

Abstract
Molecular targets to reduce muscle weakness and atrophy due to oxidative stress have been elusive. Here we show that activation of Sarcoplasmic Reticulum (SR) Ca2+ ATPase (SERCA) with CDN1163, a novel small molecule allosteric SERCA activator, ameliorates the muscle impairment in the CuZnSOD deficient (Sod1-/-) mouse model of oxidative stress. Sod1-/- mice are characterized by reduced SERCA activity, muscle weakness and atrophy, increased oxidative stress and mitochondrial dysfunction. Seven weeks of CDN1163 treatment completely restored SERCA activity and reversed the 23% reduction in gastrocnemius mass and 22% reduction in specific force in untreated Sod1-/- versus wild type mice. These changes were accompanied by restoration of autophagy protein markers to the levels found in wild-type mice. CDN1163 also reversed the increase in mitochondrial ROS generation and oxidative damage in muscle tissue from Sod1-/- mice. Taken together our findings suggest that the pharmacological restoration of SERCA is a promising therapeutic approach to counter oxidative stress-associated muscle impairment.
Keywords: CDN1163, Skeletal muscle, CuZnSod1, SERCA, Oxidative stress
Graphical abstract
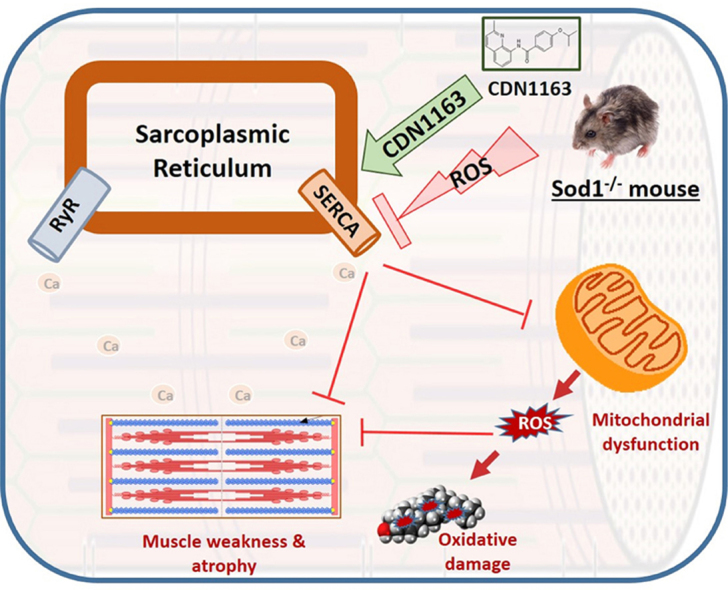
Highlights
-
•
Oxidative stress is related to SERCA dysfunction and muscle defect in Sod1-/- mice.
-
•
CDN1163 restores SERCA and prevents oxidative stress and muscle defect in Sod1-/- mice.
-
•
CDN1163 offers a promising therapy for muscle defect related to oxidative stress.
1. Introduction
Oxidative stress is commonly defined as the imbalance between pro-oxidants and anti-oxidants in tissues which manifests itself by an accumulation of oxidized molecules [1]. Characteristically, the levels of oxidative stress are increased in a wide range of conditions associated with muscle impairment including disuse, cancer cachexia, age-associated muscle loss or sarcopenia, muscular dystrophies and nerve crush injury [2]. Despite the association between oxidative stress and muscle impairment, the therapeutic role(s) of antioxidant supplements to combat muscle impairment is still controversial [3], [4]. Here we show our exciting findings supporting a pharmacologic intervention that is effective in reversing the muscle impairment in a mouse model of oxidative stress, mice deficient in CuZnSOD (Sod1-/- mice). Sod1-/- mice exhibit a number of phenotypes associated with oxidative stress-induced muscle impairment, including high levels of oxidative stress and damage, mitochondrial dysfunction and generation of reactive oxygen species (ROS), loss of neuromuscular junction integrity and accelerated loss of muscle mass and weakness. These mice have the added advantage that the majority of changes occur in mice less than 12 months of age [5], [6], [7], [8], [9]. Thus, the Sod1-/- mouse is an excellent model to test potential interventions for muscle impairment due to oxidative stress in relatively young mice.
