

Abstract
Enzymatic core components from trans‐acyltransferase polyketide synthases (trans‐AT PKSs) catalyze exceptionally diverse biosynthetic transformations to generate structurally complex bioactive compounds. Here we focus on a group of oxygenases identified in various trans‐AT PKS pathways, including those for pederin, oocydins, and toblerols. Using the oocydin pathway homologue (OocK) from Serratia plymuthica 4Rx13 and N‐acetylcysteamine (SNAC) thioesters as test surrogates for acyl carrier protein (ACP)‐tethered intermediates, we show that the enzyme inserts oxygen into β‐ketoacyl moieties to yield malonyl ester SNAC products. Based on these data and the identification of a non‐hydrolyzed oocydin congener with retained ester moiety, we propose a unified biosynthetic pathway of oocydins, haterumalides, and biselides. By providing access to internal ester, carboxylate pseudostarter, and terminal hydroxyl functions, oxygen insertion into polyketide backbones greatly expands the biosynthetic scope of PKSs.
Keywords: Baeyer–Villiger oxidation, biosynthesis, natural products, polyketides, polyketide synthases
Complex polyketides are natural products that exhibit remarkable structural diversity and play important roles in microbial interactions and as a source of drug candidates.1 In bacteria, they are usually biosynthesized by multimodular polyketide synthases (PKSs), giant proteins that assemble polyketides by stepwise incorporation and modification of short acyl building blocks.1a In textbook PKSs, each elongation step is catalyzed by an enzymatic module that typically generates either a β‐keto, β‐hydroxy, α,β‐unsaturated, or completely reduced moiety, depending on the catalytic domains present in the module. The resulting product is a contiguous carbon chain with a succession of moieties that is collinear with the order of modules. This model2 generally applies well to cis‐acyltransferase (cis‐AT) PKSs, that is, enzymes, in which each module contains an AT domain responsible for building block selection.1a Besides these well‐studied systems, trans‐AT PKSs are a second large family of modular PKSs that diverge from the canonical biosynthetic model.3 Trans‐AT PKS systems can feature a broad array of module architectures, including split modules distributed on two proteins, unusual domains within modules, and various trans‐acting accessory components that operate during elongation, such as the eponymous ATs. This enzymatic diversity enables trans‐AT PKSs to introduce a wide range of unusual features into polyketide structures.4
One of the first trans‐AT PKSs with an assigned natural product is the pederin PKS, identified in an as‐yet uncultivated bacterial symbiont of Paederus fuscipes beetles.1b, 5 Pederin (1, Figure 1), a highly toxic defensive agent,6 features among other structural peculiarities an O‐methyl instead of the conventional ester or carboxylic acid polyketide terminus. Closely related but elongated congeners, such as onnamide A (2),7 are known from another symbiotic producer, present in the marine sponge Theonella swinhoei.8 In spite of the distinct polyketide termini, the PKSs for 1 and 2 exhibit strikingly similar module architectures, suggesting that a cleavage event accounts for the shortened, noncanonical, methoxy‐group‐bearing terminus of pederin. Intriguingly, the pederin PKS cluster contains as a unique feature the gene pedG (positioned between pedF and pedH), which encodes a predicted flavin‐dependent oxygenase.1b, 9 The location of pedG might be the result of an integration event that interrupted the PKS assembly line, leading PedG to release a stalled intermediate that cannot be elongated further. Degradation of the downstream gene pedH would support this mechanism, but this was not evident.1b In another scenario, the location of pedG is coincidental, and cleavage occurs at the post‐PKS stage after biosynthesis of an onnamide‐like, fully elongated polyketide.3b A third option is that collinearity at the genetic and metabolic level reflects the sequence of biosynthetic steps (Figure 1 A), suggesting that a masked terminus is introduced during polyketide elongation by PedG acting in trans.
Figure 1.
