

Abstract
Severe and recurrent cisplatin-induced acute kidney injury (AKI) as part of standard cancer therapy is a known risk factor for development of chronic kidney disease (CKD). The specific role of superoxide (O2•-)-mediated disruption of mitochondrial oxidative metabolism in CKD after cisplatin treatment is unexplored. Cisplatin is typically administered in weekly or tri-weekly cycles as part of standard cancer therapy. To investigate the role of O2•- in predisposing patients to future renal injury and in CKD, mice were treated with cisplatin and a mitochondrial-specific, superoxide dismutase (SOD) mimetic, GC4419. Renal function, biomarkers of oxidative stress, mitochondrial oxidative metabolism, and kidney injury markers, as well as renal histology, were assessed to evaluate the cellular changes that occur one week and one month (CKD phase) after the cisplatin insult. Cisplatin treatment resulted in persistent upregulation of kidney injury markers, increased steady-state levels of O2•-, increased O2•--mediated renal tubules damage, and upregulation of mitochondrial electron transport chain (ETC) complex I activity both one week and one month following cisplatin treatment. Treatment with a novel, clinically relevant, small-molecule superoxide dismutase (SOD) mimetic, GC4419, restored mitochondrial ETC complex I activity to control levels without affecting complexes II–IV activity, as well as ameliorated cisplatin-induced kidney injury. These data support the hypothesis that increased mitochondrial O2•- following cisplatin administration, as a result of disruptions of mitochondrial metabolism, may be an important contributor to both AKI and CKD progression.
Keywords: Cisplatin, Kidney injury, Mitochondrial metabolism, Superoxide, Superoxide dismutase mimetic
Graphical abstract
Scheme for potential disruption in ETC chain activity leading to mitochondrial dysfunction and increased levels of superoxide (O2•-).

Highlights
-
•
Cisplatin-induced AKI and CKD have a negative impact in long-term renal function.
-
•
Cisplatin-induced CKD disrupts mitochondrial metabolism and increases O2•- levels.
-
•
SOD mimetic, GC4419 mitigates renal damage and mitochondrial metabolism disruptions.
1. Introduction
At any given time, approximately 22% of all cancer patients are receiving chemotherapy [1] either alone or in combination with other cancer therapies. One of the most common chemotherapies that has been used since the 1970s is cisplatin. Cisplatin is still utilized to treat a variety of solid tumors, including head and neck, lung, ovarian, breast, brain, renal, and testicular cancers [2]. Cisplatin's mechanism of action is via the formation of covalent cross linkages between purine DNA bases and the platinum [2]. Unfortunately, cisplatin treatment is associated with several serious toxicities including nephrotoxicity, hepatotoxicity, and cardiotoxicity.
The major route of cisplatin excretion is by the genitourinary system. Cisplatin is removed from the blood by both glomerular filtration and tubular secretion [3]. As a result, cisplatin concentration in the proximal tubular epithelial cells is approximately five times the concentration in serum [4].
Cisplatin-induced acute kidney injury (AKI) incidence ranges from 31% to 68% with most patients experiencing a significant decline in their renal function up to 5 years after treatment [5], [6], [7], [8]. Because platinum-based chemotherapeutic agents are so frequently utilized and because platinum-induced kidney injury is a frequently associated toxicity, there is a critical need to develop therapeutic approaches for the prevention and treatment of platinum-induced AKI and chronic kidney disease (CKD) without reducing treatment efficacy.
AKI is a significant complication of platinum-based cancer treatment, and it is characterized by tubular damage and renal function decline [3]. AKI may cause missed cancer therapies thereby limiting treatment efficacy, prolonging hospital admissions, and increasing medical costs and mortality [9]. Severe and recurrent AKI episodes are known risk factors for the development of chronic kidney disease (CKD) [10], [11], with persistent renal dysfunction, development of fibrosis, and inflammation [12].
In clinical practice, patients receive multiple cisplatin doses that can be administered either weekly or monthly. To date, there are no studies evaluating the effects of cisplatin in renal oxidative metabolism during the repair phase (one week after injury), before subsequent cisplatin is administered; or one month after the injury, which could contribute to development of CKD. Previous studies have evaluated the early effects of a single cisplatin dose treatment in renal mitochondrial function in vivo [13], [14], [15], [16], [17]. It is known that the major site of cisplatin-induced nephrotoxicity is in proximal tubule cells [3], where cisplatin forms glutathione (GSH) and cysteine conjugates, which undergo metabolic activation to form reactive thiols [18]. Cisplatin accumulates in the mitochondria of tubular epithelial cells, causes mitochondrial structural damage, increases in steady-state levels of reactive oxygen species (ROS), and decreases in total GSH and superoxide dismutase (SOD) activity [16], [19], [20], [21]. Impaired mitochondrial metabolism reduces mitochondrial electron transport chain (ETC) efficiency, increases ROS—more specifically superoxide (O2•-)—and reduces ATP generation [13], [15], [21], [22]. Increases in ROS cause tubular epithelial cell apoptosis, necrosis, and increased oxidative/nitrative stress–mediated damage during cisplatin-induced AKI (at 1–3 days) [13], [15], [17], [21], [22].
The specific role of O2•- in the repair phase (one week following cisplatin treatment) and in cisplatin-induced CKD (one month after injury) that could contribute to increased susceptibility to kidney injury is unexplored. O2•- scavenging by manganese superoxide dismutase (MnSOD) overexpression, treatment with orgotein (a copper-zinc SOD pharmaceutical preparation), synthesized cationic SOD enzymes, and non-specific O2•- scavengers have been shown to provide some measure of protection in cisplatin-induced AKI in animal models [13], [17], [23], [24], [25], [26].
