

Abstract
Enzymes from the histone deacetylase (HDAC) family are highly regulated by different mechanisms. However, only very limited knowledge exists about the regulation of HDAC8, an established target in multiple types of cancer. A previous dedicated study of HDAC class I enzymes identified no redox-sensitive cysteinyl thiol in HDAC8. This is in contrast to the observation that HDAC8 preparations show different enzyme activities depending on the addition of reducing agents. In the light of the importance of HDAC8 in tumorigenesis a possible regulation by redox signaling was investigated using biochemical and biophysical methods combined with site directed mutagenesis. The occurrence of a characteristic disulfide bond under oxidizing conditions is associated with a complete but reversible loss of enzyme activity. Cysteines 102 and 153 are the integral components of the redox-switch. A possible regulation of HDAC8 by redox signal transduction is suggested by the observed relationship between inhibition of reactive oxygen species generating NOX and concomitant increased HDAC8 activity in neuroblastoma tumor cells. The slow kinetics for direct oxidation of HDAC8 by hydrogen peroxide suggests that transmitters of oxidative equivalents are required to transfer the H2O2 signal to HDAC8.
Keywords: HDAC8 stability, Redox kinetics, Redox signaling, NOX, Disulfide bond, ROS, Hydrogen peroxide
Graphical abstract
Highlights
-
•
The enzyme activity of HDAC8 is regulated by a redox-switch.
-
•
The structure of the inactive oxidized state is thermodynamically stabilized.
-
•
C102 and C153 are the integral components of the redox-switch in HDAC8.
-
•
NOX inactivation lowers intracellular H2O2 and increases HDAC8 activity.
-
•
Slow oxidation kinetics suggests indirect transduction of the H2O2 signal in cells.
1. Introduction
Histone deacetylases (HDACs) have emerged as promising targets for chemotherapeutic intervention in cancer, neurodegenerative and immune disorders [1], [2], [3]. The HDAC enzyme family is subdivided into class I (HDAC1–3 and 8), class IIa (HDAC4,5,7 and 9), class IIb (HDAC6 and 10), class III (sirtuins SIRT1–7) and class IV (HDAC11). The zinc dependent HDACs of class I, IIa/b and IV operate with a different mechanism than the sirtuins in class III which require NAD+ for catalysis. HDAC8 is an established target for T-cell lymphoma and neuroblastoma and overexpressed in other tumors [4]. HDAC8 is found in the nucleus as well as the cytoplasm which is in line with an increasing number of identified substrates (SMC3, ERR-α(SMC3, E, RAI1, MLL2, p53, Cortactin) [5], [6], [7] and interaction partners (CREB/PP1, hEST1B, Inv(16) fusion protein, DEC-1, Hsp20, α-actin) [8], [9], [10], [11], [12] in both compartments. Consequently, the physiological role of HDAC8 in cells is complex and needs further elucidation but it is clearly linked to relevant cancer mechanisms. HDAC8 activity is negatively regulated upon phosphorylation by cyclic AMP dependent protein kinase at position serine 39 [13]. Moreover, Fierke et al. suggested that HDAC8 could be also regulated by metal switching in vivo [14]. Redox control of epigenetic processes has been proposed as a general principle to allow the cell to adapt to a changing environment. During the last years, it became clear that redox-sensitive proteins like peroxiredoxins serve not only as cellular self-defense against oxidative stress but can also act as redox relay for specific H2O2 signaling in cells [15]. Several HDACs were shown to be redox-regulated. The class IIa HDAC4 has a redox-switch that controls shuttling between the nucleus and the cytoplasm [16]. Moreover, HDAC6 has been shown to interact with peroxiredoxins [17]. In another study, class I HDACs -1, -2 and -3 but not HDAC8 were proposed to be redox-sensitive and a putative redox-switch was identified using chemical probes [18]. However, we and others observed that HDAC8 activity depends on the enzyme preparation conditions [19]. Inspired by the additional finding that inactive oxidized HDAC8 can be retransformed into active enzyme by the addition of Tris(2-carboxyethyl)phosphine (TCEP) or β-mercaptoethanol (β–ME), we initiated a comprehensive study to elucidate the mechanism of the redox-switching behavior of HDAC8. We provide unambiguous evidence that HDAC8 has a reversible thiol/disulfide redox-switch involving C153 and C102 and that the oxidized state of HDAC8 is thermodynamically more stable than the corresponding reduced state. The potential physiological relevance of redox-regulation of HDAC8 is demonstrated by the inhibition of endogenously produced H2O2 in neuroblastoma cells.
2. Materials and methods
2.1. Materials
All reagents and solvents were purchased from Sigma, Bachem, Roth, and used with further purification only if necessary. 9,9,9-trifluoro-8-oxo-N-phenyl-nonanamide (SATFMK) was prepared as described [20]. To minimize artificial oxidation of free thiols all measurements were performed in degassed buffer solutions. To prevent oxidation of enzyme during storage, enzymes were stored in the presence of 1 mM TCEP. TCEP was removed immediately before measurements by gel permeation chromatography with Zeba Spin Desalting Columns 7K MWCO (Thermo Scientific).
2.2. HDAC8 mutant variants
Mutant HDAC8 variants were generated using splicing by overlap extension PCR (SOE-PCR) with the following Primers:
HD8_BamHI_rev: 5′-AGGTGGATCCTTAAACAACGTGCTTCAGATTGCC-3′,
HD8_NdeI_for: 5′-GCGCATATGGAGGAGCCGGAGGAG-3′,
HD8_C102S_for: 5′-GGGCTAGGTTATGACTCCCCAGCCACTGAAGGGATA-3′,
HD8_C102S_rev: 5′-TATCCCTTCAGTGGCTGGGGAGTCATAACCTAGCCC-3′,
HD8_C153S_for: 5′-GATGAAGCATCTGGTTTTTCTTATCTCAATGATGCT-3′,
HD8_C153S_rev: 5′-AGCATCATTGAGATAAGAAAAACCAGATGCTTCATC-3′.
DNA sequencing was performed at the sequencing service at the LMU Munich with the cycle, clean and run (BigDye v3.1) protocol.