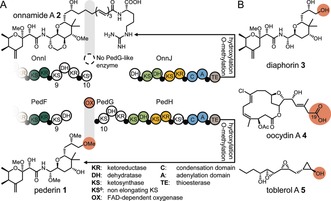
Oxygen insertion during polyketide biosynthesis. Moieties putatively derived from oxygen insertion are highlighted in orange. A) Polyketide structures and corresponding PKS architecture for onnamide A and pederin. Small filled circles denote ACP domains. Numbers below KS domains refer to their positions in the assembly lines. B) The trans‐AT PKS derived compounds diaphorin (3), oocydin A (4), and toblerol A (5). Shared colors indicate orthologous modules in the compared PKS architectures.
Interestingly, PedG‐like proteins appear in other PKS systems, but the polyketides do not necessarily feature hydroxy termini like in 1. To investigate the prevalence of oxygenation in these systems and their corresponding products, we phylogenetically analyzed sequences of PedG‐like proteins and related homologues of flavin‐dependent monooxygenases (Figure 2). In this analysis, PedG forms a distinct clade with 29 oxygenases, including those from trans‐AT PKSs for the pederin‐like compound diaphorin (3) from a psyllid symbiont,10 for oocydin A (4) from S. plymuthica 4Rx13,11 and for the methylobacterial toblerol A (5).12 Another member of the clade is the myxobacterial AmbI from the cis‐AT PKS ambruticin (6) pathway, an enzyme suggested to be involved in a decarboxylative hydroxylation.13 In addition, the clade contains at least five homologues from as‐yet uncharacterized and unrelated trans‐AT PKS pathways from diverse bacteria (Figure 2).
Figure 2.
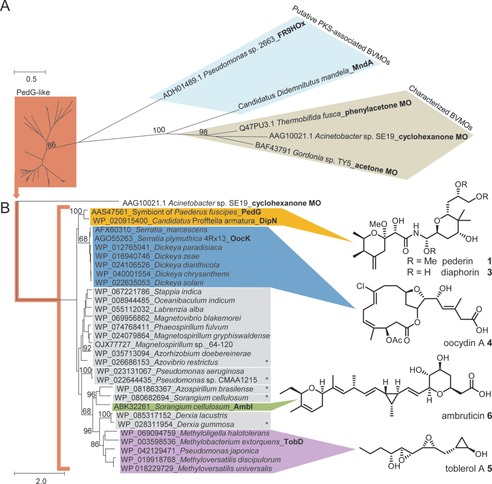
Phylograms of various flavin adenine dinucleotide (FAD)‐dependent monooxygenases (MO). A) PedG‐like enzymes, characterized Baeyer–Villiger monooxygenases (BVMOs), and putative PKS‐associated BVMOs. B) Expansion of the branch containing PedG‐like enzymes from various bacteria. Tip labels: accession number, organism, and protein name (if it exists). Outgroup: cyclohexanone monooxygenase from Acinetobacter sp. SE19. Characterized polyketides are indicated. Asterisks refer to PedG‐like enzymes from uncharacterized PKS clusters.
If oxygenation occurs during chain extension for pederin, a likely candidate for accepting the resulting intermediate would be the downstream ped KS10. Since trans‐AT PKS ketosynthases (KSs) tend to clade according to the polyketide moiety close to the thioester in the accepted intermediate,3 ped KS10 would likely be phylogenetically distinct. To test this scenario, a phylogenetic analysis was conducted for ped KS10, using an alignment containing 580 KSs from all known trans‐AT PKSs (status Jan. 2017, Figure S1 in the Supporting Information). This analysis grouped ped KS10 exclusively with functionally unassigned KSs from various pathways, including 3 and 4. Interestingly, 4 lacks a hydroxylated terminus, but rather harbors carboxylated moieties at both ends. We therefore hypothesized that these related KS domains accept an ester function as a common progenitor moiety, introduced by oxygen insertion into the polyketide chain. One carboxylate terminus of oocydin A (4) would therefore result from hydrolytic cleavage of an extended precursor.
