

Abstract
The in vitro micronucleus (MN) assay is a well‐established test for evaluating genotoxicity and cytotoxicity. The use of manual microscopy to perform the assay can be laborious and often suffers from user subjectivity and interscorer variability. Automated methods including slide‐scanning microscopy and conventional flow cytometry have been developed to eliminate scorer bias and improve throughput. However, these methods possess several limitations such as lack of cytoplasmic visualization using slide‐scanning microscopy and the inability to visually confirm the legitimacy of MN or storage of image data for re‐evaluation using flow cytometry. The ImageStreamX® MK II (ISX) imaging flow cytometer has been demonstrated to overcome all of these limitations. The ISX combines the speed, statistical robustness, and rare event capture capability of conventional flow cytometry with high resolution fluorescent imagery of microscopy and possesses the ability to store all collected image data. This paper details the methodology developed to perform the in vitro MN assay in human lymphoblastoid TK6 cells on the ISX. High resolution images of micronucleated mono‐ and bi‐nucleated cells as well as polynucleated cells can be acquired at a high rate of capture. All images can then be automatically identified, categorized and enumerated in the data analysis software that accompanies the ImageStream, allowing for the scoring of both genotoxicity and cytotoxicity. The results demonstrate that statistically significant increases in MN frequency when compared with solvent controls can be detected at varying levels of cytotoxicity following exposure to well‐known aneugens and clastogens. This work demonstrates a fully automated method for performing the in vitro micronucleus assay on the ISX imaging flow cytometry platform. © 2018 The Author. Cytometry Part A published by Wiley Periodicals, Inc. on behalf of ISAC.
Keywords: In vitro micronucleus assay, genotoxicity, cytotoxicity, TK6 cells, cytochalasin B, automation, imaging flow cytometry, image analysis, ImageStream®
INTRODUCTION
The in vitro micronucleus (MN) assay is one of the most widely used tests to determine genotoxicity and cytotoxicity, especially as a screening tool in the development of chemicals and pharmaceuticals as well as for biomonitoring of human populations exposed to various lifestyle, occupational, or environmental factors 1, 2, 3. MN are formed from whole chromosomes or chromosome fragments that lag behind during the metaphase–anaphase transition and are not included inside one of the two main nuclei following division 2. Instead, MN form into small, rounded bodies surrounded by their own nuclear envelope. As a result of their mechanism of formation, MN represent chromosomal mutations and can be used as an endpoint in genotoxicity testing 4. The preferred method for measurement of MN is the cytokinesis‐block micronucleus (CBMN) assay in which the frequency of MN in binucleated (BN) cells is quantified following the addition of Cytochalasin‐B (Cyt‐B) to halt cellular division. The use of Cyt‐B limits scoring of MN to once‐divided BN cells and avoids the scoring of spurious MN that may appear from a number of confounding factors such as less than optimal cell culture conditions and altered cell division kinetics 5. However, it has been demonstrated that noncytokinesis‐blocked versions of the assay can identify statistically significant increases in MN among aneugens which arise from the phenomenon of mitotic slippage 6, 7, 8. Therefore, it is often necessary to perform the assay both in the presence and absence of Cyt‐B, which increases the number of samples that must be evaluated. In recent years, several protocols and exposure schedules to perform the MN assay have been developed and validated in a number of recommended cell types 9, 10, 11, 12. Current forms of the assay are able to detect the presence of clastogens and aneugens in both rodent and human derived cell lines as well as in human peripheral blood lymphocytes 13, 14, 15, 16, 17.