2.3. HDAC8 expression and purification
pET14b vector (Novagen, EMD Millipore) containing codon-optimized human HDAC8, fused to a His6 SUMO tag, was used to express HDAC8 in E. coli (BL21) DE3 pLysS. Cells were harvested by centrifugation for 10 min at 8000g and 4 °C and resuspended in lysis buffer (pH 8.0) containing 150 mM KCl, 50 mM Tris, 5 mM imidazole, 5 mM DTT, 1x HALT protease inhibitor cocktail (Thermo Scientific) and 5 µg/mL DNAseI. The cell suspension was sonicated and lysates were clarified by centrifugation at 18,000g for 30 min at 4 °C and sterile filtration. The filtrate was subsequently added to a 5 mL column volume of cOmplete His tag purification resin (Roche), equilibrated with immobilized metal affinity chromatography (IMAC) buffer A (pH 8.0) containing 150 mM KCl, 50 mM Tris and 5 mM imidazole. After washing with 50 mL of the same buffer His6-SUMO-HDAC8 was eluted with IMAC buffer B (pH 8.0) containing 150 mM KCl, 50 mM Tris and 100 mM imidazole. Subsequently 10 µg/mL His6 tagged SUMO-Protease was added to the eluted HDAC8 fusion protein. Cleavage of His6-SUMO tag occurred overnight whilst dialyzing against 25 mM Tris, 50 mM NaCl and 5 mM ß-ME (pH 8.0) at 4 °C. Then His6-SUMO tag and SUMO-Protease were removed by a second IMAC with AIC buffer A (pH 8.0) containing 25 mM Tris and 50 mM NaCl and 5 mM imidazole. HDAC8 containing flow through was concentrated and further purified on a strong anion exchanger (Bio-Scale Mini Macro-Prep High Q 5 mL Cartridge, Biorad). After a washing step using AIC buffer A HDAC8 was eluted using AIC buffer B (pH 8.0) containing 25 mM Tris and 1 M NaCl. 5 mM DTT was added to prevent oxidation and remove possible ß-ME cysteine adducts. The final purification step included size exclusion chromatography with a HiLoad Superdex 75 material (GE) equilibrated with GPC Puffer (pH 8.0) containing 150 mM KCl and 50 mM Tris. The protein containing fractions were collected and concentrated. Glycerol and TCEP were added to final concentrations of 25% and 1 mM and protein was stored at −20 °C. We typically obtained 3–5 mg HDAC8 from 1 L culture.
2.4. Enzyme activity assays
The activity of all HDAC8 variants was determined in black half area 96-well microplates (Greiner bio-one, Germany) by a colorimetric assay described by Wegener et al. [21]. HDAC8 (10 nM) was incubated with indicated concentrations of H2O2 for 1 h at 30 °C in HDAC8 assay buffer containing 25 mM Tris-HCl, 75 mM KCl and 0.001% Pluoronic F-127 pH 8.0. Excess H2O2 was quenched by the addition of 5.6 µg/mL freshly dissolved catalase. The reaction was initiated by the addition of 10 µM of the substrate Boc-Lys(tri-fluoroacetyl)-7-amino-4-methylcoumarin (Boc-Lys(TFA)-AMC). After incubation for 60 min, the reaction was stopped by the addition of 1.67 µM SATFMK and the deacetylated substrate was converted into a fluorescent dye (AMC) by the addition of 0.42 mg/mL trypsin. Measurements were performed in a fluorescence microplate reader (PHERAstar FS, BMG LABTECH). The data was analyzed with GraphPad Prism version 6.01.
2.5. Electrophoretic mobility shift assay (EMSA)
For the analysis of disulfide bond formation via migration change on non-reducing SDS-PAGE 5 µM of the respective HDAC8 variant was treated with increasing concentrations of H2O2 (0–10 mM) in redox buffer containing 20 mM NaH2PO4, 20 mM Na2HPO4, 150 mM NaCl and 5 mM EDTA pH 7.0. After 1 h incubation at room temperature excess H2O2 was quenched by the addition of 10 µg/mL catalase and free thiole groups were blocked by the addition of 8.3 mM NEM to prevent unwanted rearrangements of disulfide bonds followed by an incubation period of 30 min at room temperature. Finally, 4x non-reducing sample buffer was added containing 8% SDS, 250 mM Tris-HCL (pH 6.8), 40% Glycerol and 0.02% Bromophenol blue. The samples were denaturated for 5 min at 95 °C and cooled on ice. Subsequently, SDS-PAGE was performed on 12.5% gels at 200 V. Gels were stained with Coomassie brilliant blue solution.
2.6. Determination of the redox-potential between Cys102 and Cys153
A codon optimized gene was purchased, with every cysteine (C28, C125, C131, C244, C275, C287, C314 and C352) changed to serine except Cys102 and Cys153. This HDAC8lowC variant was expressed and purified as described above. At first a 2-fold serial dilution of 20 mM GSH was performed by keeping GSSG constant at 2 mM in a volume of 20 µL in buffers with three different pH-values (HEPES 100 mM, EDTA 100 µM, pH 7.0; Tris 100 mM, EDTA 100 µM, pH 8.0; CHES 100 mM, EDTA 100 µM, pH 9.0). Immediately after preparing the solutions 20 µL of the mutant HDAC8 was added to each mixture and kept overnight under nitrogen atmosphere to prevent oxygen oxidation. After reaching the equilibrium 5 µL TCA (100% (w/v)) was added to each sample and protein was precipitated for 20 min at −20 °C followed by 10 min centrifugation at 18,000g at 4 °C. The supernatant was removed, and the pellet resuspended by shaking in 30 µL redox-buffer containing 1 mM NEM for 30 min at 30 °C. After alkylation of nascent thiols 10 µL 4x non-reducing sample buffer was added to each sample followed by denaturation for 5 min at 95 °C. Samples were subjected to non-reducing SDS-PAGE and blotted on a PVDF Membrane (Merck, Millipore). Protein was detected using primary antibody for HDAC8 (E5, Santa Cruz biotechnology) and dye-labelled IRDye 800CW goat anti-mouse antibody (LiCor). Band intensities were quantified using the Image Studio Lite Software (LiCor). The redox-potential was determined at pH 7.0, 8.0 and 9.0. The dependency of the standard redox-potential from pH is described by the following general equation involving two protons:
2.7. Differential scanning fluorimetry to determine protein stability
Protein melting points were determined using a Prometheus NT.48 instrument from Nanotemper. 12 µM of the respective HDAC8 variant was treated with increasing concentrations of H2O2 (ranging from 0 to 5 mM) in redox buffer containing 20 mM NaH2PO4, 20 mM Na2HPO4, 150 mM NaCl and 5 mM EDTA pH 7.0. After 1 h incubation at room temperature, 10 µg/mL catalase was added to eliminate excess H2O2 and stop the oxidation process. The measurement was performed by increasing the temperature to 95 °C with a heating rate of 1 °C per minute and simultaneous detection of the fluorescence ratio of 350 nm/330 nm. The melting points were determined from the minimum of the first derivative of the function.
