

Summary
Macrophage plasticity is essential for innate immunity, but in-depth signaling mechanism(s) regulating their functional phenotypes are ill-defined. Here we report that interferon (IFN) γ priming of naive macrophages induces store-mediated Ca2+ entry and inhibition of Ca2+ entry impairs polarization to M1 inflammatory phenotype. In vitro and in vivo functional analyses revealed ORAI1 to be a primary contributor to basal Ca2+ influx in macrophages, whereas IFNγ-induced Ca2+ influx was mediated by TRPC1. Deficiency of TRPC1 displayed abrogated IFNγ-induced M1 inflammatory mediators in macrophages. In a preclinical model of peritonitis by Klebsiella pneumoniae infection, macrophages showed increased Ca2+ influx, which was TRPC1 dependent. Macrophages from infected TRPC1−/− mice showed inhibited expression of M1-associated signature molecules. Furthermore, in human patients with systemic inflammatory response syndrome, the level of TRPC1 expression in circulating macrophages directly correlated with M1 inflammatory mediators. Overall, TRPC1-mediated Ca2+ influx is essential for the induction/shaping of macrophage polarization to M1 inflammatory phenotype.
Subject Areas: Biological Sciences, Immune Response, Immunology
Graphical Abstract
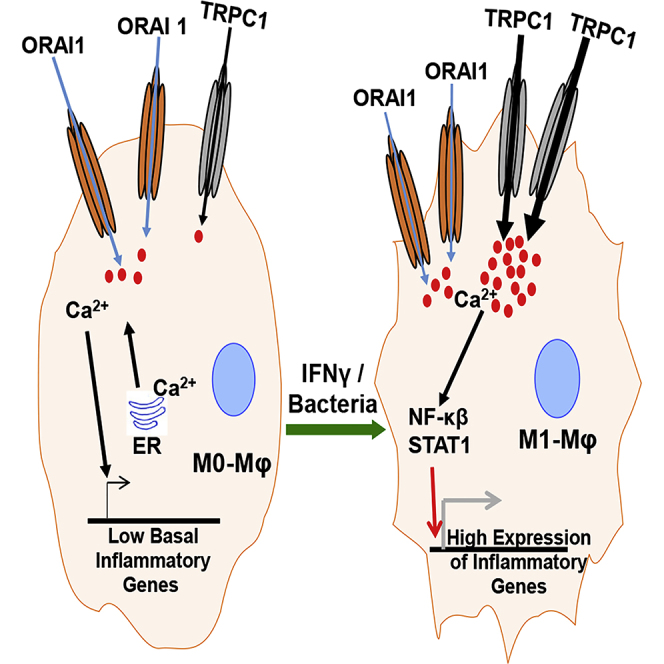
Highlights
-
•
TRPC1 mediates sterile or infection-induced Ca2+ influx and M1 phenotype in macrophages
-
•
ORAI1 mediates the basal Ca2+ influx in macrophages
-
•
In patients with SIRS, the TRPC1 level correlates with M1 inflammatory mediators in macrophages
Biological Sciences; Immune Response; Immunology
Introduction
The functional phenotypes of macrophage vary in response to the external stimuli that they receive through a wide variety of surface receptors (Murray and Wynn, 2011). Macrophage with the M1, or classical activation phenotype, release multiple pro-inflammatory molecules (Su et al., 2015). M1 macrophages are critical effectors of inflammation and innate immunity as well as orchestrators of adaptive immunity (Glass and Natoli, 2016, Locati et al., 2013). On the contrary, macrophage with M2, or alternatively activated phenotype, produces factors involved in anti-inflammatory and tissue remodeling functions (Murray et al., 2014, Ruckerl and Allen, 2014, Wynn, 2015, Wynn and Vannella, 2016). Although originally macrophage functions were defined in the context of host defense against infections, this dynamic continuum of macrophage functional phenotypes is now appreciated as being important in various acute and chronic disease conditions (Labonte et al., 2014, McNelis and Olefsky, 2014, Moore et al., 2013). Thus, clearly, the mechanistic underpinnings of macrophage activation to critical functional phenotypes will have important therapeutic implications. In this regard, environmental signals such as interferon (IFN) γ or Toll-like receptor ligands upon interactions with their specific cell surface receptors activate signal transduction pathways involving phosphorylation of STAT1 and nuclear factor (NF)-κB (Murray et al., 2014). The cascade of events that follow, including the integration of these signals culminates in the production of M1-associated inflammatory molecules (Ivashkiv, 2013, Lawrence and Natoli, 2011). However, the upstream factors interacting with these endogenous signaling pathways culminating in the development of M1 inflammatory macrophages are poorly understood.
Cellular activation requires the collaboration of many pathways to cause a precise stimulus-specific induction of gene expression. In stimulated cells, divalent cations serve as second messengers and regulate the function of many enzymes and transcription factors (Clapham, 2007). Intracellular calcium (Ca2+ is an important divalent cation that controls many cellular functions such as cell plasticity, development, cell proliferation, and differentiation (Feske et al., 2015, Hogan et al., 2010). Increases in intracellular Ca2+ levels also enhance immune functions (Cahalan and Chandy, 2009, Rao and Hogan, 2009, Vig and Kinet, 2009). In activated T cells, Ca2+ signaling regulates the cytokine and chemokine gene expression in a calcineurin-NFAT-dependent manner (Feske, 2007, Hogan et al., 2003, Macian, 2005, Oh-Hora et al., 2008, Oh-Hora et al., 2013, Watanabe et al., 1996), highlighting its role in adaptive immune cell activation. In innate immune functions, inhibition of intracellular Ca2+ signaling was shown to reduce phagocytosis, and production of tumor necrosis factor (TNF)-α, and nitric oxide (NO) in J774 cells, a macrophage cell line (Chen et al., 1998, Watanabe et al., 1996). However, the identity of the specific channels involved in Ca2+ entry to modulate the macrophage phenotype remains obscure.
In non-excitable cells, the cytosolic Ca2+ changes occur mainly by the release of Ca2+ from intracellular endoplasmic reticulum (ER) stores, followed by influx through the plasma membrane (PM) Ca2+ channels, which is termed as store-operated Ca2+ entry (SOCE) (Pani et al., 2012). The PM Ca2+ channels could be broadly divided into two groups: the CRAC (Ca2+ release-activated Ca2+/SOCE channels and the non-CRAC/non-SOCE channels (Feske et al., 2012). The ORAI1 gene encodes functions of a CRAC channel (Feske et al., 2006, McNally et al., 2012, Prakriya et al., 2006, Zhou et al., 2013), whereas the TRPC (transient receptor potential canonical) channels have been shown to be activated upon store depletion as well as through receptor-mediated activation, which requires second messengers (Feske et al., 2015, Vaeth and Feske, 2018, Yuan et al., 2007). The majority of the studies reporting the role of Ca2+ influx in immune cell function have been focused on CRAC channels. For instance, patients with CRAC channelopathy develop lethal pathological outcome upon infection with bacteria, fungi, and viruses (Byun et al., 2010, Feske, 2010). A loss-of-function mutation in ORAI1 is associated with a low humoral response against vaccination or infection with various pathogens in affected humans (Le Deist et al., 1995, Picard et al., 2009). In preclinical experimental models, mice lacking STIM1, the ER Ca2+ sensor responsible for the assembly of ORAI1-based Ca2+ entry and an activator of TRPC1-based Ca2+ entry, display an increased susceptibility to bacterial infection, but a diminished disease severity in inflammatory bowel disease or experimental autoimmune encephalomyelitis (EAE) (referenced in Feske et al., 2015). Although other studies described T cell-specific functions of CRAC channels (Jairaman et al., 2015, Oh-Hora et al., 2013), their role in macrophage responses to modulate immune pathology is limited.
Besides ORAI and TRPC channels, TRPV, TRPM, P2RX, and voltage-gated channels are also expressed in immune cells and could also modulate immune function (Ramirez et al., 2018, Tomilin et al., 2016, Vaeth et al., 2016, Vaeth et al., 2017, Vaeth and Feske, 2018, Wang et al., 2016). Cav channels are found to modulate T cell responses and development of experimental asthma and EAE (Wang et al., 2016). P2RX7 was shown to modulate the inflammatory response of macrophage in the development of autoimmune disease (Fernandes-Alnemri et al., 2009, Junger, 2011), as well as in controlling bacterial infection (Idzko et al., 2014). Among the TRP channel proteins, direct evidence suggests that in the absence of TRPM2 or TRPV2, mice display lower inflammatory responses and are resistant to autoimmune disorders such as obesity-induced insulin resistance or EAE, respectively (Feske et al., 2015). In contrast, these mice displayed increased susceptibility to Listeria monocytogenes, presumably because of lower inflammatory response to the infection (Knowles et al., 2011). Some TRP channels seem to play a role in innate immunity/macrophage functions, e.g., chemotaxis, and phagocytosis (Link et al., 2010). Despite this mounting evidence for the importance of Ca2+ signaling in the pathogenesis of infections and autoimmune disorders, little is known about the identity or function of Ca2+ channel(s) directly responsible for induction of the M1 functional phenotype.
The goal of this study was to identify the PM Ca2+ channel involved during naive macrophage polarization to the M1 inflammatory phenotype using well-characterized stimuli such as IFNγ and bacterial infection. In addition to the ORAI1-based CRAC channel, the best documented TRPC-based Ca2+ influx channel that is activated upon store depletion is TRPC1 (Liu et al., 2007, Yuan et al., 2007, Zitt et al., 1996) (Abplanalp et al., 2009, Huang et al., 2006, Kim et al., 2009). Studying TRPC1−/− and wild-type (WT) macrophages or after knocking down TRPC1 transiently using small interfering RNA (siRNA), in vitro, ex vivo, and in vivo, we found that TRPC1 mediates induction of Ca2+ influx during polarization from naive macrophage to M1 macrophage as seen by the level of expression of M1-associated inflammatory cytokines, chemokines, surface maturation markers, and signaling pathways. Importantly, our findings with the preclinical mouse model were translatable to human disease condition wherein analysis of circulating monocyte/macrophages from human patients with systemic inflammatory response syndrome (SIRS) exhibited direct correlation between high TRPC1 expression and M1 inflammatory mediators. These data identify, for the first time, TRPC1 as a specific PM Ca2+ channel that regulates M1 inflammatory functions in macrophage.