A number of methods have been developed to perform the in vitro MN assay. Each of these has advantages and limitations concerning throughput, accuracy and visual confirmation of MN. The assay is most commonly performed through manual fluorescent microscopy which has the benefit of visual confirmation of MN; however, this method is time consuming and prone to scorer subjectivity due to both interscorer variability and fatigue when many samples must be scored 18 Automated microscopy methods have been developed using software algorithms that are able to identify and capture images of fluorescently‐labeled nuclei and MN 19, 20, 21, 22. While these automated methods remove the tedious nature of manual slide‐scoring, they do not typically allow visualization of the cytoplasm and therefore it can be difficult to associate one (or multiple) MN to a particular cell. Also, many of these methods currently do not support automatic scoring of polynucleated (POLY) cells, a required parameter for cytotoxicity calculations when the assay is performed with Cyt‐B 12. Furthermore, some inherent slide‐making concerns, such as ensuring high slide quality and optimal cell density, can be difficult to optimize 23. Laser scanning cytometry (LSC) methods were introduced in the early 2000s and have been recently improved with the advent of new generation instrumentation. LSC systems allow for the identification and automated enumeration of MN as well as mononucleated (MONO), BN, and POLY cells. While LSC can differentiate MN from other DNA positive objects through the use of multiple stains, many systems are still hindered by the need to create high quality microscope slides with adequate cell separation 24, 25, 26. Finally, conventional flow cytometry methods have been developed to perform the in vitro MN assay. One of the original publications by Nüsse and Marx described a method based on ethidium bromide fluorescence and forward and side scatter intensities to differentiate between debris, nuclei, and MN 27. Although this method agreed well with microscopy, difficulties in distinguishing MN from other DNA positive events persisted 28, 29. Through the addition of ethidium monoazide bromine (EMA) which stains only the chromatin of apoptotic or necrotic bodies, MN can now be more confidently identified and differentiated from the main nuclei and other DNA positive events. As a result, the current traditional flow cytometry methodology possesses the ability to acquire data from thousands of cells in just a few minutes and allows automatic analysis of all data. The speed of this method and the need for minimal user intervention are attractive features for toxicology testing. Over the last decade, significant improvements were made to the methodology by Avlasevich et al. 30 and Bryce et al. 31 to incorporate additional fluorescent dyes in order to remedy the difficulties encountered when differentiating MN, cellular debris, and free chromosomes from mitotic cells. This method was successful and configured into a commercially available kit by Litron Laboratories and several publications employing many well‐known clastogens and aneugens have demonstrated that statistically significant increases in MN frequency can be detected when compared with controls 32, 33. The observed MN frequencies also compared well with those obtained through visual microscopy. Most recently, the method has been optimized and miniaturized, such that all required processing steps (e.g., cell processing, sampling and data acquisition) can be performed in 96‐well plate format 34, 35. However, conventional flow cytometry methods suffer from several drawbacks, the most important being the lack of cellular visualization. In the absence of this visual information it is impossible to verify that MN identified by the cytometer are indeed genuine. Another limitation is the requirement to lyse all cell membranes to liberate both the main nuclei and MN which creates a suspension that may contain debris such as individual apoptotic bodies, whole chromosomes or chromosome aggregates in addition to the main nuclei and MN. As a result, despite the recent advances in methodology, a definitive distinction between MN and DNA positive debris cannot be reliably obtained 24. In addition, removal of the cell membrane eliminates the possibility to associate MN to a particular cell or to identify events in which multiple MN are contained within one cell 36. Finally, due to the requirement to lyse cells, Cyt‐B cannot be used in the conventional flow cytometry method. This eliminates the ability to accurately quantify the frequency of MONO, BN, and POLY cells which can introduce difficulties when demonstrating that cells have indeed completed nuclear division. This is of particular importance when examining chemicals or agents that are less potent 37.
In light of these limitations, the next logical step in the development of automating the in vitro MN assay would be the use of a system that can combine the high‐throughput nature of conventional flow cytometry with the high resolution imagery obtained by microscopy, such as the ImageStream® imaging flow cytometer. The ImageStream (ISX) combines the visual information content obtained by fluorescent microscopy with the speed, statistical robustness and rare event capture capability of conventional flow cytometry. Cells in suspension enter into a fluidics system and are hydrodynamically focused into the center of a flow cell cuvette. Cells are then orthogonally illuminated by a brightfield (BF) light‐emitting diode (LED), side scatter laser and at least one laser to induce fluorescence. Emitted photons are then collected by a high numerical aperture objective lens, pass through a spectral decomposition element and are focused onto a charge‐coupled device (CCD) camera. In the ISX, the spectral decomposition element allows for wavelengths between ∼400 and 800 nm to be separated into specific ranges and projected onto different sections of the CCD. This in turn allows for up to 10 fluorescent images to be captured simultaneously from every cell that passes through the system. This generates up to twelve images per object (2 BF, 10 fluorescent), allowing images of several different structures in the cell to be captured simultaneously. All images are stored in a data file that allows analysis to be performed at any time post‐acquisition in the Image Data Exploration and Analysis Software (IDEAS®). All data files retain the linkage between cell images and bivariate plots; that is, any dot on any bivariate plot can be selected and its corresponding images (BF and fluorescent) will be displayed in the image gallery 38, 39.