We selected the oocydin pathway in S. plymuthica 4Rx13 as a culturable representative containing both a PedG‐like protein (OocK) and the downstream ooc KS3 that clades with the ped KS10 (Figure S1). For knock‐out studies, we introduced a kanamycin resistance cassette through homologous recombination to disrupt oocK, yielding the mutant S. plymuthica 4Rx13ΔoocK. Analysis of culture extracts by ultra‐high‐performance liquid chromatography/high‐resolution data‐dependent mass spectrometry (UHPLC‐HRMS2) and molecular networking15 revealed no detectable oocydin‐related metabolites. Oocydin production was restored when we introduced a plasmid harboring the gene oocK into 4Rx13ΔoocK (Figure S2–S4), this suggesting that OocK is essential for polyketide core assembly.
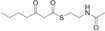
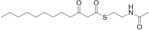
For biochemical studies, we cloned oocK, tobD, and pedG into a pET28a plasmid for E. coli expression as N‐terminally His6‐tagged proteins (Table S1 and Figure S5). Only OocK was obtained as a soluble yellow protein. Analysis of ethyl acetate extracts of the denatured enzyme by UHPLC‐HRMS was consistent with FAD as the cofactor (Figure S6). In both the ped and ooc pathways, the genes preceding pedG/oocK encode modules that contain a KS, a pseudo‐dehydratase lacking the catalytic histidine residue within the active‐site motif (HX3GX4P),15 and an acyl carrier protein (ACP) domain, thus suggesting that the substrate for oxygenation is a β‐keto thioester. To test this hypothesis, we synthesized β‐keto thioesters 7–10 (Table 1) using various routes. These compounds harbor an N‐acetylcysteamine (SNAC) units as simplified surrogates of the ACP‐bound 4′‐phosphopantetheinyl intermediates.16 Assay mixtures with test substrate, OocK, and NADPH were incubated for 90 min at room temperature and then extracted with ethyl acetate. For substrates 7 and 8 with medium or long aliphatic chains, UHPLC‐HRMS (Figure 3 A and Figures S7,S8) analysis showed the formation of a new product with a mass difference of +16 Da (Figure 3 and Figure S7), which is consistent with substrate oxygenation. No such product was detected for assays with the boiled enzyme or with test compounds 9–17.
Table 1.
Substrates used in the assay with OocK.
| Compound | Structure | Product detected |
|---|---|---|
| 7 |
|
Yes |
| 8 |
|
Yes |
| 9 |
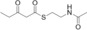
|
No |
| 10 |
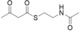
|
No |
| 11 |
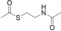
|
No |
| 12 |
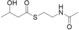
|
No |
| 13 |
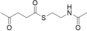
|
No |
| 14 |
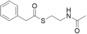
|
No |
| 15 |
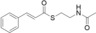
|
No |
| 16 |
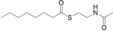
|
No |
| 17 |
|
No |
Figure 3.
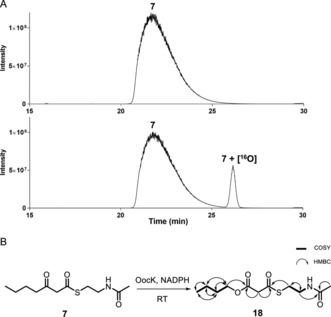
Enzymatic assay with OocK and 7. A) Representative UHPLC‐HRMS data showing the extracted ion chromatograms (EIC) of the assay of 7 with OocK, including the boiled‐enzyme negative control (upper) and test reaction using all components (lower). EIC for 7 and 7+[16O] (calculated for [M+H]+ as 246.1158 and 262.1108, respectively). Mass spectra of the corresponding compounds are shown in Figures S7, S8. B) Schematic representation of the enzymatic assay, including COSY and HMBC key correlations for the product 18.