Results
Calcium Influx Is Required for IFNγ−Induced Polarization of Macrophages to the M1 Phenotype In Vitro
To address whether Ca2+ influx has a role in the induction of M0 to M1 macrophage phenotype, bone marrow-derived (BM) macrophage cells were incubated with IFNγ for various times (Figure 1). The amount of Ca2+ influx in unstimulated (M0) macrophages cultured for 2, 6, or 24 hr (Figures 1B′, 1C′, S1A, S1A′, and S1A″) were largely similar. Ca2+ imaging revealed a significant increase in internal ER Ca2+ released by thapsigargin (Tg) (first peak), followed by enhanced Ca2+ influx (second peak) in IFNγ-activated macrophages (for 2 and 6 hr) when compared with mock-treated control cells (M0) cultured in medium alone for corresponding duration (Figures 1B, 1B′, S1A, and S1A′). Importantly, prolonged IFNγ treatment (for 24 hr) further increased SOCE, suggesting that IFNγ treatment modulates Ca2+ influx (Figures 1C and 1C′). To address whether the increased Ca2+ influx in IFNγ-primed macrophage is specific, cells were incubated with a known SOCE Ca2+ channel inhibitor 2APB (Sukumaran et al., 2015). 2APB significantly inhibited Ca2+ influx, but not the ER release of Ca2+ in IFNγ-primed BM macrophage (Figures S1A and S1A′). Next, electrophysiological recordings were performed to measure the currents associated with the Ca2+ influx in IFNγ-primed BM macrophages and mock-treated control cells. Currents recorded using voltage-clamp conditions showed that application of Tg in the presence of external Ca2+ (2 mM) caused a significant increase of non-selective inward currents that reversed between 0 and −5 mV in IFNγ-primed BM macrophages cultured for 2, 6, or 24 hr compared with the mock-treated control cells cultured in medium alone for corresponding duration (Figures 1D and S1A‴; data not shown). Together, these data indicate that IFNγ-primed macrophage displayed an increased Ca2+ influx, which likely influences their function in vitro.
Figure 1.

IFNγ Induces Ca2+ Influx and Shapes M1 Functional Phenotype Development in Macrophage In Vitro
(A) BM macrophages were generated in vitro (20 ng/mL GMCSF) and cultured in the presence or absence of 20 ng/mL IFNγ (-phenotype inducer). Whole-cell patch-clamp and imaging analysis on these cells were performed to measure IFNγ-induced effects on Ca2+ release and influx. M1-associated mediators were measured in cells cultured in the presence or absence of 50 μM 2APB (Ca2+ entry inhibitor) by western blot, RT-PCR, and colorimetric assay.
(B and C) BM macrophages were pulsed with medium alone (M0) or IFNγ (M1) for 2 and 24 hr and loaded with Fura-2AM. 1 μM Tg was added (first arrow) to the Fura-2AM-loaded cells bathed in Ca2+-free medium to measure the internal Ca2+ release (first peak); thereafter 2 mM external Ca2+ was added (second arrow) to measure Ca2+ entry/influx through PM (second peak). Average analog plots of the fluorescence ratio (340/380 nm) from an average of 40–50 cells are shown. (B′ and C′) The corresponding bar graphs represent the mean ± SEM of Ca2+ release (first peak) and store-operated Ca2+ entry (SOCE) (second peak) under these conditions.
(D) Representative time course of Ca2+ current at −80 mV with 0 mV holding potential from BM macrophages pulsed with medium alone (M0) or IFNγ. Whole-cell patch-clamp was performed with Tg in the pipette solution.
(E) Comparison of NOS2 mRNA expression by qPCR analysis of BM macrophages cultured in medium alone (M0) or with IFNγ (M1) in the presence or absence of 2APB. The bars are representative of three independent experiments.
(F) Comparison of NO levels in culture supernatant of BM macrophages cultured with medium alone (M0) or IFNγ (M1) in the presence or absence of 2APB. Data shown are Mean ± SEM.
(G) The level of pNF-κB p65 (pp65) (Cell Signaling, 3033S), pSTAT1 (Cell Signaling, 9167S), GAPDH, p65, or STAT1 in BM macrophages cultured with medium alone (M0) or IFNγ (M1) in the presence or absence of 2APB by immunoblot. Data shown are representative of three independent experiments with similar results. The average pixel intensity of pSTAT1 or pp65 bands was measured and expressed in bar graphs as mean ± SEM (G′).
*p ≤ 0.05, ***p ≤ 0.001 (Student's t test).
See also Figure S1.
IFNγ interaction with the cell surface receptor(s) on macrophages activates specific polarizing signal transduction pathways (e.g., STAT1, NF-κB) to develop the M1 functional phenotype (Ginhoux et al., 2016, Murray et al., 2014). In this regard, production of NO and/or induction of inducible nitric oxide synthase (NOS2) enzyme that regulates NO production are considered to be the signature responses associated with the M1 phenotype. IFNγ treatment induced BM macrophages to produce significant amounts of NO (Figure 1F). This IFNγ-induced NO production was inhibited upon treatment with 2APB, the non-specific Ca2+ channel inhibitor (Figure 1F). Moreover, 2APB treatment significantly inhibited IFNγ-induced increase in NOS2 gene expression (Figure 1E). Moreover, immunoblot analysis revealed that exposure of BM macrophage to IFNγ leads to a time-dependent increase in the level of pSTAT1 and pNF-κB p65 (pp65), whereas addition of 2APB caused a reduction when compared with cells exposed to IFNγ alone (Figures 1G, 1G′, and S1B), which was statistically significant (Figure 1G′). As expected, macrophages exhibited similar levels of no significant effect on total p65, STAT1, and GAPDH with or without treatment. Together, these data suggest that in in vitro conditions Ca2+ influx may be required for M1 phenotype function.
ORAI1 and TRPC1 Channels Differentially Mediate Ca2+ Influx in M0/Naive and M1 Macrophage In Vitro
The identity of the Ca2+ channel-regulating M1 macrophages is unknown. Thus electrophysiological recordings were performed to identify specific PM Ca2+ channel(s) contributing to Ca2+ influx during polarization of naive to M1 inflammatory macrophages in vitro. The Ca2+ currents were recorded at a holding potential of −80 mV, and I-V curves were developed using a ramp protocol where current density was evaluated at various membrane potentials and plotted. Our results revealed that unstimulated BM macrophages (naive/M0 macrophages) cultured for 2, 6, or 24 hr in medium alone displayed basal currents that appeared to be rectifying inward with ICRAC- like properties, which were similar to those previously observed for ORAI1 channels (Figure 2A; data not shown). To address whether ORAI1 plays a role in basal Ca2+ influx and hence contributes to the observed ICRAC- like current in naive macrophage, cells were transfected with non-targeting control siRNA (M0-siC) or ORAI1 siRNA (M0-siO1) (reduced ORAI1 expression was verified [Figure S2B]). Electrophysiological recordings on these M0-siO1 cells showed a significant decrease in the inward-rectifying ICRAC- like currents (∼2-fold decrease in ORAI1-silenced cells versus control siRNA-treated cells) (Figures 2C and 2C′). In contrast, IFNγ exposure induced a progressive increase in current with properties similar to Isoc (Figure 2A). In addition, 6 and 24 hr post-IFNγ exposure, the resulting macrophage displayed a non-selective Ca2+ current that reversed between 0 and 5 mV with Isoc-like properties that have been reported for the TRPC1 channel (Figure 2A) (Selvaraj et al., 2012). To test whether TRPC1 is required for the M1 phenotype, BM macrophages derived from TRPC1−/− mice were used. Exposure to IFNγ displayed an attenuation of IFNγ-increased Isoc-like current (∼2.0- to 3.0-fold decrease) in TRPC1−/− cells versus WT cells (Figure 2B). Likewise, BM macrophage transfected with TRPC1 siRNA (reduced TRPC1 expression was verified [Figure S2B]) displayed significantly reduced Isoc-like currents after exposure to IFNγ (Figures S2A and S2A′). In contrast, IFNγ-primed macrophage displayed largely intact Ca2+ current upon ORAI1-silencing (reduced ORAI1 expression was verified [Figure S2B]), albeit at a marginally lower level compared with the cells transfected with control siRNA (Figures 2C and 2C′). However, silencing of both TRPC1 and ORAI1 significantly decreased Ca2+ current in both naive and IFNγ-induced M1-BM macrophages (Figures 2D and 2D′). As the magnitude of the decrease in Ca2+ current in ORAI1-silenced BM macrophage with or without exposure to IFNγ was similar (Figures 2C and 2C′), it suggested that TRPC1 could compensate for IFNγ-induced Ca2+ currents associated with BM macrophage polarization from naive/M0 to M1 phenotype. Together these data suggest that under in vitro conditions ORAI1 functions as a dominant contributor to the basal Ca2+ influx in naive macrophage, whereas TRPC1 is the major contributor to the Ca2+ influx in IFNγ-induced M1 macrophages.
Figure 2.

PM Ca2+ Influx Channels in M1 Macrophages In Vitro
(A) BM macrophages were pulsed with medium alone (M0) or IFNγ (M1) for the indicated times and subjected to whole-cell patch clamp recordings. The I-V curves display presence of signature current for TRPC1 channels in M1 macrophages and for ORAI1 channels in M0 macrophages. Averages of 8–10 recordings at −80 mV and corresponding statistics are shown in bar graph (A′).
(B) Comparison of IV curves of WT and TRPC1−/− macrophages cultured with medium alone (M0) for 24 hr, or IFNγ (M1) for 24 hr. The signature current for TRPC1 channels increased in IFNγ-exposed WT macrophages, but not in TRPC1−/− macrophages. Average current density recordings from 8 to 10 cells at −80 mV and corresponding statistics are shown in bar graph (B′).
(C) BM macrophages transfected with control siRNA (siC) or siRNA specific for ORAI1 (siO1) were cultured under M0 and M1 conditions for 24 hr. I-V curves were compared in control and ORAI1 knockdown cells by whole-cell patch-clamp recordings. Statistics from 8–10 recordings are shown in bar graph (C′).