Recently, an imaging flow cytometry method to perform the CBMN assay specifically for use in triage radiation biodosimetry was developed 40, 41, 42, 43, 44, 45. Results from this method demonstrated that cellular images of both the cytoplasm and DNA content could be captured at higher throughput than other automated methods and stored in a data file, eliminating the need to create microscope slides 40. Candidate BN cells with and without MN were automatically identified in IDEAS through the use of mathematical algorithms to implement the scoring criteria developed by Fenech et al. 9, 46. The ISX‐based version of the CBMN assay was shown to automatically score more BN cells in a fraction of the time compared with manual microscopy, therefore enhancing the statistical robustness and speed of the assay. A dose response calibration curve to evaluate the frequency of MN per BN cell was created from 0 to 4 Gy and was similar in magnitude to other calibration curves published in the literature 43. Finally, through the use of blinded samples it was verified that the method could generate dose estimates to within ±0.5 Gy of the true dose, sufficient for triage radiation biodosimetry 44.
This article describes for the first time, the adaptation of the in vitro MN assay to an automated imaging flow cytometry‐based method for the purposes of estimating genotoxicity and cytotoxicity in human lymphoblastoid TK6 cells. Using the ISX, BF and fluorescent images of MONO, BN, and POLY cells as well as MN were captured and automatically identified, classified and enumerated in IDEAS. The work described here details a data analysis template for the rapid calculation of both genotoxicity and cytotoxicity following the exposure of TK6 cells to known MN‐inducing clastogens and aneugens, in the presence and absence of Cyt‐B. This work demonstrates a fully automated method for performing the in vitro MN assay on an imaging flow cytometry platform.
Materials and Methods
Cell Line and Culture Conditions
Human lymphoblastoid TK6 cells were purchased from MilliporeSigma (MilliporeSigma, Billerica, MA, USA; cat. 95111735) and were grown in HyClone RPMI‐1640 media (GE Healthcare Life Sciences, Logan, Utah, USA; cat. SH30027.01) supplemented with 10% fetal bovine serum (GE Healthcare Life Sciences, Logan, UT; cat. SH30071.03), 1% nonessential amino acid (Lonza, Allendale, NJ, USA; cat. 13–114E), 1% sodium pyruvate (GE Healthcare Life Sciences, Logan, UT; cat. SH30239.01), and 1% penicillin–streptomycin (Gibco, Thermo Fisher Scientific, Waltham, MA, USA; cat. 15070063). The TK6 cells were routinely passaged to ensure that they were kept in the exponential growth phase (1 × 104–1.5 × 106 cells/ml) for all experiments and their doubling time was approximately 15 h.
Test Chemicals
The tested chemicals were purchased from MilliporeSigma and were selected since they are well known clastogens and aneugens, and there is significant literature demonstrating their genotoxic and cytotoxic behavior. The chemicals used were mitomycin C (MMC; CAS no. 50‐07‐7), methyl methanesulfonate (MMS; CAS no. 66‐27‐3), colchicine (CAS no. 64–86‐8), and vinblastine sulfate (VS; CAS no. 143‐67‐9). Mannitol (CAS no. 69‐65‐8) was used as a negative control. All test chemicals were dissolved in sterile water except for VS which was dissolved in dimethyl sulfoxide (DMSO). For all chemicals, a range of doses was selected such that the top dose achieved a cytotoxicity of 55 ± 5% as recommended by the Organization for Economic Co‐operation and Development (OECD) Test Guideline 487 for the in vitro MN assay 12.
Exposure to Test Chemicals
Test chemicals were introduced into T25 culture flasks containing 10 mL of media and TK6 cells at a concentration of approximately 1–3 × 105 cells/mL (absence of Cyt‐B) and 7–8 × 105 cells/mL (presence of Cyt‐B). All test chemicals dissolved in sterile water were added to each culture at 10% (v/v) of the final culture volume to achieve the desired final concentrations; VS which was dissolved in DMSO, was added to each culture at 1% (v/v). All cultures were exposed to the test chemicals for 3 h at 37°C, 5% CO2 as suggested by the OECD Guideline 12. Following the exposure time, all cultures were washed with fresh media to remove the test chemical and returned to culture in 10 mL of fresh media for 24 h. For exposures in the presence of Cyt‐B, an appropriate volume of Cyt‐B was added to each culture to obtain a final concentration of 3 μg/ml. Solvent controls (sterile water or DMSO) were used as unexposed negative controls in each experiment. All experiments were performed in duplicate.