A number of potential factors contributing to the underlying mechanisms of muscle defects related to increased oxidative stress have been proposed, including, mitochondrial dysfunction, defects in muscle regeneration and impairments in calcium regulation and the muscle excitation-contraction (EC) coupling system [10], [11]. EC coupling involves a series of molecular events that convert membrane depolarization into muscle contraction by releasing Ca2+ from the sarcoplasmic reticulum (SR) Ca2+ stores via the ryanodine receptors (RyRs). The subsequent reuptake of Ca2+ after contraction is executed by the SERCA pumps. Impaired function of the SERCA pump is associated with many chronic pathologies including aging [12], denervation [13] and muscular dystrophies [14]. One potential molecular mechanism underlying this phenomenon may involve oxidative modification of SERCA and/or associated proteins. For example, treating isolated SR vesicles with peroxides causes oxidation and partial inactivation of SERCA pumps [15]. SERCA protein oxidation and reduced activity have also been shown to occur in biological aging [16]. Reduced SERCA function can result in cytoplasmic Ca2+ buildup leading to reduced muscle quality and/or quantity via activation of Ca2+ dependent proteases and mitochondrial dysfunction and ROS generation [17]. Restoration of SERCA activity is a promising target to counter muscle pathologies in conditions of sarcopenia and oxidative stress. Overexpression of SERCA [18] or deletion of the SERCA inhibitory protein sarcolipin [14]1 increases SERCA ATPase activity and mitigates the disease phenotype in mouse models of Duchenne muscular dystrophy (DMD). Specifically, increased SERCA activity in the DMD mouse model is associated with improved muscle regeneration, reduced muscle necrosis, improved mitochondrial morphology, extended lifespan and protection from contraction-induced injuries and Ca2+ – driven necrosis in the gastrocnemius muscle. While these genetic approaches to increasing SERCA function are encouraging, here we tested the ability of a pharmacological intervention to enhance muscle SERCA function using CDN1163, an allosteric activator of the SERCA pump. We show for the first time that CDN1163 can increase muscle mass, restore muscle force and reduce mitochondrial ROS and oxidative stress in a mouse model of oxidative stress, the Sod1-/- mice. Our findings suggest that pharmacological activation of the SERCA pump may represent a promising therapy for muscle impairment associated with oxidative stress [5], [7].
2. Materials and methods
2.1. Animals and CDN1163 information
The generation and characterization of the Sod1-/- mice is described in detail elsewhere [5], [19]. Female ≈ 2 months old C57BL/6J wild-type (WT; n = 12–18) and Sod1-/- mice (n = 12–18) were divided into four groups and treated with either vehicle (10% DMSO, 10% Tween-80 in PBS) or CDN1163 (50 mg/kg) by intraperitoneal injection three times per week for 7 weeks. Care and management of the mice was executed according to The Guide for the Care and Use of Laboratory Animals and approved by the institutional Animal Care and Use Committee at the Oklahoma Medical Research Foundation (OKC, OK, USA). CDN1163 was purchased from Tocris Bioscience (Minneapolis, MN, USA). CDN1163 and its derivatives show acceptable pharmacokinetics in mice. It has a plasma half-life of 1.17 h and plasma stability of 94% at 1 h. The compound also shows high permeability across membranes and is not inhibited by cytochrome C450 enzymes of the liver. It is also remarkably selective over potential off-target effects. CDN1163 was screened for 160 targets and did not show significant activity at any of the off-targets [20], [21].
2.2. Protein preparation and western blot
Gastrocnemius muscle was homogenized in RIPA buffer containing 50 mM Tris (pH 7.4), 150 mM NaCl, and protease inhibitors. Protein was quantified using the Bio-Rad kit (Sigma-Aldrich, Poole, UK) and transferred to nitrocellulose membrane after electrophoresis using 8–15% polyacrylamide gels. Bands were scanned and quantified using Gene Tool system (SynGene – Frederick, MD, USA). Primary antibodies used include rabbit anti-SERCA1 (Cell Signaling technology; #12293, Danvers, MA, USA), rabbit anti-SERCA2 (Cell Signaling technology; #4388, Danvers, MA, USA) and rabbit anti-LC3 (Cell Signaling technology; #2775, Danvers, MA, USA) at 1:1000 dilution each. Secondary antibody used was HRP-linked anti-rabbit IgG (Cell Signaling technology; #7074, Danvers, MA, USA) at 1:10,000 dilution. All image intensities were normalized to protein intensity based on the ponceau stain.
2.3. Analysis of oxidative damage by F2-isoprostane level
The levels of F2-isoprostanes in quadriceps muscle were measured using thin layer chromatography and GC- mass spectrometry as described previously [22].
2.4. Quantification of mRNA levels using real-time PCR
Total RNA was extracted from gastrocnemius muscles using TRI reagent and the cDNA was prepared from 1 mg of the total RNA using iScriptTM cDNA Synthesis kit (Bio-Rad, Hercules, CA, USA). 2.5 ng of cDNA samples were amplified using specific primers along with fast SYBR green master mix (Applied Biosystems, Grand Island, NY, USA). The data were analyzed using the ΔΔCt method.
2.5. Contractile force measurements
Contractile force generation was measured in isolated EDL muscle using a 1200A in vitro test system (Aurora Scientific Inc. ON, Canada) as described elsewhere [9]. Briefly, muscles were individually tied to a model 300C servomotor (Aurora Scientific Inc.) and fixed within a water bath containing an oxygenated (95% O2, 5% CO2) Krebs-Ringer solution (in mM: 137 NaCl, 5 KCl, 1 MgSO4, 1 NaH2PO4, 24 NaHCO3, 2 CaCl2) maintained at 32 °C. Computer controlled stimulation was applied through a model 701C stimulator (Aurora Scientific Inc.). Force frequency curves were generated with stimulation frequencies between 1 and 300 Hz. All data were recorded and analyzed using commercial software (DMC and DMA, Aurora Scientific, ON, Canada). Specific force (N/cm2) was measured using muscle length and mass.