(D) I-V curves were compared between BM macrophage transfected with control siRNA (siC) and cells transiently transfected with siRNA against TRPC1 and ORAI1 together (siT1+siO1) and cultured under M0 and M1 conditions for 24 hr. Averages (8–10 recordings at −80 mV) and statistics are shown in bar graph (D′).
(E) Representative time course of TRPC1 and ORAI1 protein expression in response to IFNγ by BM macrophages after immunoprecipitation with anti-STIM1 antibody (Cell Signaling, 4916S), followed by immunoblotting as seen upon subjecting 30 μg protein to SDS-PAGE and anti-TRPC1 (Abcam, ab192031), anti-ORAI1 (Alamone Lab, ACC-060), or anti-STIM1 (Cell Signaling, 4916S; used as loading control). The bar graphs (E′) represent average pixel intensity of the respective protein bands from three independent experiments.
(F) TRPC1 and ORAI1 protein expression in BM macrophage pulsed with IFNγ for 0 min, 2 hr, or 6 hr by western blot as seen subjecting 30 μg protein to SDS-PAGE and using anti-TRPC1 (Abcam, ab192031), anti-ORAI1 (Alamone Lab, ACC-060), or β-actin (Cell Signaling, 4970S). The bar graphs (F′) represent the average pixel intensity of the respective TRPC1 and ORAI1 protein bands from three independent experiments.
*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (Student's t test).
See also Figures S2 and S4.
STIM1 has been shown to be the critical regulator for both ORAI1 and TRPC1 channels (Huang et al., 2006). Thus association of STIM1 with ORAI1 and TRPC1 in naive versus M1 macrophages was evaluated (Figures 2E, 2F, and S4). Cells exposed to IFNγ exhibited increased TRPC1-STIM1 association (Figure 2E), which was statistically significant compared with the basal level (Figure 2E′). On the other hand, these cells displayed a loss in ORAI1-STIM1 association compared with the association in naive macrophages (Figure 2E). Moreover, ORAI1 expression at protein level was detected at a significantly higher level compared with TRPC1 in naive macrophages (Figures 2F and 2F′). Remarkably, IFNγ exposure induced a progressive increase in TRPC1 expression (Figure 2F), which was statistically significant (Figure 2F′). In contrast, ORAI1 expression remained unchanged compared with basal levels (at 0 hr) in BM macrophage (Figures 2F and 2F′). Together, the results revealed that IFNγ stimulation specifically induces an increased TRPC1 expression, along with increased TRPC1-STIM1 interaction, which increases TRPC1 channel activity in M1 macrophage in vitro.
TRPC1 Mediates the Elevated Ca2+ Influx in BM Macrophage Polarized to M1 Phenotype In Vivo
To test the physiological relevance of our in vitro findings, we next used a murine peritonitis model to determine if TRPC1 had a similar effect in M1 activation in vivo (Figure 3). Lipofectamine complexed with ORAI1-siRNA or TRPC1-siRNA was administered i.p. into the thioglycolate-injected mice to transiently knock down the pertinent channels in peritoneal macrophages in vivo (Figure S3). To determine if TRPC1 contributes to the elevated Ca2+ influx in M1 cells generated upon IFNγ exposure in vivo, Ca2+ imaging was performed. TRPC1-deficient peritoneal macrophages (M1-siT1) from mice that received TRPC1-siRNA and IFNγ displayed a significantly decreased Ca2+ influx when compared with the peritoneal macrophages from mice that were given IFNγ and control siRNA (M1-siC) (Figures 3C and 3C′). In contrast, ORAI1-deficient peritoneal macrophages from mice that received ORAI1- siRNA and IFNγ (M1-siO1) displayed only a marginal decrease in Ca2+ influx that was not significant when compared with PM1-siC cells (Figures 3C and 3C′). Importantly, peritoneal macrophages from TRPC1−/− mice that received IFNγ (M1-TRPC1−/−) displayed a robust decrease in Ca2+ influx when compared with the peritoneal macrophages from WT mice with IFNγ (M1-WT) (Figures 3B and 3B′). Furthermore, electrophysiological recordings showed that peritoneal macrophages from the WT mice that were treated with IFNγ displayed a robust ∼3.0-fold increase in non-selective Ca2+ current compared with the M0-WT (Figures 3D and 3D′). In addition, the current reversed at 0 mV and had similar properties to that of TRPC1-dependent Isoc (Figure 3D) as observed earlier. Indeed, IFNγ-primed peritoneal macrophages from TRPC1−/− mice (M1-TRPC1−/−) failed to display the induced Isoc-like Ca2+ current compared with the cells from WT mice (Figures 3D and 3D′). Moreover, peritoneal macrophages from WT (M0-WT) and TRPC1−/− (M0-TRPC1−/−) mice that received PBS only displayed a Ca2+ current with properties similar as ORAI1-dependent ICRAC (Figure 3D).
Figure 3.

TRPC1 Mediates IFNγ-Induced Ca2+ Influx in Peritoneal Macrophages In Vivo
(A) Schematic showing calcium imaging, electrophysiological recordings, and biochemical analysis performed on peritoneal macrophages (peritoneal macrophages) from IFNγ i.p. injected WT and TRPC1−/− mice or mice injected with TRPC1 siRNA or ORAI1 siRNA to transiently knock down these proteins in vivo before the animals received IFNγ.
(B) Ca2+ entry triggered by Tg in peritoneal macrophages from WT or TRPC1−/− mice that received IFNγ i.p. (M1). Analog plots of the fluorescence ratio (340/380 nm) from an average of 40–50 cells are shown. The bar graph (B′) indicates means ± SEM of the Ca2+ release (left peak in B) and store-operated Ca2+ entry (SOCE) (right peak in B).
(C) Ca2+ entry triggered by Tg in IFNγ-exposed peritoneal macrophages transiently deficient in TRPC1 (M1-siT1) or ORAI1 (M1-siO1), or control cells (M1-siC) obtained from mice that received non-targeting siRNA. Analog plots of the fluorescence ratio (340/380 nm) from an average of 40–50 cells are shown. The bar graph (C′) indicates means ± SEM of the Ca2+ release.
(D) I-V curves in peritoneal macrophages from WT or TRPC1−/− mice that received IFNγ (M1) or vehicle (M0) i.p. Average of 8–10 recordings for current intensity at −80 mV is presented in the bar graph (D′).
(E) IFNγ-exposed peritoneal macrophages transiently deficient in TRPC1 (M1-siT1) or ORAI1 (M1-siO1), or control cells (M1-siC) were subjected to whole-cell patch-clamp recordings. Average of 8–10 recordings used for I-V relationships are shown in bar graph (E′).
(F) TRPC1-STIM1 and ORAI1-STIM1 complex formation in peritoneal macrophages from C57BL/6 mice i.p. injected with PBS (M0-PBS), thioglycolate (M0-Thio) and PBS, or thioglycolate and IFNγ (M1). Immunoprecipitation was performed on 250 μg of protein extracts from approx. 5 × 106 cells using anti-STIM1 antibodies (Cell Signaling, 4916S) as seen after subjecting the immunoprecipitates to SDS-PAGE followed by immunoblot detection with anti-TRPC1 (Abcam, ab192031) and anti-ORAI1(Alamone Lab, ACC-060). Anti-STIM1 (Cell Signaling, 4916S) was used for loading control. Bar graph (F′) represents the average pixel intensity of the respective protein bands from three independent experiments.
(G) TRPC1 and ORAI1 protein expression in peritoneal macrophages from C57BL/6 mice i.p. injected with PBS (M0-PBS), thioglycolate and PBS (M0-Thio), or thioglycolate and IFNγ (M1) as seen subjecting 30 μg protein to SDS-PAGE and using anti-TRPC1 (Abcam, ab192031), anti-ORAI1 (Alamone Lab, ACC-060), or anti-GAPDH for western blot. Bar graph (G′) represents the average pixel intensity of the respective protein bands from three independent experiments.
*p ≤ 0.05, ***p ≤ 0.001 (Student's t test).
See also Figure S3.
Next, peritoneal macrophages from mice that received TRPC1-siRNA, ORAI1-siRNA, or control siRNA before i.p. injection with IFNγ or vehicle (PBS) were further analyzed. TRPC1-deficient peritoneal macrophages from mice that received TRPC1-siRNA and IFNγ (M1-siT1), but not ORAI1-siRNA and IFNγ (M1-siO1), displayed complete inhibition of the IFNγ-induced Isoc with compared with M1-siC cells (Figures 3E and 3E′). Indeed, similar to the M1-TRPC1−/−, M1-siT1, the IFNγ-primed peritoneal macrophages that are transiently deficient in TRPC1, displayed a low amount of residual current, which is similar in magnitude and characteristic (inwardly rectifying like ORAI1- mediated ICRAC) to the basal current detected in control macrophage (Figures 3D and 3E). These data further suggest that ORAI1-dependent Ca2+ influx is present in naive macrophage, whereas TRPC1 mediates the IFNγ-induced Ca2+ influx in M1 macrophage in vivo.
To confirm the in vivo role of TRPC1 in IFNγ-induced macrophage polarization to M1 phenotype, the interaction between STIM1 and TRPC1/ORAI1 was also analyzed (Figures 3F and S4). Mice that received PBS or thioglycolate only, or thioglycolate and IFNγ, were sacrificed for analysis of STIM1-TRPC1 and STIM1-ORAI1 interactions. Co-immunoprecipitation using anti-STIM1 on cell lysate followed by western blot analysis with relevant antibodies were performed. Peritoneal macrophages from control mice that received either PBS (M0-PBS) or thioglycolate + PBS (M0-Thio) displayed a scarce presence of TRPC1, while exhibiting strong interaction between Orai1 and STIM1, as evident by robust detection of ORAI1 (Figures 3F and 3F′). STIM1 interaction with ORAI1 was unchanged in peritoneal macrophages from mice that received IFNγ (Figures 3F and 3F′), whereas cells from the IFNγ-treated mice displayed increased STIM1-TRPC1 interaction (∼4-fold) compared with the cells from mice given PBS or thioglycolate only (Figures 3F and 3F′). Consistent with the ability of IFNγ to elevate TRPC1 expression in vitro, peritoneal macrophages from mice given thioglycolate and IFNγ displayed significantly induced expression of TRPC1 (increased by ∼6-fold), whereas the ORAI1 level remains unchanged in these cells compared with the peritoneal macrophages from mice that received thioglycolate only (Figures 3G and 3G′).