Cell Harvesting, Processing, and Staining
At the completion of culture, all samples were centrifuged at 200 X g for 8 min at 20°C. The supernatant was aspirated and the cell pellets were resuspended. A cytoplasmic swelling step was performed by slowly adding 5 mL of 75 mM KCl (stored at 4°C), mixing three times gently by inversion and incubating for 7 min at 4°C. Following this, 2 mL of 4% formalin (Polysciences, Warrington, PA, USA; cat. 04018‐1) was added and cells were incubated for an additional 10 min at 4°C. Cells were centrifuged at 200 X g for 8 min at 20°C, the supernatant was aspirated and the cells were resuspended in 100 μL of 4% formalin and incubated at 4°C for 20 min. Following this incubation, 5 mL of 1× PBS containing 0.5% FBS was added and cells were centrifuged at 200 X g for 8 min at 20°C. The supernatant was aspirated and the cells were resuspended in 100 μL of 1X PBS containing 0.5% FBS and transferred to a 1.5 mL Eppendorf tube. RNase (MilliporeSigma, Billerica, MA, USA; CAS‐9001‐99‐4) was added to each sample at a final concentration of 50 μg/ml. Finally, Hoechst 33342 (Thermo Fisher Scientific, Waltham, MA; cat. H3570) was added to each sample at a final concentration of 10 μg/ml. All samples were incubated for 30 min at 37°C and then micro‐centrifuged at 150 X g for 8 min at 20°C. The supernatant was carefully removed such that approximately 25–30 μL of sample remained; this ensured that all samples were highly concentrated to achieve the maximum possible speed of data acquisition on the ISX.
Data Acquisition on the ISX and Analysis in IDEAS
All samples were run on an ISX MKII (MilliporeSigma, Seattle, WA) dual CCD camera system equipped with the MultiMag option (20×, 40×, and 60× magnification), 405, 488, 561, 592, and 642 nm lasers. Channels 1 and 9 were used to capture cytoplasmic images from the BF LED and the 405 nm laser was set to 15 mW to capture Hoechst images (nuclei and MN) in channel 7. All other channels were disabled during data acquisition. Unlike with other conventional flow cytometers, no other information was required for this study (e.g., scatter) and as such, all other lasers were turned off. For all experiment samples, 20,000 events were collected at 60× magnification using a data acquisition template created in the INSPIRE (MilliporeSigma, Seattle, WA) software that controls the ISX, described below. In previous studies, DRAQ5 was used as the nuclear stain 40, 41, 42, 43, 44. In this work, Hoechst 33342 was selected as the nuclear stain due to the very sharp gradient it creates at the edge of the main nuclei and minimal cytoplasmic background staining, especially in the presence of RNase. These properties, when combined with features to collect highly focused events, allowed for the acquisition of sharp images. This reduced the possibility of the MN mask highlighting staining artifacts during data analysis. All data was saved as raw image files and analyzed using a data analysis template created in IDEAS v6.2 that is described below.
Evaluation of Genotoxicity and Cytotoxicity
The use of Cyt‐B permits nuclear division but blocks cellular division. In this work, cells were not synchronized before exposure to the test chemicals and are and therefore are at different stages of the cell cycle when exposed to Cyt‐B. Thus, upon harvest, some cells may contain two (binucleated), three (trinucleated) or four (quadranucleated) nuclei. To evaluate genotoxicity in samples not exposed to Cyt‐B, MN were scored in MONO cells and in samples exposed to Cyt‐B, MN were scored in BN cells. In both cases, to determine statistical significance, the number of cells with MN in the exposed samples were compared by a Yates corrected chi‐squared test to the number of cells with MN in the control samples. Fold increases in MN frequency were determined by dividing the MN frequency in MN‐containing cells by the spontaneous MN frequency in control samples. To evaluate cytotoxicity in samples exposed to Cyt‐B the following relation was used 12:
| (1) |
where T indicates chemically exposed cultures, C indicates control cultures and CBPI is the cytokinesis‐block proliferation index calculated as 12:
| (2) |
All parameters in the CBPI formula were scored automatically in IDEAS using masks, features and gating strategies described in this article. The total number of POLY cells was calculated by summing the number of trinucleated and quadranucleated cells scored. In samples not exposed to Cyt‐B, cell counts were obtained by hemocytometer following completion of culture but prior to processing. Using these cell counts, the relative increase in cell counts (RICC) and the relative population doubling (RPD) parameters were calculated 12:
| (3) |
| (4) |
Where population doubling (PD) is calculated as:
| (5) |
Cytotoxicity estimates through calculation of relative cell count (RCC) were not determined since it has been observed that RCC can underestimate cytotoxicity and its use is no longer recommended 12.