To pinpoint the location of oxygen in the converted products, a scaled‐up assay with 7 afforded sufficient amounts of product for NMR characterization. HMBC and HSQC experiments were consistent with structure 18 (Figure 3 B, Table S2 and Figure S9–S13). This result demonstrates that OocK accepts thioesters bearing β‐keto groups and acts as a Baeyer–Villiger monooxygenase (BVMO) to generate malonyl esters. The data also suggest that for oocydin A (4), the terminal carboxylate at C19 is not derived from a malonyl starter unit as previously proposed,17 but from an oxygen insertion and ester hydrolysis event involving an extended precursor.
Oocydin‐type polyketides, which include haterumalides and biselides (Table 2), are known from diverse biological sources comprising various marine ascidians,18 sponges,19 and bacteria.11, 17, 20 Several of the invertebrate‐derived haterumalides (4, 19, 20)18c, 19, 21 and biselides (21–22)18a, 18b, 21 feature ester or amide moieties instead of the proposed OocK‐derived carboxylate unit at C19 of oocydin A (4). In light of our results, we speculated that some of these congeners might be uncleaved precursors. To predict the precursor of 4, we analyzed the domain architecture of OocJ, which has no counterpart in the polyketide structure (Figure 4).17 The first two modules, OocJ1–2, have the identical domain set to that found in the phormidolide PKS.22 OocJ1 exhibits an architecture that is, with variations, found in PKSs incorporating 1,3‐bisphosphoglycerate‐derived starter units.23 In phormidolide biosynthesis, this first module generates an unusual 2‐methoxyacrylyl starter, which is putatively elongated by a second module that introduces a methylene β‐branch. The next module, distributed among OocJ, OocK, and OocL (OocJKL), is the proposed oxygen‐insertion module. This module is absent in the phormidolide PKS.22 Combination of these individual module actions suggests compound 23 (Figure 4) as an oocydin precursor. Based on this analysis, we suspected that haterumalide B (19) and biselide B (21) are uncleaved PKS products with a non‐hydrolyzed methoxyvinyl moiety. Since uncleaved congeners have not been reported from the oocydin A producer S. plymuthica 4Rx13, we reexamined the metabolites produced by this strain. HPLC‐based purification from culture extracts allowed structure elucidation by NMR of a new congener (24; Figure 4, Table S3 and Figures S14–S19). A related compound, haterumalide B (19), was reported in the patent literature from another S. plymuthica strain (A 153), for which the genome is not available.24 Linking the uncleaved compound 24 to the ooc gene cluster suggests the occurrence of highly similar PKS assembly lines in tunicates, sponges, and free‐living bacteria, and supports the existence of an oxygen insertion module that includes OocK. It is therefore likely that the structurally related haterumalides and biselides from marine invertebrates are generated by highly similar routes, likely from bacterial symbionts.
Table 2.
Selected oocydin, haterumalide, and biselide congeners and their biological sources.
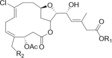
| Names (source) | R1 | R2 |
|---|---|---|
| haterumalide NA/oocydin A/FR177391 (4) (bacterium, sponge) |
OH | H |
| haterumalide B (19) (tunicate, sponge) |
|
H |
| haterumalide NB (20) (sponge) |
O‐nBu | H |
| biselide B (21) (tunicate) |
|
OAc |
| biselide D/taurohaterumalide NA (22) (tunicate) |
|
H |
Figure 4.
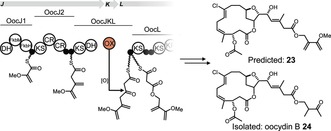
Proposed early steps of oocydin biosynthesis in which the third ooc module is a split module involving OocJ, OocK, and OocL. The hypothetical ester 23 was predicted based on the PKS domain architecture. Differences to the isolated congener 24 might result from additional enzymatic activities in the oocydin producer.