2.6. SERCA activity
The measurement of SERCA ATPase activity was performed in the muscle homogenates at 37 °C using a spectrophotometric assay as previously described [23].
2.7. Mitochondrial function
Mitochondrial isolation and assays for H2O2 generation were performed in gastrocnemius muscle as described previously by our laboratory [24]. Freshly isolated mitochondria were used for H2O2 release assay using the Amplex red-HRP method [25]. HRP (1 U/ml) catalyzes the H2O2-dependent oxidation of nonfluorescent Amplex red (80 µM) (Molecular Probes, Eugene, OR) to fluorescent resorufin red. Fluorescence was followed at an excitation wavelength of 544 nm and emission wavelength of 590 nm using a Fluoroskan Ascent type 374 multiwell plate reader (Labsystems, Helsinki, Finland). The slope of the increase in fluorescence is converted to the rate of H2O2 production with a standard curve. All assays were performed at 37C in 96-well plates. Substrates used were 10 mM succinate plus rotenone and 5 mM glutamate plus malate. For each assay, one reaction well contained buffer only, and another contained buffer with mitochondria, to estimate the background oxidation rates of Amplex red and to estimate the rate of peroxide release in mitochondria without substrate (state 1). The reaction buffer consisted of 125 mM KCl, 10 mM HEPES, 5 mM MgCl2, 2 mM K2HPO4, pH 7.44 and 37.5 U/ml Sod [24].
2.8. Statistical analysis
All numerical values are presented as mean ± SEM and the comparisons among the four groups were performed by one-way analysis of variance (ANOVA) and Turkey's multiple comparison test, with a single pooled variance. Data were analyzed using GraphPad Prism 7 and the p values of less than 0.05 were considered statistically significant.
3. Results
3.1. CDN1163 rescues the SERCA Ca+2-ATPase activity in the Sod1-/- mice
The effect of CDN1163 on SERCA activity was determined by measuring the decrease in NADH absorbance at 340 nm in gastrocnemius muscle homogenates (Fig. 1A). The maximum change in Ca2+ dependent Ca2+-ATPase is significantly reduced (≈ 29%; p < 0.05) in muscle homogenates from the Sod1-/- mice when compared to WT mice (Fig. 1B). The reduction in SERCA activity is completely restored with 7 weeks of CDN1163 treatment (Fig. 1A and B). Levels of SERCA1 and SERCA2 mRNA and protein were not changed in Sod1-/- mice and were not altered by CDN1163 treatment (Fig. 1C and D).
Fig. 1.

SERCA Ca2+ – dependent ATPase activity as measured by the change in NADH absorbance and the expression of SERCA in the extracts from gastrocnemius muscles of ≈ 4 month old WT and Sod1-/- mice treated with CDN1163 or vehicle for 7 weeks. CDN1163 treatment restored the SERCA ATPase activity in the Sod1-/- mice (A) as indicated by an increase in the maximum change in the NADH absorbance (B). CDN1163 had no effect on the expression of SERCA isoforms at the mRNA (C) and protein (D) levels. Values are expressed as Mean ± SEM. (WT-DMSO; n = 4, WT-CDN1163; n = 6, Sod1-/--DMSO; n = 4, Sod1-/--CDN1163; n = 6).; One-way ANOVA. * P ≤ 0.05.
3.2. CDN1163 prevents gastrocnemius muscle atrophy in the Sod1-/- mice
At 2 months of age (the starting point for this study) the Sod1-/- mice have a significantly lower body mass (~19% less than age matched WT mice; 20.6 g ± 0.4 versus 16.6 g ± 0.4) as we have previously reported [5], [9] (Fig. 2A). CDN1163 treatment for 7 weeks did not alter body weight in WT or Sod1-/- mice, and the Sod1-/- mice remained approximately 14% smaller than the WT mice at the end of the 7 week treatment period. At 2 months of age, neither absolute (Fig. 2B) nor normalized (Fig. 2C) gastrocnemius muscle mass is statistically lower in Sod1-/- versus WT female mice. However, at the end of the 7 week period (~ 4 months of age) there is a significant decrease in both absolute and normalized gastrocnemius muscle mass in the untreated Sod1-/- mice compared to untreated WT mice (Fig. 2B and C) consistent with our previous reports [5]. Remarkably, 7 weeks of CDN1163 treatment completely prevented this atrophy in the Sod1-/- mice and the normalized gastrocnemius mass of CDN treated Sod1-/- mice is 23% greater than Sod1-/- untreated mice (Fig. 2C). This protective effect of CDN1163 on muscle mass may in part be due to enhancement of autophagy in the Sod1-/- mice as evident by increase in protein expression of LC3-II and the ratio of LC3-II / LC3-I (Fig. 2D).
Fig. 2.