Results
Optimization of Region Boundaries during Data Acquisition
In order to determine the optimal settings for region boundaries in all plots of the data acquisition template in the INSPIRE software, positive and negative truth populations for all cell types were first identified (Fig. 1). Thirty thousand events from each dose point of one replicate of a MMC exposure in the presence of Cyt‐B was collected with only very small cellular debris not collected during acquisition. These data files were merged into a single file containing approximately 200,000 cellular images which was used to optimize gating. A similar file with the same number of total events was created from a MMC exposure in the absence of Cyt‐B. This ensured that representative cell imagery across the full dose range of at least one test chemical would be included. In IDEAS, positive and negative truth populations can be hand‐tagged and saved as separate populations. These populations can then be used to set appropriate region boundaries on all bivariate plots 43. Using the merged files, truth populations consisting of 50 positive and 50 negative hand‐tagged cells were created for all cell types of interest (MONO, BN, and POLY cells). As demonstrated in Figure 1, the positive cell populations (middle column) have visible cytoplasmic material, highly circular and in‐focus nuclei and, in the case of multinucleated cells, nuclei that are similar in size and staining intensity relative to one another as per the scoring criteria described by Fenech et al. 9, 46. On the other hand, the negative population (right hand column) contains cells with abnormal nuclear morphology, overlapping nuclei or nuclei of differing intensity, area, circularity and aspect ratios. Using the truth populations, the acquisition region boundaries were adjusted to minimize the inclusion of negative events and to ensure inclusion of at least 85% of positive events.
Figure 1.
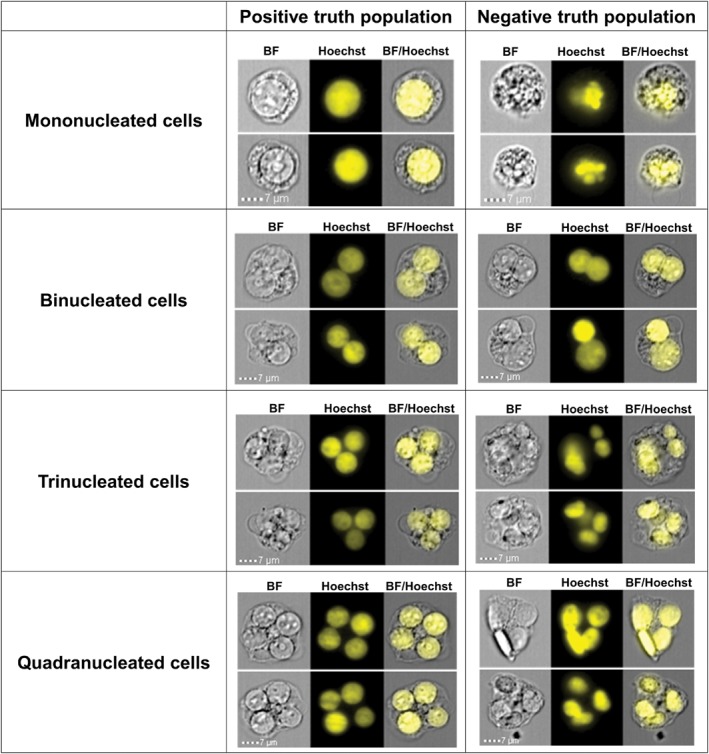
Example imagery from positive and negative mono‐, bi‐, tri‐, and quadranucleated cell truth populations. The middle column contains representative images of positive truth populations that were created by selecting cells with visible cytoplasmic material and possessing nuclei of normal circular morphology and uniform intensity. The right hand column contains representative images of negative truth populations that were created by selecting cells with abnormal nuclear morphology, overlapping nuclei or nuclei of differing intensity, area, circularity, and aspect ratios.
