aAA biosynthesized from aLA. Elongation products of aLA to aAA were measured in aLA-incorporated RAW264.7 macrophages. Quantities of each intermediate are the mean ± standard deviation of triplicate analyses. (B) KLA activation induces lipid electrophile protein adduction. Lipid electrophiles are generated during normal physiological processes of the cell (+aLA/-KLA), and increased adduction is measured across the proteome by endogenously formed lipid electrophiles in activated macrophages (+aLA/+KLA).
Consistent with the results in Figure S1, during aLA incorporation into RAW264.7 cell phospholipids, the amount of LA decreased, but the total aLA + LA pool did not change, suggesting that aLA displaced LA. aAA was also incorporated into phospholipids following its biosynthesis from aLA, but aAA did not displace AA Instead, as shown in Figure S4, it increased the total pool size of aAA + AA-containing phospholipid species in agreement with the data in Figure S2.
Taken in totality, we have shown that alkynyl fatty acids are incorporated into, metabolized, and released from the phospholipid bilayer in a manner similar to that of native fatty acids. As noted above, we previously demonstrated that the autoxidation of these alkynyl PUFAs is also equivalent to that of the native species.17 Together, these findings validate the use of alkynyl fatty acid surrogates to study lipid electrophile generation and protein adduction in the larger context of the cellular milieu. The remaining studies described here focus on cells that were preincubated with aLA, as it results in the incorporation of both aLA and aAA. The expectation was that, under conditions of oxidative stress, alkynyl lipid electrophiles would be generated from both lipids, thereby increasing the diversity of the alkynyl electrophile pool and making the alkynyl electrophile panel generated more physiologically relevant.
Activation of RAW264.7 Macrophages Induces Lipid Electrophile-Protein Adducts.
To evaluate protein targets susceptible to adduction by lipid-derived electrophilic species, macrophages were preincubated in the presence of aLA, treated with or without KLA, and lysed, and a protein extract was prepared and treated with NaBH4 to stabilize protein- electrophile conjugates. Adducted proteins containing an alkyne tag were conjugated to a biotin derivative using click chemistry. Proteins were separated by SDS-PAGE, and biotin- containing bands were visualized using a streptavidin-based fluorophore. As shown in Figure 1B, activation with KLA resulted in a substantial increase in measurable protein adduction in aLA-incorporated macrophages when compared to unactivated controls or macrophages with no aLA incorporation. Low levels of protein adduction can also be seen in unactivated, aLA-incorporated macrophages, as lipid oxidation and electrophile generation occur during normal cellular metabolism.
SILAC Quantification of Lipid Electrophile-Adducted Proteins Reveals Nearly Half of the Detectable Proteome Is Subject to Adduction.
KLA-mediated activation of RAW264.7 macrophages resulted in marked changes in both protein expression and lipid electrophile adduction of proteins. To quantify these changes, we employed a well-characterized SILAC model (Figure 2A).27 Three biological replicates of the input (proteome) samples were analyzed via multidimensional protein identification technology tandem mass spectrometry (MudPIT-MS/MS; Tables S1, S2, and S3). This analysis yielded the reference proteome, a total of 2406 input proteins that were common across the three replicates (Figure 3A). The reference proteome includes all proteins detectable before affinity enrichment for electrophile- adducted proteins, and the ratio of heavy peptides/light peptides for each protein provides a relative quantification of expression changes resulting from KLA activation of the macrophages. Our data provided an experimental power of 0.9 to detect a 1.5-fold change, with P < 0.05. In total, we observed 192 proteins with heavy/light > 1.5 (Table S7), including proteins known to be induced during TLR-4-mediated macrophage activation such as inflammatory response proteins [e.g., cyclooxygenase-2 (COX-2; heavy/light = 18.9),28 tumor necrosis factor-alpha (TNFa; heavy/light = 5.6),28 immune- responsive gene 1 (Irg1; heavy/light = 16.4)29] and anti-inflammatory response proteins [e.g., superoxide dismutase 2 (Sod2; heavy/light = 4.3),30 and heme oxygenase-1 (Hmox1; heavy/light = 1.7)31].
Figure 2.