CDN1163 restored muscle mass but not the body mass in the Sod1-/- mice. Body mass (A), absolute gastrocnemius muscle mass (B) and the gastrocnemius mass normalized to body mass (C) from the WT and Sod1-/- mice treated with CDN1163 or vehicle. CDN1163 induced autophagy in the Sod1-/- mice as shown by increase in protein expression of LC3-II and the ratio of LC3-II / LC3-I (D). Values are expressed as Mean ± SEM. (2 month old; WT; n = 7, Sod1-/-; n = 6, 4 months old; WT-DMSO; n = 10, WT-CDN1163; n = 8, Sod1-/--DMSO; n = 6, Sod1-/--CDN1163; n = 8). One-way ANOVA. * P ≤ 0.05, **** P ≤ 0.001.
3.3. CDN1163 prevents contractile dysfunction in the Sod1-/- mice
In agreement with our previous findings, we measured a significant decline in the in-vitro specific force measured in EDL muscle of Sod1-/- mice (~ 19%, p < 0.05) when compared to WT mice (Fig. 3A). Consistent with the maintenance of muscle mass, 7 weeks of CDN1163 treatment completely restored the specific force in the Sod1-/- mice. Importantly, these changes occur in vitro independent of muscle mass and innervation status suggesting a beneficial effect of CDN1163 on intrinsic force-generating properties of the EDL muscle. On the other hand, CDN1163 has no effect on the TTP and RT1/2 in the EDL muscle during twitch contractions (Fig. 3B and C). CDN1163 had no effect on force-frequency curve when force was plotted as percentage of maximum force (Fig. 3D) or as specific force (Fig. 3E).
Fig. 3.

Contractile properties of the EDL muscles measured in the WT and Sod1-/- mice treated with CDN1163 or vehicle. CDN1163 restored the specific force (A) in the Sod1-/- mice without affecting the time to twitch peak tension (TTP) (B) and half relaxation time (1/2 R) (C). CDN1163 had no effect on force-frequency curve when force was plotted as percentage of maximum force (D) or as specific force (E).; Values are expressed as Mean ± SEM. (WT-DMSO; n = 9, WT-CDN1163; n = 5, Sod1-/--DMSO; n = 5, Sod1-/--CDN1163; n = 5).; One-way ANOVA. * P ≤ 0.05, ** P ≤ 0.01, *** P ≤ 0.001.
3.4. CDN1163 attenuates mitochondrial dysfunction in the Sod1-/- mice
It is well documented that elevated levels of cytosolic Ca2+ lead to increased mitochondrial ROS production [26]. We have previously shown that muscle mitochondria from the Sod1 -/- mice show structural and functional defects [6], [7]. To test whether the activation of SERCA pump function improves mitochondrial function, we measured mitochondrial ROS production as peroxide emission using isolated mitochondria from the gastrocnemius muscle. In accordance with our previous findings, mitochondria from Sod1-/- mice showed significantly greater (≈ 340%, p < 0.001) H2O2 production in State-1 respiration (mitochondria respiring without addition of external substrate), than mitochondria from gastrocnemius muscle from WT mice (Fig. 4A). In contrast, isolated mitochondria from CDN1163 treated muscle from Sod1-/- mice do not show elevated levels of peroxide production, i.e., levels are similar to mitochondria from WT mice. When respiratory substrates glutamate/malate are added to stimulate electron flow through Complex I, mitochondrial peroxide production is still significantly higher in the Sod1-/- mice (≈ 56%, p < 0.05) compared to WT mice. CDN1163 treatment in the Sod1-/- mice returns glutamate/malate stimulated H2O2 generation to WT levels (Fig. 4A).
Fig. 4.

Markers of oxidative stress in the WT and Sod1-/- mice treated with CDN1163 or vehicle. CDN1163 reduces the state-1 and state-2 complex 1-linked (glutamate malate) mitochondrial peroxide production (A) in the gastrocnemius muscles and the F2-isoprostanes as a marker of lipid peroxidation (B) in the quadriceps muscles in the in the Sod1-/- mice when compared to the vehicle treatment. Values are expressed as Mean ± SEM (WT-DMSO; n = 4, WT-CDN1163; n = 6, Sod1-/--DMSO; n = 5, Sod1-/--CDN1163; n = 5).; One-way ANOVA. * P ≤ 0.05.
3.5. CDN1163 reduces the oxidative damage in the Sod1-/- mice
We have previously reported increased oxidative damage in the skeletal muscles of Sod1-/- mice [8] that might be contributing to muscle atrophy and weakness. CDN1163 was shown to reduce plasma levels of malondialdehyde, a marker of oxidative stress, in the ob/ob mouse model [27]. Therefore we investigated whether CDN1163 can prevent oxidative stress-induced damage by measuring the levels of F2-isoprostanes, an indicator of lipid peroxidation, in the quadriceps muscles. As shown in Fig. 4B, the F2-isoprostanes levels in quadriceps muscle are significantly higher (≈ 76%, p < 0.001) in the Sod1-/- mice compared to WT mice. However, CDN1163 treatment prevented this increase in the Sod1-/- mice.