Figure 2 shows the bivariate plots and histograms applied sequentially during data acquisition in INSPIRE to ensure that the best focused single cell images were captured from each sample. The first bivariate plot of BF Aspect Ratio versus BF Area was created to select for single cells and to exclude small debris, doublets and other large events (Fig. 2A). The Aspect Ratio is the ratio of the semi‐minor axis to the semi‐major axis based on a specific mask and allows for the separation of circular objects from elongated objects (e.g. doublets). Next, using the single cell population from Figure 2A, a bivariate plot of Hoechst Gradient RMS versus BF Gradient RMS was created. The Gradient RMS feature measures the sharpness of an image by detecting changes in adjacent pixel values in the image and is useful for the selection of focused objects. The region in Figure 2B selects for highly focused cells and eliminates unfocused events. Here, the advantage of using image‐based truth populations to define region boundaries is demonstrated. While there is a large population of events below the region, the majority of these events are out of focus. Collecting these events would make it difficult for the mathematical algorithms used during data analysis in IDEAS to correctly identify appropriate cell populations. Finally, a histogram of Hoechst Intensity was created to eliminate very dimly stained cells (Fig. 2C). For each sample, 20,000 events were collected from the final Hoechst‐positive population in Figure 2C. Table 1A shows the percentage of each truth population remaining following the application of each region in the data acquisition template and Table 1B lists all region parameters and positions. This data acquisition template was used to collect all data from all experimental samples.
Figure 2.

INSPIRE® data acquisition template used to collect single, well‐focused, DNA‐positive cells. (A) Bivariate plot of BF Aspect Ratio versus BF Area allows for the selection of single cells and the removal of doublets and small and large debris. (B) Using the single cell population from panel A, a bivariate plot of Hoechst Gradient RMS versus BF Gradient RMS allows for the selection of well‐focused cells. (C) Using the well‐focused single cell population from panel B, a histogram of Hoechst Intensity allows for the selection of DNA positive cells and the elimination of very dimly stained events. The data acquisition template in INSPIRE® applies these regions sequentially and 20,000 events were collected from each sample from the Hoechst+ region in panel C. The percentages shown in each panel are taken from control samples and may vary slightly in samples that have been exposed to chemical agents.
Table 1.
Demonstration of the effectiveness of the gating strategy to retain positive events while removing negative events during data acquisition in INSPIRE®. In addition, the features, region boundaries and purpose for using each feature in the data acquisition template are shown. (A) The percentage of events remaining from the data acquisition positive and negative truth populations for mono‐, bi‐, tri‐, and quadranucleated cells following sequential application of each region. Region boundaries were adjusted to minimize the inclusion of negative events and to ensure inclusion of at least 85% of positive events. (B) Summary of region boundaries and all features used to create the data acquisition template in INSPIRE as described in Figure 2. The purpose for the use of each feature is also shown.
| (A) | ||
|---|---|---|
| Region | % Positive tagged events remaining following application of each region | % Negative tagged events remaining following application of each region |
| Mononucleated cells | ||
| Single cells | 100 | 94 |
| BF and Hoechst focus | 92 | 2 |
| Hoechst+ | 90 | 2 |
| Binucleated cells | ||
| Single cells | 98 | 88 |
| BF and Hoechst focus | 92 | 0 |
| Hoechst+ | 92 | 0 |
| Trinucleated cells | ||
| Single cells | 100 | 92 |
| BF and Hoechst focus | 92 | 0 |
| Hoechst+ | 92 | 0 |
| Quadranucleated cells | ||
| Single cells | 100 | 100 |
| BF and Hoechst focus | 94 | 2 |
| Hoechst+ | 94 | 2 |
| (B) | ||||
|---|---|---|---|---|
| Figure panel | Features | Region boundaries (min/max) | Purpose of feature/region | % of total events |
| 2A |
BF Aspect Ratio BF Area |
0.75/1 100/900 |
Selects for single cells | 63.3 |
| 2B |
Hoechst Gradient RMS BF Gradient RMS |
10/14 45/70 |
Selects for the most well‐focused single, DNA positive cells | 4.9 |
| 2C | Hoechst Intensity | 2×105/2×106 | Eliminates dimly stained events | 4.2 |
Binucleated Cell Masking and Gating Strategy
In previous publications on this work, a data analysis template was developed in IDEAS to identify candidate BN cell events 40, 41, 43. The analysis in this work, including masking and gating strategy, is similar but has been slightly modified since the sample preparation methods have been improved; KCl was used to swell the cells and create better separation between the main nuclei, and Hoechst was used as the DNA stain rather than DRAQ5. Furthermore, cellular images were collected at 60× magnification instead of 40× since MN induced by some chemicals have been shown to be smaller than radiation induced MN 47. As a result, the cellular images are larger and the main nuclei in BN cells can be more easily masked by the algorithms in IDEAS. This slight modification to the BN Cell Mask components allowed the region boundaries to be stricter, resulting in more specific identification of BN cells. Additional truth populations (50 BN cells and 50 non‐BN cells) were hand‐tagged from the DNA positive population in Figure 2C and were used to optimize the new data analysis template for the selection of BN cells.