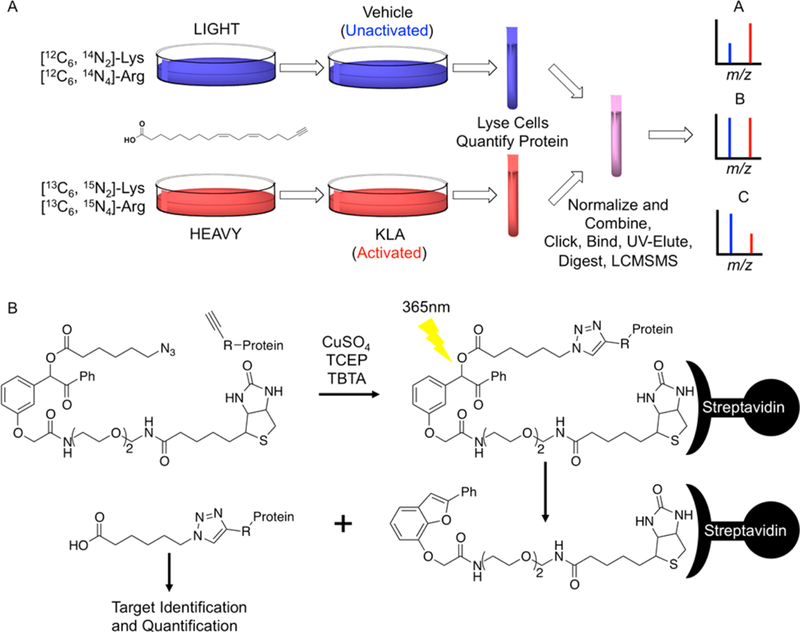
(A) SILAC workflow. Both heavy and light cell lines were incorporated with aLA. The light cell line was unactivated (vehicle control), while the heavy cell line was activated with KLA. After activation, the heavy and light cells were harvested and lysed, and the two samples were then combined in equal amounts of protein. Adducts were stabilized by reduction with NaBH4 and then attached to (B) UV-biotin by copper-mediated click chemistry for affinity enrichment of adducted proteins. MudPIT-MS/MS was used to analyze adducted proteins. (A) Three distinct protein classes can be observed that are described by different activated/unactivated ratios. Theoretical spectra for the three potential activated/unactivated ratios are seen for protein A where activated/unactivated > 1, protein B where activated/unactivated = 1, and protein C where activated/unactivated < 1.
Figure 3.
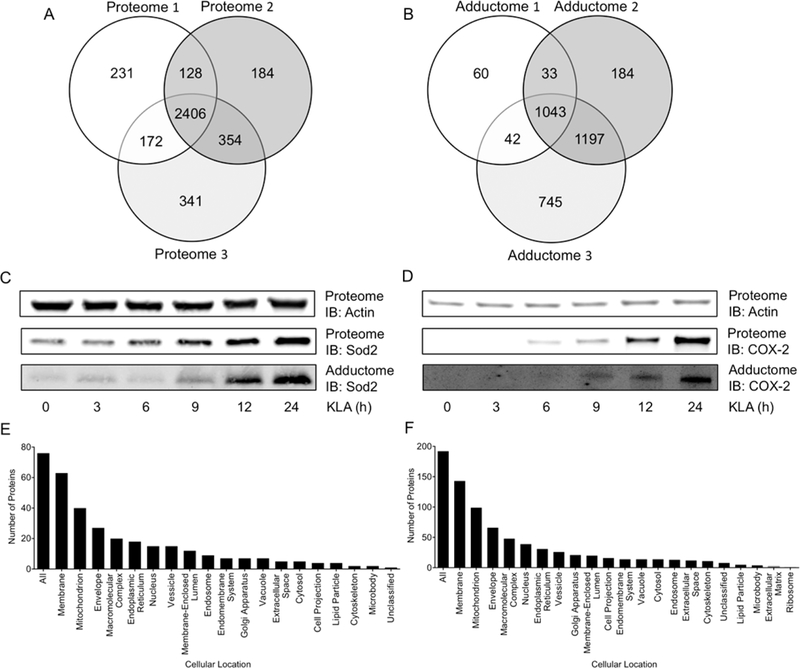
(A) Venn diagram depicting common proteins across three proteome replicates. In total, 3816 proteins were detected, with 2406 proteins common across all three proteome replicates. (B) Venn diagram depicting common proteins across three adductome replicates. In total, 3304 proteins were detected as adducted, with 1043 proteins commonly adducted across all three adductome replicates. (C) Affinity purification and Western blotting were used to confirm Sod2 as adducted by lipid electrophiles during macrophage activation. Both adduction and induction of Sod2 increase with time and at approximately the same rate. (D) Affinity purification and Western blotting were also used to confirm COX-2 as adducted by lipid electrophiles during macrophage activation. Both adduction and induction of COX-2 increase with time and at approximately the same rate. These blots show that induction is not driven by adduction for these candidate proteins. (E) Cellular compartment enrichment of the 192 most differentially expressed proteins during macrophage activation (P < 0.05, heavy/light > 1.5). The membrane and mitochondrion are the most heavily enriched cellular locations of proteins demonstrating increased expression. (F) Cellular compartment enrichment of the 76 most differentially adducted proteins by lipid electrophiles in activated macrophages (P < 0.05, heavy/light > 1.5). The membrane and mitochondrion are the most heavily enriched cellular locations of protein targets for lipid electrophiles.