To our knowledge, OocK is the first characterized modular PKS component that inserts atoms other than carbon into growing backbones (except for thioester alcoholysis).25 Some distantly related oxygenases may have similar oxygen‐insertion functions. An oxygenase from FR901464 biosynthesis was suggested to catalyze oxygen insertion during elongation, but at the thioester rather than the β‐keto moiety, resulting in a thiocarbonate intermediate.26 However, later work concluded that this oxygenase is not a BVMO, but rather an epoxidase.27 Another putative BVMO, CalD from the calyculin pathway, was proposed to accept an α‐ketothioester intermediate and account for a net one‐carbon elongation following decarboxylation.28 Furthermore, recent work suggests that the putative monooxygenase domain in the protein MndA is responsible for Baeyer–Villiger oxidative cleavage, giving rise to mandelalides.29 This biosynthetic model provides an α‐alkoxyacetyl, rather than a malonate derivative. MndA and CalD, which do not fall into the PedG/OocK clade, remain to be functionally characterized (Figure 2).
The presence of genes encoding close OocK and PedG homologues in other PKS clusters suggests that oxygen insertion is a recurring PKS feature to install a terminal hydroxylcarboxylate pseudostarter and internal ester moieties. Oxygenases of the OocK/PedG clade were identified in five additional, as yet chemically unassigned trans‐AT PKS systems, which could possibly give rise to even higher polyketide diversity.
In this work, we provide insights into the timing and mechanism of polyketide cleavage using biochemical experiments and bioinformatic analyses. The data suggest that for oocydin, oxygen is inserted during polyketide elongation through a Baeyer–Villiger‐type reaction catalyzed by an integral component of trans‐AT PKSs, which occurs in various unrelated polyketide pathways. Oxygen insertion accounts for a moiety previously assumed to be carboxylated starter unit in oocydin, suggesting common biosynthetic features among oocydin‐like polyketides isolated from bacteria, sponges, and tunicates. Oxygen incorporation into polyketide backbones expands the basic functional repertoire of PKSs and offers attractive opportunities for biosynthetic diversification.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank F. Hemmerling, P. Moosmann and A. L. Vagstad for discussions on experiments and A. F. Canovas Martinez and F. Severi for technical assistance. We are grateful for funding from the EU (ERC Advanced Project SynPlex to J.P.).
R. A. Meoded, R. Ueoka, E. J. N. Helfrich, K. Jensen, N. Magnus, B. Piechulla, J. Piel, Angew. Chem. Int. Ed. 2018, 57, 11644.
References
- 1.
- 1a. Hertweck C., Angew. Chem. Int. Ed. 2009, 48, 4688–4716; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4782–4811; [Google Scholar]
- 1b. Piel J., Proc. Natl. Acad. Sci. USA 2002, 99, 14002–14007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Keatinge-Clay A. T., Chem. Rev. 2017, 117, 5334–5366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Helfrich E. J. N., Piel J., Nat. Prod. Rep. 2016, 33, 231–316; [DOI] [PubMed] [Google Scholar]