2.8. CD-spectroscopy
Spectra were collected by using a Jasco j-815cd spectropolarimeter (Easton, MD) equipped with a temperature controlling device in a quartz cell with a path length of 0.1 cm. Before experiments, HDAC8 was desalted by GPC with Zeba Spin Desalting Columns 7K MWCO (Thermo Scientific) equilibrated with CD buffer (pH 8.0) containing 5 mM Tris, 0.5% glycerol, 15 mM KCl and 0.1 mM DTT. Oxidized HDAC8 was generated by the addition of 5 mM H2O2 for 1 h at room temperature and desalted against CD buffer without DTT. The final protein concentration was 5 µM. Spectra were recorded in the far-UV from 260 to 195 nm and were the average of five scans.
2.9. Kinetics of HDAC8wt oxidation
To evaluate the kinetics of disulfide bond formation between the amino acids C102 and C153, 2.5 µM HDAC8wt was treated with various concentrations of H2O2 (ranging from 0 to 2.5 mM) and 200 µM Fluor-de-Lys HDAC substrate in potassium phosphate buffer (pH 7.6) containing 30 mM potassium phosphate, 100 mM KCl and 5% glycerol at 20 °C. Aliquots were removed at the indicated time periods and the reaction was quenched by the addition of 100 µM N‐hydroxy‐N′‐phenyloctanediamide (SAHA) and 0.04 mg/mL catalase after oxidation, 0.8 mg/mL Trypsin was added to generate fluorescent AMC for 15 min at 30 °C. The data was fitted using an exponential function in Graph Pad Prism and the observed pseudo first order rate constants were plotted against the different H2O2 concentrations. The second order rate constant of oxidation was determined from the slope of the plot.
2.10. Reversible redox modulation of HDAC8wt activity
H2O2 and TCEP were added successively to a solution of fully reduced HDAC8wt (2.5 mg/ml in 25 mM Tris-HCL, 75 mM KCl and 0.001% Pluronic) to achieve alternating reducing and oxidizing conditions. HDAC8wt was incubated with H2O2 for 45 min at 25 °C and TCEP for 60 min at 4 °C, respectively, while shaking at 650 rpm in a Thermomixer from Eppendorf. The oxidation process was stopped by adding 0.1 mg/mL catalase from bovine liver (Sigma-Aldrich). As control untreated HDAC8wt was supplemented by corresponding volumes of buffer. Aliquots were taken from each mixture to measure HDAC8wt activity. The samples for the enzyme activity assay (see above) of HDAC8wt were pretreated as follows: TCEP was removed and the buffer renewed using Zeba™ Spin Desalting Columns, 7K MWCO (Thermo Scientific). The redox modulation of HDAC8C102S/C153S activity was determined under the same conditions.
2.11. Western blot analysis
Western blot analysis was performed as described previously. [22] The following antibodies were used for detection: anti-acetyl-SMC3 (provided by Prof. K Shirahige, University of Tokyo, Tokyo, Japan) and anti-β-actin (clone AC-15; Sigma).
2.12. Determination of the Michaelis-Menten-parameters
10 nM (HDAC8wt, HDAC8C102S) and 100 nM (HDAC8C153S, HDAC8C102S/C153S) were incubated with increasing concentrations of Boc-Lys(TFA)-AMC for 5 min at 21 °C in HDAC8 assay buffer containing 25 mM Tris-HCl, 75 mM KCl and 0.001% Pluronic F-127 pH 8.0. After incubation 0.5 mg/mL Trypsin and 50 µM SAHA was added to stop enzyme reaction and generate fluorescent AMC signal. Additionally, a series of varying concentrations AMC in the buffer described above was measured and RFU was plotted against AMC concentration. Resulting fluorescent signal per µM AMC was observed by the slope of linear regression.
2.13. NOX inhibition
BE(2)-C cells were incubated for 6 h with 5 and 10 µM NADPH oxidase (NOX) inhibitor VAS2870 [23]. Whole cell lysates were blotted against SMC3 and normalized by the actin concentration of the same sample.
3. Results and discussion
3.1. Enzyme activity of HDAC8 correlates with its redox state
There is a large number of publications about the modulation of HDAC8 enzyme activity by inhibitors or incorporated metal ions [14], [24]. In particular, the identification of N-acetylthiourea compounds as putative activators of HDAC8 raised considerable attention [25]. However, Toro et al. demonstrated that the activation effect was essentially due to enzyme preparations with inherently low activity [19]. Toro et al. suspected that the preparation conditions in the study of Singh et al. might have led to partially metal-depleted HDAC8 or stabilized a particular inactive conformation of the enzyme, which could be possibly activated by the N-acetylthiourea compounds. However, the authors did not identify the specific mechanism of activation, nor did they take into consideration possible redox effects as a causal factor for the postulated preparation effects.
We and others observed that the enzyme activity of HDAC8 decreases rapidly even if stored refrigerated at 4 °C. We also found that part of the activity of HDAC8wt can be recovered by the addition of reducing agents like DTT or TCEP. To elucidate a putative redox-sensitivity of HDAC8, we initiated a comprehensive study combining a variety of complementary biochemical, biophysical and cell-based approaches. Analysis of HDAC8wt under non-reducing conditions revealed that enzyme activity is lost depending on hydrogen peroxide (H2O2) concentration (Fig. 1A). The oxidized form of HDAC8wt with disulfide bonds adopts a more constrained structure after denaturation and thus runs significantly faster in a non-reducing SDS-PAGE than the reduced form of HDAC8wt (lower panel of Fig. 1A). Increasing amounts of H2O2 shift the redox-equilibrium progressively to the oxidized state of HDAC8wt. The percentage of the reduced state of HDAC8wt correlates perfectly with enzyme activity (Fig. 1A).
Fig. 1.

Enzyme activity (upper panel) and EMSA (lower panel) of (A) HDAC8wt and (B) double-mutant HDAC8C102S/C153S in the presence of increasing amounts of H2O2. The enzyme activity test exploited the conversion of Boc-Lys(TFA)-AMC in the first step followed by the addition of trypsin to release fluorescent AMC. Gels were stained with Coomassie brilliant blue. The most left gel slot contains a ruler with indicated molecular weights. The upper band represents the reduced and the lower band the oxidized form of HDAC8wt. The mean and standard deviations were calculated from four independent experiments each performed in triplicates.
In contrast, the double mutant HDAC8C102S/C153S is essentially insensitive to H2O2 (Fig. 1B). The enzyme activity remains unchanged in the presence of up to 10 mM H2O2 and no additional band indicating a disulfide bridge becomes visible in the corresponding non-reducing gel.