4. Discussion
This is the first study suggesting that direct pharmacological activation of skeletal muscle SERCA by the SERCA activator CDN1163 rescues muscle atrophy and weakness in a mouse model of oxidative stress. Here we report that SERCA activation is associated with the restoration of muscle mass and force and prevention of mitochondrial ROS generation and oxidative damage in the Sod1-/- mice. These findings suggest that the pharmacological restoration of SERCA may be a powerful intervention to prevent oxidative stress- mediated muscle impairment during aging and diseases involving muscular degeneration.
SERCA plays a critical role in cellular maintenance of calcium levels. This is especially important in skeletal muscle where calcium levels are elevated following muscle contraction and need to be repeatedly restored to resting levels to avoid deleterious effects of high calcium. There are two major SERCA isoforms in mouse hind limb muscles, SERCA1a the more abundant fast twitch isoform and SERCA2a the relatively low abundance slow twitch isoform. Together they account for ≥ 70% of Ca2+ removal from the cytosol [28]. Reduced SERCA ATPase activity is associated with aging muscle [12], denervation [13] and muscular dystrophies [29]. Thus, restoration of SERCA function is an attractive therapeutic target to prevent muscle defects. Indeed, previous studies have shown that stabilizing SERCA activity with elevated levels of Hsp72 [30], deletion of the SERCA inhibitor sarcolipin [14] or overexpression of SERCA protein itself [18] all work to ameliorate muscle pathology in the mouse models of Duchenne muscular dystrophy.
SERCA has been shown to be inactivated by elevated oxidative stress in aging muscle and denervation [12], [13] thus it is possible that the reduction in SERCA activity in the Sod1-/- mice is related to oxidative inactivation of the enzyme. In support of this, peroxides are shown to selectively oxidize cysteine residues of SERCA and partially inactivate the pump in the isolated SR preparations [15]. Thus it is plausible that the restoration of oxidative balance by the CDN1163 (Fig. 4) has prevented oxidative inactivation of the SERCA. Because high levels of oxidative stress and mitochondrial dysfunction have previously been linked to elevated cytosolic calcium levels [17], we hypothesized that activation and maintenance of SERCA activity by CDN1163 would maintain physiologic calcium levels and prevent or reduce muscle atrophy and weakness in the Sod1-/- mice. Interestingly, CDN1163 was recently shown to prevent ER stress and have beneficial effects in hepatic [27], pancreatic [31] and neuronal [20] tissues. In support of our hypothesis, CDN1163 restored SERCA activity to levels found in wild type mice, reversed mitochondrial ROS generation, prevented oxidative damage and protected from muscle atrophy and loss of contractile force generation.
The protective effects of CDN1163 on muscle mass and function may be due to the maintenance of calcium levels and/or an indirect effect of the compound on mitochondrial function and downstream oxidative stress that can propagate muscle degenerative processes. Mitochondria are an important source of ROS in the skeletal muscle, and the local structural and functional communications between SR and mitochondria are well characterized [26]. A correlation exists between the levels of cytosolic calcium transient and the amount of mitochondrial calcium uptake so that an enhanced cytosolic calcium transient leads to enhanced mitochondrial calcium uptake [32]. Accordingly, defect in SR calcium handling and elevated cytosolic calcium promote mitochondrial calcium uptake and ROS production in various muscle diseases [33]. Restoration of SERCA activity has been shown to reduce mitochondrial swelling and free radical production in the gastrocnemius muscles of mice with muscular dystrophy [29]. While it is difficult to align these observations along a disease causing pathway, they support our notion that the reduced SERCA pump activity and the increased mitochondrial ROS production facilitate or induce muscle impairment in the Sod1-/- mice. The reduced emission of mitochondrial peroxides we measured in the CDN1163 treated Sod1-/- mice is consistent with this idea.
Proper maintenance of autophagy is fundamental for the homeostasis of skeletal muscle during stress. The inhibition as well as over activation of autophagy exerts detrimental effects on muscle health [34]. The defect in autophagic flux in the Sod1-/- mice (Fig. 2D) is potentiallypathogenic and is consistent with the mouse models of ALS [35] and the age-decline of autophagy system in rodents [36], [37] and humans [38]. These studies indicate that the reduced autophagic capacity to eliminate damaged and dysfunctional proteins and organelles contribute to muscle impairment. Hence the restoration of autophagy by CDN1163 to the wild-type levels could be an additional mechanism to counter oxidative stress- induced muscle impairment (Fig. 2D).
Apart from inducing mitochondrial damage, SERCA pump dysfunction can also disrupt EC coupling machinery leading to reduced muscle strength as the mice with decrease in SERCA pump activity show reduced force-generating capacity [29]. We can propose at least two ways that the disrupted EC coupling and the associated oxidative stress might contribute to muscle weakness in the Sod1-/- mice. It is possible that the Ca+2 dysregulation and increased ROS production reduce Ca2+ sensitivity of the contractile apparatus. In support of this, a direct coupling between increased ROS and myofibrillar Ca2+ sensitivity is proposed leading to reduced force production in the conditions of increased oxidative stress [39]. Alternatively, increased ROS may damage SR Ca2+ release channels RyRs, resulting in reduced Ca release during contraction and contributing to muscle weakness. Hence the SERCA restoration-mediated improved Ca2+ handling and reduced ROS production in the CDN1163 treated Sod1 -/- mice might be contributing to restored force generating capacity.