TRPC1-Deficient Macrophage Are Impaired in Their Ability to Develop IFNγ-Induced M1 Inflammatory Response In Vitro
To determine the effect of TRPC1 on the induction of the M1 functional phenotype, a series of experiments were performed to directly compare signaling, transcriptional responses, and surface expression of maturation markers. Unstimulated BM macrophages that were transfected with non-targeting siRNA (M0-siC) or TRPC1 siRNA (M0-siT1) displayed a low basal pSTAT1 and pNF-κB (pp65) (Figures 4A and 4A′). However, when stimulated with IFNγ, only BM macrophages that were transfected with non-targeting siRNA (M1-siC) displayed induced elevated amounts of pSTAT1 and pp65 (Figures 4A and 4A′). Importantly, IFNγ-induced pSTAT-1 and pp65 levels were also significantly reduced by severalfold in BM macrophages that were transfected with TRPC1 siRNA (BM1-siT1) at all time points tested (Figures 4A′ and S6A). Next, the effects of IFNγ on TRPC1-deficient and TRPC1-positive cells were compared in vitro for the production of signature M1 inflammatory molecules: anti-microbial effectors (e.g., NO and NOS2), cytokines (e.g., TNF-ᾳ, interleukin [IL]-6, IL-23), Th1-recruiting chemokines (e.g., CXCL9, CXCL10), and factors involved in promoting cytotoxic adaptive immunity (e.g., major histocompatibility complex class II [MHC-II] and costimulatory molecules CD80, CD86) (Ivashkiv, 2013). M2 functional phenotypic signature factors (e.g., Arg1, CCL22) were also analyzed to have better clarity about the biological significance of the role of TRPC1. IFNγ treatment resulted in the presence of large amounts of NO by 18 hr post-exposure in the supernatants of control siRNA-transfected BM macrophages (Figure 4B) or BM macrophages that are isolated from WT mice (Figure S5). In contrast, NO level was barely detected in supernatants of IFNγ-stimulated macrophage transfected with TRPC1-siRNA (Figure 4B) or BM macrophage isolated from TRPC1−/− mice (Figure S5). IFNγ exposure of TRPC1-sufficient BM macrophages that either are isolated from WT mice or were transfected with non-targeting siRNA (M1-siC) displayed induced expression of NOS2, TNF-α, IL-6, IL-23, CXCL9, and CXCL10 (Figures 4C and S7). In contrast, in the absence of TRPC1 BM macrophages that are isolated from TRPC1−/− mice (M1-TRPC1−/−) or BM macrophages that were transfected with TRPC-siRNA (M1-siT1), IFNγ exposure resulted in significantly lower expression of M1 phenotype factors (Figures 4C, 4D, and S7). Among the two M2 anti-inflammatory mediators analyzed, CCL22 and arginase-1 (ARG-1) expression were found to be unaffected by IFNγ exposure in TRPC1-positive and TRPC1-deficient BM macrophages in vitro (Figures 4D and S7). Next, flow cytometry was performed to analyze the effect of TRPC1 on the surface expression of maturation markers MHC-II, CD80, and CD86. BM macrophage transfected with control siRNA exposed to IFNγ showed increased surface expression of MHC-II, CD80, and CD86 when compared with mock cells cultured in the presence of the medium alone (Figures 4E, 4E′, and S6B). However, BM macrophage transfected with TRPC1-siRNA exposed to IFNγ exhibited an inhibition of IFNγ-induced upregulation of MHC-II and costimulatory molecules CD80 and CD86, which was statistically significant (Figures 4E, 4E′, and S6B). Together these results demonstrate that the absence of TRPC1 efficiently prevents the development of the IFNγ-induced M1 functional phenotype in macrophages in vitro.
Figure 4.

Effect of TRPC1 Deficiency on the Ability of IFNγ to Induce M1 Macrophages In Vitro
To analyze the effect of TRPC1 deficiency on M1 macrophage functions, BM macrophages from WT and TRPC1−/− mice were generated in vitro. In addition, BM macrophages from C57BL/6 mice were transfected with non-targeting siRNA or TRPC1 siRNA to transiently knock down TRPC1. Cells were cultured in the presence or absence of IFNγ, and the level of M1-associated signature immune mediators and transcriptions factors were measured by western blot, RT-PCR, and colorimetric assay.
(A) BM macrophages transfected with non-targeting siRNA (siC), or TRPC1 siRNA (siT1) were pulsed with medium alone (M0-siC, M0-siT1) or IFNγ (M1-siC, M1-siT1). Immunoblot analysis were performed using anti-pSTAT1, anti-pNFκB p65 (pp65), STAT1, and p65. The average pixel intensity of the pSTAT1 and pp65 protein bands from three independent experiments is shown in A′.
(B) NO was assessed by colorimetric assay in supernatants collected at 24 hr from SiC and siT1 cells treated as described in (A).
(C) The relative mRNA expression of M1 inflammatory mediators, NOS2, IL-23, CXCL9, and CXCL10 in BM macrophages transfected with control siRNA (siC) or TRPC1 siRNA (siT1) and pulsed for 24 hr with IFNγ (M1) versus medium only (M0).
(D) The relative mRNA levels of M1 inflammatory mediators, NOS2, TNF-ᾳ, IL-6, IL-23, CXCL9, and CXCL10, and M2 anti-inflammatory mediators, CCL22 and arginase-1 (ARG-1), were analyzed in BM macrophages from WT and TRPC1−/− mice and pulsed for 24 hr with IFNγ (M1) versus medium only (M0).
(E) BM macrophages transfected with control siRNA or TRPC1 siRNA and pulsed for 24 hr with medium only (M0-siC, M0-siT1) or IFNγ (M1-siC, M1-siT1). The surface expression of costimulatory molecule CD86 was measured by flow cytometry. Plots in (E′) depict the mean ± SEM of M0-siC, M0-siT1, M1-siC, and M1-siT1 cells expressing MHC-II, CD80, or CD86 as measured by flow cytometry (density plots shown in E and Figure S4B).
*p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (Student's t test).
See also Figures S4–S7.
TRPC1 Is Critical for Induction of M1 Inflammatory Response in IFNγ-Primed Peritoneal Macrophages In Vivo
To determine if TRPC1 participates in the induction of the M1 inflammatory response in vivo, we used the previously described murine peritonitis model. Peritoneal macrophages from mice that were given IFNγ and control siRNA (M1-siC) displayed upregulated amounts of pSTAT1 and pp65 when compared with cells from mice that received PBS only (M0-WT) (Figures 5A and 5A′). In contrast, peritoneal macrophages from mice that received TRPC1-siRNA and IFNγ (M1-siT1) had negligible amounts of pSTAT1 or pp65 (Figures 5A and 5A′). Likewise, peritoneal macrophages from WT mice that received IFNγ (M1-WT) displayed increased pSTAT1 and pp65 when compared with peritoneal macrophages from PBS-injected mice (M0-WT) (Figures 5B and 5B′). On the contrary, peritoneal macrophages from TRPC1−/− mice that received IFNγ (M1-TRPC1−/−) presented no change in pSTAT1 and pp65 levels when compared with the peritoneal macrophages from TRPC1−/− mice that received PBS only (M0-TRPC1−/−) (Figures 5B and 5B′). Moreover, peritoneal macrophages from mice that were given control siRNA, similar to the cells from WT mice, exhibited robust increase in the expression of NOS2, TNF-α, IL-6, IL-23, CXCL9, and CXCL10 in response to IFNγ compared with vehicle/PBS (Figures 5D and S8). On the contrary, peritoneal macrophages from mice that were given TRPC1-siRNA (Figure 5D), similar to the cells from TRPC1−/− mice, had impaired upregulation of NOS2, TNF-α, IL-6, IL-23, CXCL9, and CXCL10 gene expression while exhibiting no effect on the expression of M2 molecules CCL22 and ARG-1 (Figure 5C). Gene expression of all these inflammatory mediators was detected at a significantly lower level in TRPC1-deficient peritoneal macrophages in response to IFNγ when compared with the TRPC1-positive macrophages (Figure 5C). These results suggest that TRPC1 is required for in vivo IFNγ-activated M1 inflammatory macrophage responses.
Figure 5.

TRPC1 Deficiency Results in Reduced IFNγ-Induced Phosphorylation of STAT1 and NF-κB as well as Impaired Production of M1 Inflammatory Mediators in Peritoneal Macrophages In Vivo
Immunoblotting and qRT-PCR analysis were performed on peritoneal macrophages from IFNγ i.p.-injected WT and TRPC1−/− mice. IFNγ-induced effect was also analyzed in peritoneal macrophages in which TRPC1 was transiently knocked down in vivo by i.p. injection of TRPC1 siRNA before the animals received IFNγ.
(A) Peritoneal macrophages transiently deficient in TRPC1 or control cells from mice that received siRNA specific for TRPC1 or non-targeting siRNA were harvested 24 hr after i.p. injection with vehicle (M0-siC, M0-siT1) or IFNγ (M1-siC, M1-siT1). Western blot analysis using anti-pSTAT1, pp65, p65, STAT1, and anti-GAPDH was performed on equal amount of the respective cell lysates. The bar graph (A′) depicts average ± SEM of pixel intensity of the pSTAT1 and pp65 protein bands.
(B) Immunoblots of pSTAT1 and pp65 levels in peritoneal macrophages from PBS- (M0) or IFNγ (M1)-injected WT and TRPC1−/−mice. The bar graph (B′) depicts averages ±SEM of pixel intensity of the pSTAT1 and pp65 protein bands.
(C) The expression of M1-associated inflammatory mediators and M2-specific anti-inflammatory mediators measured by qRT-PCR in peritoneal macrophages from PBS- (M0) or IFNγ (M1)-injected WT and TRPC1−/−mice.