3.2. The redox state-dependent enzyme activity of HDAC8 is reversible
The enzyme activity of oxidized HDAC8wt can be reversed repeatedly by adding reducing agent (Fig. 2A). However, overdosing and long exposure to H2O2 produces overoxidation of HDAC8wt that cannot be reversed. During this process, thiol groups are in general irreversibly oxidized further to sulfinic and sulfonic acid [26], [27]. Overoxidation was prevented to a great extent by the addition of catalase immediately after H2O2 mediated oxidation. Nevertheless, the maximum enzyme activity decreased significantly (p < 0.0001) after each redox-cycle presumably due to unavoidable residual overoxidation (Fig. 2A). To determine whether the progressive loss of enzyme activity was due to oxidation elsewhere in the protein or specific for the C102/C153 redox switch, we repeated the experiment with HDAC8C102S/C153S double mutant protein (Fig. 2B). The progressive loss of enzyme activity during the redox cycles was clearly less pronounced for the HDAC8 C102S/C153S variant lacking the redox switch than for HDAC8wt (71% remaining activity for HDAC8 C102S/C153S and 27% remaining activity for HDAC8wt). Therefore, the postulated overoxidation process appears to be predominantly specific for the redox switch. However, other less dominant irreversible deactivation pathways cannot be completely ruled out.
Fig. 2.

Reversibility of redox modulation of the enzyme activity of (A) HDAC8wt and (B) HDAC8C102S/C153S. The enzyme activity of the same HDAC8 sample after subsequent alternating addition of slight stoichiometric excesses of TCEP (red.) and H2O2 (ox.) is shown, respectively. The data represent means and standard deviations (HDAC8wt: n = 15, HDAC8C102S/C153S: n = 6). Statistical significance was determined by an unpaired t-test. ns: not significant (p > 0.05), *: 0.001 < p < 0.05 and ****: p < 0.0001.
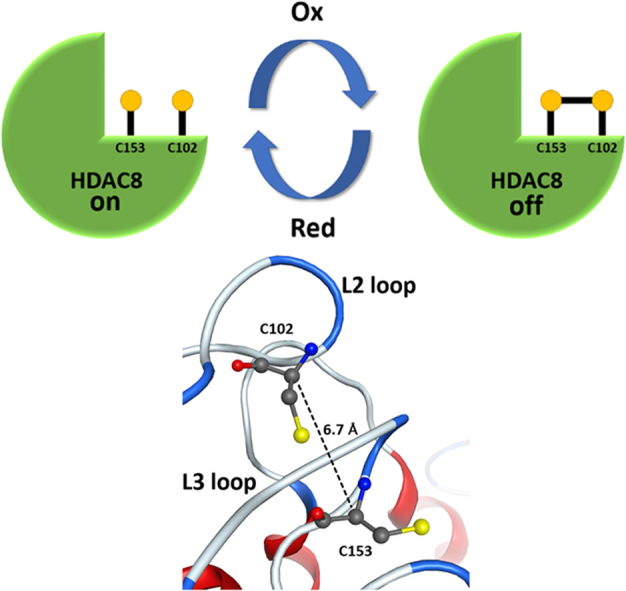
The redox switch of HDAC8 consists of C102 and C153. In a previous study, no redox-sensitive cysteines were identified in HDAC8 using cyclopentenone prostaglandins as tool compounds to identify redox-sensitive cysteine residues by alkylation [18]. In contrast, HDAC1,-2 and -3 were shown to contain redox-responsive cysteine residues in the same study. The alkylated redox-sensitive cysteines in HDAC1 were found at two different positions, where HDAC8 showed no homologous cysteine. After that study, several X-ray structures of class I HDACs 1, -2 and -3 were published. A closer look for example at the crystal structures of HDAC1 (PDB-ID: 4BKX) reveals that C273 sits on the surface of HDAC1 and is more than 16 Å away from the closest other cysteine. However, this cysteine might be involved in the formation of dimers upon oxidation. The second cysteine, C261, is buried in the interior of the properly folded protein and appears virtually inaccessible for larger alkylating agents such as cyclopentenone prostaglandins. It appears also reasonable that these bulky alkylating agents could have failed to modify the redox-sensitive C102 and C153 in HDAC8 due to sterical hindrance. The analysis of the X-ray structure of HDAC8 (PDB-ID: 1T64) reveals that C153 and C102 are in close proximity to each other (distance between Cα-atoms: 6.7 Å) (Fig. 3) and in contact with the active site.
Fig. 3.

A) Scheme of redox-switch: HDAC8wt is inactivated by H2O2 induced reversible formation of a disulfide bridge between C153 and C102. B) Arrangement of C102 and C153 in two adjacent flexible loops, L2-loop containing C102 and L3-loop containing C153, showing a distance that enables the formation of a disulfide bond (PDB-ID: 1T64).
According to the crystal structure (PDB-ID: 1T64) at least one of these cysteines, located in the L3-loop lining the major active site binding pocket (C153) or the neighboring L2-loop (C102), should be accessible by H2O2 and eventually form a disulfide bond with the other cysteine. Single exchange of C102 against serine has no significant influence on the catalytic efficiency of about 13,000 M−1 s−1 in HDAC8wt (Table 1, Fig. S1). In contrast, the HDAC8C153S and HDAC8C102S/C153S mutants show a 4–9 fold lower catalytic efficiency than the wildtype enzyme indicating that C153 but not C102 plays a certain role in the catalytic mechanism of HDAC8 (Table 1, Fig. S1). However, substitution of C153 by serine does not abolish catalytic activity completely.
Table 1.
Michaelis-Menten-parameters of HDAC8 variants.
| Enzyme | KM (µM) | kcat (s−1) | kcat KM−1 (M−1 s−1) |
|---|---|---|---|
| HDAC8wt | 96 ± 23 | 1.22 ± 0.14 | 13,000 ± 4400 |
| HDAC8C153S | 179 ± 42 | 2.08 ± 0.24 | 3200 ± 1200 |
| HDAC8C102S | 116 ± 27 | 0.58 ± 0.08 | 18,000 ± 6200 |
| HDAC8C102S/C153S | 193 ± 32 | 0.38 ± 0.04 | 2000 ± 520 |
Means and standard deviations are provided, n = 3.