Although we did not investigate the off-target effects of the compound in this short-term study, we did not see a change in kidney, brain and liver weights in CDN1163-treated vs untreated Sod1 -/- mice (data not shown). The compound also showed remarkable safety against off-target effects when screened for 160 potential targets [20], [21]. Further, while the impact of chronic pharmacological activation of SERCA ATPase pump are unknown, genetic activation of SERCA ATPase by overexpressing SERCA [18], [29] or deleting sarcolipin [14] shows no chronic side effects. Therefore we are confident about the safety profile of CDN1163 in long-term.
In conclusion, we validate for the first time that the pharmacological activation of SERCA is a powerful intervention to prevent muscle impairment associated with the increased oxidative stress. Importantly, CDN1163 restores oxidative balance, force-generating capacity and the muscle mass by reversing the loss of SERCA function in the Sod1-/- mice. Currently, there are no known drugs to effectively offsetting muscle weakness and atrophy. This study provides an important proof-of-concept that the CDN1163 has the potential to become an effective therapy for age- and oxidative stress- related muscle diseases.
Funding
This work was supported by an Irene Diamond Fund/AFAR Postdoctoral Transition Award in Aging to RQ, and NIH grant AG051442-02O to Dr. Van Remmen a VA. HVR is also supported by a Senior VA Research Career Scientist Award from the Department of Veterans Affairs.
Footnotes
SERCA, Sarcoplasmic Reticulum Ca-ATPase; TTP, Time To Peak tension; RT1/2, half Relaxation Time.
References
- 1.Breitenbach M., Eckl P. Introduction to oxidative stress in biomedical and biological research. Biomolecules. 2015;5:1169–1177. doi: 10.3390/biom5021169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arthur P.G., Grounds M.D., Shavlakadze T. Oxidative stress as a therapeutic target during muscle wasting: considering the complex interactions. Curr. Opin. Clin. Nutr. Metab. Care. 2008;11:408–416. doi: 10.1097/MCO.0b013e328302f3fe. [DOI] [PubMed] [Google Scholar]
- 3.Brocca L., Pellegrino M.A., Desaphy J.F., Pierno S., Camerino D.C., Bottinelli R. Is oxidative stress a cause or consequence of disuse muscle atrophy in mice? A proteomic approach in hindlimb-unloaded mice. Exp. Physiol. 2010;95:331–350. doi: 10.1113/expphysiol.2009.050245. [DOI] [PubMed] [Google Scholar]
- 4.Powers S.K., Kavazis A.N., McClung J.M. Oxidative stress and disuse muscle atrophy. J. Appl. Physiol. (1985) 2007;102:2389–2397. doi: 10.1152/japplphysiol.01202.2006. [DOI] [PubMed] [Google Scholar]
- 5.Muller F.L., Song W., Liu Y., Chaudhuri A., Pieke-Dahl S., Strong R., Huang T.T., Epstein C.J., Roberts L.J., 2nd, Csete M., Faulkner J.A., Van Remmen H. Absence of CuZn superoxide dismutase leads to elevated oxidative stress and acceleration of age-dependent skeletal muscle atrophy. Free Radic. Biol. Med. 2006;40:1993–2004. doi: 10.1016/j.freeradbiomed.2006.01.036. [DOI] [PubMed] [Google Scholar]
- 6.Jang Y.C., Liu Y., Hayworth C.R., Bhattacharya A., Lustgarten M.S., Muller F.L., Chaudhuri A., Qi W., Li Y., Huang J.Y., Verdin E., Richardson A., Van Remmen H. Dietary restriction attenuates age-associated muscle atrophy by lowering oxidative stress in mice even in complete absence of CuZnSOD. Aging Cell. 2012;11:770–782. doi: 10.1111/j.1474-9726.2012.00843.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jang Y.C., Lustgarten M.S., Liu Y., Muller F.L., Bhattacharya A., Liang H., Salmon A.B., Brooks S.V., Larkin L., Hayworth C.R., Richardson A., Van Remmen H. Increased superoxide in vivo accelerates age-associated muscle atrophy through mitochondrial dysfunction and neuromuscular junction degeneration. FASEB J. 2010;24:1376–1390. doi: 10.1096/fj.09-146308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sakellariou G.K., Pye D., Vasilaki A., Zibrik L., Palomero J., Kabayo T., McArdle F., Van Remmen H., Richardson A., Tidball J.G., McArdle A., Jackson M.J. Role of superoxide-nitric oxide interactions in the accelerated age-related loss of muscle mass in mice lacking Cu,Zn superoxide dismutase. Aging Cell. 2011;10:749–760. doi: 10.1111/j.1474-9726.2011.00709.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Larkin L.M., Davis C.S., Sims-Robinson C., Kostrominova T.Y., Van Remmen H., Richardson A., Feldman E.L., Brooks S.V. Skeletal muscle weakness due to deficiency of CuZn-superoxide dismutase is associated with loss of functional innervation. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011;301:R1400–R1407. doi: 10.1152/ajpregu.00093.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ali S., Garcia J.M. Sarcopenia, cachexia and aging: diagnosis, mechanisms and therapeutic options – a mini-review. Gerontology. 2014;60:294–305. doi: 10.1159/000356760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boncompagni S., d'Amelio L., Fulle S., Fano G., Protasi F. Progressive disorganization of the excitation-contraction coupling apparatus in aging human skeletal muscle as revealed by electron microscopy: a possible role in the decline of muscle performance. J. Gerontol. A Biol. Sci. Med. Sci. 2006;61:995–1008. doi: 10.1093/gerona/61.10.995. [DOI] [PubMed] [Google Scholar]
- 12.Thomas M.M., Khan W., Betik A.C., Wright K.J., Hepple R.T. Initiating exercise training in late middle age minimally protects muscle contractile function and increases myocyte oxidative damage in senescent rats. Exp. Gerontol. 2010;45:856–867. doi: 10.1016/j.exger.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Dufresne S.S., Dumont N.A., Boulanger-Piette A., Fajardo V.A., Gamu D., Kake-Guena S.A., David R.O., Bouchard P., Lavergne E., Penninger J.M., Pape P.C., Tupling A.R., Frenette J. Muscle RANK is a key regulator of Ca2+ storage, SERCA activity, and function of fast-twitch skeletal muscles. Am. J. Physiol. Cell Physiol. 2016;310:C663–C672. doi: 10.1152/ajpcell.00285.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voit A., Patel V., Pachon R., Shah V., Bakhutma M., Kohlbrenner E., McArdle J.J., Dell'Italia L.J., Mendell J.R., Xie L.H., Hajjar R.J., Duan D., Fraidenraich D., Babu G.J. Reducing sarcolipin expression mitigates Duchenne muscular dystrophy and associated cardiomyopathy in mice. Nat. Commun. 2017;8:1068. doi: 10.1038/s41467-017-01146-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dremina E.S., Sharov V.S., Davies M.J., Schoneich C. Oxidation and inactivation of SERCA by selective reaction of cysteine residues with amino acid peroxides. Chem. Res. Toxicol. 2007;20:1462–1469. doi: 10.1021/tx700108w. [DOI] [PubMed] [Google Scholar]
- 16.Sharov V.S., Dremina E.S., Galeva N.A., Williams T.D., Schoneich C. Quantitative mapping of oxidation-sensitive cysteine residues in SERCA in vivo and in vitro by HPLC-electrospray-tandem MS: selective protein oxidation during biological aging. Biochem. J. 2006;394:605–615. doi: 10.1042/BJ20051214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vallejo-Illarramendi A., Toral-Ojeda I., Aldanondo G., Lopez de Munain A. Dysregulation of calcium homeostasis in muscular dystrophies. Expert Rev. Mol. Med. 2014;16:e16. doi: 10.1017/erm.2014.17. [DOI] [PubMed] [Google Scholar]
- 18.Mazala D.A., Pratt S.J., Chen D., Molkentin J.D., Lovering R.M., Chin E.R. SERCA1 overexpression minimizes skeletal muscle damage in dystrophic mouse models. Am. J. Physiol. Cell Physiol. 2015;308:C699–C709. doi: 10.1152/ajpcell.00341.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Elchuri S., Oberley T.D., Qi W., Eisenstein R.S., Jackson Roberts L., Van Remmen H., Epstein C.J., Huang T.T. CuZnSOD deficiency leads to persistent and widespread oxidative damage and hepatocarcinogenesis later in life. Oncogene. 2005;24:367–380. doi: 10.1038/sj.onc.1208207. [DOI] [PubMed] [Google Scholar]
- 20.Dahl R. A new target for Parkinson's disease: small molecule SERCA activator CDN1163 ameliorates dyskinesia in 6-OHDA-lesioned rats. Bioorg. Med. Chem. 2017;25:53–57. doi: 10.1016/j.bmc.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 21.Krajnak K., Dahl R. A new target for Alzheimer's disease: a small molecule SERCA activator is neuroprotective in vitro and improves memory and cognition in APP/PS1 mice. Bioorg. Med. Chem. Lett. 2018;28:1591–1594. doi: 10.1016/j.bmcl.2018.03.052. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y., Ikeno Y., Bokov A., Gelfond J., Jaramillo C., Zhang H.M., Liu Y., Qi W., Hubbard G., Richardson A., Van Remmen H. Dietary restriction attenuates the accelerated aging phenotype of Sod1(-/-) mice. Free Radic. Biol. Med. 2013;60:300–306. doi: 10.1016/j.freeradbiomed.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu M.H., Tupling A.R. Protective effects of Hsp70 on the structure and function of SERCA2a expressed in HEK-293 cells during heat stress. Am. J. Physiol. Heart Circ. Physiol. 2009;296:H1175–H1183. doi: 10.1152/ajpheart.01276.2008. [DOI] [PubMed] [Google Scholar]