Despite previous work with ROS scavengers, the use of O2•--specific SOD mimics that are safe in humans have not been tested in the prevention of recurrent cisplatin-induced AKI or transition to CKD. The current study assessed the role of O2•- following cisplatin-induced kidney injury and the pre-disposition to future renal injuries thereby transitioning to CKD. Herein, we evaluate the effects of cisplatin one week and one month after injury using an O2•--specific SOD mimetic (GC4419) currently in clinical trials (Fig. 1). Recent clinical trials have shown that GC4419 is well tolerated and does not decrease tumor response to therapy [27]. We hypothesize that O2•- mediates cisplatin-induced AKI and that scavenging steady-state levels of O2•- with GC4419 can provide protection in the repair phase of renal injury and prevent CKD burden. To test this hypothesis, we used a relevant model administrating a lower cisplatin dose and weekly cisplatin to characterize the effects of O2•- on mitochondrial oxidative metabolism and oxidative/nitrosative stress in the repair phase and in CKD.
Fig. 1.
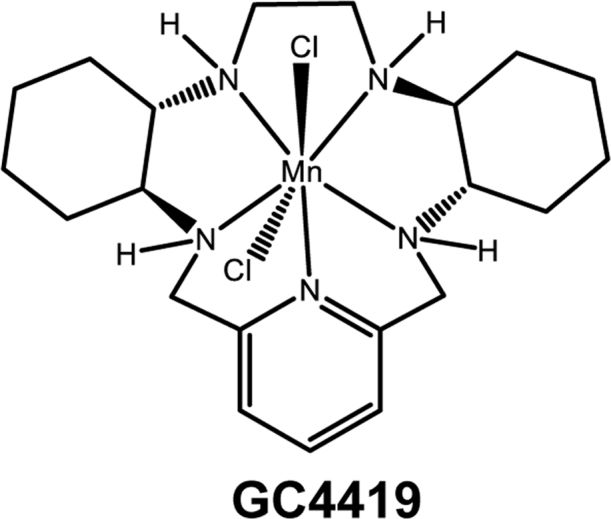
GC4419 chemical structure. Small molecular weight superoxide dismutase (SOD) mimetic (previously known as M40419), molecular weight 483 (49).
2. Material and methods
2.1. Animal maintenance and care
Ten-week-old C57BL/6 J male mice were obtained from The Jackson Laboratory and maintained in accordance with ACURF approval #4121235 at the University of Iowa Animal Care facility. Mice were maintained on normal diets and water ad libitum through the course of the experiment. Animals were randomly assigned to experimental groups which included Vehicle control, Cisplatin only, GC4419 only, and Cisplatin+GC4419. Ten- to 12-week-old mice received GC4419 at 10 mg/kg body weight by i.p. injection vs. vehicle starting 5 days before cisplatin injection and until day of harvest in Cisplatin 1X group, and 5 days after second cisplatin injection in Cisplatin 2X group. GC4419 (Galera Therapeutics, molecular weight 483) was prepared at a stock concentration of 4.83 mg/mL in saline with 10 mmol/L sodium bicarbonate (pH 7.1–7.4). GC4419 (previously known as M40419) is a small-molecule SOD mimetic specific for dismutation of superoxide with a rate constant of (1 × 107 mol s–1) [28], comparable with the native SOD enzyme (1.2 × 109 mol s–1) [29]. Cisplatin-induced kidney injury was generated by injecting cisplatin 10 mg/kg (1 mg/mL; molecular weight 300) purchased from Hospira, Inc or 0.9% saline by i.p. injection at day 0 in Cisplatin 1X group and at day 0 and day 7 in Cisplatin 2X group. Mice were weighted daily and received normal saline (NS) 10 mL/kg i.p. if they had more than 10% body weight loss until they maintained weight. Mice were euthanized 7 days after cisplatin injection (Cisplatin 1 ×) and 31 days after the second cisplatin injection (Cisplatin 2 ×).
2.2. Measurement of renal function
Whole blood BUN was measured using an i-STAT handheld clinical analyzer purchased from Abbott-Point of Care (Princeton, NJ) and single-use i-STAT test cartridges (Chem8 +).
2.3. Histopathology
For histological analysis, kidneys were fixed in 4% PFA and evaluated for tubular necrosis and tubulointerstitial inflammation with Periodic acid-Schiff (PAS) stain. Semi-quantitative evaluation was performed as previously described by two blinded pathologists [30]. For evaluation of renal fibrosis, sections were stained with Trichrome stain. Sections were scanned using a Leica Ariol Slide Scanner. Five random 20X cortical areas were sampled per kidney section using OlyVIA software. The percentage of fibrosis was calculated by measuring fibrosis-positive areas over the total cortex using ImageJ software. Paraffin-embedded tissues were used to detect changes in 3-nitrotyrosine (3-NT, 06–285; Millipore Sigma at 1:1000) using immunohistochemistry. See supplemental section for more details.
2.4. RNA isolation and real-time PCR
RNA was isolated from one quarter of a mouse kidney and complementary DNA was synthesized using the High Capacity cDNA Reverse Transcription Kit from Applied Biosystems (436814) according to the manufacturer's protocol. Real-time polymerase chain reaction (PCR) was performed using SYBR Green ER (Thermo Fisher Scientific). Expression was normalized to ribosomal U36B4, which was used as a housekeeping gene.
2.5. Mitochondrial ETC complex activities
Measurements of the ETC complex activities were done as described previously [31], [32]. Kidney tissues were homogenized in 20 mM phosphate buffer. The assays were performed on a Beckman DU 800 spectrophotometer (Brea, CA). Complex I activity was assayed as the rate of rotenone-inhibitable NADH oxidation. Complex II activity was assayed as the rate of reduction of 2,6-dichloroindophenol by coenzyme Q in the presence and absence of 0.2 M succinate. Complex III activity was assayed as the rate of cytochrome c reduction by coenzyme Q2. Complex IV activity was assayed as the rate of cytochrome c oxidation. For more details, please see the supplemental section.
2.6. Aconitase activity
Total aconitase activity was measured using the protocol previously described [33]. Protein was quantified using the Lowry assay [34]. Rates were determined from slopes determined by regression analysis of data.
2.7. Citrate synthase activity
Citrate synthase activity was measured using the protocol previously described [35]. For more details, please see the supplemental section.
2.8. Glutathione assay
Kidneys were homogenized in 5% 5-sulfosalicylic acid (Sigma Aldrich). The samples were then assayed for total glutathione (GSH) content by the method of Griffith [36]. The protein levels determined using the BCA Assay Kit (Thermo Scientific, Rockford, IL). Values were normalized to the protein content of whole homogenates. Percent of total glutathione as glutathione disulfide was calculated. For more details, see the supplemental section.