Furthermore, if the cysteines (C102, C153) involved in the putative redox-switch are exchanged with serine, the residual enzyme activity of the double mutant HDAC8C102S/C153S becomes insensitive to treatment with H2O2 (Fig. 1B). Consistently, no second band that is indicative for the oxidative state of HDAC8 occurs in the non-reducing SDS-PAGE gel upon treatment with hydrogen peroxide. Taken together, these results clearly prove that C102 and C153 constitute a redox-switch in HDAC8.
3.3. The reduced form of HDAC8wt is thermodynamically stabilized upon oxidation
The overall structure of oxidized HDAC8 shows no major deviations from its reduced form. The CD-spectra of the non-functional oxidized and the functional reduced form of HDAC8wt show no significant differences (Fig. S2). Similarly, the spectra of mutant proteins HDAC8C102S, HDAC8C153S and HDAC8C102S/C153S resemble that of wildtype HDAC8. Thus, oxidation of HDAC8 is not associated with major rearrangements of its overall structure. To investigate the effect of oxidation on protein stability, Differential Scanning Fluorimetry analyses of HDAC8wt were performed. Increasing H2O2 concentrations cause a continuous shift of melting temperature from 42.27 °C of the reduced form to 44.14 °C (Table 2, Fig. S3A/B) indicating that the oxidized form of the protein is considerably more stable than the reduced form. This is in agreement with the formation of a stabilizing disulfide bond between C102 and C153. Consistently, the functional reduced HDAC8wt and mutant variants of HDAC8 with at least one of the cysteines C102 or C153 exchanged against serine show similar melting temperatures significantly distinct from that of inactive oxidized HDAC8wt (Table 2, Fig. S3C/D). The L2-loop is highly unordered in crystal structures of unbound HDAC8 and becomes more ordered upon binding to small molecule ligands [28]. Moreover, ligand binding and the enzyme activity of HDAC8 largely depend on the conformation and flexibility of the L1- and L2-loops [29], [30], [31], [32]. In fact, major movements of these loops and the side chain of F152 in the L3-loop are responsible for a transition from a wide-open state (PDB-ID: 1VKG) of the active site pocket into a sub-open state with a second transient binding pocket (PDB-ID: 1T64) and then into a closed state (PDB-ID: 1T69) [33]. These three representative crystal structures of HDAC8 were analyzed using the TRAnsient Pockets in Proteins (TRAPP) software platform developed in the laboratory of Rebecca Wade. The results confirmed that the L2- and L3-loop of HDAC8 are regions of increased flexibility (Fig. S4), which is in agreement with the experimentally observed large structural variations around the active site binding pocket of HDAC8. In a structurally highly related HDAC homolog HDAH from Bordetella/Alcaligenes, the L2- and L3-loop are interconnected and stabilized by a network of hydrogen bonds involving the pivotal amino acid T101. Substitution of T101 by alanine interrupts the strong interaction between both loops and leads to a dramatically increased flexibility of the L2-loop changing the mode of ligand interaction [34]. The formation of a disulfide bridge between C153 (L3-loop) and C102 (L2-loop) in HDAC8 introduces a covalent link between the L2- and L3-loop, which is also supposed to reduce the intrinsic flexibility of these loops adjacent to the catalytic site pocket. Wang et al. as well as Whitehead et al. postulated a mechanism of acetate release involving major conformational changes in the acetate release channel with R37 and W141 (numbering refers to HDAC8) acting as gate keepers [35], [36]. The crucial importance of R37 for catalysis and function of the internal acetate release cavity of HDAC8 was experimentally confirmed by Haider et al. [37]. In summary, it can be concluded that the enzyme activity of HDACs and in particular HDAC8 is closely related with conformational flexibility at the active site binding pocket as well as the internal acetate release channel. The decreased flexibility at the active site upon oxidation of the redox-switch and covalent linkage of the L2- and L3-loop is suggested to contribute to the observed concomitant total loss in enzyme activity.
Table 2.
Melting temperatures of HDAC8wt and mutant enzymes in the absence or in the presence of indicated concentrations of H2O2. Melting temperatures (Tm)are shown as means with standard deviations, n = 3.
| HDAC8wt | ||
|---|---|---|
| H2O2 (mM) | Tm (°C) | |
| 5.00 | 44.14 ± 0.33 | |
| 2.50 | 43.96 ± 0.02 | |
| 1.25 | 43.60 ± 0.08 | |
| 0.63 | 43.26 ± 0.06 | |
| 0.31 | 42.92 ± 0.03 | |
| 0.16 | 42.57 ± 0.07 | |
| 0.08 | 42.42 ± 0.05 | |
| 0 | 42.27 ± 0.08 | |
| HDAC8C102 | ||
| 0 | 42.91 ± 0.05 | |
| HDAC8C153 | ||
| 0 | 42.38 ± 0.01 | |
| HDAC8C102S/C153S | ||
| 0 | 42.26 ± 0.03 | |
3.4. The amino acids C102 and C153 are conserved in class I HDACs
C153 in HDAC8 is conserved throughout all members of human HDACs from classes I, IIa and IIb, whereas C102 is only conserved in class I HDACs (Fig. S5). In class IIa HDACs 4, 5, and 9 this cysteine is replaced by serine with the exception of HDAC7, where cysteine is replaced by alanine. Although the multiple sequence alignment suggests also a cysteine in the second domain of HDAC6 at the position of C102 (HDAC8), this is clearly an artifact of the heuristic alignment algorithm, because an alignment of crystal structures of HDAC1, HDAC8 and HDAC6 proves that I569 and no cysteine of HDAC6 matches C102 of HDAC8 (Fig. S6). Therefore, other HDAC class I members are also potentially regulated through a redox-switch corresponding to C102/C153 in HDAC8.
3.5. Standard redox-potential of C102/C153 switch
Since the ratio of the oxidized and reduced form of HDAC8 can be easily obtained from a non-reducing gel, the redox-potential of the molecular switch of HDAC8 could be determined from redox titrations (Fig. S7). The redox-potential of the C102/C153 redox-switch shows a linear dependency on pH and decreases from −218 mV at pH 7.0 to −340 mV at pH 9.0. ese redox-potential differences per pH-unit are in excellent agreement with 60.2 mV calculated from the corresponding equation under Materials and Methods at 30 °C involving two protons. The very good correlation between the experimental and calculated pH-dependency of the redox-potentials in HDAC8lowC confirms that the C102/153 couple is exclusively responsible for redox-switching of HDAC8. Typical redox-potentials of the GSH/GSSG couple observed in cells range from −170 mV [38], [39] to −258 mV [40].