Affinity purified, electrophile-adducted proteins from the three biological replicates (adductome) were analyzed via MudPIT-MS/MS (Figure 2B, Tables S4, S5, and S6). This analysis identified a total of 1043 unique targets common to all three replicates. These proteins, detected after affinity enrichment for electrophile-adducted proteins, constitute the adductome (Figure 3B). Individual proteins fell into three distinct classes based on their heavy/light ratios, defined as follows: (A) the protein is more adducted in the activated sample; (B) the protein is equally adducted in the activated and unactivated samples; (C) the protein is less adducted in the activated sample (Figure 2A). A heavy/light >1.5 was applied, as our data provide an experimental power of 0.9 to detect a 1.5-fold change. These filters resulted in a list of 76 unique proteins (Table S8), including proteins involved in inflammatory signaling as well as oxidant defense [e.g., COX-2 (heavy/ light = 18.6), TNFα (heavy/light = 9.5), and Sod2 (heavy/light = 4.6)].
Adduction and Expression Are Correlated.
The reference proteome and adductome were compared for each replicate individually to explore the relationship between protein expression and electrophile adduction. Figure S5 and Table S9 show that proteins in the proteome and adductome exhibit similar heavy/light ratios, as evidenced by their high Spearman coefficients. Two possible nonexclusive explanations exist for this correlation: (l) protein expression increases as electrophile generation increases, and therefore, adduction increases because there is more protein to be adducted; (2) proteins are adducted and lose function, and activity loss triggers increased expression.
Immunoblotting Confirms Targets of Adduction.
To confirm targets of adduction and to determine if adduction and subsequent inactivation drives induction, RAW264.7 cells were incorporated with aLA and then exposed to KLA for 0, 3, 6, 9, 12, or 24 h. Adducted proteins from the cells were enriched by adsorption to streptavidin beads and then probed with antibodies against Sod2 or COX-2, two proteins with high heavy/light ratios in the adductome, to validate their status as targets of adduction. As seen in Figure 3C, Sod2 adduction appeared first at the 9 h time point and increased between 9 and 24 h. COX-2 adduction was also confirmed by immunoblotting, with adduction first appearing at the 9 h time point, and the majority occurring between 12 and 24 h (Figure 3D). We expect that substantial oxidative stress occurs during the peak KLA response period, given prior reports demonstrating that ROS levels reach a maximum at 3–6 h post KLA exposure in J774 macrophages.32 However, others have shown that ROS levels remain elevated in macrophages for as long as 16 h following KLA treatment.21, 33 Both proteins exhibited increases in expression at the same time points as the measured adduction increases, confirming the correlation between expression changes and adduction changes, and indicating that, for the investigated proteins, adduction does not trigger induction.
Membrane and Mitochondrial Proteins Show the Highest Increase in Both Expression and Adduction during Macrophage Activation.
We evaluated the cellular location of the 192 proteins that showed the greatest increase in expression and the 76 proteins that showed the greatest increase in adduction using Webgestalt,34 a web-based gene set analysis tool (Figure 3E and F, respectively). Membrane proteins (74.5%) and mitochondrial proteins (51.6%) constituted the largest proportions of the 192 induced proteins as well as the largest proportions of the 76 adducted proteins at 82.9% and 52.6%, respectively. (It should be noted that a protein may reside in more than one cellular component category.) In the reference proteome, membrane and mitochondrial proteins, respectively, represent 42.7% and 19.1% of the quantifiable proteins. Therefore, we observe a 1.7-fold and 1.9-fold enrichment for membrane protein expression and adduction respectively, and a 2.7-fold and 2.8- fold enrichment for mitochondrial protein expression and adduction, respectively, above what would be expected if protein expression changes were randomly distributed across the detectable proteome. Not all cellular locations were enriched for adduction. For example, nuclear proteins represent 19.7% of the most differentially adducted proteins but 38.7% of the reference proteome, indicating that nuclear proteins are not heavily adducted by the alkynyl lipid electrophiles generated here. The broad induction of mitochondrial protein expression may be explained by induction of Hmoxl in response to KLA. Hmoxl produces CO, promoting mitochondrial biogenesis,35 a mechanism by which macrophages restore degraded mitochondrial function following inflammation-associated oxidative damage.36 This evolutionary phenomenon of mitochondrial biogenesis to restore damaged protein potentially links the correlation between the adduction and induction we observe.