(D) The expression of M1-associated inflammatory mediators in M0 and M1 peritoneal macrophages from PBS- (M0) or IFNγ (M1)-injected mice measured by qRT-PCR after in vivo treatment with siC or siT1 RNA, detailed in (A).
***p ≤ 0.001 (Student's t test).
See also Figure S8.
Macrophages in TRPC1-Deficient Mice Display Impaired Ca2+ Influx and Ability to Induce the M1 Inflammatory Protective Response during Klebsiella pneumoniae Infection
To determine whether TRPC1-mediated Ca2+ influx is involved in the development of the M1 inflammatory phenotype in a clinically relevant infection, a murine model of peritonitis was used (Figure 6A). WT and TRPC1−/− mice were i.p. infected with Klebsiella pneumoniae (KPn) or PBS (mock) for 24 hr before collection of peritoneal macrophages for Ca2+ imaging and electrophysiological analysis ex vivo. Peritoneal macrophages from mock and KPn-infected WT and TRPC1−/− mice in response to Tg in a Ca2+-free buffer show similar internal Ca2+ release (Figures 6A1 and 6A2) and a similar Ca2+ influx upon addition of external Ca2+ to Tg-treated cells. In contrast, Tg stimulation of peritoneal macrophages from KPn-infected WT mice in a Ca2+-free buffer resulted upon subsequent Ca2+ addition in a significantly increased influx of Ca2+ compared with that seen in mock peritoneal macrophages (second peak, Figure 6A1; quantitative data are shown as bar graph, Figure 6A2). In contrast, TRPC1−/− peritoneal macrophages from KPn-infected mice failed to show any significant increase in Ca2+ influx when compared with mock control (Figure 6A1). Moreover, peritoneal macrophages from infected TRPC1−/− mice exhibited significantly lower Ca2+ influx compared with the cells from the WT animals (Figure 6A2). Indeed, electrophysiological recordings in these cells when stimulated with Tg in the presence of 2 mM external Ca2+ display a rapid and significant increase in Ca2+ currents only in peritoneal macrophages from WT-infected mice (Figure 6A3). Consistent with the above results, Ca2+ currents were also greatly attenuated in TRPC1−/− peritoneal macrophages from KPn-infected mice (Figure 6A3).
Figure 6.

TRPC1 Mediates Ca2+ Influx in Macrophage during Preclinical Peritonitis due to Klebsiella pneumoniae (KPn) Infection and in Human Patients with SIRS
(A) Calcium imaging and electrophysiological recordings were performed on peritoneal macrophage from WT and TRPC1−/− mice i.p. infected with KPn for 24 hr.
(A1) Peritoneal macrophages from WT and TRPC1−/− KPn-infected mice loaded with Fura-2AM and Tg added in Ca2+-free medium to measure the internal Ca2+ release (first peak) before addition of external Ca2+ as indicated to measure Ca2+ entry/influx through the plasma membrane (second peak). Analog plot of the fluorescence ratio (340/380 nm) from an average of 40–50 cells is shown. The bar graph (A2) indicates average ratio ± SEM of the Ca2+ release (first peak) and store-operated Ca2+ entry (SOCE) (second peak). (A3) Representative Ca2+ currents at −80 mV from a 0 mV holding potential of peritoneal macrophages from WT and TRPC1−/−KPn-infected mice by whole-cell patch-clamp analysis with 1 μM Tg in the pipette solution. (A4) I-V curves showing the TRPC1 channel-associated signature Isoc in peritoneal macrophages from KPn-infected WT, but not in TRPC1−/− mice. The signature current for ORAI1 channels (Icrac) was present in peritoneal macrophages from mock control WT and KPn-infected TRPC1−/− mice. Average current density recordings from 8 to 10 cells at −80 mV and corresponding statistics are shown in the bar graph (A5). (A6) From the KPn-infected WT and TRPC1−/− mice, peritoneal lavage, and liver and blood samples were collected. Liver was homogenized. Blood or liver homogenate or peritoneal lavage samples were serially diluted and plated on LB plates. Bacterial burden was enumerated after incubating the plates overnight at 37°C. Results shown here are mean ± SE from three experimental animals (n = 3). *p < 0.05**, p ≤ 0.01, ***p ≤ 0.001 (Student's t test).
(B) M1 inflammatory activation phenotype in circulating monocytes/macrophage in humans with SIRS correlated with TRPC1 expression. Blood samples were collected from patients with SIRS every 24 hr for up to 10 days or until discharged from the ICU. PBMCs from patient and healthy donors (HC) were isolated and circulating monocytes/macrophages purified by positive magnetic selection. (B1) Western blot analysis using anti-TRPC1 and anti-ORAI1 was performed on cell lysates. GAPDH was used as loading control. The bar graph (B2) depicts averages ±SEM of pixel intensity of the TRPC1 and ORAI1 protein bands. Representative of n = 4 healthy donors and patients with SIRS. (B3) The expressions of M1-associated inflammatory mediators, CXCL9 and CXCL10, and M2 anti-inflammatory mediator, CCL22, were measured by qRT-PCR. *p < 0.05, **p ≤ 0.01 (Student's t test).
Electrophysiological recordings showed that peritoneal macrophages from KPn-infected WT mice developed a non-selective Ca2+ current that reversed between 0 and 5 mV, which is consistent with TRPC1-dependent ISOC currents (Figures 6A4 and 6A5). However, peritoneal macrophages from KPn-infected TRPC1−/− mice failed to show an increase in Ca2+ current (Figures 6A4 and 6A5), suggesting the important role played by TRPC1 in SOCE in activated macrophages during bacterial infection of the host. Indeed, macrophage from KPn-infected TRPC1−/− mice displayed slightly inward-rectifying Ca2+ currents (ORAI1-dependent Ca2+ current) (Figure 6A4) as observed in naive/M0 macrophage from WT and TRPC1−/− mice (Figures 2 and 3). Thus results from this study clearly demonstrate that the KPn infection in mice induced an increased Ca2+ influx specifically through the TRPC1 channels on the PM of macrophage.
Macrophages from KPn-Infected TRPC1−/− Mice Exhibit Reduced STAT1 and NFκB p65 Activation and Express Lower Levels of M1 Inflammatory Mediators
Because macrophages activate specific polarizing signal transduction pathways (STAT1, p65, and NF-κB in M1 inflammatory and STAT6 in M2 anti-inflammatory macrophages), we performed western blot analysis on cell homogenates to measure the levels of pSTAT1 and pSTAT6 in peritoneal macrophages from KPn-infected WT and TRPC1−/− mice. In both strains, peritoneal macrophages from mock-infected mouse peritoneum displayed low basal levels of pSTAT1 and pp65 (Figures 7A and 7A′). Peritoneal macrophages from KPn-infected WT mice exhibited increased levels of pSTAT1 and pp65 (Figures 7A and 7A′). In contrast, peritoneal macrophages from infected TRPC1−/− mice failed to display an increase in pSTAT1 and pp65 (Figures 7A and 7A′). This effect was selective for M1-associated signaling as pSTAT6 levels remained undetectable in peritoneal macrophages from both the WT and TRPC1−/− mock and infected mice (data not shown). Thus the data suggest that induction of M1 macrophage-specific signal transduction pathways was completely inhibited in the absence of TRPC1 in mice undergoing bacterial infection.
Figure 7.

Peritoneal Macrophage from TRPC1−/− Mice Exhibit Reduced M1 Inflammatory Responses during Experimental Peritonitis due to Klebsiella pneumoniae Infection
(A) Peritoneal macrophages from mock control (M) and KPn-infected WT and TRPC1−/− mice were harvested. Western blot analysis using anti-pSTAT1, pp65, p65, STAT1, and GAPDH was performed on equal amount of the respective cell lysates. GAPDH, p65, and STAT1 were used as loading control. The bar graph (A′) depicts averages ± SEM of pixel intensity of the pSTAT1 and pp65 protein bands.
(B) Peritoneal macrophages from mock control (M) and KPn-infected WT and TRPC1−/− mice were harvested, and the relative mRNA expression of M1 inflammatory mediators, NOS2, TNF-ᾳ, IL-6, IL-23, CXCL9, and CXCL10, and M2 anti-inflammatory mediators, CCL22 and ARG-1, was measured by qRT-PCR.
**p ≤ 0.01, ***p ≤ 0.001 (Student's t test).
To assess the functional relevance of TRPC1 on the induction of M1 inflammatory response in a clinically relevant infection model, RT-PCR was performed to measure key inflammatory mediators in peritoneal macrophages from mock and KPn-infected WT and TRPC1−/− mice. In both strains, peritoneal macrophages from mock-infected mice displayed low basal levels of M1 signature factors, NOS2, TNF-α, IL-6, IL-23, CXCL9, and CXCL10 (Figure 7B). On the contrary, peritoneal macrophages upon KPn infection in WT mice exhibited significantly robust increase in expression of all these signature M1 inflammatory mediators at 24 hr post-infection (Figure 7B). In contrast, the transcript levels of all these M1 inflammatory mediators were detected at a significantly lower level in peritoneal macrophages from KPn-infected TRPC1−/− mice (p < 0.001) (Figure 7B). We also performed flow cytometry analysis to examine the differences in the surface expression of maturation markers, MHC-II, CD80, and CD86, on macrophages from TRPC1−/− and WT mock and KPn-infected mice. The increased expression of MHC-II, CD80, and CD86, in KPn-infected WT mice was partially inhibited in macrophage from KPn-infected TRPC1−/− mice when compared with WT mice (data not shown). This impaired M1 inflammatory response was correlated with a significantly higher bacterial load in the peritoneal lavage, liver, as well as blood (Figure 6A6) of these animals when compared with the WT mice. Taken together, the data clearly demonstrate that absence of TRPC1-dependent Ca2+ influx leads to significant inhibition of M1 inflammatory responses in macrophages during KPn infection of mice, which likely contributes to the increased bacteremia of TRPC1 mice to this infection.
Correlation between Expression of TRPC1 and M1 Inflammatory Mediators in Macrophages of Patients with SIRS
We next assessed if the results obtained from the mouse models could be extrapolated to human disease condition. To this end, peripheral blood mononuclear cells (PBMCs) of a prospective cohort of individuals exhibiting symptoms of SIRS admitted to the intensive care unit (ICU) were isolated from blood samples obtained every 24 hr for 10 days or until discharge. Monocytes/macrophages purified from patients' samples as well as healthy age- and sex-matched controls by positive selection followed by kinetic expression of TRPC1 and ORAI1 by western blot. In line with our results from murine studies, TRPC1 protein was barely detectable in circulating monocytes/macrophages of healthy control (HC) humans (Figure 6B1). Patients with SIRS exhibited 20- to 40-fold higher levels of TRPC1 expression at all post-admission times tested (Figure 6B2). Interestingly, the increase in TRPC1 protein expression correlated with the severity of SIRS (Figures 6B1 and 6B2). A concomitant higher transcript level of M1 inflammatory mediators CXCL9 and CXCL10, but not CCL22 (M2 anti-inflammatory mediator) was observed in monocytes/macrophages from these patients (Figure 6B3). The baseline expression of ORAI1 was found to be higher than that of TRPC1 in HCs (Figures 6B1 and 6B2). Patients with SIRS, however, did not show any change in expression of ORAI1 (Figures 6B1 and 6B2), reproducing our findings in peritonitis mice model or in in vitro after IFNγ stimulation. Overall, analysis of human patient samples correlated TRPC1, but not ORAI1, with M1 inflammatory phenotypic activation in macrophages.
Discussion
In the last decade, there has been an increased appreciation of the Ca2+-mediated regulation of inflammation in autoimmunity and immune response against pathogens. Previous studies have clearly identified an association of mutations in Ca2+ signaling pathways with severe immune deficiency in humans (Oh-Hora et al., 2013). Several experimental models complemented these studies by showing a defect in Ca2+ signaling as the cause of impaired T cell and B cell responses (Feske et al., 2015, Hogan et al., 2010). However, the role of Ca2+ and the identity of specific Ca2+ channels that regulate innate immune functions involving macrophage M1 activation phenotype have been largely elusive. The data presented here show an essential function of agonist-induced Ca2+ influx in the activation of functional phenotype-determining transcription factors and gene expression of signature M1 inflammatory molecules. Importantly, these studies identify a novel function of TRPC1 in the induction of Ca2+ influx during macrophage polarization to the M1-activated phenotype in multiple experimental models in vitro and in vivo.
Macrophage responses to different pattern recognition receptor (PRR) ligands have been linked to Ca2+ signals (Goodridge et al., 2007, Zanoni et al., 2009, Zanoni et al., 2011), but direct inhibition studies of Ca2+ elevations on the functions of these cells are disparate. The discrepancies are particularly evident among studies reporting the role of SOCE in macrophage functions (Demaurex and Nunes, 2016). For example, studies using stim1−/− mice display abrogated SOCE and markedly reduced Ca2+ elevations upon FcγR cross-linking in peritoneal macrophages (Braun et al., 2009). The observed reduced FcγR-dependent phagocytosis in these settings, and the fact that the chimeric stim1−/− mice were resistant to IgG-dependent autoimmune hemolytic anemia (AIHA), as well as autoimmune thrombocytopenia and anaphylaxis, provide support for the idea that Ca2+ elevations by SOCE are an important contributor to macrophage functions. Moreover, studies involving stim2−/− mice showed a reduced production of inflammatory mediators correlated with increased survival during lipopolysaccharide (LPS)-induced sepsis (Sogkas et al., 2015). Although these stim2−/− mice were not protected from AIHA, it provides additional support for the idea that Ca2+ plays a role in macrophage functions. In contrast, a study using conditional deletion of both the Stim genes (Stim1fl/fl; Stim2fl/fl) alone or together in macrophages shows that despite a reduced SOCE, immune functions such as phagocytosis as well as the production of several cytokines in response to several PRR ligands were unaffected (Vaeth et al., 2015). Interestingly, in this setting, abolishing Ca2+ signaling by chelation of intracellular Ca2+ with BAPTA (1,2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid) or 2APB reduced macrophage functions in terms of antigen presentation or IL-1β secretion, respectively, following PRR activation. Thus, despite discrepancies in the role of SOCE, there is a strong indication for Ca2+ signals contributing to macrophage function(s). In this regard, Ca2+ imaging studies presented here showed that IFNγ exposure increases Ca2+ influx. Moreover, 2APB treatment inhibits this IFNγ -induced Ca2+ influx, which correlated with an attenuated M1 pro-inflammatory response. Thus our findings provide strong support for the functional role of Ca2+ entry in macrophage polarization to M1 phenotype.
The finding from electrophysiological studies on M0-like cells from both unstimulated BM macrophages in vitro and in vivo peritoneal macrophages from vehicle-injected control mice show a highly Ca2+-selective inward-rectifying current. This phenotype of Ca2+ current typically corresponds to the ORAI channel. Indeed, our results show that ORAI1 is highly expressed, and interacts with STIM1, indicating the functional relevance of this channel in naive/M0 macrophage. In contrast, TRPC1, which at the protein level is barely detected in M0 macrophages, increases gradually upon IFNγ treatment. Since ORAI1-siRNA-treated naive/M0 macrophages displayed a massive reduction in ICRAC currents, it provides additional support for a fundamentally important function of ORAI1 in basal Ca2+ influx in macrophage. However, although ORAI1-siRNA-treated BM macrophage in vitro or peritoneal macrophages ex vivo exposed to IFNγ display reduced Ca2+ currents, there was no difference observed in macrophage lacking ORAI1 under M1-polarizing conditions. If IFNγ exposure activates ORAI1-dependent Ca2+ influx, we would expect to observe a larger decrease in Ca2+ current in IFNγ-activated ORAI-siRNA-treated macrophage in vitro or in vivo. Moreover, western blot analysis should have shown an increased ORAI1 level and/or increased ORAI-STIM1 interaction during IFNγ-activated polarization to M1 phenotype when compared with the M0/naive macrophages, which we did not observe. Instead, we found a massive increase in Ca2+ current with channel properties typically linked to TRPC1-dependent Ca2+ influx in the IFNγ-activated cells polarized to the M1 phenotype, both in in vitro and in in vivo settings. Importantly, the IFNγ-induced increase in Ca2+ current was abolished in TRPC1-deficient macrophages from mice that received TRPC1-siRNA or macrophages from TRPC1−/− mice. This was also correlated with an inhibition of the IFNγ-activation-induced increase in intracellular Ca2+ levels in TRPC1-deficient macrophage. The IFNγ-induced increase in TRPC1 protein in BM macrophage in vitro or in peritoneal macrophages from mice injected with IFNγ further support a central role of TRPC1 in Ca2+ influx during polarization of naive macrophage to M1 functional phenotype.
Our findings raised the fundamental question of whether the M1 activation-induced TRPC1-dependent Ca2+ influx is functional and regulates the induction of inflammatory molecules in macrophages. Indeed, the expression of various key M1-associated inflammatory molecules, e.g., the enzyme NOS2, the Th1-recruiting chemokines CXCL9 and CXCL10, as well as the inflammatory cytokines TNF-α and IL-6, were inhibited in TRPC1-deficient BM macrophage following stimulation with the M1 polarizing stimulus IFNγ. In addition, activation of transcription factors STAT1 and NF-κB induced by IFNγ was attenuated in these TRPC1-deficient cells. Similarly, in IFNγ-injected TRPC1−/− mice or WT mice that received TRPC1-siRNA, the expression of these M1 inflammatory mediators was impaired in peritoneal macrophages at the site of inflammation, again highlighting the critical function of the TRPC1-dependent Ca2+ influx activated by IFNγ exposure in mediating the M1 functional phenotype.
Among the known TRP channel proteins, the role of TRPM2, TRPV4, TRPC3, and TRPC4 has been shown in macrophage functions (Di et al., 2017, Hamanaka et al., 2010, Scheraga et al., 2016). The absence of TRPC3 is linked to a reduced ER stress-induced apoptosis/necrosis in M1 macrophages present in advanced atherosclerotic plaques (Smedlund et al., 2015, Solanki et al., 2014, Solanki et al., 2017). TRPM2, was shown to promote macrophage-associated inflammation, which was thought to provide protection from L. monocytogenes infection (Knowles et al., 2011) while exacerbating obesity-induced insulin resistance (Zhang et al., 2012). In contrast, TRPM4 was found to mediate Na+ influx to antagonize Ca2+ influx (Serafini et al., 2012) and inhibit macrophage function in a sepsis model (Serafini et al., 2012). These findings directly support the relevance of TRP proteins in innate immune function, and it is possible that other TRP members could promote the development of specialized functions in macrophages. Our findings show the specific involvement of TRPC1 in mediating intracellular Ca2+ increase in a murine bacterial peritonitis model thereby substantiating that this mechanism is relevant in in vivo immunopathological situations. Similar to our findings using IFNγ-activated macrophages, macrophages from the peritoneum of KPn-infected TRPC−/- mice produced less M1 pro-inflammatory cytokines, chemokines, signature enzymes, and surface maturation markers than those of WT mice. These findings are directly relevant to human disease, and it is notable that we recently reported that immune suppressive helminth molecules specifically inhibit LPS (M1 stimuli)-induced and Tg-induced TRPC1 channel activity in macrophages (Chauhan et al., 2014, Sun et al., 2014). In a recent report, we have also shown that deficiency of TRPC1 impairs host defense and pro-inflammatory responses to bacterial Infection (Zhou et al., 2015). However, it is difficult to ascertain the exact clinical significance of the observed high TRPC1 level in macrophages with M1, but not M2, phenotype during SIRS, as M1 inflammatory response plays protective role in killing microbes, whereas an exuberant inflammatory response has been linked to the development of severe pathology in sepsis. Nevertheless, these results coupled with our data showing uncontrolled bacteremia in Kpn-infected TRPC1−/− mice leads us to posit that TRPC1-mediated M1 inflammatory response by macrophage is important in providing immunity to microbial infection. On the other hand, as persistent inflammation is linked to a majority of chronic immune/autoimmune disorders, we anticipate TRPC1-mediated M1 inflammatory response may function in exacerbating chronic inflammatory disease conditions, a hypothesis we are currently investigating. In toto, the finding that induction of TRPC1-mediated Ca2+ influx occurs in macrophage polarization to M1 phenotype caused by IFNγ activation or bacterial infection, and that absence of TRPC1 completely inhibits M1 cell activation, may offer important mechanisms to target in inflammatory disease conditions.