HDAC8 can exist in both redox forms under these conditions. Interestingly, the standard redox potential of rapidly proliferating cells appears to be more negative than for differentiated, slowly proliferating cells [40]. Therefore, the ratio of the reduced active and the oxidized inactive form of HDAC8 in faster proliferating cancer cells (E = −258 mV) would be about 20:1 according to the Nernst equation. In contrast, this ratio is inverted (ca. 1:40) for differentiated cells (E = −170 mV). This is in agreement with previously referenced independent data proving that active (reduced) HDAC8 is involved in cancer. Moreover, a gene expression analysis found genes involved in antioxidant defense overexpressed associated with clinical resistance in a vorinostat phase 1 trial [41] and drug resistance can be overcome by a combination with redox-modulating compounds in leukemia cell lines and primary leukemia cells [42]. These findings highlight a significant role for the redox environment in cancer cells and particularly drug resistance. Moreover, our data suggest a positive relationship between cancer and enzyme activity of HDAC8. However, differences in redox-potentials of whole cells do not display the real redox conditions in particular subcellular compartments and are unsufficient to predict the rate of interconversion between different redox partner molecules.
3.6. Oxidation kinetics of HDAC8
Having shown that redox-switching of HDAC8 involves amino acids C102 and C153, is reversible and characterized by a more stable and enzymatically inactive oxidized form, we analyzed the redox properties, kinetics and mechanism of the redox-switch in more detail (Fig. 4). For these experiments it was of utmost importance to stop the oxidation process after the indicated time by neutralizing excess H2O2 by catalase. The observed oxidation rates, kobs, were obtained from exponential fits to the time-dependent enzyme activity of HDAC8 and analyzed as a function of H2O2 concentration. From the linear relationship, the specific rate constant of HDAC8 oxidation by H2O2 was calculated to be 0.51 ± 0.01 M−1 s−1 (Fig. 4). This rate constant is much slower than for the antioxidants peroxiredoxins, glutathione peroxidase 1 or catalase (105–107 M−1 s−1) and also slower than known redox-switches like PTP1B, SHP-2 or Keap1 (20–140 M−1 s−1) [43]. The small oxidation rate requires the molecular interaction with a redox transfer molecule such as a redox-protein to enable efficient redox regulation of HDAC8 under physiological conditions. On the other side, such a mechanism ensures selectivity through molecular recognition. H2O2 is a signaling messenger that can selectively modify cysteines in redox-regulated proteins, which react rather slowly with the free oxidant. Although thermodynamically favored, the oxidation of cysteines by H2O2 is kinetically hindered. In principle, the oxidation reaction can be accelerated catalytically, e.g. by thiol peroxidases [15], [44], [45]. It is currently widely accepted that peroxiredoxins are not only key protectors against oxidative stress [44], [46], [47]. There is growing evidence that peroxiredoxins are major sensors and transmitters of H2O2 signals [48]. It was shown that peroxiredoxins are able to transfer oxidizing equivalents to kinases, transcription factors and other redox-regulated proteins through specific protein-protein interactions [15], [45], [48], [49].
Fig. 4.

Oxidation kinetics of HDAC8wt: A) Enzyme kinetics of HDAC8wt in the absence (filled down triangles) and in the presence of indicated concentrations of H2O2. Slow oxidation kinetics by H2O2 causes a time-dependent decrease of enzyme activity. Smooth curved lines represent mono-exponential fits. B) The observed pseudo-first order rate constants (kobs) obtained from A) are plotted versus different indicated H2O2 concentrations (ranging from 0 to 2.5 mM) used to oxidize HDAC8. The second order rate constant of the oxidation reaction is 0.51 ± 0.01 M−1 s−1. Data means and standard deviations are shown, n = 3.
3.7. Physiological relevance of redox-regulation of HDAC8
To analyze the cellular impact of redox regulation of HDAC8, we used the BE(2)-C neuroblastoma tumor cell line known to depend on HDAC8 for cell growth and survival [22], [50], [51]. We investigated the putative redox-regulation of HDAC8 under physiological conditions by modulating the concentration of endogenous H2O2. NOX family members are reactive oxygen species (ROS)-generating enzymes that regulate redox-sensitive signaling pathways involved in cancer development and progression [52]. They produce high levels of superoxide and H2O2 in various cancer cell lines [52].
NOX activation and ROS production is also linked to the activation of oncogenes, such as RTKs (receptor tyrosine kinases) and their downstream signaling cascades (e.g. PI3K, mTOR, AKT) leading to tumor growth and survival [52]. In neuroblastoma, NOX enzymes are involved in retinoic acid-induced neuroblastoma cell differentiation [53]. Treatment of the BE(2)-C cells with NOX inhibitor VAS2870 reduces the intracellular production of H2O2 and concomitantly the acetylation of SMC3 [5] (Fig. 5). This demonstrates a relationship between the intracellular levels of ROS including H2O2 and HDAC8 activity. Together, these data suggest that HDAC8 could be redox-regulated under physiological conditions despite its very slow reaction kinetics with free H2O2. We hypothesize that the mechanism of redox regulation involves at least one H2O2 sensor and transmitter enzyme to accelerate specific oxidation of HDAC8. Interestingly, peroxiredoxin 6 has been described as a putative interaction partner and substrate of HDAC8 using a proteomic approach [54]. This raises the possibility that both proteins may form a redox relay, where oxidative equivalents are transmitted from peroxiredoxin 6 to HDAC8. However, the exact mechanism of intracellular ROS/H2O2 signaling to HDAC8 is unknown and currently under investigation.
Fig. 5.

Inhibition of NOX in BE(2)-C cells by different indicated concentrations of NOX inhibitor VAS2870 leads to a lower intracellular H2O2-level and decreased acetylation of SMC3. Immuno-blot results (A) and their respective quantification (B) are shown. The solvent (DMSO) control is indicated with a hyphen sign. Data here are the average ± standard deviation of five independent repeats. Statistical significance was determined by Student's t-test. *: p-value = 0.019 and ***: p-value = 0.0002.
4. Conclusions
We provide evidence that the enzyme activity of HDAC8 depends strongly on its redox state. The occurrence of a characteristic disulfide bond under oxidizing conditions is associated with a transition into a significantly more stable protein resulting in a complete but reversible loss of enzyme activity. C102 and C153 are clearly identified to be the integral components of the redox-switch in HDAC8. The physiologically relevant regulation of HDAC8 by redox signal transduction is suggested by a clear connection between the activities of ROS/H2O2 generating NOX and HDAC8 in neuroblastoma tumor cells. The redox-switch is only conserved among the members of HDAC class I suggesting that HDACs 1, 2 and 3 could also be regulated by a homologous switch. Further studies are required to obtain a full understanding of the redox-regulation network of human class I but also class II HDACs.