The Electron Transport Chain Is the Major Mitochondrial Pathway Both Induced and Targeted by Lipid Electrophiles during Macrophage Activation.
A pathway enrichment analysis of the 192 most differentially expressed and 76 most differentially adducted proteins during macrophage activation was performed in Webgestalt using the Wikipathway database. As expected from the cellular component analyses described above, mitochondrial pathways were heavily represented in the pathway enrichment analyses. Table 1 shows pathways that contained three or more proteins with increased expression during activation (adjusted P < 0.05). The electron transport chain (ETC) was the most significantly increased for expression, exhibiting 6.4-fold enrichment when compared to the reference proteome. Expression changes of the ETC occurred mainly in complexes II, III, and V (Figure S6). The tricarboxylic acid cycle (TCA), amino acid metabolism, and oxidative damage pathways also showed increased protein expression during macrophage activation.
Table 1.
Wikipathway Enrichment for the 192 Proteins in the Most Differentially Expressed Classa
| pathway | proteins | fold enrichment |
adjP value | protein identifications |
|---|---|---|---|---|
| electron | 25 | 6.4 | 1.7 × 10−14 | Atp5a1, Atp5b, Atp5c1, Atp5d, Atp5f1, Atp5h, Atp5j, Atp5j2, Atp5k, Atp5l, Atp5o, Atp8, Cox17, Uqcrc1, |
| transport | U1crc2, Uqcr10, Uqcrfs1, Uqcrq, Ndufab1, Ndufb10, Sdha, Sdhb, Sdhc, Slc25a4, Slc25a5 | |||
| chain | ||||
| TCA cycle | 8 | 4.3 | 0.0030 | Cs, Dld, Dlst, Mdh2, Ogdh, Sdha, Sdhb, Sdhc |
| amino acid | 11 | 2.4 | 0.032 | Arg2, Cs, Dld, Dlst, Glud1, Got2, Hibadh, Mdh2, Oat, Ogdh, Sdha |
| metabolism | ||||
| oxidative | 3 | 7.1 | 0.032 | Cycs, Tnf, Tnfrsf1b |
| damage | ||||
Data are provided for pathways containing greater than three proteins with an adjusted P (adjP) value < 0.05 as determined by Webgestalt.
The ETC was also the most significantly enriched pathway for adduction, exhibiting a 7.1-fold enrichment in the adductome as compared to the reference proteome (Table 2). Adduction in the ETC occurred predominately in complex V (ATP synthase) as can be seen by the pathway enrichment for oxidative phosphorylation (Figure S7). Within ATP synthase, a majority of the adduction (four out of five proteins) occurred in the F1 subunit, the catalytic subunit responsible for the conversion of ADP to ATP. Previous studies demonstrated that macrophages activated with LPS shift energy generation from oxidative phosphorylation to glycolysis.37,38 Our findings identify oxidative damage to ETC proteins as a potential mechanism to explain that shift. Enzymes involved in central carbon metabolism were also heavily adducted, with the TCA cycle, amino acid metabolism, glycolysis, and gluconeogenesis represented in the most heavily adducted pathways. In addition, many enzymes that play important roles in ROS detoxification were in this class, including Sod2, Hmox1, and peroxiredoxin 5 (Prdx5).
Table 2.
Wikipathway Enrichment for the 76 Proteins in the Most Differentially Adducted Classa
| pathway | proteins | fold enrichment |
adjP value | protein identifications |
|---|---|---|---|---|
| electron transport chain | 11 | 7.1 | 2.7 × 10−6 | Atp5a1, Atp5b, Atp5c1, Atp5d, Atp5h, Atp5j, Atp5j2, Uqcrc1, Uqcrc2, Ndufab1, Sdha |
| amino acid metabolism | 6 | 5 | 5.0 × 10−4 | Cs, Mdh2, Got2, Dlst, Hibadh, Glud1, Oat, Dld, Sdha |
| TCA cycle | 5 | 6.8 | 2.6 × 10−3 | Cs, Mdh2, Dlst, Dld, Sdha |
| glycolysis and gluconeogenesis | 4 | 4.8 | 0.02 | Slc2a1, Mdh2, Got2, Dld |
Data are provided for pathways containing greater than three proteins with an adjusted P (adjP) value < 0.05 as determined by Webgestalt.