Limitations of Study
We show that the function of TRPC1 is necessary, but the study design does not address if it is sufficient for the development of M1 inflammatory phenotypic response in macrophages. Sensing of M1-inducing stimuli by surface receptors needs integration of many signaling events into a coherent pattern of gene transcription reprogramming. In this regard, other PM calcium channel(s), despite not contributing to M1 stimuli-induced calcium influx, may in a parallel manner control selective physiological processes underlying phenotype-specific gene expression. Finally, our data with TRPC1 expression and M1 inflammatory mediators in circulating monocytes/macrophages in patients with SIRS is only correlative and does not directly address the precise function of TRPC1 in development/sustenance of M1 inflammatory macrophages in these patients.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by National Institutes of Health (United States) grants (R21DE024300 to B.B.S. and B.B.M.; P20GM113123 to B.B.M., J.S., and B.B.S.; R21AI107457, R01AI121804 to J.S.; R01DE017102 to B.B.S.; and Z01-ES-101684 to L.B.). The Flow Cytometry core facility was supported by the National Institutes of Health COBRE Grant 5P20GM113123 and INBRE Grant 5P20GM103442.
Author Contributions
A.C., Y.S., P.S., AS, F.O.Q., P.C., and C.N.J. performed the experiments. R.E.S., D.L.E., and M.O.A. collected and provided human blood samples. A.C., Y.S., B.B.S., and B.B.M. contributed to the study design and data analyses. B.B.S., J.S., and B.B.M., contributed to the interpretation of results. L.B., B.B.S., J.S., and B.B.M. wrote the manuscript.
Declaration of Interests
The authors have declared that no conflict of interest exists.
Published: October 26, 2018
Footnotes
Supplemental Information includes Transparent Methods, eight figures, and one table and can be found with this article online at https://doi.org/10.1016/j.isci.2018.09.014.
Supplemental Information
References
- Abplanalp A.L., Morris I.R., Parida B.K., Teale J.M., Berton M.T. TLR-dependent control of Francisella tularensis infection and host inflammatory responses. PLoS One. 2009;4:e7920. doi: 10.1371/journal.pone.0007920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun A., Gessner J.E., Varga-Szabo D., Syed S.N., Konrad S., Stegner D., Vogtle T., Schmidt R.E., Nieswandt B. STIM1 is essential for Fcgamma receptor activation and autoimmune inflammation. Blood. 2009;113:1097–1104. doi: 10.1182/blood-2008-05-158477. [DOI] [PubMed] [Google Scholar]
- Byun M., Abhyankar A., Lelarge V., Plancoulaine S., Palanduz A., Telhan L., Boisson B., Picard C., Dewell S., Zhao C. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J. Exp. Med. 2010;207:2307–2312. doi: 10.1084/jem.20101597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahalan M.D., Chandy K.G. The functional network of ion channels in T lymphocytes. Immunol. Rev. 2009;231:59–87. doi: 10.1111/j.1600-065X.2009.00816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan A., Sun Y., Pani B., Quenumzangbe F., Sharma J., Singh B.B., Mishra B.B. Helminth induced suppression of macrophage activation is correlated with inhibition of calcium channel activity. PloS One. 2014;9:e101023. doi: 10.1371/journal.pone.0101023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.C., Chou C.F., Lin W.W. Pyrimidinoceptor-mediated potentiation of inducible nitric-oxide synthase induction in J774 macrophages. role of intracellular calcium. J. Biol. Chem. 1998;273:29754–29763. doi: 10.1074/jbc.273.45.29754. [DOI] [PubMed] [Google Scholar]
- Clapham D.E. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Demaurex N., Nunes P. The role of STIM and ORAI proteins in phagocytic immune cells. Am. J. Physiol. Cell Physiol. 2016;310:C496–C508. doi: 10.1152/ajpcell.00360.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di A., Kiya T., Gong H., Gao X., Malik A.B. Role of the phagosomal redox-sensitive TRP channel TRPM2 in regulating bactericidal activity of macrophages. J. Cell Sci. 2017;130:735–744. doi: 10.1242/jcs.196014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes-Alnemri T., Yu J.W., Datta P., Wu J., Alnemri E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature. 2009;458:509–513. doi: 10.1038/nature07710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007;7:690–702. doi: 10.1038/nri2152. [DOI] [PubMed] [Google Scholar]
- Feske S. CRAC channelopathies. Pflugers Arch. Eur. J. Physiol. 2010;460:417–435. doi: 10.1007/s00424-009-0777-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S., Gwack Y., Prakriya M., Srikanth S., Puppel S.H., Tanasa B., Hogan P.G., Lewis R.S., Daly M., Rao A. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature. 2006;441:179–185. doi: 10.1038/nature04702. [DOI] [PubMed] [Google Scholar]
- Feske S., Skolnik E.Y., Prakriya M. Ion channels and transporters in lymphocyte function and immunity. Nat. Rev. Immunol. 2012;12:532–547. doi: 10.1038/nri3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feske S., Wulff H., Skolnik E.Y. Ion channels in innate and adaptive immunity. Annu. Rev. Immunol. 2015;33:291–353. doi: 10.1146/annurev-immunol-032414-112212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginhoux F., Schultze J.L., Murray P.J., Ochando J., Biswas S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Glass C.K., Natoli G. Molecular control of activation and priming in macrophages. Nat. Immunol. 2016;17:26–33. doi: 10.1038/ni.3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge H.S., Simmons R.M., Underhill D.M. Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J. Immunol. 2007;178:3107–3115. doi: 10.4049/jimmunol.178.5.3107. [DOI] [PubMed] [Google Scholar]
- Hamanaka K., Jian M.Y., Townsley M.I., King J.A., Liedtke W., Weber D.S., Eyal F.G., Clapp M.M., Parker J.C. TRPV4 channels augment macrophage activation and ventilator-induced lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2010;299:L353–L362. doi: 10.1152/ajplung.00315.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan P.G., Chen L., Nardone J., Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003;17:2205–2232. doi: 10.1101/gad.1102703. [DOI] [PubMed] [Google Scholar]
- Hogan P.G., Lewis R.S., Rao A. Molecular basis of calcium signaling in lymphocytes: STIM and ORAI. Annu. Rev. Immunol. 2010;28:491–533. doi: 10.1146/annurev.immunol.021908.132550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G.N., Zeng W., Kim J.Y., Yuan J.P., Han L., Muallem S., Worley P.F. STIM1 carboxyl-terminus activates native SOC, I(crac) and TRPC1 channels. Nat. Cell Biol. 2006;8:1003–1010. doi: 10.1038/ncb1454. [DOI] [PubMed] [Google Scholar]
- Idzko M., Ferrari D., Eltzschig H.K. Nucleotide signalling during inflammation. Nature. 2014;509:310–317. doi: 10.1038/nature13085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B. Epigenetic regulation of macrophage polarization and function. Trends Immunol. 2013;34:216–223. doi: 10.1016/j.it.2012.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jairaman A., Yamashita M., Schleimer R.P., Prakriya M. Store-operated Ca2+ release-activated Ca2+ channels regulate PAR2-activated Ca2+ signaling and cytokine production in airway epithelial cells. J. Immunol. 2015;195:2122–2133. doi: 10.4049/jimmunol.1500396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junger W.G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 2011;11:201–212. doi: 10.1038/nri2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.S., Zeng W., Yuan J.P., Shin D.M., Worley P.F., Muallem S. Native store-operated Ca2+ influx requires the channel function of Orai1 and TRPC1. J. Biol. Chem. 2009;284:9733–9741. doi: 10.1074/jbc.M808097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles H., Heizer J.W., Li Y., Chapman K., Ogden C.A., Andreasen K., Shapland E., Kucera G., Mogan J., Humann J. Transient receptor potential melastatin 2 (TRPM2) ion channel is required for innate immunity against Listeria monocytogenes. Proc. Natl. Acad. Sci. U S A. 2011;108:11578–11583. doi: 10.1073/pnas.1010678108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labonte A.C., Tosello-Trampont A.C., Hahn Y.S. The role of macrophage polarization in infectious and inflammatory diseases. Mol. Cells. 2014;37:275–285. doi: 10.14348/molcells.2014.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence T., Natoli G. Transcriptional regulation of macrophage polarization: enabling diversity with identity. Nat. Rev. Immunol. 2011;11:750–761. doi: 10.1038/nri3088. [DOI] [PubMed] [Google Scholar]
- Le Deist F., Hivroz C., Partiseti M., Thomas C., Buc H.A., Oleastro M., Belohradsky B., Choquet D., Fischer A. A primary T-cell immunodeficiency associated with defective transmembrane calcium influx. Blood. 1995;85:1053–1062. [PubMed] [Google Scholar]
- Link T.M., Park U., Vonakis B.M., Raben D.M., Soloski M.J., Caterina M.J. TRPV2 has a pivotal role in macrophage particle binding and phagocytosis. Nat. Immunol. 2010;11:232–239. doi: 10.1038/ni.1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., Cheng K.T., Bandyopadhyay B.C., Pani B., Dietrich A., Paria B.C., Swaim W.D., Beech D., Yildrim E., Singh B.B. Attenuation of store-operated Ca2+ current impairs salivary gland fluid secretion in TRPC1(-/-) mice. Proc. Natl. Acad. Sci. U S A. 2007;104:17542–17547. doi: 10.1073/pnas.0701254104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locati M., Mantovani A., Sica A. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol. 2013;120:163–184. doi: 10.1016/B978-0-12-417028-5.00006-5. [DOI] [PubMed] [Google Scholar]
- Macian F. NFAT proteins: key regulators of T-cell development and function. Nat. Rev. Immunol. 2005;5:472–484. doi: 10.1038/nri1632. [DOI] [PubMed] [Google Scholar]