Acknowledgement
We acknowledge support by the Open Access Publishing Fund of Hochschule Darmstadt – University of Applied Sciences. We would also like to thank Michael Schröder for very helpful technical support. Additionally, we like to thank AG Fessner at the TU Darmstadt for the opportunity to use the Prometheus for the determination of the melting points and AG Hellmann at the University Mainz for support in acquiring the CD spectra. The kind gift of the anti-Ac-SMC3 antibody by Prof. K. Shirahige at the University of Tokyo is gratefully acknowledged.
Acknowledgments
Funding sources
Center for research and development (ZFE) of University of Applied Sciences.
Supporting information
-
•
Determination of Michaelis-Menten Parameters of HDAC8 variants
-
•
CD-spectra of reduced and oxidized form of HDAC8
-
•
Differential scanning fluorimetric curves showing HDAC8wt stability in the presence of increasing hydrogen peroxide concentrations
-
•
Dynamics simulations to demonstrate flexibility in HDAC8
-
•
Multiple sequence alignment of L2- and L3-loop.
-
•
Structural alignment of HDACs1, 6 and 8.
-
•
Redox titration of HDAC8.
This material is available free of charge via the Internet at http://pubs.acs.org.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.09.013.
Appendix A. Supplementary material
Supplementary material
References
- 1.Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 2.Didonna A., Opal P. The promise and perils of HDAC inhibitors in neurodegeneration. Ann. Clin. Transl. Neurol. 2015;2:79–101. doi: 10.1002/acn3.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang L. Trend of histone deacetylase inhibitors in cancer therapy: isoform selectivity or multitargeted strategy. Med. Res. Rev. 2015;35:63–84. doi: 10.1002/med.21320. [DOI] [PubMed] [Google Scholar]
- 4.Chakrabarti A. Targeting histone deacetylase 8 as a therapeutic approach to cancer and neurodegenerative diseases. Future Med. Chem. 2016;8:1609–1634. doi: 10.4155/fmc-2016-0117. [DOI] [PubMed] [Google Scholar]
- 5.Deardorff M.A. HDAC8 mutations in Cornelia de Lange syndrome affect the cohesin acetylation cycle. Nature. 2012;489:313–317. doi: 10.1038/nature11316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilson B.J., Tremblay A.M., Deblois G., Sylvain-Drolet G., Giguère V. An acetylation switch modulates the transcriptional activity of estrogen-related receptor/alpha. Mol. Endocrinol. 2010;24:1349–1358. doi: 10.1210/me.2009-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olson D.E. An unbiased approach to identify endogenous substrates of "histone" deacetylase 8. ACS Chem. Biol. 2014 doi: 10.1021/cb500492r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao J., Siddoway B., Huang Q., Xia H. Inactivation of CREB mediated gene transcription by HDAC8 bound protein phosphatase. Biochem. Biophys. Res. Commun. 2009;379:1–5. doi: 10.1016/j.bbrc.2008.11.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee H., Sengupta N., Villagra A., Rezai-Zadeh N., Seto E. Histone deacetylase 8 safeguards the human ever-shorter telomeres 1B (hEST1B) protein from ubiquitin-mediated degradation. Mol. Cell. Biol. 2006;26:5259–5269. doi: 10.1128/MCB.01971-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durst K.L., Lutterbach B., Kummalue T., Friedman A.D., Hiebert S.W. The inv (16) fusion protein associates with corepressors via a smooth muscle myosin heavy-chain domain. Mol. Cell. Biol. 2003;23:607–619. doi: 10.1128/MCB.23.2.607-619.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Qian Y., Zhang J., Jung Y.S., Chen X. DEC1 coordinates with HDAC8 to differentially regulate TAp73 and DeltaNp73 expression. PLoS One. 2014;9:e84015. doi: 10.1371/journal.pone.0084015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karolczak-Bayatti M. Acetylation of heat shock protein 20 (Hsp20) regulates human myometrial activity. J. Biol. Chem. 2011;286:34346–34355. doi: 10.1074/jbc.M111.278549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H., Rezai-Zadeh N., Seto E. Negative regulation of histone deacetylase 8 activity by cyclic amp-dependent protein kinase A. Mol. Cell. Biol. 2004;24:765–773. doi: 10.1128/MCB.24.2.765-773.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castaneda C.A. Active site metal identity alters histone deacetylase 8 substrate selectivity: a potential novel regulatory mechanism. Biochemistry. 2017;56:5663–5670. doi: 10.1021/acs.biochem.7b00851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sobotta M.C. Peroxiredoxin-2 and STAT3 form a redox relay for H2O2 signaling. Nat. Chem. Biol. 2015;11:64–70. doi: 10.1038/nchembio.1695. [DOI] [PubMed] [Google Scholar]
- 16.Ago T. A redox-dependent pathway for regulating class II HDACs and cardiac hypertrophy. Cell. 2008;133:978–993. doi: 10.1016/j.cell.2008.04.041. [DOI] [PubMed] [Google Scholar]
- 17.Parmigiani R.B. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA. 2008;105:9633–9638. doi: 10.1073/pnas.0803749105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle K., Fitzpatrick F.A. Redox signaling, alkylation (carbonylation) of conserved cysteines inactivates class I histone deacetylases 1, 2, and 3 and antagonizes their transcriptional repressor function. J. Biol. Chem. 2010;285:17417–17424. doi: 10.1074/jbc.M109.089250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Toro T.B. KDAC8 with high basal velocity is not activated by N-acetylthioureas. PLoS One. 2016;11:e0146900. doi: 10.1371/journal.pone.0146900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Frey R.R. Trifluoromethyl ketones as inhibitors of histone deacetylase. Bioorg. Med. Chem. Lett. 2002;12:3443–3447. doi: 10.1016/s0960-894x(02)00754-0. [DOI] [PubMed] [Google Scholar]
- 21.Wegener D., Hildmann C., Riester D., Schwienhorst A. Improved fluorogenic histone deacetylase assay for high-throughput-screening applications. Anal. Biochem. 2003;321:202–208. doi: 10.1016/s0003-2697(03)00426-3. [DOI] [PubMed] [Google Scholar]
- 22.Rettig I. Selective inhibition of HDAC8 decreases neuroblastoma growth in vitro and in vivo and enhances retinoic acid-mediated differentiation. Cell Death Dis. 2015;6:e1657. doi: 10.1038/cddis.2015.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Altenhofer S. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol. Life Sci. 2012;69:2327–2343. doi: 10.1007/s00018-012-1010-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dowling D.P., Gattis S.G., Fierke C.A., Christianson D.W. Structures of metal-substituted human histone deacetylase 8 provide mechanistic inferences on biological function. Biochemistry. 2010;49:5048–5056. doi: 10.1021/bi1005046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Singh R.K. Histone deacetylase activators: N-acetylthioureas serve as highly potent and isozyme selective activators for human histone deacetylase-8 on a fluorescent substrate. Bioorg. Med. Chem. Lett. 2011;21:5920–5923. doi: 10.1016/j.bmcl.2011.07.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sobotta M.C. Exposing cells to H2O2: a quantitative comparison between continuous low-dose and one-time high-dose treatments. Free Radic. Biol. Med. 2013;60:325–335. doi: 10.1016/j.freeradbiomed.2013.02.017. [DOI] [PubMed] [Google Scholar]