A clear and striking discovery resulting from this work is the susceptibility of mitochondrial and membrane proteins to adduction by endogenous lipid electrophiles. In particular, proteins of the electron transport chain and oxidative phosphorylation were enriched for adduction. This finding is dramatically different from the results obtained in our recent survey of protein targets of exogenously provided alkynyl-4- hydroxy-2-nonenal and alkynyl-4-oxo-2-nonenal. In those experiments, proteins that were adducted at a very low electrophile concentration were primarily associated with cytoskeletal regulation, while those that were only adducted at high concentrations were involved in protein synthesis and turnover. Remarkably, proteins of the electron transport chain and oxidative phosphorylation fell into the never-adducted category, even though mitochondrial matrix proteins were observed to be adducted.12 These data indicate that the adduction pattern resulting from exogenously added electro- philes is distinctly different from that of endogenously generated electrophiles. It is likely that a major cause of these differences is the electrophile source. Exogenous electrophiles enter the cell through the plasma membrane and immediately encounter defense mechanisms such as cytosolic glutathione. In contrast, endogenous electrophiles are most likely generated in membrane phospholipids and may immediately damage nearby membrane-associated proteins. Regardless of the exact mechanisms, these observations highlight the importance of evaluating electrophile damage under physiological conditions if realistic conclusions are to be made concerning the impact of such damage on cellular structure and function.
MitoTEMPO Modulates Lipid Electrophile Adduction in Activated Macrophages.
Superoxide is generated in the mitochondrion as an oxidative phosphorylation byproduct. Our SILAC adductome data showed significant enrichment for mitochondrial proteins, leading to the hypothesis that mitochondrial superoxide is the source of ROS that leads to electrophile generation and protein adduction. To test this hypothesis, we employed two superoxide-scavenging agents to evaluate the effects of quenching mitochondrial superoxide on protein adduction: MitoTEMPO, which localizes to the mitochondrion, and TEMPOL, which is ubiquitously dispersed throughout the cell.39 MitoTEMPO or TEMPOL was added simultaneously with KLA to RAW264.7 macrophages that had been incorporated with aLA, and adduction across the proteome was assessed. Figure 4A demonstrates that MitoTEMPO reduced measurable lipid electrophile adduction to nearly basal levels in KLA-activated cells. Additionally, MitoTEMPO reduced basal electrophile adduction in unactivated macrophages. TEMPOL did not modulate protein adduction in either activated or unactivated macrophages. The differential effects of MitoTEMPO and TEMPOL suggest that mitochondrial superoxide is a precursor to both physiological and pathophysiological lipid electrophile generation. These data are consistent with a recent study that showed a specific class of LA-derived lipid electrophiles is generated in the mitochondrion. 40
Figure 4.
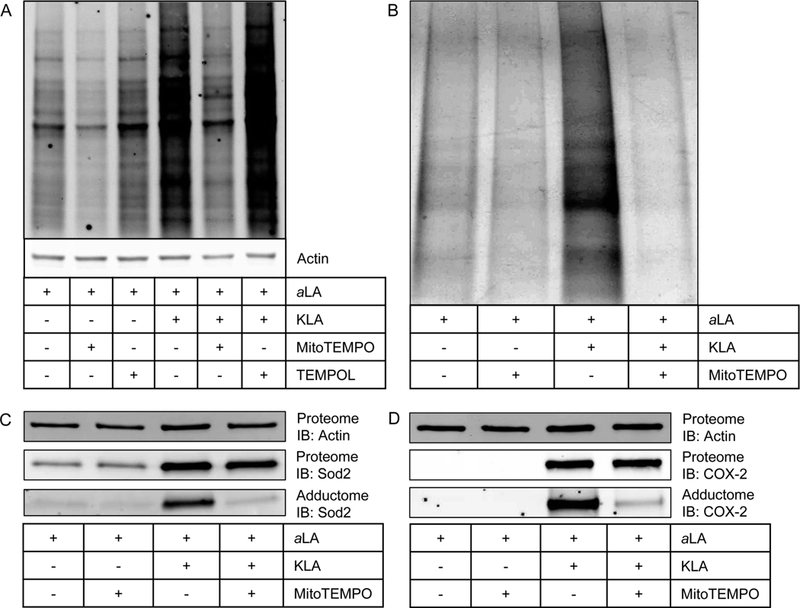
(A) Results of modulation of KLA-induced protein adduction by MitoTEMPO and TEMPOL. MitoTEMPO, a mitochondrially targeted superoxide scavenger, was able to reduce both physiological and pathophysiological protein adduction by lipid electrophiles compared to vehicle and KLA, respectively. TEMPOL, a ubiquitously dispersed superoxide scavenger, did not modulate electrophile formation in either physiological or pathophysiological settings. (B) Consistent with previous results, affinity-purified adducted protein is greatest in the KLA-activated macrophages. MitoTEMPO reduces total adducted protein in both the vehicle-treated and KLA-activated macrophages. (C) MitoTEMPO does not change the KLA-induced expression (Proteome) of Sod2 but does decrease the amount of Sod2 adduction (Adductome). (D) MitoTEMPO does not change the KLA-induced expression (Proteome) of Sod2 but does decrease the amount of Sod2 adduction (Adductome). These data indicate that both mitochondrial and nonmitochondrial targets are adducted by lipid electrophiles generated through a process that involves mitochondrial superoxide.