- 24.Chen L., Na R., Gu M., Salmon A.B., Liu Y., Liang H., Qi W., Van Remmen H., Richardson A., Ran Q. Reduction of mitochondrial H2O2 by overexpressing peroxiredoxin 3 improves glucose tolerance in mice. Aging Cell. 2008;7:866–878. doi: 10.1111/j.1474-9726.2008.00432.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Muller F.L., Song W., Jang Y.C., Liu Y., Sabia M., Richardson A., Van Remmen H. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007;293:R1159–R1168. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 26.Eisner V., Csordas G., Hajnoczky G. Interactions between sarco-endoplasmic reticulum and mitochondria in cardiac and skeletal muscle – pivotal roles in Ca(2)(+) and reactive oxygen species signaling. J. Cell Sci. 2013;126:2965–2978. doi: 10.1242/jcs.093609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kang S., Dahl R., Hsieh W., Shin A., Zsebo K.M., Buettner C., Hajjar R.J., Lebeche D. Small molecular allosteric activator of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) attenuates diabetes and metabolic disorders. J. Biol. Chem. 2016;291:5185–5198. doi: 10.1074/jbc.M115.705012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Periasamy M., Kalyanasundaram A. SERCA pump isoforms: their role in calcium transport and disease. Muscle Nerve. 2007;35:430–442. doi: 10.1002/mus.20745. [DOI] [PubMed] [Google Scholar]
- 29.Goonasekera S.A., Lam C.K., Millay D.P., Sargent M.A., Hajjar R.J., Kranias E.G., Molkentin J.D. Mitigation of muscular dystrophy in mice by SERCA overexpression in skeletal muscle. J. Clin. Investig. 2011;121:1044–1052. doi: 10.1172/JCI43844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gehrig S.M., van der Poel C., Sayer T.A., Schertzer J.D., Henstridge D.C., Church J.E., Lamon S., Russell A.P., Davies K.E., Febbraio M.A., Lynch G.S. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature. 2012;484:394–398. doi: 10.1038/nature10980. [DOI] [PubMed] [Google Scholar]
- 31.Tong X., Kono T., Anderson-Baucum E.K., Yamamoto W., Gilon P., Lebeche D., Day R.N., Shull G.E., Evans-Molina C. SERCA2 deficiency impairs pancreatic beta-cell function in response to diet-induced obesity. Diabetes. 2016;65:3039–3052. doi: 10.2337/db16-0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brini M., Manni S., Pierobon N., Du G.G., Sharma P., MacLennan D.H., Carafoli E. Ca2+ signaling in HEK-293 and skeletal muscle cells expressing recombinant ryanodine receptors harboring malignant hyperthermia and central core disease mutations. J. Biol. Chem. 2005;280:15380–15389. doi: 10.1074/jbc.M410421200. [DOI] [PubMed] [Google Scholar]
- 33.Zhou J., Dhakal K., Yi J. Mitochondrial Ca(2+) uptake in skeletal muscle health and disease. Sci. China Life Sci. 2016;59:770–776. doi: 10.1007/s11427-016-5089-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sandri M. Autophagy in skeletal muscle. FEBS Lett. 2010;584:1411–1416. doi: 10.1016/j.febslet.2010.01.056. [DOI] [PubMed] [Google Scholar]
- 35.Lee J.K., Shin J.H., Lee J.E., Choi E.J. Role of autophagy in the pathogenesis of amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2015;1852:2517–2524. doi: 10.1016/j.bbadis.2015.08.005. [DOI] [PubMed] [Google Scholar]
- 36.Sebastian D., Sorianello E., Segales J., Irazoki A., Ruiz-Bonilla V., Sala D., Planet E., Berenguer-Llergo A., Munoz J.P., Sanchez-Feutrie M., Plana N., Hernandez-Alvarez M.I., Serrano A.L., Palacin M., Zorzano A. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016;35:1677–1693. doi: 10.15252/embj.201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Joseph A.M., Adhihetty P.J., Wawrzyniak N.R., Wohlgemuth S.E., Picca A., Kujoth G.C., Prolla T.A., Leeuwenburgh C. Dysregulation of mitochondrial quality control processes contribute to sarcopenia in a mouse model of premature aging. PLoS One. 2013;8:e69327. doi: 10.1371/journal.pone.0069327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Drummond M.J., Addison O., Brunker L., Hopkins P.N., McClain D.A., LaStayo P.C., Marcus R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: a cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014;69:1040–1048. doi: 10.1093/gerona/glu004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Moopanar T.R., Allen D.G. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J. Physiol. 2005;564:189–199. doi: 10.1113/jphysiol.2005.083519. [DOI] [PMC free article] [PubMed] [Google Scholar]