2.9. Dihydroethidium (DHE) oxidation
OCT frozen mouse kidney tissues were sectioned at 10 µm and stained with 10 mmol/L DHE for 10 min at 37 °C in DPBS containing 5 mmol/L sodium pyruvate prior to analysis by confocal microscopy using the Olympus Fluoview FV1000 confocal microscope. All sections were imaged at 40X magnification, 37 °C using Cy3 as the fluorochrome. Antimycin A, an electron transport chain blocker, was used as a positive control at 10 μmol/L for 10 min. Each image obtained was quantified in a blinded fashion by quantifying the mean fluorescence intensity of at least 150 cell nuclei and normalized to control.
2.10. Statistical analysis
All statistics were done using the GraphPad prism software. Statistical significance was determined using one-way analysis of variance (ANOVA) for non-parametric measurements. Tukey post-hoc test was performed wherever appropriate. Unless otherwise specified in the figure legend, error bars represent ± standard error of mean (SEM) and statistical significance was defined as * * = p < 0.05.
3. Results
3.1. Cisplatin-induced AKI is inhibited by a small-molecule SOD mimetic, GC4419
To evaluate the short-term effects of O2•- scavenging following cisplatin exposure, 10- to 12-week-old C57BL6/J male mice were pre-treated with daily intraperitoneal (i.p.) injections of GC4419 (10 mg/kg) or vehicle for four days prior to cisplatin i.p. injection (10 mg/kg) and continued daily GC4419 until the time of harvest, 6–7 days following cisplatin injection (Cisplatin 1X) (Fig. 2A). Cisplatin administration caused weight loss in mice, indicative of systemic toxicity that frequently occurs in humans receiving cisplatin treatment [37]. Mice treated with GC4419 partially mitigated weight loss (Fig. 2B) and demonstrated significant improvements in blood urea nitrogen (BUN) three days following cisplatin exposure (Fig. 2C), as well as reduction of tubular necrosis six days after cisplatin treatment (Fig. 2D–E). Cisplatin increased mRNA expression of the kidney injury markers neutrophil gelatinase-associated lipocalin (NGAL) and kidney injury molecule-1 (KIM1) six days after cisplatin exposure, and this was significantly reduced by treatment with GC4419 (Fig. 2F–G). These data suggest that O2•- contributes to cisplatin-induced AKI, including persistent histological abnormalities and increases in injury markers up to 6–7 days after injection.
Fig. 2.

Cisplatin-induced AKI is inhibited by GC4419. Representative schematic of cisplatin-induced AKI model in combination with the SOD mimetic, GC4419 (A). Mice received a single dose of intraperitoneal (i.p.) cisplatin (10 mg/kg) vs. vehicle on day 0 and were treated with i.p. GC4419 (10 mg/kg) vs. vehicle 4 days prior to until day of harvest at 6–7 days post-cisplatin injection. Weight loss in cisplatin-and/or GC4419-treated mice (B) (n = 5 per group). Data expressed as mean ± SEM, * = p < 0.05 comparing Cisplatin 1X and Cisplatin 1X+ GC4419 compared to control and * * = p < 0.05 comparing Cisplatin 1Xvs. Cisplatin 1 X+ GC4419 as analyzed by two-way ANOVA, post-hoc Tukey multiple comparison test. Blood urea nitrogen (BUN) measured in whole blood at 3 days following treatment with cisplatin (C) (control n = 11, GC4419 n = 5, Cisplatin 1X n = 11, Cisplatin 1X + GC4419 n = 5). Error bars represent SEM, * = p < 0.05 Cisplatin 1X vs. vehicle control as analyzed by two-way ANOVA, post-hoc Tukey. Acute tubular necrosis score quantified in kidney sections using periodic acid shift (PAS) staining (40X magnification) (D and E). mRNA expression of the kidney injury markers KIM1 (F) and NGAL (G) was measured by qRT-PCR using SYBR Green ER (Thermo Fisher Scientific) in kidney tissue harvested 6 days after cisplatin injection. mRNA values were normalized relative to the ribosomal U36B4 mRNA levels. KIM1 (F) and NGAL (G) mRNA expression were significantly reduced in cisplatin-induced AKI mice treated with GC4419 (n = 5). Data represent SEM, * = p < 0.05 using one-way ANOVA, post-hoc Tukey.
3.2. Superoxide mediates cisplatin-induced injury and disruptions in mitochondrial oxidative metabolism during the repair phase
Cisplatin-induced AKI in the first three days following injury has been shown to significantly increase steady-state O2•- levels [17], [38], leading to downregulation of electron transport chain (ETC) complexes I–IV activity [13] and ROS/reactive nitrogen species (RNS)–mediated renal damage [17]. In the current study, cisplatin-induced AKI resulted in a persistent 2-fold increase in SOD-inhibitable dihydroethidium (DHE) oxidation (as a measure of tissue O2•- levels) in fresh frozen tissue sections, relative to control and GC4419 treated groups, 6 days after cisplatin injection (Fig. 3A–B).
Fig. 3.

Superoxide mediates cisplatin-induced injury and disruptions in mitochondrial oxidative metabolism in the repair phase. DHE oxidation levels in mouse kidney sections 7 days following a single dose of cisplatin (10 mg/kg) alone or in combination with the SOD mimetic, GC4419 (10 mg/kg) (A –B). Briefly, 8-micron sections were treated with 10 μM DHE for 15 min and imaged using confocal microscopy at 20X magnification. At least 100 nuclei were quantified in a blinded fashion per mouse. The electron transport chain blocker Antimycin A was used as positive control at 10 μM (n = 3). Spectrophotometric analysis of mitochondrial ETC complex activities was measured as described previously (49) in kidney homogenates. Treatment with a single dose of cisplatin resulted in significant increase in complex I activity (rate of rotenone-inhibitable NADH oxidation) (C) (control n = 11, GC4419 n = 5, Cisplatin 1X n = 11, Cisplatin 1X + GC4419 n = 5); no significant change was observed in complex II (rate of reduction of 2, 6-dichloroindophenol by coenzyme Q in the presence and absence of 0.2 M succinate) (D) (control n = 11, GC4419 n = 5, Cisplatin 1X n = 11, Cisplatin 1X + GC4419 n = 5), complex III (rate of cytochrome c reduction by coenzyme Q2) (E), complex IV (rate of cytochrome c oxidation) (F) (n = 5). Treatment with a single dose of cisplatin also resulted in significant increase in citrate synthase (G) with no change in aconitase activity (H) measured in kidney homogenates (control n = 11, GC4419 n = 5, Cisplatin 1X n = 11, Cisplatin 1X + GC4419 n = 5). Mice receiving a combination of cisplatin and GC4419 treatment had significant reduction of complex I activity. Peroxynitrate-mediated damage, formed by the reaction of superoxide with nitric oxide, was significantly ameliorated by GC4419 in cisplatin-treated mice (I and J). 3-Nitrotyrosine immunostaining-positive area (20X magnification) was measured in kidney sections using ImageJ software (control n = 5, GC4415 n = 5, Cisplatin 1X n = 7, Cisplatin 1X + GC4419 n = 5). Data is expressed as mean and SEM, * = p < 0.05 as analyzed by one-way ANOVA, post-hoc Tukey multiple comparison test.
Mitochondrial ETC complexes I and III are thought to be the major sites of O2•- production, and under pathological conditions, disruption of the flow of electrons is associated with increased steady-state O2•- [39], [40], [41], [42], [43]. We found that the cisplatin-induced AKI repair phase is accompanied by significant upregulation of complex I activity, with no change in complexes II–IV activity (Fig. 3C–F). These changes suggest the presence of increased one electron reductions of O2 to form O2•- by complex I.
The citric acid cycle plays a pivotal role in mitochondrial bioenergetics by generating NADH and FADH2 for ATP synthesis and NADPH for reduction of ROS. The activity of two citric acid cycle enzymes, citrate synthase [a marker of mitochondrial abundance [35]] and aconitase [an iron-sulphur cluster protein susceptible to oxidation [44]] were assessed. Citrate synthase activity was found to be increased during AKI repair phase, and this increase was inhibited by GC4419 treatment (Fig. 3G). No significant change in aconitase activity was noted in the cisplatin-induced AKI repair phase (Fig. 3H).
Cisplatin-induced, ROS-mediated damage was assessed by measuring 3-nitotyrosine (3-NT) adducts on proteins, as the diffusion-limited reaction of nitric oxide with O2•- yields peroxynitrite, which nitrates proteins to form 3-NT [45]. Treatment with cisplatin resulted in a significant increase in peroxynitrite-mediated damage as measured by 3-NT immunohistochemical (IHC) staining, which was significantly reduced by treatment with GC4419 (Fig. 3I–J). Taken together, the results in Fig. 3 indicate that cisplatin-induced AKI results in altered mitochondrial metabolism during the repair phase and is accompanied by persistent increases to steady-state levels of O2•- and 3-NT. Finally, treatment with the SOD mimic, GC4419, reduced O2•- steady-state levels, restored mitochondrial metabolism, and protected mice from ROS-mediated damage in the cisplatin AKI repair phase.
3.3. GC4419 protects mice from recurrent cisplatin treatments and ameliorates cisplatin-induced CKD
To evaluate the specific role of superoxide in the setting of recurrent cisplatin treatments, 10- to 12-week-old C57BL6/J mice were treated with cisplatin i.p. on day 0 and day 7 (10mg/kg each dose). Mice were pre-treated with daily i.p. GC4419 (10 mg/kg) or vehicle for four days prior to the initial cisplatin i.p. injection and continued daily GC4419 until 5 days after the second cisplatin i.p. injection (day −4 to day 12, 10 mg/kg). Mice were sacrificed 38 days after first cisplatin injection (31 days after the second cisplatin injection) (Fig. 4A). As shown in Fig. 4B, mice treated with cisplatin developed significant weight loss; however, it was ameliorated by GC4419. In cisplatin-treated mice, GC4419 resulted in significant improvement of BUN at 21 and 38 days following cisplatin injection (Fig. 4C). Two treatments of cisplatin (Cisplatin 2 ×) resulted in persistent increases in mRNA expression of kidney injury markers, NGAL and KIM1, one month after cisplatin treatment, which were significantly reduced by treatment with GC4419 (Fig. 4D–E). Histological analysis demonstrated that Cisplatin 2X resulted in a significant increase of tubular necrosis and interstitial inflammation (Fig. 4F and H–I). Mice treated with Cisplatin 2X and GC4419 did not show significant increase in inflammation (Fig. 4F and I). Moreover, the cisplatin treatment group had significant increases in the fibrotic area that decreased with GC4419 treatment (Fig. 4G and J). These data suggest that O2•- scavenging with GC4419 ameliorates tubular damage in recurrent cisplatin-induced kidney injury, prevents renal function decline, and ameliorates renal fibrosis and inflammation present in cisplatin-induced CKD phenotype.
Fig. 4.

The SOD mimetic GC4419 protects mice from recurrent cisplatin-induced kidney injury and ameliorates cisplatin-induced CKD. Representative schematic of cisplatin-induced CKD model using the small-molecule SOD mimetic GC4419 (A). Briefly, mice received intraperitoneal (i.p.) cisplatin (10 mg/kg) vs. vehicle on day 0 and day 7. Mice were treated with GC4419 (10 mg/kg) i.p. vs. vehicle from day –4 until day 12 (5 days post–second cisplatin injection). All mice were euthanized on day 38 post–cisplatin injection 1 (day 31 post–cisplatin injection 2). Cisplatin-induced weight loss is ameliorated by GC4419 (B) (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8). Data expressed in mean and SEM, * = p < 0.05 Cisplatin 1X and Cisplatin 1X + GC4419 compared to control, and * *p < 0.05 Cisplatin 1 ×vs. Cisplatin 1X + GC4419 by two-way ANOVA, post-hoc Tukey multiple comparison test. BUN measured in whole blood at 7, 21, and 38 days following cisplatin treatment (C). There was a significant reduction of BUN in Cisplatin 2X + GC4419 compared to Cisplatin 2X mice at 21 and 38 days after cisplatin (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8). Data expressed in mean and SEM, * * = p < 0.05 Cisplatin 2X vs. Cisplatin 2X + GC4419 by two-way ANOVA, post-hoc Tukey multiple comparison test. mRNA expression of the kidney injury markers NGAL (D) and KIM1 (E) was measured by qRT-PCR using SYPBR Green ER (Thermo Fisher Scientific) in kidney tissue 38 days post–cisplatin injection. mRNA values were normalized relative to the ribosomal U36B4 mRNA levels. There was significant increase of NGAL (D) and KIM1 (E) mRNA expression 38 days after injury and it was significantly reduced by GC4419 treatment (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8). Periodic acid shift (PAS) (F) and Trichrome (G) staining in kidney sections (magnification 20X). PAS staining used to quantify tubular necrosis (H) and interstitial inflammation (I) in a blinded fashion (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8). Trichrome staining used to quantify fibrosis (J) in kidney tissues using ImageJ software (control n = 6, GC4419 n = 5, cisplatin 2X n = 6, cisplatin 2X+ CG4419 n = 8). Data expressed in mean and SEM, * = p < 0.05 by one-way ANOVA, post-hoc Tukey multiple comparison test.
3.4. GC4419 ameliorates cisplatin-induced oxidative damage and disruptions in mitochondrial oxidative metabolism in the CKD model
It has previously been demonstrated that mice treated with multiple, weekly cisplatin injections have significant increases in DHE staining, lipid peroxidation, and ROS-mediated DNA damage in kidney tissue one month after the first cisplatin injection [46]. We found that treatment with GC4419 prevented peroxynitrite-mediated damage as seen by greatly diminished 3-NT (Fig. 5A and B) and preserved redox status of the GSH/GSSG redox couple in the Cisplatin 2X group (Fig. 5C). Moreover, consistent with our single treatment of cisplatin model, there was a significant increase in mitochondrial complex I activity, which was restored by treatment with GC4419 (Fig. 5D), and no change in complex II activity (Fig. 5E).
Fig. 5.

The SOD mimetic GC4419 ameliorates cisplatin-induced mediated oxidative damage and disruptions in mitochondrial oxidative metabolism in the CKD model. Cisplatin-induced CKD resulted in increased superoxide-mediated damage evaluated by quantification of 3-Nitrotyrosine (3NT) immunostaining-positive area (20X magnification) in kidney sections quantified using ImageJ software (A–B) (n = 6) and determination of percent of total glutathione (GSH + GSSG) as glutathione disulfide (GSSG) measured in whole kidney (C) (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8), which was prevented by GC4419. Spectrophotometric analysis of mitochondrial ETC complex activities was measured as described previously [49] in kidney homogenates. Cisplatin-induced CKD resulted in significant increase in complex I activity (rate of rotenone-inhibitable NADH oxidation) (D) whereas, no significant change was observed in complex II (rate of reduction of 2, 6-dichloroindophenol by coenzyme Q in the presence and absence of 0.2 M succinate) (E), and citrate synthase (F) activity, and there was a significant decrease in aconitase activity (G) measured in whole kidney tissue. Treatment with GC4419 resulted in significant reduction of complex I and citrate synthase activity (D and F) (control n = 6, GC4419 n = 7, Cisplatin 2X n = 6, Cisplatin 2X + CG4419 n = 8). Data expressed in mean and SEM, * = p < 0.05 by one-way ANOVA, post-hoc Tukey multiple comparison test.
Contrary to our results in Cisplatin 1X model, mice treated with Cisplatin 2X and evaluated one month after injection had no change in citrate synthase activity, and treatment with GC4419 resulted in significant decrease in citrate synthase activity in both control and cisplatin-treated groups (Fig. 5F). Aconitase activity was significantly downregulated in both Cisplatin 2X groups, suggesting that despite accomplishing amelioration of cisplatin-induced CKD with GC4419 treatment, even mild CKD phenotype could result in persistent increase of O2•-, causing decreased aconitase activity (Fig. 5G). Together, these data in Fig. 5 suggest that cisplatin-induced CKD results in persistent increased steady-state levels of O2•- and associated oxidative damage and that mitochondrial reverse electron transport into complex I could be a source of damaging O2•- in cisplatin-induced CKD.
4. Discussion
Mitochondrial structural damage, increased ROS levels, and decreased thiol levels have all been implicated in cisplatin-induced injury [16], [19], [20], [21], potentially leading to tubular epithelial cell apoptosis and necrosis in the acute phase of AKI [13], [14], [15], [17], [23], [24], [26], [47]. However, a clear understanding of the relationship between O2•--mediated oxidative stress and mitochondrial oxidative metabolism occurring after cisplatin-induced AKI and how they contribute to increase susceptibility to future renal insults and CKD progression have not been well studied.
In clinical practice, cisplatin is administered in weekly or monthly infusions. Recurrent AKI increases susceptibility to further renal insults and CKD progression [11], [12]. In the current study, we used a non-lethal, murine, cisplatin-induced kidney injury model that allowed us to administer weekly cisplatin injections. Our model resulted in persistent upregulation of kidney injury markers and increased steady-state levels of O2•-, consistent with previous studies using the same strategy [46]. We went on to demonstrate that cisplatin-induced kidney injury leads to changes in mitochondrial oxidative metabolism one week and up to 38 days (CKD phase) following cisplatin treatment.
We used a clinically relevant SOD mimetic, GC4419, to elucidate the role of O2•- in increasing susceptibility to further renal insults and CKD after cisplatin injury. Strengths in our study includes the use of clinically relevant cisplatin dose and a specific SOD mimetic that could be used in humans. It has previously been demonstrated that ROS scavenging protects mice from severe cisplatin-induced AKI (doses between 20 and 25 mg/kg) in the first 3 days after injury using antioxidants that are non-specific for O2•- scavenging [13], [17], [26]. Theses cisplatin doses were lethal and prevented the study of late effects. The main limitations of using native SOD enzymes have been large size (>30 kDa), high manufacturing cost of recombinant enzymes, bell-shape dose-response curves, low cell permeability, antigenicity, and short circulating half-life [48]. Additionally, drawbacks of the non-specific SOD mimics include off-target effects, making it challenging to understand the specific role of O2•- in cisplatin-induced AKI. Finally, there is concern about the potential antagonistic effects of antioxidants on cancer therapy response from using ROS scavengers, and treatment with GC4419 did not result in lower chemotherapy response when used in humans [27].
In our single-dose, cisplatin-induced AKI model, mice had mild but significant renal function decline by 3 days and demonstrated that despite improvement in renal function one week after injury, there is histological evidence of tubular necrosis and upregulation of kidney injury makers with increased steady-state levels of O2•-, which were ameliorated by the SOD mimic, GC4419. Moreover, mice that were treated with GC4419 and recurrent cisplatin injections (to model CKD) had significant improvement of renal functional markers during treatment and reduction of fibrosis, tubular injury markers, and ROS/RNS-mediated damage compared to mice that received cisplatin and vehicle control. These data support the hypothesis that persistent increase in O2•- steady-state levels after cisplatin AKI may have a significant contribution in increasing susceptibility to recurrent AKI and CKD progression.
It has previously been shown that an increase in ROS could be associated with alterations in the stoichiometry of mitochondrial ETC complexes [49]. Data in Fig. 3, Fig. 5 clearly show alterations in mitochondrial ETC complex I activity that could potentially contribute to increased steady-state levels of O2•- and the ROS- and RNS-mediated damage seen in cisplatin-induced kidney injury. One week after cisplatin injection, there was a significant increase of complex I activity, but no change in complex II–IV activity, suggesting the buildup of electrons in complex I in the presence of slower electron transport in complexes II-IV. In animal models of ischemia reperfusion–induced AKI, succinate accumulates during the ischemic injury phase and then is rapidly metabolized following reperfusion, driving extensive ROS generation by reverse electron transport (RET) from complex II to complex I [42]. We hypothesize that both RET and electrons entering from NADH dehydrogenase might be significantly contributing to increased steady-state levels of O2•- after cisplatin injury. This disruption in mitochondrial metabolism was more pronounced in the Cisplatin 2X model with a 2-fold change of complex I activity and no changes in complex II activity. Additionally, there was a significant increase in percentage of total glutathione (GSH+GSSG) in the form of GSSG and a reduction of aconitase activity, which may be a consequence of chronic oxidative stress [50], [51]. Reductions in aconitase activity have been shown to decrease mitochondrial membrane potential and H2O2 production, possibly as a control mechanism to prevent O2•- and H2O2 formation by the respiratory chain [50]. Treatment with GC4419 also reduced complex I and citrate synthase activities in vehicle and cisplatin-treated mice in cisplatin 2X group.
In conclusion, cisplatin treatment results in persistent alterations in mitochondrial oxidative metabolism for up to one month after cisplatin treatment. Mitochondrial electron leak via complex I may represent a potential source of O2•- production that increases susceptibility to recurrent AKI and CKD progression. GC4419 could represent a novel therapeutic strategy for treating and preventing cisplatin-induced AKI and CKD by reducing O2•--mediated disruptions in mitochondrial oxidative metabolism.
Acknowledgements
The authors would like to thank Dr. Michael L. McCormick from the Radiation and Free Radical Research Core at the University of Iowa for technical assistance. The authors thank Gareth Smith for assistance with graphics. The authors thank Katherine N. Gibson-Corley from the Comparative Pathology Laboratory in the Department of Pathology at the University of Iowa for her assistance in paraffin-embedded sample processing, sectioning, and staining. The authors would also like to thank the Kathy Walters and Chantal Allamargot at the University of Iowa for assistance with imaging processing.
Acknowledgments
Financial support details
This work is supported by NICHD K12 HD027748, The Stead Family Department of Pediatrics at the University of Iowa, University of Iowa Dance Marathon, Research Program of Excellence in Redox Biology and Medicine at Iowa, the Holden Comprehensive Cancer Center Oberley Award, P30 CA086862, R01 CA182804, and the Department of Radiation Oncology.
Conflicts of interest
Dr. Dennis Riley is the Chief Scientific Officer at Galera Therapeutics, Inc. which provided the SOD mimetic, GC4419, used in this study. Drs. Spitz and Allen have Sponsored Research Agreements supported by Galera Therapeutics, Inc. to study GC4419 in preclinical and clinical studies of cancer therapy. No other author has competing financial interests in this work.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.redox.2018.09.020.
Appendix A. Supplementary material
Supplementary material
References
- 1.Kolodziej M., Hoverman J.R., Garey J.S., Espirito J., Sheth S., Ginsburg A. Benchmarks for value in cancer care: an analysis of a large commercial population. J. Oncol. Pract. 2011;7(5):301–306. doi: 10.1200/JOP.2011.000394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dasari S., Tchounwou P.B. Cisplatin in cancer therapy: molecular mechanisms of action. Eur. J. Pharmacol. 2014;740:364–378. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miller R.P., Tadagavadi R.K., Ramesh G., Reeves W.B. Mechanisms of cisplatin nephrotoxicity. Toxins. 2010;2(11):2490–2518. doi: 10.3390/toxins2112490. (Basel) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuhlmann M.K., Burkhardt G., Kohler H. Insights into potential cellular mechanisms of cisplatin nephrotoxicity and their clinical application. Nephrol. Dial. Transplant. 1997;12(12):2478–2480. doi: 10.1093/ndt/12.12.2478. [DOI] [PubMed] [Google Scholar]
- 5.Bhat Z.Y., Cadnapaphornchai P., Ginsburg K., Sivagnanam M., Chopra S., Treadway C.K. Understanding the risk factors and long-term consequences of cisplatin-associated acute kidney injury: an observational cohort study. PLoS One. 2015;10(11):e0142225. doi: 10.1371/journal.pone.0142225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latcha S., Jaimes E.A., Patil S., Glezerman I.G., Mehta S., Flombaum C.D. Long-term renal outcomes after cisplatin treatment. Clin. J. Am. Soc. Nephrol. 2016;11(7):1173–1179. doi: 10.2215/CJN.08070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arga M., Oguz A., Pinarli F.G., Karadeniz C., Citak E.C., Emeksiz H.C. Risk factors for cisplatin-induced long-term nephrotoxicity in pediatric cancer survivors. Pediatr. Int. 2015;57(3):406–413. doi: 10.1111/ped.12542. [DOI] [PubMed] [Google Scholar]
- 8.Jones D.P., Spunt S.L., Green D., Springate J.E. Children's oncology G. Renal late effects in patients treated for cancer in childhood: a report from the Children's Oncology Group. Pediatr. Blood Cancer. 2008;51(6):724–731. doi: 10.1002/pbc.21695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lam A.Q., Humphreys B.D. Onco-nephrology: aki in the cancer patient. Clin. J. Am. Soc. Nephrol. 2012;7(10):1692–1700. doi: 10.2215/CJN.03140312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thakar C.V., Christianson A., Himmelfarb J., Leonard A.C. Acute kidney injury episodes and chronic kidney disease risk in diabetes mellitus. Clin. J. Am. Soc. Nephrol. 2011;6(11):2567–2572. doi: 10.2215/CJN.01120211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chawla L.S., Amdur R.L., Amodeo S., Kimmel P.L., Palant C.E. The severity of acute kidney injury predicts progression to chronic kidney disease. Kidney Int. 2011;79(12):1361–1369. doi: 10.1038/ki.2011.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Basile D.P., Bonventre J.V., Mehta R., Nangaku M., Unwin R., Rosner M.H. Progression after AKI: understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016;27(3):687–697. doi: 10.1681/ASN.2015030309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ahmed L.A., Shehata N.I., Abdelkader N.F., Khattab M.M. Tempol. a superoxide dismutase mimetic agent, ameliorates cisplatin-induced nephrotoxicity through alleviation of mitochondrial dysfunction in mice. PLoS One. 2014;9(10):e108889. doi: 10.1371/journal.pone.0108889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dutta R.K., Kondeti V.K., Sharma I., Chandel N.S., Quaggin S.E., Kanwar Y.S. Beneficial effects of myo-inositol oxygenase deficiency in cisplatin-induced AKI. J. Am. Soc. Nephrol. 2017;28(5):1421–1436. doi: 10.1681/ASN.2016070744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galgamuwa R., Hardy K., Dahlstrom J.E., Blackburn A.C., Wium E., Rooke M. Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J. Am. Soc. Nephrol. 2016;27(11):3331–3344. doi: 10.1681/ASN.2015070827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Husain K., Morris C., Whitworth C., Trammell G.L., Rybak L.P., Somani S.M. Protection by ebselen against cisplatin-induced nephrotoxicity: antioxidant system. Mol. Cell. Biochem. 1998;178(1–2):127–133. doi: 10.1023/a:1006889427520. [DOI] [PubMed] [Google Scholar]
- 17.Mukhopadhyay P., Horvath B., Zsengeller Z., Zielonka J., Tanchian G., Holovac E. Mitochondrial-targeted antioxidants represent a promising approach for prevention of cisplatin-induced nephropathy. Free Radic. Biol. Med. 2012;52(2):497–506. doi: 10.1016/j.freeradbiomed.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Townsend D.M. Metabolism of cisplatin to a nephrotoxin in proximal tubule cells. J. Am. Soc. Nephrol. 2003;14(1):1–10. doi: 10.1097/01.asn.0000042803.28024.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Singh G. A possible cellular mechanism of cisplatin-induced nephrotoxicity. Toxicology. 1989;58(1):71–80. doi: 10.1016/0300-483x(89)90105-4. [DOI] [PubMed] [Google Scholar]
- 20.Sadzuka Y., Shoji T., Takino Y. Effects of cisplatin on the activities of enzymes which protect agains lipid peroxidation. Biochem. Pharmacol. 1992;15(43):1872–1875. doi: 10.1016/0006-2952(92)90725-x. [DOI] [PubMed] [Google Scholar]
- 21.Kruidering M., Van de Water B., De Heer E., Mulder G.J., Nagelkerke F. Cisplatin-induced nephrotoxicity in procine proximal tubular cells: mitochondrial dysfunction by inhibition of complexes I to IV of the respiratory chain. J. Pharmacol. Exp. Ther. 1997;280(2):638–649. [PubMed] [Google Scholar]
- 22.Suzuki T., Yamaguchi H., Kikusato M., Hashizume O., Nagatoishi S., Matsuo A. Mitochonic Acid 5 binds mitochondria and ameliorates renal tubular and cardiac myocyte damage. J. Am. Soc. Nephrol. 2016;27(7):1925–1932. doi: 10.1681/ASN.2015060623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis C.A., Nick H.S., Agarwal A. Manganese superoxide dismutase attenuates cisplatin-induced renal injury: importance of superoxide. J. Am. Soc. Nephrol. 2001;12(12):1683–1690. doi: 10.1681/ASN.V12122683. [DOI] [PubMed] [Google Scholar]
- 24.McGinness J.E., Proctor P.H., Demopoulos H.B., Hokanson J.A., Kirkpatrick D.S. Amelioration of cis-platinum nephrotoxicity by orgotein (superoxide dismutase) Physiol. Chem. Phys. 1978;10(3):267–277. [PubMed] [Google Scholar]
- 25.Nishikawa M., Nagatomi H., Nishijima M., Ohira G., Change B.J., Sato E. Targeting superoxide dismutase to renal proximal tubule cells inhibits nephrotoxicity of cisplatin and increases the survival of cancer-bearing mice. Cancer Lett. 2001;171(2):133–138. doi: 10.1016/s0304-3835(01)00591-2. [DOI] [PubMed] [Google Scholar]
- 26.Pan H., Shen K., Wang X., Meng H., Wang C., Jin B. Protective effect of metalloporphyrins against cisplatin-induced kidney injury in mice. PLoS One. 2014;9(1):e86057. doi: 10.1371/journal.pone.0086057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anderson C.M., Sonis S.T., Lee C.M., Adkins D., Allen B.G., Sun W. Phase 1b/2a trial of the superoxide dismutase mimetic GC4419 to reduce chemoradiotherapy-induced oral mucositis in patients with oral cavity or Oropharyngeal Carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2018;100(2):427–435. doi: 10.1016/j.ijrobp.2017.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuder R.M., Zhen L., Cho C.Y., Taraseviciene-Stewart L., Kasahara Y., Salvemini D. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am. J. Respir. Cell Mol. Biol. 2003;29(1):88–97. doi: 10.1165/rcmb.2002-0228OC. [DOI] [PubMed] [Google Scholar]
- 29.Forman H.J., Fridovich I. Superoxide dismutase: a comparison of rate constants. Arch. Biochem. Biophys. 1973;158(1):396–400. doi: 10.1016/0003-9861(73)90636-x. [DOI] [PubMed] [Google Scholar]
- 30.Hur E., Garip A., Camyar A., Ilgun S., Ozisik M., Tuna S. The effects of vitamin d on gentamicin-induced acute kidney injury in experimental rat model. Int. J. Endocrinol. 2013;2013:313528. doi: 10.1155/2013/313528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pullman M.E., Penefsky H.S., Datta A., Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J. Biol. Chem. 1960;235:3322–3329. [PubMed] [Google Scholar]
- 32.Birch-Machin M.A., Briggs H.L., Saborido A.A., Bindoff L.A., Turnbull D.M. An evaluation of the measurement of the activities of complexes I-IV in the respiratory chain of human skeletal muscle mitochondria. Biochem. Med. Metab. Biol. 1994;51(1):35–42. doi: 10.1006/bmmb.1994.1004. [DOI] [PubMed] [Google Scholar]
- 33.Morton R.L., Ikle D., White C.W. Loss of lung mitochondrial aconitase activity due to hyperoxia in bronchopulmonary dysplasia in primates. Am. J. Physiol. 1998;274(1 Pt 1):L127–L133. doi: 10.1152/ajplung.1998.274.1.L127. [DOI] [PubMed] [Google Scholar]
- 34.Lowry O.H., Rosebrough N.J., Farr A.L., Randall R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951;193(1):265–275. [PubMed] [Google Scholar]
- 35.Barrientos A. In vivo and in organello assessment of OXPHOS activiites. Methods. 2002;24(4):307–316. doi: 10.1016/S1046-2023(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 36.Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal. Biochem. 1969;27(3):502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- 37.Szturz P., Wouters K., Kiyota N., Tahara M., Prabhash K., Noronha V. Weekly low-dose versus three-weekly high-dose cisplatin for concurrent chemoradiation in locoregionally advanced non-nasopharyngeal head and neck cancer: a systematic review and meta-analysis of aggregate data. Oncologist. 2017;22(9):1056–1066. doi: 10.1634/theoncologist.2017-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Trujillo J., Molina-Jijon E., Medina-Campos O.N., Rodriguez-Munoz R., Reyes J.L., Barrera D. Superoxide anion production and expression ofgp91(phox) and p47(phox) are increased in glomeruli and proximal tubules of cisplatin-treated rats. J. Biochem. Mol. Toxicol. 2015;29(4):149–156. doi: 10.1002/jbt.21679. [DOI] [PubMed] [Google Scholar]
- 39.Slane B.G., Aykin-Burns N., Smith B.J., Kalen A.L., Goswami P.C., Domann F.E. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Res. 2006;66(15):7615–7620. doi: 10.1158/0008-5472.CAN-06-0833. [DOI] [PubMed] [Google Scholar]
- 40.Treberg J.R., Quinlan C.L., Brand M.D. Evidence for two sites of superoxide production by mitochondrial NADH-ubiquinone oxidoreductase (complex I) J. Biol. Chem. 2011;286(31):27103–27110. doi: 10.1074/jbc.M111.252502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Quinlan C.L., Orr A.L., Perevoshchikova I.V., Treberg J.R., Ackrell B.A., Brand M.D. Mitochondrial complex II can generate reactive oxygen species at high rates in both the forward and reverse reactions. J. Biol. Chem. 2012;287(32):27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chouchani E.T., Pell V.R., Gaude E., Aksentijevic D., Sundier S.Y., Robb E.L. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. 2014;515(7527):431–435. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Holmstrom K.M., Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014;15(6):411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- 44.Yarian C.S., Toroser D., Sohal R.S. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech. Ageing Dev. 2006;127(1):79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beckman J.S., Koppenol W.H. Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and the ugly. Am. J. Physiol. 1996;271(5):C1424–C1437. doi: 10.1152/ajpcell.1996.271.5.C1424. [DOI] [PubMed] [Google Scholar]
- 46.Katagiri D., Hamasaki Y., Doi K., Negishi K., Sugaya T., Nangaku M. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016;89(2):374–385. doi: 10.1038/ki.2015.327. [DOI] [PubMed] [Google Scholar]
- 47.Pabla N., Dong G., Jiang M., Huang S., Kumar M.V., Messing R.O. Inhibition of PKCdelta reduces cisplatin-induced nephrotoxicity without blocking chemotherapeutic efficacy in mouse models of cancer. J. Clin. Investig. 2011;121(7):2709–2722. doi: 10.1172/JCI45586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bonetta R. Potential therapeutic applications of MnSODs and SOD-Mimetics. Chemistry. 2017 doi: 10.1002/chem.201704561. [DOI] [PubMed] [Google Scholar]
- 49.Mapuskar K.A., Flippo K.H., Schoenfeld J.D., Riley D.P., Strack S., Hejleh T.A. Mitochondrial Superoxide increases age-associated susceptibility of human dermal fibroblasts to radiation and chemotherapy. Cancer Res. 2017;77(18):5054–5067. doi: 10.1158/0008-5472.CAN-17-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Scandroglio F., Tortora V., Radi R., Castro L. Metabolic control analysis of mitochondrial aconitase: influence over respiration and mitochondrial superoxide and hydrogen peroxide production. Free Radic. Res. 2014;48(6):684–693. doi: 10.3109/10715762.2014.900175. [DOI] [PubMed] [Google Scholar]
- 51.Buettner G.R., Wagner B.A., Rodgers V.G. Quantitative redox biology: an approach to understand the role of reactive species in defining the cellular redox environment. Cell Biochem. Biophys. 2013;67(2):477–483. doi: 10.1007/s12013-011-9320-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material