We used click chemistry, affinity purification, and Western blotting to test the effect of MitoTEMPO on the KLA- mediated adduction of the specific targets COX-2 and Sod2 in aLA-labeled macrophages. Consistent with the results described above, KLA-activation was associated with an increase in total affinity-purified protein, and MitoTEMPO reduced the amount of affinity-purified protein in both the vehicle and KLA-treated samples (Figure 4B). MitoTEMPO did not affect the KLA- induced expression of Sod2 (Figure 4C) or COX-2 (Figure 4D) but drastically reduced the amount of lipid electrophile adduction of both proteins. These data show that the expression changes observed during macrophage activation are dependent upon signaling during activation, and that lipid electrophile adduction is dependent upon mitochondrial oxidant production. Additionally, lipid electrophiles arise from mitochondrial superoxide adduct proteins that are not located in the mitochondrion as seen with COX-2.
CONCLUSION
Global profiling of proteins adducted during KLA activation by endogenously formed lipid electrophiles is an important step in understanding the potential pathological mechanisms of inflammatory signaling. Increased protein adduction has been measured in patients with inflammation-related diseases, including in the brains of patients with Alzheimer’s disease,5 in the spinal cord motor neurons of patients with amyotrophic lateral sclerosis,3 and in the arteries of patients with atherosclerosis.4 The inability to comprehensively enrich for endogenously generated electrophile-adducted proteins due to the complexities of both the electrophile pool and the targeted proteome has hindered their global identification and quantification. Thus, we used aLA incorporation and KLA activation of RAW264.7 macrophages to generate diverse alkynyl lipid electrophiles in a physiologically relevant setting, thereby enabling capture and identification of the adducted proteins. We identify and quantify any protein adducted by any electrophile that retains the terminal alkyne tag. Our current study combined with these previous observations suggests that stimulated mitochondrial ROS generation and the resulting mitochondrial protein damage by lipid electrophiles may be an initiating and propagating factor in inflammatory disease pathogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health R37 CA087819 (L.J.M.), the American Heart Association 13PRE17270009 (W.N.B.), and the National Center for Advancing Translational Sciences UL1TR000445. Mass spectrometry data were acquired using an instrument supported by National Institutes of Health grant S10RR027714. The authors wish to thank S. Milne and D. Myers for expert technical assistance with the lipidomic analysis.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Nathan C, and Ding A (2010) Nonresolving inflammation. Cell 140, 871–882. [DOI] [PubMed] [Google Scholar]
- (2).West JD, and Marnett LJ (2006) Endogenous reactive intermediates as modulators of cell signaling and cell death. Chem. Res. Toxicol. 19, 173–194. [DOI] [PubMed] [Google Scholar]
- (3).Kondo M, Shibata T, Kumagai T, Osawa T, Shibata N, Kobayashi M, Sasaki S, Iwata M, Noguchi N, and Uchida K (2002) 15-Deoxy-Delta(12,14)-prostaglandin J(2): the endogenous electrophile that induces neuronal apoptosis. Proc. Natl. Acad. Sci. U. S. A. 99, 7367–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, and Uchida K (2004) A lipid peroxidation-derived inflammatory mediator: identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. J. Biol. Chem. 279, 48389–48396. [DOI] [PubMed] [Google Scholar]
- (5).Markesbery WR, and Lovell MA (1998) Four- hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 19, 33–36. [DOI] [PubMed] [Google Scholar]