- 3b. Piel J., Nat. Prod. Rep. 2010, 27, 996–1047. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Calderone C. T., Iwig D. F., Dorrestein P. C., Kelleher N. L., Walsh C. T., Chem. Biol. 2007, 14, 835–846; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4b. Jenner M., Kosol S., Griffiths D., Prasongpholchai P., Manzi L., Barrow A. S., Moses J. E., Oldham N. J., Lewandowski J. R., Challis G. L., Nat. Chem. Biol. 2018, 14, 270; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4c. Moldenhauer J., Chen X. H., Borriss R., Piel J., Angew. Chem. Int. Ed. 2007, 46, 8195–8197; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2007, 119, 8343–8345; [Google Scholar]
- 4d. Moldenhauer J., Götz D. C. G., Albert C. R., Bischof S. K., Schneider K., Süssmuth R. D., Engeser M., Gross H., Bringmann G., Piel J., Angew. Chem. Int. Ed. 2010, 49, 1465–1467; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1507–1509; [Google Scholar]
- 4e. Kusebauch B., Busch B., Scherlach K., Roth M., Hertweck C., Angew. Chem. Int. Ed. 2010, 49, 1460–1464; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 1502–1506; [Google Scholar]
- 4f. Pöplau P., Frank S., Morinaka B. I., Piel J., Angew. Chem. Int. Ed. 2013, 52, 13215–13218; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 13457–13460. [Google Scholar]
- 5. Piel J., Wen G. P., Platzer M., Hui D. Q., ChemBioChem 2004, 5, 93–98. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Borroni G., Brazzelli V., Rosso R., Pavan M., Am. J. Dermatopathol. 1991, 13, 467–474; [DOI] [PubMed] [Google Scholar]
- 6b. Wan S., Wu F., Rech J. C., Green M. E., Balachandran R., Horne W. S., Day B. W., Floreancig P. E., J. Am. Chem. Soc. 2011, 133, 16668–16679; [DOI] [PubMed] [Google Scholar]
- 6c. Kellner R. L. L., Dettner K., Oecologia 1996, 107, 293–300. [DOI] [PubMed] [Google Scholar]
- 7. Sakemi S., Ichiba T., Kohmoto S., Saucy G., Higa T., J. Am. Chem. Soc. 1988, 110, 4851–4853. [Google Scholar]
- 8.
- 8a. Piel J., Hui D. Q., Wen G. P., Butzke D., Platzer M., Fusetani N., Matsunaga S., Proc. Natl. Acad. Sci. USA 2004, 101, 16222–16227; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Wilson M. C., Mori T., Ruckert C., Uria A. R., Helf M. J., Takada K., Gernert C., Steffens U. A., Heycke N., Schmitt S., Rinke C., Helfrich E. J., Brachmann A. O., Gurgui C., Wakimoto T., Kracht M., Crüsemann M., Hentschel U., Abe I., Matsunaga S., Kalinowski J., Takeyama H., Piel J., Nature 2014, 506, 58–62. [DOI] [PubMed] [Google Scholar]
- 9. Piel J., Höfer I., Hui D. Q., J. Bacteriol. 2004, 186, 1280–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nakabachi A., Ueoka R., Oshima K., Teta R., Mangoni A., Gurgui M., Oldham N. J., Van Echten-Deckert G., Okamura K., Yamamoto K., Inoue H., Ohkuma M., Hongoh Y., Miyagishima S. Y., Hattori M., Piel J., Fukatsu T., Curr. Biol. 2013, 23, 1478–1484. [DOI] [PubMed] [Google Scholar]
- 11. Matilla M. A., Leeper F. J., Salmond G. P., Environ. Microbiol. 2015, 17, 2993–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ueoka R., Bortfeld-Miller M., Morinaka B. I., Vorholt J. A., Piel J., Angew. Chem. Int. Ed. 2018, 57, 977–981; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 989–993. [Google Scholar]
- 13. Julien B., Tian Z. Q., Reid R., Reeves C. D., Chem. Biol. 2006, 13, 1277–1286. [DOI] [PubMed] [Google Scholar]
- 14. Yang J. Y., Sanchez L. M., Rath C. M., Liu X., Boudreau P. D., Bruns N., Glukhov E., Wodtke A., de Felicio R., Fenner A., Wong W. R., Linington R. G., Zhang L., Debonsi H. M., Gerwick W. H., Dorrestein P. C., J. Nat. Prod. 2013, 76, 1686–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.