- McNally B.A., Somasundaram A., Yamashita M., Prakriya M. Gated regulation of CRAC channel ion selectivity by STIM1. Nature. 2012;482:241–245. doi: 10.1038/nature10752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNelis J.C., Olefsky J.M. Macrophages, immunity, and metabolic disease. Immunity. 2014;41:36–48. doi: 10.1016/j.immuni.2014.05.010. [DOI] [PubMed] [Google Scholar]
- Moore K.J., Sheedy F.J., Fisher E.A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Wynn T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-Hora M., Komatsu N., Pishyareh M., Feske S., Hori S., Taniguchi M., Rao A., Takayanagi H. Agonist-selected T cell development requires strong T cell receptor signaling and store-operated calcium entry. Immunity. 2013;38:881–895. doi: 10.1016/j.immuni.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh-Hora M., Yamashita M., Hogan P.G., Sharma S., Lamperti E., Chung W., Prakriya M., Feske S., Rao A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008;9:432–443. doi: 10.1038/ni1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pani B., Bollimuntha S., Singh B.B. The TR (i)P to Ca(2)(+) signaling just got STIMy: an update on STIM1 activated TRPC channels. Front. Biosci. 2012;17:805–823. doi: 10.2741/3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C., McCarl C.A., Papolos A., Khalil S., Luthy K., Hivroz C., LeDeist F., Rieux-Laucat F., Rechavi G., Rao A. STIM1 mutation associated with a syndrome of immunodeficiency and autoimmunity. N. Engl. J. Med. 2009;360:1971–1980. doi: 10.1056/NEJMoa0900082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakriya M., Feske S., Gwack Y., Srikanth S., Rao A., Hogan P.G. Orai1 is an essential pore subunit of the CRAC channel. Nature. 2006;443:230–233. doi: 10.1038/nature05122. [DOI] [PubMed] [Google Scholar]
- Ramirez G.A., Coletto L.A., Sciorati C., Bozzolo E.P., Manunta P., Rovere-Querini P., Manfredi A.A. Ion channels and transporters in inflammation: special focus on TRP channels and TRPC6. Cells. 2018;7 doi: 10.3390/cells7070070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao A., Hogan P.G. Calcium signaling in cells of the immune and hematopoietic systems. Immunol. Rev. 2009;231:5–9. doi: 10.1111/j.1600-065X.2009.00823.x. [DOI] [PubMed] [Google Scholar]
- Ruckerl D., Allen J.E. Macrophage proliferation, provenance, and plasticity in macroparasite infection. Immunol. Rev. 2014;262:113–133. doi: 10.1111/imr.12221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheraga R.G., Abraham S., Niese K.A., Southern B.D., Grove L.M., Hite R.D., McDonald C., Hamilton T.A., Olman M.A. TRPV4 mechanosensitive ion channel regulates lipopolysaccharide-stimulated macrophage phagocytosis. J. Immunol. 2016;196:428–436. doi: 10.4049/jimmunol.1501688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Sun Y., Watt J.A., Wang S., Lei S., Birnbaumer L., Singh B.B. Neurotoxin-induced ER stress in mouse dopaminergic neurons involves downregulation of TRPC1 and inhibition of AKT/mTOR signaling. J. Clin. Invest. 2012;122:1354–1367. doi: 10.1172/JCI61332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serafini N., Dahdah A., Barbet G., Demion M., Attout T., Gautier G., Arcos-Fajardo M., Souchet H., Jouvin M.H., Vrtovsnik F. The TRPM4 channel controls monocyte and macrophage, but not neutrophil, function for survival in sepsis. J. Immunol. 2012;189:3689–3699. doi: 10.4049/jimmunol.1102969. [DOI] [PubMed] [Google Scholar]
- Smedlund K.B., Birnbaumer L., Vazquez G. Increased size and cellularity of advanced atherosclerotic lesions in mice with endothelial overexpression of the human TRPC3 channel. Proc. Natl. Acad. Sci. U S A. 2015;112:E2201–E2206. doi: 10.1073/pnas.1505410112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogkas G., Stegner D., Syed S.N., Vogtle T., Rau E., Gewecke B., Schmidt R.E., Nieswandt B., Gessner J.E. Cooperative and alternate functions for STIM1 and STIM2 in macrophage activation and in the context of inflammation. Immun. Inflamm. Dis. 2015;3:154–170. doi: 10.1002/iid3.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki S., Dube P.R., Birnbaumer L., Vazquez G. Reduced necrosis and content of apoptotic M1 macrophages in advanced atherosclerotic plaques of mice with macrophage-specific loss of Trpc3. Sci. Rep. 2017;7:42526. doi: 10.1038/srep42526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki S., Dube P.R., Tano J.Y., Birnbaumer L., Vazquez G. Reduced endoplasmic reticulum stress-induced apoptosis and impaired unfolded protein response in TRPC3-deficient M1 macrophages. Am. J. Physiol. Cell Physiol. 2014;307:C521–C531. doi: 10.1152/ajpcell.00369.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su X., Yu Y., Zhong Y., Giannopoulou E.G., Hu X., Liu H., Cross J.R., Ratsch G., Rice C.M., Ivashkiv L.B. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat. Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumaran P., Sun Y., Vyas M., Singh B.B. TRPC1-mediated Ca(2)(+) entry is essential for the regulation of hypoxia and nutrient depletion-dependent autophagy. Cell Death Dis. 2015;6:e1674. doi: 10.1038/cddis.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y., Chauhan A., Sukumaran P., Sharma J., Singh B.B., Mishra B.B. Inhibition of store-operated calcium entry in microglia by helminth factors: implications for immune suppression in neurocysticercosis. J. Neuroinflammation. 2014;11:210. doi: 10.1186/s12974-014-0210-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomilin V.N., Cherezova A.L., Negulyaev Y.A., Semenova S.B. TRPV5/V6 channels mediate Ca(2+) influx in Jurkat T cells under the control of extracellular pH. J. Cell Biochem. 2016;117:197–206. doi: 10.1002/jcb.25264. [DOI] [PubMed] [Google Scholar]
- Vaeth M., Eckstein M., Shaw P.J., Kozhaya L., Yang J., Berberich-Siebelt F., Clancy R., Unutmaz D., Feske S. Store-operated Ca(2+) entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity. 2016;44:1350–1364. doi: 10.1016/j.immuni.2016.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M., Feske S. Ion channelopathies of the immune system. Curr. Opin. Immunol. 2018;52:39–50. doi: 10.1016/j.coi.2018.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M., Maus M., Klein-Hessling S., Freinkman E., Yang J., Eckstein M., Cameron S., Turvey S.E., Serfling E., Berberich-Siebelt F. Store-operated Ca(2+) entry controls clonal expansion of T cells through metabolic reprogramming. Immunity. 2017;47:664–679.e6. doi: 10.1016/j.immuni.2017.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaeth M., Zee I., Concepcion A.R., Maus M., Shaw P., Portal-Celhay C., Zahra A., Kozhaya L., Weidinger C., Philips J. Ca2+ signaling but not store-operated Ca2+ entry is required for the function of macrophages and dendritic cells. J. Immunol. 2015;195:1202–1217. doi: 10.4049/jimmunol.1403013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vig M., Kinet J.P. Calcium signaling in immune cells. Nat. Immunol. 2009;10:21–27. doi: 10.1038/ni.f.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H., Zhang X., Xue L., Xing J., Jouvin M.H., Putney J.W., Anderson M.P., Trebak M., Kinet J.P. Low-voltage-activated CaV3.1 calcium channels shape T helper cell cytokine profiles. Immunity. 2016;44:782–794. doi: 10.1016/j.immuni.2016.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe N., Suzuki J., Kobayashi Y. Role of calcium in tumor necrosis factor-alpha production by activated macrophages. J. Biochem. 1996;120:1190–1195. doi: 10.1093/oxfordjournals.jbchem.a021540. [DOI] [PubMed] [Google Scholar]
- Wynn T.A. Type 2 cytokines: mechanisms and therapeutic strategies. Nat. Rev. Immunol. 2015;15:271–282. doi: 10.1038/nri3831. [DOI] [PubMed] [Google Scholar]
- Wynn T.A., Vannella K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J.P., Zeng W., Huang G.N., Worley P.F., Muallem S. STIM1 heteromultimerizes TRPC channels to determine their function as store-operated channels. Nat. Cell Biol. 2007;9:636–645. doi: 10.1038/ncb1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I., Ostuni R., Capuano G., Collini M., Caccia M., Ronchi A.E., Rocchetti M., Mingozzi F., Foti M., Chirico G. CD14 regulates the dendritic cell life cycle after LPS exposure through NFAT activation. Nature. 2009;460:264–268. doi: 10.1038/nature08118. [DOI] [PubMed] [Google Scholar]
- Zanoni I., Ostuni R., Marek L.R., Barresi S., Barbalat R., Barton G.M., Granucci F., Kagan J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z., Zhang W., Jung D.Y., Ko H.J., Lee Y., Friedline R.H., Lee E., Jun J., Ma Z., Kim F. TRPM2 Ca2+ channel regulates energy balance and glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2012;302:E807–E816. doi: 10.1152/ajpendo.00239.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Ye Y., Sun Y., Li X., Wang W., Privratsky B., Tan S., Zhou Z., Huang C., Wei Y.Q. Transient receptor potential channel 1 deficiency impairs host defense and proinflammatory responses to bacterial infection by regulating protein kinase c alpha signaling. Mol. Cell. Biol. 2015;35:2729–2739. doi: 10.1128/MCB.00256-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y., Srinivasan P., Razavi S., Seymour S., Meraner P., Gudlur A., Stathopulos P.B., Ikura M., Rao A., Hogan P.G. Initial activation of STIM1, the regulator of store-operated calcium entry. Nat. Struct. Mol. Biol. 2013;20:973–981. doi: 10.1038/nsmb.2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitt C., Zobel A., Obukhov A.G., Harteneck C., Kalkbrenner F., Luckhoff A., Schultz G. Cloning and functional expression of a human Ca2+-permeable cation channel activated by calcium store depletion. Neuron. 1996;16:1189–1196. doi: 10.1016/s0896-6273(00)80145-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.