- (6).Connor RE, Marnett LJ, and Liebler DC (2011) Protein- selective capture to analyze electrophile adduction of hsp90 by 4- hydroxynonenal. Chem. Res. Toxicol. 24, 1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Liu Q, Ullery J, Zhu J, Liebler DC, Marnett LJ, and Zhang B (2013) RNA-seq data analysis at the gene and CDS levels provides a comprehensive view of transcriptome responses induced by 4-hydroxynonenal. Mol. BioSyst. 9, 3036–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Zhao Y, Miriyala S, Miao L, Mitov M, Schnell D, Dhar SK, Cai J, Klein JB, Sultana R, Butterfield DA, Vore M, Batinic- Haberle I, Bondada S, and Clair DK St. (2014) Redox proteomic identification of HNE-bound mitochondrial proteins in cardiac tissues reveals a systemic effect on energy metabolism after doxorubicin treatment. Free Radical Biol. Med. 72, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Codreanu SG, Zhang B, Sobecki SM, Billheimer DD, and Liebler DC (2009) Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol. Cell. Proteomics 8, 670–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Galligan JJ, Smathers RL, Fritz KS, Epperson LE, Hunter LE, and Petersen DR (2012) Protein carbonylation in a murine model for early alcoholic liver disease. Chem. Res. Toxicol. 25, 1012–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP, and Marnett LJ (2014) Stable histone adduction by 4-oxo-2-nonenal: a potential link between oxidative stress and epigenetics. J. Am. Chem. Soc. 136, 11864–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Codreanu SG, Ullery JC, Zhu J, Tallman KA, Beavers WN, Porter NA, Marnett LJ, Zhang B, and Liebler DC (2014) Alkylation damage by lipid electrophiles targets functional protein systems. Mol. Cell. Proteomics 13, 849–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wang C, Weerapana E, Blewett MM, and Cravatt BF (2013) A chemoproteomic platform to quantitatively map targets of lipid-derived electrophiles. Nat. Methods 11, 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).McGrath CE, Tallman KA, Porter NA, and Marnett LJ (2011) Structure-activity analysis of diffusible lipid electrophiles associated with phospholipid peroxidation: 4-hydroxynonenal and 4- oxononenal analogues. Chem. Res. Toxicol. 24, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Vila A, Tallman KA, Jacobs AT, Liebler DC, Porter NA, and Marnett LJ (2008) Identification of protein targets of 4- hydroxynonenal using click chemistry for ex vivo biotinylation of azido and alkynyl derivatives. Chem. Res. Toxicol. 21, 432–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Kim HY, Tallman KA, Liebler DC, and Porter NA (2009) An azido-biotin reagent for use in the isolation of protein adducts of lipid-derived electrophiles by streptavidin catch and photorelease. Mol. Cell. Proteomics 8, 2080–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Beavers WN, Serwa R, Shimozu Y, Tallman KA, Vaught M, Dalvie ED, Marnett LJ, and Porter NA (2014) omega- Alkynyl lipid surrogates for polyunsaturated fatty acids: free radical and enzymatic oxidations. J. Am. Chem. Soc. 136, 11529–11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Raetz CR, Garrett TA, Reynolds CM, Shaw WA, Moore JD, Smith DC Jr., Ribeiro AA, Murphy RC, Ulevitch RJ, Fearns C, Reichart D, Glass CK, Benner C, Subramaniam S, Harkewicz R, Bowers-Gentry RC, Buczynski MW, Cooper JA, Deems RA, and Dennis EA (2006) Kdo2-Lipid A of Escherichia coli, a defined endotoxin that activates macrophages via TLR-4. J. Lipid Res. 47, 1097–1111. [DOI] [PubMed] [Google Scholar]
- (19).Ong SE, and Mann M (2007) A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat. Protoc 1, 2650–2660. [DOI] [PubMed] [Google Scholar]
- (20).Rouzer CA, Ivanova PT, Byrne MO, Milne SB, Marnett LJ, and Brown HA (2006) Lipid profiling reveals arachidonate deficiency in RAW264.7 cells: Structural and functional implications. Biochemistry 45, 14795–14808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Maitra U, Singh N, Gan L, Ringwood L, and Li L (2009) IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes. J. Biol. Chem. 284, 35403–35411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Robichaud PP, Poirier SJ, Boudreau LH, Doiron JA, Barnett DA, Boilard E, and Surette ME (2016) On the cellular metabolism of the click chemistry probe 19-alkyne arachidonic acid. J. Lipid Res. 57, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Milne S, Tallman K, Serwa R, Rouzer C, Armstrong M, Marnett L, Lukehart C, Porter N, and Brown H (2010) Capture and release of alkyne-derivatized glycerophospholipids using cobalt chemistry. Nat. Chem. Biol. 6, 205–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mead JF, and Howton DR (1957) Metabolism of essential fatty acids. VII. Conversion of gamma-linolenic acid to arachidonic acid. J. Biol. Chem. 229, 575–582. [PubMed] [Google Scholar]
- (25).Rouzer CA, and Marnett LJ (2003) Mechanism of free radical oxygenation of polyunsaturated fatty acids by cyclooxygenases. Chem. Rev. 103, 2239–2304. [DOI] [PubMed] [Google Scholar]
- (26).Steinberg G, Slaton WH Jr., Howton DR, and Mead JF (1957) Metabolism of essential fatty acids. V. Metabolic pathway of linolenic acid. J. Biol. Chem. 224, 841–849. [PubMed] [Google Scholar]
- (27).Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, and Mann M (2002) Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 1, 376–386. [DOI] [PubMed] [Google Scholar]
- (28).Rouzer CA, Jacobs AT, Nirodi CS, Kingsley PJ, Morrow JD, and Marnett LJ (2005) RAW264.7 cells lack prostaglandin-dependent autoregulation of tumor necrosis factor-alpha secretion. J. Lipid Res. 46, 1027–1037. [DOI] [PubMed] [Google Scholar]
- (29).Michelucci A, Cordes T, Ghelfi J, Pailot A, Reiling N, Goldmann O, Binz T, Wegner A, Tallam A, Rausell A, Buttini M, Linster CL, Medina E, Balling R, and Hiller K (2013) Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. U. S. A. 110, 7820–7825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sharif O, Bolshakov VN, Raines S, Newham P, and Perkins ND (2007) Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Camhi SL, Alam J, Otterbein L, Sylvester SL, and Choi AM (1995) Induction of heme oxygenase-1 gene expression by lipopolysaccharide is mediated by AP-1 activation. Am. J. Respir. Cell Mol. Biol. 13, 387–398. [DOI] [PubMed] [Google Scholar]
- (32).Menon D, Coll R, O’Neill LA, and Board PG (2014) Glutathione transferase omega 1 is required for the lipopolysaccharide- stimulated induction of NADPH oxidase 1 and the production of reactive oxygen species in macrophages. Free Radical Biol. Med. 73, 318–327. [DOI] [PubMed] [Google Scholar]
- (33).Zhao G, Yu R, Deng J, Zhao Q, Li Y, Joo M, van Breemen RB, Christman JW, and Xiao L (2013) Pivotal role of reactive oxygen species in differential regulation of lipopolysaccharide- induced prostaglandins production in macrophages. Mol. Pharmacol. 83, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhang B, Kirov S, and Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res. 33, W741–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).MacGarvey NC, Suliman HB, Bartz RR, Fu P, Withers CM, Welty-Wolf KE, and Piantadosi CA (2012) Activation of mitochondrial biogenesis by heme oxygenase-1-mediated NF-E2- related factor-2 induction rescues mice from lethal Staphylococcus aureus sepsis. Am. J. Respir. Crit. Care Med. 185, 851–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36).Piantadosi CA, Withers CM, Bartz RR, MacGarvey NC, Fu P, Sweeney TE, Welty-Wolf KE, and Suliman HB (2011) Heme oxygenase-1 couples activation of mitochondrial biogenesis to anti-inflammatory cytokine expression. J. Biol. Chem. 286, 16374–16385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, Tourlomousis P, Dabritz JH, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, and O’Neill LA (2016) Succinate Dehydrogenase Supports Metabolic Repurposing of Mitochondria to Drive Inflammatory Macrophages. Cell 167, 457–470, e413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Rodriguez-Prados JC, Traves PG, Cuenca J, Rico D, Aragones J, Martin-Sanz P, Cascante M, and Bosca L (2010) Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol. 185, 605–614. [DOI] [PubMed] [Google Scholar]
- (39).Dikalov S (2011) Cross talk between mitochondria and NADPH oxidases. Free Radical Biol. Med. 51, 1289–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Tyurina YY, Poloyac SM, Tyurin VA, Kapralov AA, Jiang J, Anthonymuthu TS, Kapralova VI, Vikulina AS, Jung MY, Epperly MW, Mohammadyani D, Klein-Seetharaman J, Jackson TC, Kochanek PM, Pitt BR, Greenberger JS, Vladimirov YA, Bayir H, and Kagan VE (2014) A mitochondrial pathway for biosynthesis of lipid mediators. Nat. Chem. 6, 542–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.