- 27.Loi V.V., Rossius M., Antelmann H. Redox regulation by reversible protein S-thiolation in bacteria. Front. Microbiol. 2015;6:187. doi: 10.3389/fmicb.2015.00187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dowling D.P., Gantt S.L., Gattis S.G., Fierke C.A., Christianson D.W. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decroos C. Biochemical and structural characterization of HDAC8 mutants associated with cornelia de lange syndrome spectrum disorders. Biochemistry. 2015;54:6501–6513. doi: 10.1021/acs.biochem.5b00881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Decroos C. Compromised structure and function of HDAC8 mutants identified in cornelia de lange syndrome spectrum disorders. ACS Chem. Biol. 2014 doi: 10.1021/cb5003762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Somoza J.R. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure. 2004;12:1325–1334. doi: 10.1016/j.str.2004.04.012. [DOI] [PubMed] [Google Scholar]
- 32.Decroos C. Variable active site loop conformations accommodate the binding of macrocyclic largazole analogues to HDAC8. Biochemistry. 2015;54:2126–2135. doi: 10.1021/acs.biochem.5b00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deschamps N., Simões-Pires C.A., Carrupt P.-A., Nurisso A. How the flexibility of human histone deacetylases influences ligand binding: an overview. Drug Discov. Today. 2015;20:736–742. doi: 10.1016/j.drudis.2015.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Meyners C., Kramer A., Yildiz O., Meyer-Almes F.J. The thermodynamic signature of ligand binding to histone deacetylase-like amidohydrolases is most sensitive to the flexibility in the L2-loop lining the active site pocket. Biochim. Biophys. Acta. 2017;1861:1855–1863. doi: 10.1016/j.bbagen.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 35.Whitehead L. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg. Med. Chem. 2011;19:4626–4634. doi: 10.1016/j.bmc.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 36.Wang D.F., Wiest O., Helquist P., Lan-Hargest H.Y., Wiech N.L. On the function of the 14 A long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J. Med. Chem. 2004;47:3409–3417. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 37.Haider S., Joseph C.G., Neidle S., Fierke C.A., Fuchter M.J. On the function of the internal cavity of histone deacetylase protein 8: R37 is a crucial residue for catalysis. Bioorg. Med. Chem. Lett. 2011;21:2129–2132. doi: 10.1016/j.bmcl.2011.01.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cai J., Jones D.P. Superoxide in apoptosis. Mitochondrial generation triggered by cytochrome c loss. J. Biol. Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 39.Jones D.P., Maellaro E., Jiang S., Slater A.F., Orrenius S. Effects of N-acetyl-L-cysteine on T-cell apoptosis are not mediated by increased cellular glutathione. Immunol. Lett. 1995;45:205–209. doi: 10.1016/0165-2478(95)00004-o. [DOI] [PubMed] [Google Scholar]
- 40.Kirlin W.G. Glutathione redox potential in response to differentiation and enzyme inducers. Free Radic. Biol. Med. 1999;27:1208–1218. doi: 10.1016/s0891-5849(99)00145-8. [DOI] [PubMed] [Google Scholar]
- 41.Garcia-Manero G. Phase 1 study of the histone deacetylase inhibitor vorinostat (suberoylanilide hydroxamic acid [SAHA]) in patients with advanced leukemias and myelodysplastic syndromes. Blood. 2008;111:1060–1066. doi: 10.1182/blood-2007-06-098061. [DOI] [PubMed] [Google Scholar]
- 42.Hu Y. Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound beta-phenylethyl isothiocyanate. Blood. 2010;116:2732–2741. doi: 10.1182/blood-2009-11-256354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antunes F., Brito P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017;13:1–7. doi: 10.1016/j.redox.2017.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fomenko D.E. Thiol peroxidases mediate specific genome-wide regulation of gene expression in response to hydrogen peroxide. Proc. Natl. Acad. Sci. USA. 2011;108:2729–2734. doi: 10.1073/pnas.1010721108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarvis R.M., Hughes S.M., Ledgerwood E.C. Peroxiredoxin 1 functions as a signal peroxidase to receive, transduce, and transmit peroxide signals in mammalian cells. Free Radic. Biol. Med. 2012;53:1522–1530. doi: 10.1016/j.freeradbiomed.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 46.Poole L.B., Nelson K.J. Discovering mechanisms of signaling-mediated cysteine oxidation. Curr. Opin. Chem. Biol. 2008;12:18–24. doi: 10.1016/j.cbpa.2008.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veal E.A., Day A.M., Morgan B.A. Hydrogen peroxide sensing and signaling. Mol. Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 48.Stocker S., Maurer M., Ruppert T., Dick T.P. A role for 2-Cys peroxiredoxins in facilitating cytosolic protein thiol oxidation. Nat. Chem. Biol. 2018;14:148–155. doi: 10.1038/nchembio.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gutscher M. Proximity-based protein thiol oxidation by H2O2-scavenging peroxidases. J. Biol. Chem. 2009;284:31532–31540. doi: 10.1074/jbc.M109.059246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oehme I. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009;15:91–99. doi: 10.1158/1078-0432.CCR-08-0684. [DOI] [PubMed] [Google Scholar]
- 51.Shen J. A kinome-wide RNAi screen identifies ALK as a target to sensitize neuroblastoma cells for HDAC8-inhibitor treatment. Cell Death Differ. 2018 doi: 10.1038/s41418-018-0080-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Block K., Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nitti M. PKC delta and NADPH oxidase in retinoic acid-induced neuroblastoma cell differentiation. Cell Signal. 2010;22:828–835. doi: 10.1016/j.cellsig.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 54.Lopez G. HDAC8, a potential therapeutic target for the treatment of malignant peripheral nerve sheath tumors (MPNST) PLoS One. 2015;10:e0133302. doi: 10.1371/journal.pone.0133302. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material