- 15a. Wagner D. T., Zhang Z., Meoded R. A., Cepeda A. J., Piel J., Keatinge-Clay A. T., ACS Chem. Biol. 2018, 0, 0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15b. Labonte J. W., Townsend C. A., Chem. Rev. 2013, 113, 2182–2204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Franke J., Hertweck C., Cell Chem. Biol. 2016, 23, 1179–1192. [DOI] [PubMed] [Google Scholar]
- 17. Matilla M. A., Stockmann H., Leeper F. J., Salmond G. P. C., J. Biol. Chem. 2012, 287, 39125–39138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.
- 18a. Teruya T., Suenaga K., Maruyama S., Kurotaki M., Kigoshi H., Tetrahedron 2005, 61, 6561–6567; [Google Scholar]
- 18b. Teruya T., Shimogawa H., Suenaga K., Kigoshi H., Chem. Lett. 2004, 33, 1184–1185; [Google Scholar]
- 18c. Ueda K., Hu Y. M., Tetrahedron Lett. 1999, 40, 6305–6308. [Google Scholar]
- 19. Takada N., Sato H., Suenaga K., Arimoto H., Yamada K., Ueda K., Uemura D., Tetrahedron Lett. 1999, 40, 6309–6312. [Google Scholar]
- 20.
- 20a. Levenfors J. J., Hedman R., Thaning C., Gerhardson B., Welch C. J., Soil Biol. Biochem. 2004, 36, 677–685; [Google Scholar]
- 20b. Strobel G., Li J. Y., Sugawara F., Koshino H., Harper J., Hess W. M., Microbiology 1999, 145 (Pt 12), 3557–3564; [DOI] [PubMed] [Google Scholar]
- 20c. Sato B., Nakajima H., Fujita T., Takase S., Yoshimura S., Kinoshita T., Terano H., J. Antibiot. 2005, 58, 634–639. [DOI] [PubMed] [Google Scholar]
- 21. Kigoshi H., Hayakawa I., Chem. Rec. 2007, 7, 254–264. [DOI] [PubMed] [Google Scholar]
- 22. Bertin M. J., Vulpanovici A., Monroe E. A., Korobeynikov A., Sherman D. H., Gerwick L., Gerwick W. H., ChemBioChem 2016, 17, 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Hildebrand M., Waggoner L. E., Liu H. B., Sudek S., Allen S., Anderson C., Sherman D. H., Haygood M., Chem. Biol. 2004, 11, 1543–1552; [DOI] [PubMed] [Google Scholar]
- 23b. Sun Y. H., Hong H., Gillies F., Spencer J. B., Leadlay P. F., ChemBioChem 2008, 9, 150–156. [DOI] [PubMed] [Google Scholar]
- 24. Gerhardson B., Thaning C., Weissmann R., Welch C., Borowicz J., Hedman R. (Google Patents), US20030130121A1, 2003.
- 25.
- 25a. Biggins J. B., Melinda M. A., Brady S. F., J. Am. Chem. Soc. 2012, 134, 13192–13195; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Franke J., Ishida K., Hertweck C., Angew. Chem. Int. Ed. 2012, 51, 11611–11615; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 11779–11783; [Google Scholar]
- 25c.C. M. Thomas, J. Hothersall, C. L. Willis, T. J. Simpson, Nat. Rev. Microbiol 2010, 8, 281–289. [DOI] [PubMed]
- 26. Tang M. C., He H. Y., Zhang F., Tang G. L., ACS Catal. 2013, 3, 444–447. [Google Scholar]
- 27. Eustaquio A. S., Janso J. E., Ratnayake A. S., O'Donnell C. J., Koehn F. E., Proc. Natl. Acad. Sci. USA 2014, 111, 3376–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wakimoto T., Egami Y., Nakashima Y., Wakimoto Y., Mori T., Awakawa T., Ito T., Kenmoku H., Asakawa Y., Piel J., Abe I., Nat. Chem. Biol. 2014, 10, 648–655. [DOI] [PubMed] [Google Scholar]
- 29. Lopera J., Miller I. J., McPhail K. L., Kwan J. C., mSystems 2017, 2, e00096-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary