

SUMMARY
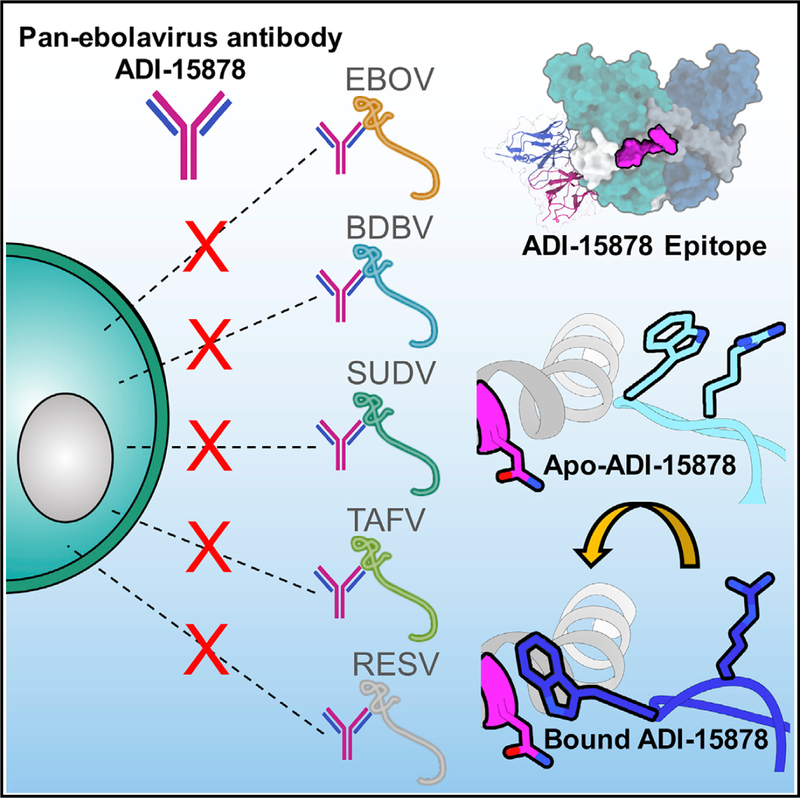
Monoclonal antibodies (mAbs) with pan-ebolavirus cross-reactivity are highly desirable, but development of such mAbs is limited by a lack of a molecular understanding of cross-reactive epitopes. The anti body ADI-15878 was previously identified from a human survivor of Ebola virus Makona variant (EBOV/ Mak) infection. This mAb demonstrated potent neutralizing activity against all known ebolaviruses and provided protection in rodent and ferret models against three ebolavirus species. Here, we describe the unliganded crystal structure of ADI-15878 as well as the cryo-EM structures of ADI-15878 in complex with the EBOV/Mak and Bundibugyo virus (BDBV) glycoproteins (GPs). ADI-15878 binds through an induced-fit mechanism by targeting highly conserved residues in the internal fusion loop (IFL), bridging across GP protomers via the heptad repeat 1 (HR1) region. Our structures provide a more complete description of the ebolavirus immunogenic landscape, as well as a molecular basis for how rare but potent antibodies target conserved filoviral fusion machinery.
In Brief
The threat of another major filoviral outbreaks looms, underlined by the current lack of approved vaccines or therapeutics. Murin et al. describe the molecular nature of neutralization by the human survivor pan-ebolavirus antibody ADI-15878. Their structures collectively provide a blueprint that can aid in the development of more potent pan- ebolavirus therapeutics.
Graphical Abstract
INTRODUCTION
There has been a resurgence of efforts to develop treatments and vaccines for Ebola virus disease (EVD) after the recent pandemic in western Africa, from 2013–2016. Monoclonal antibodies (mAbs) are at the forefront of therapeutic development since showing great promise in animal models. A tri-mAb cocktail, ZMapp, is being evaluated in clinical trials after demonstrating the ability to revert advanced EVD in non-human primates and showing modest success in a small number of patients infected in the aforementioned outbreak (Prevail II Writing Group et al., 2016; Qiu et al., 2014). One disadvantage of ZMapp and similar antibodies is their limited cross-reactivity to other ebolavirus species (Murin et al., 2014). In addition to Ebola virus (EBOV), there are four other species of ebolaviruses that are antigenically divergent, differing by at least 30% on the amino acid level, including Sudan virus (SUDV), Bundibugyo virus (BDBV), Reston virus (RESV), and Tai Forest virus (TAFV). Historically, EBOV, BDBV, and SUDV have caused highly virulent outbreaks in human populations (Burket al., 2016). Ebolaviruses are part of the larger filovirus family, which also includes Marburg virus (MARV) of the marburgvirus genus. MARV has also caused several large human outbreaks, with high lethality (Centers for Disease Control and Prevention, 2014). Given the great unpre dictability and serious nature of ebolavirus outbreaks, a more ideal therapeutic would be one that could target any filovirus with equal potency.
The primary target of anti-ebolavirus mAbs is the viral glycoprotein (GP), which is the only protein attached to the viral surface and is indispensable for the viral life cycle (Lee et al., 2008). The viral GP acts as a machine, providing the key to unlocking the host cell membrane and gaining entry into target cells. Entry is achieved by storing tightly regulated potential energy within the metastable, pre-fusion GP, which is released after interaction with the host receptor NPC1, as well as other downstream events that are not well understood (Lee and Sa- phire, 2009; Miller et al., 2012; White and Schornberg, 2012). Despite the large antigenic diversity among filoviruses, they share their mechanism of entry via structural and sequence conservation in the fusion machinery (Hunt et al., 2012; Miller et al., 2012; White and Schornberg, 2012). The conserved regions include the receptor binding site (RBS), the IFL, and the HR1 and HR2 regions. Filoviral GPs also possess a variable, unstructured, and heavily glycosylated domain called the mucin-like domain (MLD), which is thought to be loosely positioned above ebolavirus GPs and draped over the sides of marburgvirus GPs (Hashiguchi et al., 2015). Below the MLDs in ebolaviruses is the glycan cap, which is structured and inserts itself into the RBS (Lee et al., 2008), while in the marburgviruses the analogous region is unstructured, leaving the RBS exposed on GP12,13. The RBS interacts with the host receptor NPC1 during entry and is structurally conserved across all filoviruses (Wang et al., 2016). While the RBS has been shown to elicit pan-filoviral antibodies, potency and efficacy is variable because the ebolaviruses require the proteolytic removal of the MLDs and glycan cap to expose the RBS (Bale et al., 2011; Bornholdt et al., 2016a; Miller et al., 2012; Wang et al., 2016), while the marburgviruses do not (Flyak et al., 2015; Gnirss et al., 2012; King et al., 2018). The HR2 domain has also been proposed as a hotspot of filoviral vulnerability (Flyak et al., 2016, 2018; Wec et al., 2017; Ya- mayoshi and Kawaoka, 2017), but antibodies that target this region have limited cross-reactivity.
The HR1 and IFL on filoviral GPs are highly conserved in sequence and structure across genera, as demonstrated by comparing the structures of EBOV, SUDV, and MARV GPs, making this epitope an attractive target for therapeutic antibody development (Bale et al., 2012; Dias et al., 2011; Hashiguchi et al., 2015; King et al., 2018; Lee et al., 2008; Zhao et al., 2016). HR1 is composed of an alpha helix, which cradles GP1 and contains a highly conserved glycan at N563. The IFL is composed of an anti-parallel beta hairpin structure with a hydrophobic loop and is wrapped around the exterior of the GP. In the ebolaviruses, the IFL is tucked beneath the cathepsin cleavage loop, which is loosely tucked in between GP protomers. Although NPC1 is necessary for viral fusion, it is not solely responsible for the release of the IFL (Bale et al., 2011; Miller et al., 2012; Shoemaker et al., 2013; Spence et al., 2016). It is thought that once the IFL is released, it pierces the host cell membrane, and then the viral and host membranes are fused by a large structural change in the HR1 and HR2 region. There are only two known examples of antibodies that target the IFL, including the mouse antibody 6D6 (Furuyamaet al., 2016), which specifically targets the tip of the IFL and an additional region, and the non-human primate antibody CA45 (Zhao et al., 2017), which targets the base of the IFL. Both of these mAbs exhibited broad neutralizing capability and protective efficacy; however, only 6D6 was fully pan-ebolavirus reactive. The only known human survivor mAbs that target the IFL are ADI-15878 and ADI- 15742, and both provide broad cross-reactivity and neutralization (Wec et al., 2017). Interestingly, ADI-15878 binds to an epitope between GP protomers that likely contacts both the IFL and HR1 (Wec et al., 2017), which is distinct from CA45.
Here, we present three structures: unliganded ADI-15878 antigen-binding fragment (Fab), ADI-15878 Fab bound to EBOV Makona GP mucin-like domain deleted (GPΔmuc), and ADI-15878 Fab bound to BDBV GPΔmuc. These structures reveal the molecular details of a cross-reactive antibody that targets highly conserved filoviral fusion machinery. Our structure of EBOV/Mak represents the virus responsible for the last major outbreak of EBOV in western Africa, from 2013 to 2016. In addition, we provide a high-resolution model of BDBV GP, which completes a suite of available GP structures for the three major virulent ebolaviruses. Together, these structures provide a valuable tool for evaluating antibody-based responses to infection as well as a means for generating vaccines that may elicit more broadly neutralizing antibodies. Our structures of ADI-15878, in unliganded and GP-bound forms, give important insight into the molecular nature of this highly potent and cross-reactive antibody that was derived from human infection and provides a blueprint for structure-based engineering to potentially increase binding potency and/or pharmacological properties. With the unpredictable nature yet inevitable reoccurrence of filoviral outbreaks, it is paramount that we identify viable therapeutic options that would be effective against the range of virulent filoviral species that naturally exist in nature.
RESULTS
Unliganded ADI-15878 Features a Sequestered CDRH3 Loop
We first sought to determine the X-ray structure of unliganded ADI-15878 to provide an accurate input model for our electron microscopy (EM) studies of GP complexes, as well as to determine any potential structural changes that may occur upon binding to GP. We solved a 2.1-Å resolution crystal structure of the ADI-15878 Fab alone, which contained a single Fab in the asymmetric unit (Figure 1A; Table 1). The sequence of ADI-15878 is very close to that of the germline at 90% and 95% identity in its variable heavy chain (VH, VH3–23*04) and variable light chain (VL, VK1–5*01), respectively (Wec et al., 2017). The majority of changes from germline occur in the complementarity-determining regions (CDRs) H3 and L3. The contributions of both the heavy chain (HC) and light chain (LC) were previously shown to be important for GP binding, and each of these contain several key hydrophobic residues that are implicated in binding, according to kinetic analysis and neutralization assays (Wec et al., 2017).
Figure 1. Crystal Structure of Unliganded ADI-15878 Fab.
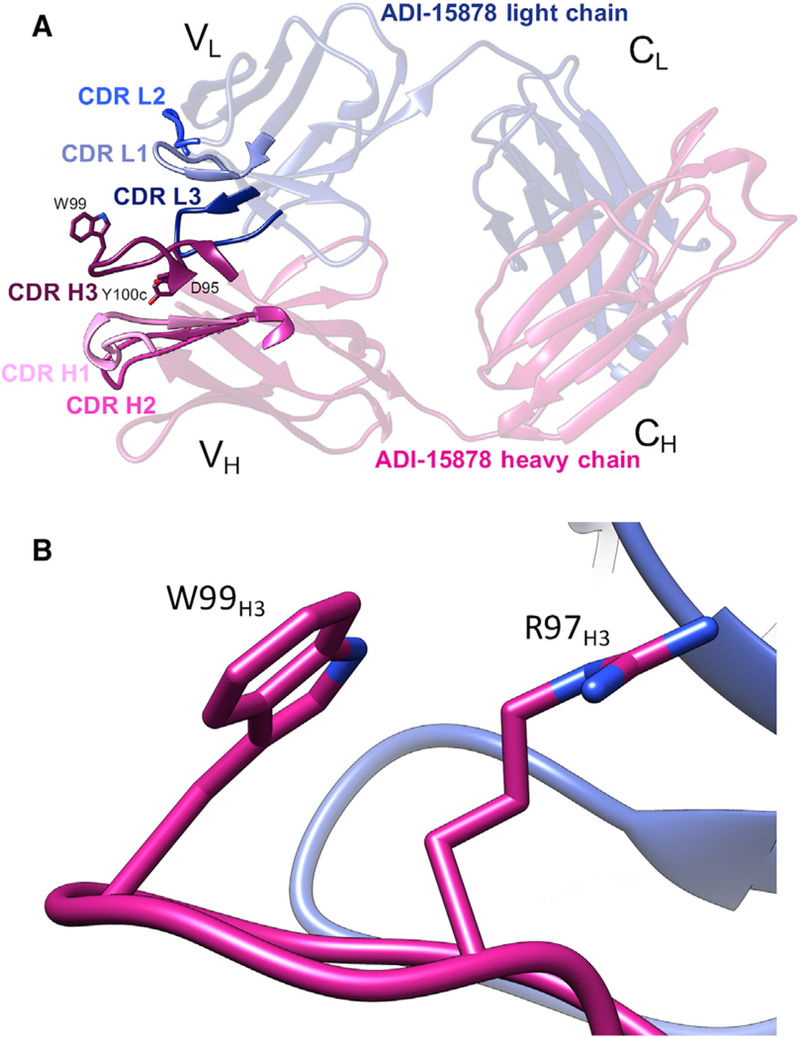
(A) Crystal structure of ADI-15878 Fab, demonstrating overall topology and complementarity-determining region (CDR) loop arrangement. Three residues shown in the CDRH3, D95, W99, and Y100c were previously identified as critical forADI-15878 binding to GP(Wec et al., 2017) and are shown as sticks. (B) Residue W99H3 forms a hydrophobic packing interaction with R97H3 in the unliganded state, sequestering W99H3 from solvent exposure.
Table 1.
Crystallographic Data Collection and Refinement Statistics
| Parameter | ADI-15878 Fab |
|---|---|
| Resolutiona (Å) | 46.45–2.10(2.14–2.10) |
| Space group | P21 |
| Unit cell (Å) | 42.06, 96.84, 54.31 |
| (°) | 90, 102.91, 90 |
| Total reflectionsa | 70,500 (3242) |
| Unique reflectionsa | 24,142 (1150) |
| Multiplicitya | 2.9 (2.8) |
| Completenessa (%) | 96.9 (96.5) |
| < I >/< σ(I) > a | 6.4 (0.9) |
| < I >/< σ(I) > in the 2.56–2.49 Å bin | 2.0 |
| Rmergea,b | 0.17(1.18) |
| Rpima,c | 0.12 (0.82) |
| CC 1/2d | 0.73(0.31) |
| Wilson B (Å2) | 29.7 |
| Reflections used for Rwork(Rfree) | 22,948(1166) |
| Rworka,e (%) | 0.22 (.36) |
| Rfreea,f(%) | 0.27 (.39) |
| RMSDg (bonds) (Å) | 0.013 |
| RMSDg (angles) (°) | 1.58 |
| Ramachandran favored (%) | 96.8 |
| Ramachandran outliers (%) | 0 |
| Average B-factor (Å2) | 32.5 |
| Macromolecules | 32.2 |
| Solvent | 35.3 |
| Ligands | 40.8 |
Values in parentheses are for the highest-resolution shell.
Rfree is the same as Rwork, with 5% of reflections chosen at random and omitted from refinement.
RMSD, root mean square deviation.
The CDRH3 contains the majority of the residues that are required for the recognition of GP (Wec et al., 2017). The length of the CDRH3 at 15 amino acids falls within a median range for the average CDRH3 length in the human repertoire (North et al., 2011; Tiller et al., 2007). The fusion loop-directed antibody CA45 also contains a CDRH3 loop of a length similar to ADI-15878 at 19 amino acids, but it recognizes an epitope distinct from that of ADI-15878 (Zhao et al., 2017). CA45 is similarly highly conserved, with its germline precursor at 86% identity in both VH and VL. While relatively little somatic hypermutation (SHM) in both ADI-15878 and CA45 may suggest that the IFL epitope is easily accessible, there are relatively few described filoviral IFL- directed antibodies and even fewer that are broadly cross-reactive. Therefore, the HC/LC pairing that is required for binding to this epitope may be more critical than SHM.
The ADI-15878 HC originates from the most common human germline HC gene, VH3–23*04, and the LC from VK1–5*01 (Born-holdt et al., 2016b; Wec et al., 2017). Given the diversity of CDRH3 loops, it was not possible to predict the germline origin of this region. As noted above, the ADI-15878 CDRH3 contains several hydrophobic residues—in particular, a tryptophan at its apex, W99H3. Our structure shows that in the unliganded state, W99H3 is sequestered away from solvent, forming a hydrophobic packing interaction with R97H3 (Figure 1B). We note, however, that the CDRH3 loop is near a crystallographic contact, possibly influencing its conformation in this unliganded structure.
The Structure of BDBV GP Reveals Similarities to EBOV/ Mak and SUDV GPs
The BDBV was first identified in Uganda during an outbreak that occurred in 2007 (Towner et al., 2008). An additional outbreak occurred in 2012, with a high mortality rate, demonstrating the prevalence and virulence of this virus (Albariño et al., 2013). Here, we present a cryo-EM structure of BDBV GPΔmuc bound to ADI-15878 Fab (Figure 2A) and confirm that this structure is highly similar to other ebolavirus GPs, despite low sequence identity; for example, 77% for EBOV GP (PDB: 5JQ3) and 66% for SUDV GP (PDB: 3VE0). The global resolution of our complex is ~4.3 Å, while the majority of the core GP structure is in the 4.0–4.2 Å resolution range (Table S1; Figure S1). Our cryo-EM map also revealed N-linked glycans associated with the following residues: N228 and N257 in GP1 and N563 and N618 in GP2. The glycan at N563 is highly conserved and resembles analogous glycans built at this position in other structures.
Figure 2. Cryo-EM Structure of BDBV GPΔmuc and Key Structural Elements Relevant to the ADI-15878 Epitope.
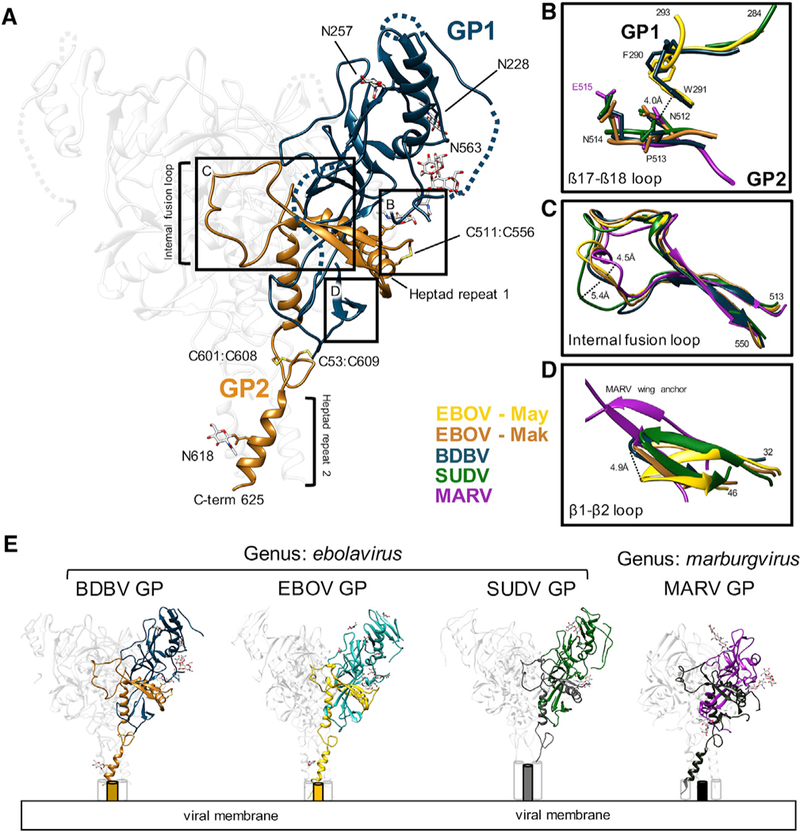
(A) Atomic model of BDBV GPΔmuc derived from the cryo-EM map of BDBV GPΔmuc bound to ADI-15878 Fab (4.3 Å), with GP1 (dark blue) and GP2 (brown) highlighted. Two predicted N-linked glycans are modeled in GP1 and two are modeled in GP2. In addition, there is clear density for three disulfides (labeled), along with a large portion of HR2 that extends to residue 625. Dashed lines represent regions that were included in our construct but we did not build in our model.
(B) Details ofthe β17-β18 loop modeled inthe BDBV structure (dark blue), demonstrating homology to previously modeled loops in SUDV GP (green; PDB: 3VE0) and EBOV/May GP (yellow; PDB: 5JQ3). This loop interacts with the base of the internal fusion loop (IFL) (orange), which shows homology across Filoviridae, including MARV (purple; PDB: 6BP2), SUDV, and EBOV/May (yellow). See also Figure 3.
(C) Comparison of the IFL across known filoviral GP structures. The tip of the IFL is shifted in SUDV and MARV in comparison to unliganded EBOV/ May GP as well as BDBV and EBOV/Mak bound to ADI-15878, although SUDV and MARV shift in opposite directions.
(D) The β1-β2 loop of BDBV, near the base of the ADI-15878 epitope, is most similar to the structure of SUDV bound to 16F6. The MARV wing anchor, although divergent in sequence, occupies this same space and has structural similarities.
(E) Side-by-sidecomparison ofthe highest-resolution structures of filoviral GPs solved to date, showing structural similarity among the ebolaviruses and divergence in the marburgviruses. See also Figure S1 and Table S1.
The majority of the core GP structure is very similar to previously solved filoviral GPs, but there are some noteworthy similarities and differences that are relevant to the binding of ADI-15878. One similarity is the presence of the β17-β18 loop from W288-E292 in BDBV, which is well conserved and resolved in EBOV Mayinga variant (EBOV/May) (Zhao et al., 2016) and SUDV GP (Dias et al., 2011) structures (Figure 2B). The β17-β18 loop is thought to mask an epitope that exists at the base of the IFL (Howell et al., 2017). We show that this hydrophobic region associates with the base of the IFL and likely contributes to its stability.
Furthermore, we were able to model the entire IFL from residues 513–550 (Figures 2A and 2C). Comparing the IFL across other viral species revealed that it most closely resembles the structure of the unliganded EBOV/May IFL; however, there are subtle changes in the conformation of this loop in our structures of GP bound to ADI-15878, suggesting that ADI-15878 influences the conformation of the IFL (Figure 2C). Finally, we noted the presence of the cathepsin cleavage loop, which is more pronounced at lower sigma values in our cryo-EM density map. This loop is close to the ADI-15878 binding site, and cleavage here has been shown to significantly increase ADI-15878 neutralization potency (Wec et al., 2017).
Some differences between BDBV GP and other GP structures that we noted were in the β1-β2 loop at the base of the GP that is shifted when compared to unliganded GP (Zhao et al., 2016) but closely resembles the conformation of SUDV GP (Dias et al., 2011), which is also bound to an antibody near this region, and is reminiscent of the MARV “wing anchor” (Figure 2D). We also modeled a large portion of HR2 down to residue 625 (Figure 2A), which adopts a three-helix bundle within the GP2 and has been previously modeled for EBOV/May (Zhao et al., 2016) and MARV (King et al., 2018). Notably, HR2in MARV is slightly different from the ebolaviruses, with a slightly wider three-helix bundle. A side- by-side comparison of known viral GP structures demonstrates the high level of similarities within the ebolavirus genus and in the core (Figure 2E). However, MARV lacks a structured glycan cap, contains an additional immunodominant epitope at the base of the GP, and displays its MLDs in a different spatial arrangement (Fusco et al., 2015; Hashiguchi et al., 2015; King et al., 2018).
ADI-15878 Recognizes HR1 and the Fusion Loop across Two Protomers
To determine the epitope of ADI-15878, we solved cryo-EM structures of ADI-15878 Fab bound to EBOV/Mak GPΔmuc at 4.1 Å resolution (Figures 3A and S1; Table S1) and BDBV GPΔmuc at 4.3 Å resolution (Figures 3B and S1; Table S1). As expected, ADI-15878 contacts the side of the GP across two protomers, making contacts at HR1 and the IFL. The structures are nearly identical in ADI-15878 binding, and therefore we only discuss the higher-resolution EBOV/Mak here. All contacts within 4.0 Å between ADI-15878 and the GP were calculated to determine the critical epitope-paratope regions for binding (Tables S2, S3, and S4), including HC contacts within GP1 at the β1-β2 loop, near the N563 glycan in HR1, as well as several LC-mediated contacts at the tip of the IFL on an adjacent proto- mer. This exercise allowed us to determine an accurate footprint for ADI-15878 on GP (Figure 4A). Comparison of the epitope across sequences of filoviruses reveals the high sequence conservation of this region within the ebolaviruses, whereas this sequence conservation is much lower within MARV GP (Figure S2).
Figure 3. Cryo-EM Structures of ADI-15878 Fab Bound to EBOV/Mak GPΔmuc and BDBV GPΔmuc.
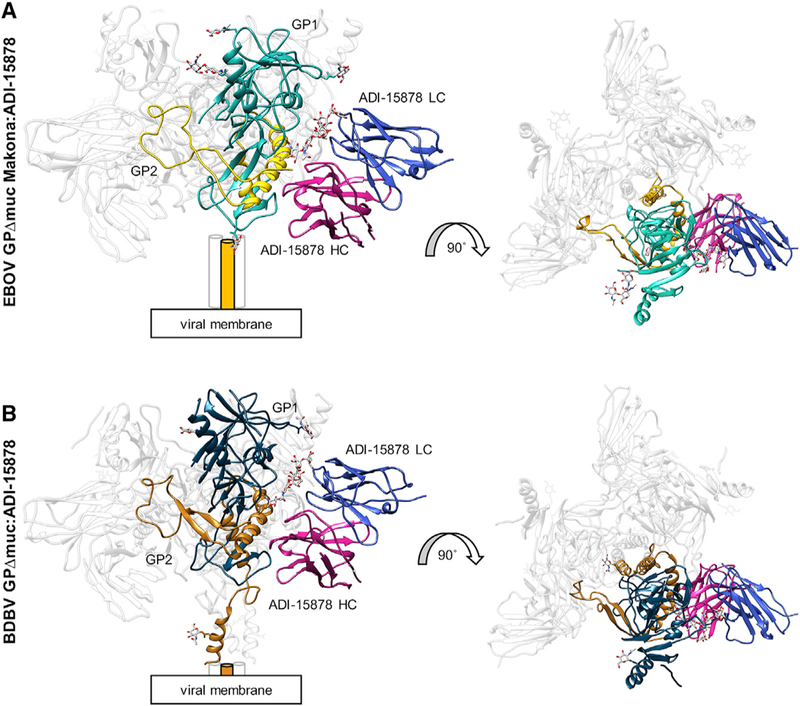
(A) Model of EBOV/Mak GPΔmuc bound to ADI- 15878 Fab. Shown are side (left) and top (right) views. We built four glycans in GP1 (cyan) and one in GP2 (yellow) and only the variable (Fv) domain of ADI-15878, with the light chain (LC) in blue and the heavy chain (HC) in magenta. Cylinders represent the HR2 domain, which were not built in this model. (B)Model of BDBV GPΔmuc bound to ADI-15878 Fab. We describe the core GP structure in Figure 2 and only built the ADI-15878 Fv domain, as described in (A).
See also Figures S1 and S2.
Figure 4. Details of the ADI-15878 Epitope in EBOV/Mak GPΔmuc.
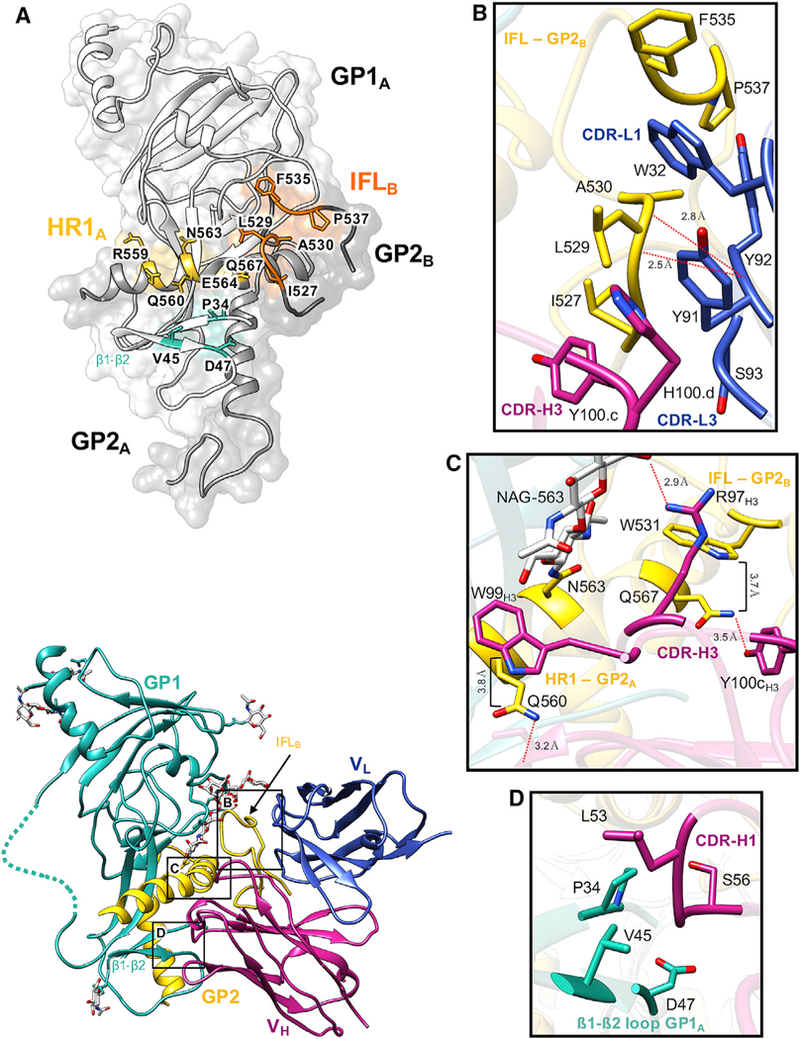
(A) The ADI-15878 epitope footprint, which spans two GP1/2 protomers, including the β1-β2 loop in GP1A (cyan), HR1 in GP1A (yellow), and the IFL in GP2B (orange). All residueswithin 4Å of ADI-15878 in our model are labeled and highlighted.
(B) Details of the ADI-15878 epitope at the IFL in GP2B. A loose hydrophobic network is formed between Y100cH3, H100dH3, Y91L3, Y92L3, W32L1, and IFL residues 527–530. The peptide backbone O from Y92L3 is also within hydrogen-bonding distance ofthe backbone N from L529 and A30 on the IFL. W32L1 inserts itself between F535 and L529 of the IFL.
(C) Details of the ADI-15878 epitope at HR1 in GP2A. W99H3 stacks against Q560HR1 at a distance of ~3.8 Å, similarto the interaction ofW531IFL with Q567. Y100cH3 and R97H3 are within hydrogen bonding distance of Q567 and NAG-563HR1, respectively. Also, NE2 of Q560 is within hydrogen bonding distance of the main chain O of S30H1.
(D) Residues L54H1 and S56H1 are within 4 Å of portions of the β1-β2 loop in GP1A and may mediate contacts. Dashed lines represent regions that we did not build in our model but were included in our construct.
See also Figures S2, S3, and S4.
The ADI-15878 HC makes the majority of critical contacts with GPs, including two CDRH3 contacts to L529 within the IFL and Q560 in HR1, with Y100cH3 and W99H3, respectively (Figures 4B and 4C). These residues on ADI-15878 were previously shown to be critical for ADI-15878 binding, although Y100cH3 was significant only for binding to SUDV (Wec et al., 2017). This may be due to a difference in sequence at L529, which is an isoleucine in SUDV, but not in any other ebolaviruses. Our structures show that ADI-15878 binding causes subtle changes in the conformation of the IFL (Figure 2C) that are accommodated by a network of hydrophobic interactions between the CDRH3, CDRL3, and a rotamer change at Y100cH3 (see below). It is possible that a disruption in this network at Y100cH3 may cause the more extended L529 to significantly clash with ADI-15878.
The main contact made on HR1 is to Q560, which is completely conserved throughout all of the ebolaviruses (Figures 4C and S2), and stacks against W99H3 similar to the stabilizing interaction that the IFL makes with HR1 further down at W531 and Q567, respectively (Figure 4C). Furthermore, NE2 of Q560 is within hydrogen-bonding distance of the main chain O of S30H1 (Figure 4C). Finally, we show that the tip of the CDRH1 appears to interact with the β1-β2 loop (Figure 4D). This interaction is not readily defined in the current lower-resolution negative stain EM model (EMD: 8700) (Wec et al., 2017).
The LC of ADI-15878, specifically the CDRL1 and CDRL3, makes contacts on the GP solely with the IFL (Figure 4B). These loops form a loose network of hydrophobic interactions within the IFL, CDRL1, CDRL3, and CDRH3, likely helping to stabilize the IFL between the GP and ADI-15878 (Figure 4B). A mutation at G528 (to glutamic acid) derived from a viral escape mutant within a recombinant vesicular stomatitis virus (rVSV) EBOV GP pseudovirion system was shown to eliminate ADI-15878 binding (Wec et al., 2017). A G528 escape mutation (to serine) that arose in ferret models with the related antibody ADI-15742 but not ADI-15878, G528S, also abolishes the binding of ADI-15878. Modeling the G528E/S mutations indicate that these residues would potentially clash with the CDRL3, providing a reason for the loss in binding by ADI-15878 to these viral escape mutants (Figure S3). Germline analysis shows a higher degree of maturation associated with the CDRL3 of both of these antibodies, but our structure indicates that residue differences between the CDRL3 of ADI-15742 (QQYNRS-P) and ADI-15878 (QQYYSS-P) may have contributed to the escape in animal models. While most contacts are driven by the HC, the contribution of the LC confirms previous observations that both chains contribute to binding (Wec et al., 2017).
Despite the significant pan-ebolavirus cross-reactivity of ADI-15878, this antibody fails to bind to the related filovirus genus of marburgviruses (Wec et al., 2017). This is likely due to a combination of a clash with the recently described MARV wing anchor at the base of the GP (King et al., 2018) (Figure S4A), as well as sequence divergence at the critical contact residue Q560, which is an arginine (R561) in MARV, a bulkier residue that likely clashes with the CDRH3(Figure S4B). It has been demonstrated that the MLDs hang over the sides of the MARV GP, in contrast to their conformation in ebolaviruses, which possibly further shields the ADI-15878 epitope in marburgviruses (Hashiguchi et al., 2015). Many LC contacts are well conserved even in MARV GP (Figures S4C and S4D) and suggests that a pan-filoviral antibody may be possible with a different HC/LC pairing and/or structure-based mAb engineering.
ADI-15878 Releases Buried Residues and Changes Conformation upon Binding to GP
We next compared our unliganded crystal structure of ADI- 15878 Fab (ADIU) to structures of BDBV GPΔmuc and EBOV/ Mak GPΔmuc bound to ADI-15878 Fab (ADIB). As noted above, the structures of ADIB in complex with EBOV/Mak and BDBV GP were nearly identical; therefore, we discuss EBOV/Mak only for the sake of clarity. Alignment of the Fab variable (Fv) domains of ADIU and ADIB revealed several key differences, indicating an induced-fit mechanism for binding. Binding of ADI-15878 to GP causes the Fv to become more open, loosening the association between the HC and LC (Figure 5A; Video S1). We measured the solvent-accessible surfaces (SASs) between the HC and LC for ADIU and ADIB using UCSF Chimera (Pettersen et al., 2004). The total SAS for the unliganded Fv is 702 Å2 and increases to 759 Å2 when ADI-15878 binds to the GP, indicating an increase in the total accessible surface area. To determine the degree of the shift in CDR loops, we aligned both structures on the Fv LCs and measured the distance between Cα residues at the tip of each HC loop, observing a 2.4- to 4.8-Å change in distance between ADIU and ADIB (Figure 5A). This change was most pronounced in CDRH3, which moved by ~4.8 Å. The CDRL3 also shifted slightly (~1.2 Å).
Figure 5. Comparison of Bound and Unbound ADI-15878 Paratopes.
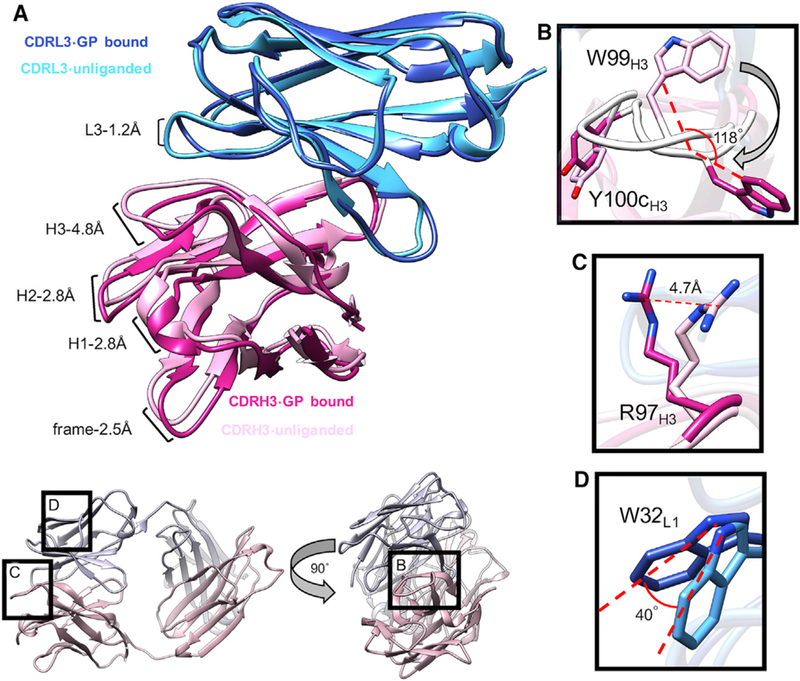
(A) Comparison of unliganded (light blue/pink) and GP-bound (dark blue/magenta) ADI-15878 Fv. Upon binding to GP, ADI-15878 opens up at the HC/LC interface, with the greatest shift occurring at CDRH3 (~4.8 Å shift). There is also a slight change in the position of CDRL3.
(B) A main-chain conformational change causes W99H3 to flip downward by ~120° upon binding to GP, causing an overall shift in the position of the CDRH3, as well as a change in the rotamer of Y100cH3, which subsequently mediates interaction with a portion of the IFL and the LC.
(C) The change in CDRH3 conformation upon GP binding shifts the position of R97H3 by 4.7 Å, allowing it to contact the glycan at N563. (D) A slight shift in the CDRL3 upon binding to GP changes the position of W32L1 by ~40°, allowing for hydrophobic interactions with the IFL.
See also Figures S4 and S5.
In the unliganded ADI-15878 structure, W99H3 is in a different orientation than when it is bound to GP, and the loop itself shifts in several places to accommodate this change (Figures 5B and 5C). In the unliganded state, W99H3 is flipped in toward the interior of the antibody and stacks against R97H3 (Figure 1B). Upon binding, W99H3 flips downward by ~120o (Figure 5B). The released R97H3 then shifts by ~4.7 Å, potentially interacting with a portion oftheconserved N563glycan (Figures 4C and 5C). The Y100cH3 position changes, making more intimate contacts with hydrophobic residues in the CDRL3 loop (Figure 5B). Overall, the root-mean-square deviation (RMSD) between the ADIu and ADIb CDRH3 (aligned on the HC) is 3.97 Å (residues 93–100). This type of large conformational change is highly unusual and is a unique attribute of ADI-15878. Another structural change that occurs within ADI-15878 upon binding to GP is a changein the W32L1 rotamer, which alters its side-chain angle by 40o (Figure 5D). This residue is important for interaction with the IFL (Figure 4B). By shifting the ADI-15878 HC toward the adjacent GP protomer upon binding, W32L1 releases its association with the HC and assumes hydrophobic packing within the IFL. Taken together, it appears that binding- induced conformational changes are important for releasing W99H3 to interact with HR1 and result in changes in the LC as well. It should be noted that W99H3 isparticipating in a crystal contact in the apo structure (Figure S5), and therefore some of the conformational differences between the bound and unbound structures may be explained by crystal packing artifacts. That being said, the extended conformation of W99H3 seen in the bound structure is likely not favored in solution without the stabilizing effects of GP, and it is likely that W99H3 is stabilized by R97H3 in the unliganded state found in solution, similar to what is seen in the crystal structure.
D95H3 was also previously shown to be critical for ADI-15878 binding and neutralization, but here we show that this residue does not make any contacts with GP. D95H3 points below the CDRH3, making contacts with portions ofthe CDRH1 (Figure 1A). Therefore, this residue is likely critical for maintaining the structural integrity of the CDRH3 and sequestering this loop within the interior of ADI-15878. W99H3 also makes minor contact with the base of the conserved glycan at N563. Despite this contact, this glycan is likely a steric barrier to binding because deglycosylation has been shown to significantly increase ADI-15878 binding to GP (Wec et al., 2017).
DISCUSSION
Here, we have described the molecular basis of viral neutralization by the potent human survivor-derived antibody ADI-15878, which has full pan-ebolavirus activity and has shown protection in three stringent animal models as a monotherapy (Bornholdt et al., 2016b; Wec et al., 2017). The basis of our analysis comes from a comparison of structures of the unliganded antibody and bound to GP derived from our newly described BDBV GP structure and the EBOV/Mak variant GP, also hitherto undescribed. In addition to providing details into the molecular nature of the interaction between ADI-15878 and GP, our atomic-level descriptions of two new viral GPs enable a better description of the immunogenic landscape of the ebolavirus genus. These data improve our ability to evaluate the antibody-based immune response to filoviral infection and enable engineering of ADI-15878 for improved pharmacological properties.
Our structures demonstrate that ADI-15878 uses a hydrophobic CDRH3 that changes conformation upon binding to GP. The existence of hydrophobic residues at the apex of CDRH3 loops has been seen for other antibodies, such as the HIV antibodies 4E10 and b12, which both use a key CDRH3 tryptophan residue in their paratopes (Rujas et al., 2015, 2017; Zhou et al., 2007). These examples indicate a conserved immunological solution for presenting hydrophobic residues on CDRH3 loops (Mian et al., 1991), although antibodies necessarily form highly unique structures to accommodate such a wide range of ligands (Regep et al., 2017), highlighting the importance of structural data to assist therapeutic development. In cases in which these antibodies suffer from solubility, for example, with the HIV antibody 10E8, it has been possible to engineer framework residues to counteract these issues (Kwon etal., 2016). For ADI-15878, however, hydrophobic residues are largely sequestered within the paratope before making contact, possibly enabling greater solubility in the unli- ganded state and generating higher specificity. Sequestering hydrophobic CDR loops in the unliganded state may offer a creative solution for engineering such antibodies for greater solubility while still retaining high affinity and specificity upon binding to the cognate antigen.
The IFL and HR1 are seemingly well exposed on the surface of the GP, similar to other enveloped viruses for which these features have been identified as sites of vulnerability for broadly neutralizing antibodies (Kallewaard et al., 2016; Kong et al., 2016. Nevertheless, antibodies that target the IFL are isolated at a low frequency. This is possibly due to the partial sequestration of the fusion machinery in the GP, decoy antigens unique to ebolaviruses such as soluble GP (sGP) (de La Vega et al., 2015; Pallesen et al., 2016), and the heavily glycosylated and immunogenic MLDs and glycan cap, which elicit the largest responses in patients (Bornholdt et al., 2016b; Flyak et al., 2016; Wec et al.,. The mature version of ADI-15878 is very close to its germline precursor, having undergone very little SHM (Wec et al., 2017). Due to the acute nature of filoviral infection, extensive SHM may not be possible, as opposed to viruses that occur seasonally such as influenza or chronically such as HIV, where a much greater degree of SHM is noted in the most broadly neutralizing antibodies (Gray et al., 2011; Klein et al., 2013). The lack of major SHM and low prevalence of cross-reactive antibodies supports the idea that their generation may be largely based on the proper LC pairing that enables the targeting of more difficult to access epitopes.
Cleaved versions of virus-like particles and GPs have been proposed as a means of eliciting more cross-reactive responses (Bornholdt et al., 2016a; Wec et al., 2017; Zhao et al., 2017). Previous binding and neutralization studies also demonstrated that cleaved versions of GPs, where the glycan cap and MLDs are removed, exposing the NPC1 binding patch, make viruses more susceptible to ADI-15878 neutralization and increase its binding affinity (Wec et al., 2017). This was also shown for the IFL-directed antibody CA45, suggesting that cleavage exposes the IFL differently from native GP (Zhao et al., 2017). The GP undergoes major remodeling throughout the entry process (Lee and Saphire, 2009; White and Schornberg, 2012), resulting in a much-reduced cleaved GP (GPCL) structure and the exposure of the NPC1 binding site (Bornholdt et al., 2016a). Removal of the bulk of GPs and associated glycans likely allows easier access to the ADI-15878 epitope and may also relax some association of the IFL with the core of the GP.
ADI-15878 demonstrates excellent cross-reactivity among ebolaviruses, but it may be difficult to readily engineer crossreactivity to the marburgviruses due to several critical structural and sequence differences (Figures S2 and S4). Our structures, however, provide a valuable tool for using structure-based design in silico to generate new starting models, such as those with increased affinity, that can be subsequently integrated into an in vitro evolution pipeline (Adolf-Bryfogle et al., 2018). Such efforts have shown promise for influenza mAbs, although it seems that increases in specificity result in diminished cross-reactivity (Wu et al., 2017; Wu and Wilson, 2017, 2018). Future work should be aimed at optimizing potent antibodies such as ADI-15878 for clinical development. Structural information is invaluable in navigating this process and provides a blueprint for engineering immunotherapeutics with optimal properties.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
| Antibodies | ||
| ADI-15878 | (Bornholdt et al., 2016b); Mapp Biopharmaceutical |
N/A |
| ADI-16061 | (Bornholdt et al., 2016b); Mapp Biopharmaceutical |
N/A |
| c13C6 | Mapp Biopharmaceutical | N/A |
| BDBV289 | (Flyak et al., 2016); J.E. Crowe | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| FreeStyle 293 expression medium | Thermo Fisher | Cat no. 12338018 |
| ExpiCHO expression medium | Thermo Fisher | Cat no. A2910001 |
| ExpiCHO feed | Thermo Fisher | Cat no. A2910002 |
| Opti-MEM | Thermo Fisher | Cat no. 31985070 |
| EBOV GPΔMuc-Makona variant | This study | N/A |
| BDBV GPΔMuc | This study | N/A |
| n-Dodecyl-beta-Maltoside | Anatrace | Part no. D310S |
| A8–35 amphipole | Anatrace | Part no. A835 |
| Deposited Data | ||
| ADI-15878 Fab complex with EBOV GPΔMuc-Makona variant cryo-EM map |
This paper | EMDB; EMD-8935 |
| ADI-15878 Fab complex with BDBV GPΔMuc variant cryo-EM map |
This paper | EMDB; EMD-8936 |
| ADI-15878 Fab complex with EBOV GPΔMuc-Makona variant model |
This paper | PDB; 6DZL |
| ADI-15878 Fab complex with BDBV GPΔMuc model |
This paper | PDB; 6DZM |
| Unliganded ADI-15878 Fab crystal structure | This paper | PDB; 6DZN |
| Experimental Models: Cell Lines | ||
| 293-FreeStyle | Thermo Fisher | R79007 |
| ExpiCHO | Thermo Fisher | A29127 |
| Oligonucleotides | ||
| pPPI4 Sequencing primer For: AGCGGCAGAAGAAGATGCAGGCAGC |
This study | N/A |
| BGH_Rev Sequencing primer: CCTCGACTGTGCCTTCTA |
Eton Bioscience | N/A |
| AbVec Sequencing primer For: AGTCTATAGGCCCACCCCCT |
This study | N/A |
| AbVec Sequencing primer Rev: AACCATTATAAGCTGCAATAAACAA |
This study | N/A |
| Recombinant DNA | ||
| pPPI4-EBOV GPΔMuc Makona variant-Ek-ddStrep | This study | N/A |
| pPPI4-BDBV GPΔMuc-Ek-ddStrep | This study | N/A |
| AbVec-ADI-15878 HC Fab | This study | N/A |
| AbVec-ADI-16061 HC Fab | This study | N/A |
| AbVec-BDBV289 HC Fab | This study | N/A |
| AbVec-ADI-15878 LC | This study | N/A |
| AbVec-ADI-16061 LC | This study | N/A |
| AbVec-BDBV289 LC | This study | N/A |
| Software and Algorithms | ||
| COOT | Emsley et al., 2010 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org |
| HKL2000 | Otwinowski and Minor, 1997 | http://www.hkl-xray.com/ |
| MotionCor2 | Zheng et al., 2017 | http://msg.ucsf.edu/em/software/motioncor2.html |
| GCTF | Zhang, 2016 | |
| DoG Picker | Voss et al., 2009 | http://emg.nysbc.org/redmine/projects/software/wiki/DoGpicker |
| Relion 2.0 | Scheres, 2012 | http://www2.mrc-lmb.cam.ac.uk/relion/index.php?title=Main_Page |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Rosetta | DiMaio et al., 2015 | https://www.rosettacommons.org/ |
| Swiss Modeler | Biasini et al., 2014 | https://swissmodel.expasy.org/ |
| Phaser | McCoy et al., 2007 | http://www.ccp4.ac.uk/html/phaser.html |
| BUSTER | Bricogne et al., 2017 | https://www.globalphasing.com/buster/ |
| Refmac5 | Murshudov et al., 2011 | http://www.ccp4.ac.uk/html/refmac5.html |
| EMRinger | Barad et al., 2015 | http://emringer.com |
| Molprobity | Chen et al., 2010 | http://molprobity.biochem.duke.edu |
| PDBcare | Lütteke and von der Lieth, 2004 | www.glycosciences.de/tools/pdb-care/ |
| Privateer | Agirre et al., 2015 | www.ccp4.ac.uk/html/privateer.html |
| Other | ||
| Titan Krios 300kV electron microscope | Thermo Fisher | https://www.fei.com/ |
| Talos Arctica 200kV electron microscope | Thermo Fisher | https://www.fei.com/ |
| K2 Summit camera | Gatan | http://www.gatan.com |
| Vitrobot | Thermo Fisher | https://www.fei.com/ |
| Gatan Solarus 950 Plasma system | Gatan | http://www.gatan.com |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Andrew Ward (andrew@scripps.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
Suspension-adapted HEK293F female human embryonic kidney fibroblast cells were obtained from Thermo Fisher and cultured in FreeStyle 293F serum-free expression media (Thermo Fisher). Cell lines were maintained in a humidified 37°C incubator supplied with 8% CO2. Cell lines were not authenticated following purchase.
METHOD DETAILS
Construct design, expression and protein purification
EBOV/Mak GPΔmuc, (including residues 32–311 and 461–640, with residues 312–460 deleted to remove the mucin-like domain) was synthesized (GenBank: KR534526.1) and subcloned into the expression vector pPPI4. An Igҡ secretion signal was included on the N-terminal end of GP. On the C terminus, an enterokinase site was engineered (DDDDK) followed by a small linker (AG) and tandem strep-tags (WSHPQFEK) separated by a linker (GGGSGGGSGGGS). BDBV GPΔmuc (including residues 1–312 and 471–640, with residues 313–470 deleted to remove the mucin-like domain) was synthesized (GenBank: ALT19772.1) and subcloned into pPPI4, using the naturally occurring signal peptide on the N terminus. The C terminus was engineered as described for EBOV/Mak. Both GPs were expressed and purified from HEK293F (FreeStyle) by transient transfection. 1L of HEK293F cells at a density of 0.8–2 × 106 cells/mL was transfected using 750 μg of DNA and 2.25 mg of polyethylenimine ‘Max’ (linear, MW 25,000, Polyscience, Inc.) mixed with 50 mL of Opti-MEM (Thermo Fisher). The solution was sterile filtered using a 50 mL 0.22 μm Steriflip sterile disposable vacuum filter (Millipore) before being added to cells. After 5 days of expression in a shaking incubator at 37°C with 80% humidity and supplemented with 8% CO2, cells were harvested using centrifugation (8,000 xg for 1hr at 4°C) and filtered with a 0.45 μm filter (Millipore) to remove cellular debris. Next, BioLock biotin blocking solution (IBA Biosciences) was added according to the manufacturer’s instructions and cells were loaded over 5 mL of Strep-Tactin Superflow Plus beads (QIAGEN) that had been incubated in 100 mM Tris-HCl, pH. 8.0, 150 mM NaCl and 1 mM EDTA (1X Strep Buffer). Beads were washed with 10 mL of 1X Strep Buffer and GP was eluted using 2.5 mM d-desthiobiotin (Sigma) in 1X Strep Buffer. GP was further purified by size exclusion chromatography (SEC) using an S200 increase (S200I) column (GE) equilibrated in 150 mM NaCL, 20 mM Tris, pH 7.4 (1X TBS) to separate trimers and monomers.
ADI-15878 IgG used for cryo-EM studies was kindly provided by Mapp Biopharmaceutical and Fab was generated by optimized papain digestion. Briefly, IgG was buffer exchanged by dilution to a concentration of 1 mg/mL into 100 mM Tris and 2mM EDTA with 10 mM L-cystein and 4% (w/v) activated papain (Sigma) and allowed to incubate at 37°C for 6 hours. Digestion was stopped with the addition of 50 mM iodacetamide (Sigma) before digests were placed over a5-mL Hi-Trap Protein A column (GE) and the flow-through was collected. Fab was further purified by SEC using an S200i column (GE) in 1XTBS, pH 7.4 For crystallography, ADI-15878 variable regions from published sequences (GenBank KU602363–64) were synthesized and cloned into the expression vector AbVec, containing either the human IgG HC constant region or the human kappa LC constant region. Fab was produced by the insertion of a double stop codon after residue 226 in the HC hinge-region. ADI-15878 Fab was then expressed in ExpiCHO cells (GIBCO/ ThermoFisher Scientific) as per the manufacturer’s “max titer” protocol. Cell supernatant was then passed over a ҡ-Select 5mL column (GE Healthcare). Fab was eluted using 0.1 M glycine, pH 3.0 and subsequently buffer-exchanged into 20 mM sodium acetate (NaOAc), pH 5.6. Fab was then loaded onto a MonoS (GE Healthcare) column that had been equilibrated in 20 mM NaOAc, pH 5.6 and eluted with a gradient of 1M KCl. The appropriate fractions were pooled and further purified by SEC using an S200I column that had been equilibrated in 1X TBS.
Preparation and crystallization of ADI-15878 Fab
ADI-15878 Fab was screened for crystallization with the Joint Center for Structural Genomics (JCSG) Rigaku CrystalMation system with the JCSG Core Suites I-IV. Protein at 4.4 mg/ml was mixed 1:1 with precipitants and crystallized using the sitting drop vapor diffusion method at room temperature and 4°C. After 4 days, crystals were obtained with a precipitant of 0.1 M citric acid pH 5.0, 1 M LiCl and 20% (w/v) polyethylene glycol 6000 grown at 4°C and grew to a maximum size at day 8. Several crystals were harvested with mother liquor and soaked in 30% ethylene glycol. Data were collected at the Stanford Synchrotron Radiation Light Source beamline 9–2 and indexed, integrated and scaled using HKL-2000 (Otwinowski and Minor, 1997) to 2.1 Å (Table S1). Crystals belonged to the space group P21 with a single Fab in the asymmetric unit.
X-ray structure determination
The X-ray crystal structure of ADI-15878 Fab was determined using molecular replacement with a homology model generated using Swiss Modeler (Biasini et al., 2014) and Phaser (McCoy et al., 2007). The structure was refined using BUSTER (Bricogne et al., 2017) and Refmac5 (Murshudov et al., 2011).
Cryo EM sample preparation
EBOV/Mak GPΔmuc was incubated overnight with a 5-fold molar excess of ADI-15878 Fab and a 2-fold molar excess of c13C6 (kindly provided by Mapp Biopharmaceutical). The IgG was included here to add bulk to the complex and to increase the angular sampling. Complexes were then purified by SEC using an S200I column equilibrated in 1X TBS, pH 7.4. Fractions containing the desired complex as determined by SDS-PAGE were concentrated to 1mg/mL using a 100-kDa concentrator (Amicon Ultra, Millipore) and mixed with 0.04% (w/v) A8–35 amphipole immediately prior to freezing. Vitrification was performed with a Vitrobot (FEI) equilibrated to 4°C and 100% humidity. 3 μL of sample was applied to a CF-1.2/1.3–4C grid (Electron Microscopy Sciences, Protochips, Inc.) that had been plasma cleaned for 5 s using a mixture of Ar/O2 (Gatan Solarus 950 Plasma system), followed by a 3.5 s blot on both sides of the grid using filter paper (Whattman No. 1).
The BDBV GPΔmuc complex was prepared in a similar manner. The complex was prepared and incubated overnight with BDBV289 Fab (kindly provided by James Crowe, Vanderbilt University), ADI-15878 Fab and ADI-16061 Fab. Both Fabs were produced from IgG as described above. The Fabs BDBV289 and ADI-16061 were added to increase sample bulk and angular distribution. After SEC on an S200I column, the complex was concentrated to 3.5 mg/mL and a final concentration of 0.3 mM n-Dodecyl-beta-Maltoside (DDM) detergent was added to a sample immediately prior to freezing. This complex was vitrified in the same manner as the EBOV/Mak complex.
Cryo-EM data collection and data processing
Cryo EM data were collected as listed in Table S2. Micrograph movie frames were aligned and dose-weighted using MotionCorr2 (Zheng et al., 2017). Whole micrograph CTF estimation was then completed using GCTF (Zhang, 2016). Particles were initially identified from aligned micrographs using DoG Picker (Voss et al., 2009). Next, these particles underwent referenc-free, 2D classification with candidate images binned by a factor of four in Relion 2.0 (Scheres, 2012). Particles that corresponded to complexes were then further classified using Relion 3D classification, to generate a final stack of homogeneous particles. Particles were re-extracted without binning and refined against a 30Å low-passed filtered 3D class without symmetry. A second round of 3D classification with a tight mask was performed to select the most stable class of particles for the final refinement round using C3 symmetry and a tight mask around just the GP-ADI-15878 portion of the structure. This mask excluded density from c13C6 in the EBOV/Mak structure, as well as the density from BDBV289 and ADI-16061 in the BDBV structure. Resolutions were calculated using soft-edged masks generated in Relion and reported using the FCS 0.143 gold-standard criterion.
Cryo-EM modeling building and refinement
For building of the EBOV/Mak GPΔmuc-ADI-15878 model, a homology model of EBOV GPΔmuc was generated using Swiss Model (Biasini et al., 2014) from a high resolution crystal structure of EBOV/May GPΔmuc (PDB 5JQ3) as a template. For building BDBV GPΔmuc-ADI-15878, a homology model was similarly generated using EBOV/May GPΔmuc as a template. We used the crystal structure of ADI-15878 as a starting model for all ADI-15878 Fabs bound to GP. Models were fit into their respective cryo EM density maps using UCSF Chimera (Pettersen et al., 2004) and initially refined using real-space refinement in Phenix (Adams etal., 2010) with NCS constraints. This refined model was then used as a template for fragment-based refinement in Rosetta (DiMaio et al., 2015) and the top five scoring models were chosen for further evaluation. The model that best fit the density, especially at the antibody interface, was then used fora second round of real-space refinement in Phenix. The model was then corrected manually in Coot (Emsley et al., 2010) and refined to maximize fit. Glycans were built by placing an idealized Man9 model into glycan densities using UCSF Chimera and manually adjusting torsion angles to achieve a good agreement between map and model. Sugar moieties without strong corresponding density were deleted. The final model was further refined using Phenix real-space refinement. Final structures were evaluated by EMRinger (Barad etal., 2015) and Molprobity (Chen etal., 2010). Glycans were validated using Privateer (Agirre etal., 2015) and PDBcare (Lutteke and von der Lieth, 2004). All figures were generated in UCSF Chimera (Pettersen et al., 2004).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical models inherent to Relion were employed in image analysis to derive 2D classes and 3D models.
Supplementary Material
Highlights.
Structures of BDBV GP and EBOV Makona GP reveal similarities to other filoviral GPs
Pan-ebolavirus antibody ADI-15878 targets conserved residues on HR1 and the IFL
Comparing apo- and bound-ADI-15878 suggests an induced- fit mechanism for binding to GP
ACKNOWLEDGMENTS
This workwas supported by NIH grant U19 AI109762. We would like to thank the Joint CenterforStructural Genomics atThe Scripps Research Instituteand Henry Tien for assistance with setting up crystal trays. We would also like to thank Zachary Bernsden for assistance with processing the cryo-EM data.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five figures, fourtables, and one video and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.009.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
The accession numbers for the structures reported in this paper are PDB: 6DZL (EBOVGPΔmuc-Mak:ADI-15878 Fab), PDB: 6DZM (BDBVGGPΔmuc:ADI-15878 Fab) and PDB: 6DZN (unliganded ADI-15878 Fab). The accession numbers for the following cryo-EM maps reported in this paper are EMD: 8935 (EBOVGPΔmuc-Mak:ADI-15878 Fab) and EMD: 8936 (BDBVGPΔmuc:ADI-15878 Fab) (see Key Resources Table for details).
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adolf-Bryfogle J, Kalyuzhniy O, Kubitz M, Weitzner BD, Hu X, Adachi Y, Schief WR, and Dunbrack RL Jr. (2018). RosettaAntibodyDesign (RAbD): a general framework for computational antibody design. PLoS Com- put. Biol. 14, e1006112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agirre J, Iglesias-Fernández J, Rovira C, Davies GJ, Wilson KS, and Cowtan KD (2015). Privateer: software for the conformational validation of carbohydrate structures. Nat. Struct. Mol. Biol. 22, 833–834. [DOI] [PubMed] [Google Scholar]
- Albariño CG, Shoemaker T, Khristova ML, Wamala JF, Muyembe JJ, Balinandi S, Tumusiime A, Campbell S, Cannon D, Gibbons A, et al. (2013). Genomic analysis of filoviruses associated with fourviral hemorrhagic feveroutbreaks in Uganda and the Democratic Republic ofthe Congo in 2012. Virology 442, 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S, Liu T, Li S, Wang Y, Abelson D, Fusco M, Woods VL Jr., and Saphire EO (2011). Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membranefusion. PLoS Negl. Trop. Dis. 5, e1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale S, Dias JM, Fusco ML, Hashiguchi T, Wong AC, Liu T, Keuhne AI, Li S, Woods VL Jr., Chandran K, et al. (2012). Structural basis for differential neutralization of ebolaviruses. Viruses 4, 447–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barad BA, Echols N, Wang RY, Cheng Y, DiMaio F, Adams PD, and Fraser JS (2015). EMRinger: side chain-directed model and map validation for 3D cryo-electron microscopy. Nat. Methods 12, 943–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biasini M, Bienert S, Waterhouse A, Arnold K, Studer G, Schmidt T, Kiefer F, Gallo Cassarino T, Bertoni M, Bordoli L, and Schwede T (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, W252–W258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA, Ndungo E, Fusco ML, Bale S, Flyak AI, Crowe JE Jr., Chandran K, and Saphire EO (2016a). Host-primed ebolavirus GP exposes a hydrophobic NPC1 receptor-binding pocket, revealing a target for broadly neutralizing antibodies. MBio 7, e02154–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornholdt ZA,Turner HL, Murin CD, Li W, Sok D, Souders CA, Piper AE, Goff A, Shamblin JD, Wollen SE, et al. (2016b). Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 351, 1078–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bricogne G, Blanc E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, and Womack TO (2017). BUSTER computer program (Cambridge, UK: Global Phasing; ). [Google Scholar]
- Burk R, Bollinger L, Johnson JC, Wada J, Radoshitzky SR, Palacios G, Bavari S, Jahrling PB, and Kuhn JH (2016). Neglected filoviruses. FEMS Microbiol. Rev. 40, 494–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (2014). Marburg hemorrhagic fever (Marburg HF). Outbreakschronology: Marburg hemorrhagicfever.https://www.cdc.gov/vhf/marburg/outbreaks/chronology.html. [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de La Vega MA, Wong G, Kobinger GP, and Qiu X (2015). The multiple roles of sGP in Ebola pathogenesis. Viral Immunol. 28, 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias JM, Kuehne AI, Abelson DM, Bale S, Wong AC, Halfmann P, Muhammad MA, Fusco ML, Zak SE, Kang E, et al. (2011). A shared structural solution for neutralizing ebolaviruses. Nat. Struct. Mol. Biol. 18, 1424–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio F, Song Y, Li X, Brunner MJ, Xu C, Conticello V, Egelman E, Marlovits T, Cheng Y, and Baker D (2015). Atomic-accuracy models from 4.5-Å cryo-electron microscopydatawithdensity-guided iterative local refinement. Nat. Methods 12, 361–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010). Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Ilinykh PA, Murin CD, Garron T, Shen X, Fusco ML, Hashi- guchi T, Bornholdt ZA, Slaughter JC, Sapparapu G, et al. (2015). Mechanism of human antibody-mediated neutralization of Marburg virus. Cell 160, 893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Shen X, Murin CD, Turner HL, David JA, Fusco ML, Lamp- ley R, Kose N, Ilinykh PA, Kuzmina N, et al. (2016). Cross-reactive and potent neutralizing antibody responses in human survivors of natural ebolavi- rus infection. Cell 164, 392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak AI, Kuzmina N, Murin CD, Bryan C, Davidson E, Gilchuk P, Gulka CP, Ilinykh PA, Shen X, Huang K, et al. (2018). Broadly neutralizing antibodies from human survivors target a conserved site in the Ebola virus glycoprotein HR2-MPER region. Nat Microbiol. 6, 670–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuyama W, Marzi A, Nanbo A, Haddock E, Maruyama J, Miyamoto H, Igarashi M, Yoshida R, Noyori O, Feldmann H, and Takada A (2016). Discovery of an antibody for pan-ebolavirus therapy. Sci. Rep. 6, 20514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco ML, Hashiguchi T, Cassan R, Biggins JE, Murin CD, Warfield KL, Li S, Holtsberg FW, Shulenin S, Vu H, et al. (2015). Protective mAbs and cross-reactive mAbs raised by immunization with engineered Marburg virus GPs. PLoS Pathog. 11, e1005016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirss K, Kühl A, Karsten C, Glowacka I, Bertram S, Kaup F, Hofmann H, and Pöhlmann S (2012). Cathepsins B and L activate Ebola but not Marburg virus glycoproteins for efficient entry into cell lines and macrophages independent of TMPRSS2 expression. Virology 424, 3–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray ES, Madiga MC, Hermanus T, Moore PL, Wibmer CK, Tumba NL, Werner L, Mlisana K, Sibeko S, Williamson C, et al. ; CAPRISA002 StudyTeam (2011).Theneutralization breadth ofHIV-1 developsincrementally over four years and is associated with CD4+ T cell decline and high viral load during acute infection. J. Virol. 85, 4828–4840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashiguchi T, Fusco ML, Bornholdt ZA, Lee JE, Flyak AI, Matsuoka R, Kohda D, Yanagi Y, Hammel M, Crowe JE Jr., and Saphire EO (2015). Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 160, 904–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell KA, Brannan JM, Bryan C, McNeal A, Davidson E, Turner HL, Vu H, Shulenin S, He S, Kuehne A, et al. (2017). Cooperativity enables non-neutralizing antibodies to neutralize ebolavirus. Cell Rep. 19, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt CL, Lennemann NJ, and Maury W (2012). Filovirusentry: a novelty in the viral fusion world. Viruses 4, 258–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallewaard NL, Corti D, Collins PJ, Neu U, McAuliffe JM, Benjamin E, Wachter-Rosati L, Palmer-Hill FJ, Yuan AQ, Walker PA, et al. (2016). Structure and function analysis of an antibody recognizing all influenza A subtypes. Cell 166, 596–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King LB, Fusco ML, Flyak AI, Ilinykh PA, Huang K, Gunn B, Kirch- doerfer RN, Hastie KM, Sangha AK, Meiler J, et al. (2018). The Marburg- virus-neutralizing human monoclonal antibody MR191 targets a conserved siteto block virus receptor binding. Cell Host Microbe 23, 101–109.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein F, Diskin R, Scheid JF, Gaebler C, Mouquet H, Georgiev IS, Pan- cera M, Zhou T, Incesu RB, Fu BZ, et al. (2013). Somatic mutations ofthe immunoglobulin framework are generally required for broad and potent HIV-1 neutralization. Cell 153, 126–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong R, Xu K, Zhou T, Acharya P, Lemmin T, Liu K, Ozorowski G, Soto C, Taft JD, Bailer RT, et al. (2016). Fusion peptide of HIV-1 as a site of vulnerability to neutralizing antibody. Science 352, 828–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YD, Georgiev IS, Ofek G, Zhang B, Asokan M, Bailer RT, Bao A, Caruso W, Chen X, Choe M, et al. (2016). Optimization ofthe solubility of HIV-1-neutralizing antibody 10E8 through somatic variation and structure- based design. J. Virol. 90, 5899–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, and Saphire EO (2009). Ebolavirus glycoprotein structure and mechanism of entry. Future Virol. 4, 621–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JE, Fusco ML, Hessell AJ, Oswald WB, Burton DR, and Saphire EO (2008). Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 454, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lütteke T, and von der Lieth CW (2004). pdb-care (PDB carbohydrate residue check): a program to support annotation of complex carbohydrate structures in PDB files. BMC Bioinformatics 5, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mian IS, Bradwell AR, and Olson AJ (1991). Structure, function and properties of antibody binding sites. J. Mol. Biol. 217, 133–151. [DOI] [PubMed] [Google Scholar]
- Miller EH, Obernosterer G, Raaben M, Herbert AS, Deffieu MS, Krishnan A, Ndungo E, Sandesara RG, Carette JE, Kuehne AI, et al. (2012). Ebola virus entry requires the host-programmed recognitionofan intracellular receptor. EMBO J. 31, 1947–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murin CD, Fusco ML, Bornholdt ZA, Qiu X, Olinger GG, Zeitlin L, Kobinger GP, Ward AB, and Saphire EO (2014). Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 111, 17182–17187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, and Vagin AA (2011). REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North B, Lehmann A, and Dunbrack RL Jr. (2011). A new clustering of antibody CDR loop conformations. J. Mol. Biol. 406, 228–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, and Minor W (1997). Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Pallesen J, Murin CD, de Val N, Cottrell CA, Hastie KM, Turner HL, Fusco ML, Flyak AI, Zeitlin L, Crowe JE Jr., et al. (2016). Structures of Ebolavirus GP and sGP in complexwith therapeutic antibodies. Nat. Microbiol. 1, 16128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera-a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- PREVAIL II Writing Group, Multi-National PREVAILII StudyTeam, Davey RT Jr., Dodd L, Proschan MA, Neaton J, Neuhaus Nordwall J, Koopmeiners JS, Beigel J, Tierney J, et al. (2016). A Randomized, Controlled Trial of ZMapp for Ebola Virus Infection. N. Engl. J. Med. 375, 1448–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Wong G, Audet J, Bello A, Fernando L, Alimonti JB, Fausther- Bovendo H, Wei H, Aviles J, Hiatt E, et al. (2014). Reversion ofadvanced Ebola virus disease in nonhuman primates with ZMapp. Nature 514, 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regep C, Georges G, Shi J, Popovic B, and Deane CM (2017). The H3 loop of antibodies shows unique structural characteristics. Proteins 85,1311 — 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujas E, Gulzar N, Morante K, Tsumoto K, Scott JK, Nieva JL, and Caaveiro JM (2015). Structural and thermodynamic basis ofepitope binding by neutralizing and nonneutralizing forms of the anti-HIV-1 antibody 4E10. J. Virol. 89, 11975–11989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rujas E, Insausti S, Garcia-Porras M, Sánchez-Eugenia R, Tsumoto K, Nieva JL, and Caaveiro JM (2017). Functional contacts between MPER and the anti-HIV-1 broadly neutralizing antibody 4E10 extend into the core of the membrane. J. Mol. Biol. 429, 1213–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres SH (2012). RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker CJ, Schornberg KL, Delos SE, Scully C, Pajouhesh H, Olinger GG, Johansen LM, andWhite JM (2013). Multiplecationicamphi- philes induce a Niemann-Pick C phenotype and inhibit Ebola virus entry and infection. PLoS One 8, e56265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence JS, Krause TB, Mittler E, Jangra RK, and Chandran K (2016). Direct visualization of ebola virus fusion triggering in the endocytic pathway. MBio 7, e01857–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiller T, Tsuiji M, Yurasov S, Velinzon K, Nussenzweig MC, and Warde- mann H (2007). Autoreactivity in human IgG+ memory B cells. Immunity 26, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Towner JS, Sealy TK, Khristova ML, Albarirño CG, Conlan S, Reeder SA, Quan PL, Lipkin WI, Downing R, Tappero JW, et al. (2008). Newly discovered ebolavirusassociated with hemorrhagicfeveroutbreak in Uganda. PLoS Pathog. 4,e1000212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss NR, Yoshioka CK, Radermacher M, Potter CS, and Carragher B (2009). DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Shi Y, Song J, Qi J, Lu G, Yan J, and Gao GF (2016). Ebola viral glycoprotein bound to itsendosomal receptor Niemann-PickC1. Cell 164, 258–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wec AZ, Herbert AS, Murin CD, Nyakatura EK, Abelson DM, Fels JM, He S, James RM, de La Vega MA, Zhu W, et al. (2017). Antibodies from a human survivor define sites ofvulnerabilityfor broad protection against ebolaviruses. Cell 169, 878–890.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White JM, and Schornberg KL (2012). A new player in the puzzle of filovirus entry. Nat. Rev. Microbiol. 10, 317–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu NC, and Wilson IA (2017). A perspective on the structural and functional constraints for immune evasion: insights from influenza virus. J. Mol. Biol. 429, 2694–2709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu NC, and Wilson IA (2018). Structural insights into the design of novel anti-influenza therapies. Nat. Struct. Mol. Biol. 25, 115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu NC, Grande G, Turner HL, Ward AB, Xie J, Lerner RA, and Wilson IA (2017). In vitro evolution of an influenza broadly neutralizing antibody is modulated by hemagglutinin receptor specificity. Nat. Commun. 8, 15371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamayoshi S, and Kawaoka Y (2017). Ebolavirus’s foibles. Cell 169, 773–775. [DOI] [PubMed] [Google Scholar]
- Zhang K (2016). Gctf: real-time CTF determination and correction. J. Struct. Biol. 193, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Ren J, Harlos K, Jones DM, Zeltina A, Bowden TA, Padilla- Parra S, Fry EE, and Stuart DI (2016). Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 535, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Howell KA, He S, Brannan JM, Wec AZ, Davidson E, Turner HL, Chiang CI, Lei L, Fels JM, et al. (2017). Immunization-elicited broadly protective antibody revealsebolavirusfusion loop asasite ofvulnerability. Cell 169, 891–904.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng SQ, Palovcak E, Armache JP, Verba KA, Cheng Y, and Agard DA (2017). MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat. Methods 14, 331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou T, Xu L, Dey B, Hessell AJ, Van Ryk D, Xiang SH, Yang X, Zhang MY, Zwick MB, Arthos J, et al. (2007). Structural definition of a conserved neutralization epitope on HIV-1 gp120. Nature 445, 732–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.