Details regarding the creation of the BN Cell Mask and the BN cell identification gating strategy have been described in previous publications 40, 41, 43 but for completeness in this work, Supporting Information Figure S1 demonstrates how each mask is sequentially applied to create the BN Cell Mask. Furthermore, Supporting Information Figure S1 also illustrates the streamlined application of bivariate plots and histograms, with stricter region boundaries than previously published, to identify BN cells. Supporting Information Table S1A shows, based on the truth populations, the percentage of the positive and negative BN cell events remaining after the sequential application of each region and Supporting Information Table S1B provides the specific parameters used in each plot. Supporting Information Table S2 contains the details of how to create the BN Cell Mask.
Mono and POLY Cell Masking and Gating Strategies
In order to identify and quantify MONO and POLY cells, modifications were made to the BN Cell Mask to create a new POLY Cell Mask. This mask is a combination of the LevelSet, Watershed, Range and Component masks allowing separation between the main nuclei within a single image to be attained (Fig. 3Ai). Using the Range mask, the lower limit of the POLY Cell Mask was increased to 135 pixels (33.75 μm2), which avoided inclusion of background artifacts or small nuclear debris, but still allowed smaller nuclei within POLY cells to be correctly masked. With masks that have multiple pieces, such as the POLY Cell mask, the Component mask allows each piece to be identified as a separate, individual piece. The Component mask, was used to create individual masks to identify each nucleus in each image (Fig. 3Aii). The gating and masking strategies to identify MONO and POLY cells was less complex than for BN cells since the scoring criteria is less stringent. Candidate MONO and POLY cells need only be viable, non‐apoptotic cells with an intact cytoplasm, normal nuclear morphology and one, three or more nuclei 9. Starting with the non‐apoptotic cell population (Supporting Information Fig. S1B), the Spot Count feature was created using the POLY Cell Mask and a histogram was generated to allow straightforward enumeration of cells containing one, three and four nuclei (Fig. 3B).
Figure 3.
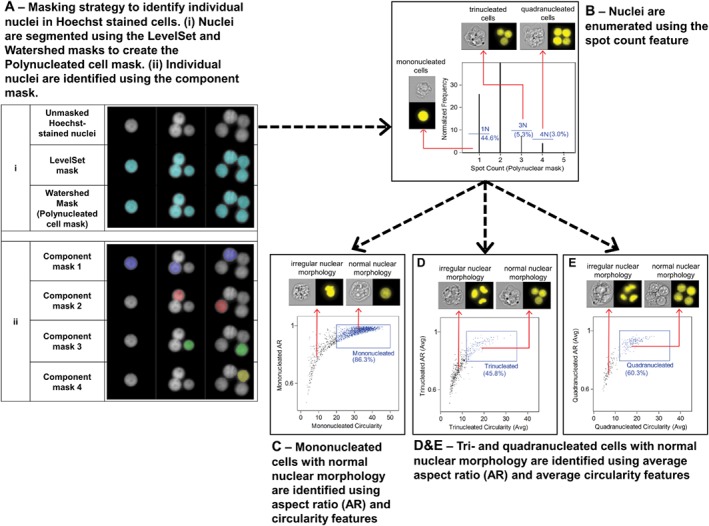
Masking and gating strategy to identify mononucleated (MONO), trinucleated (TRI), and quadranucleated (QUAD) cells containing separated nuclei with normal circular morphology. (A) Illustration of the various components used to create the Polynucleated Cell mask and the individual component masks. The Hoechst‐stained images are shown in white while the masks are shown in blue, purple, red, green and yellow. (Ai—top row) Unmasked Hoechst‐stained nuclei (Ai—rows 2 and 3) The LevelSet mask which identifies pixel homogeneities in an image used to mask all nuclei and the Watershed mask which segments any connected masks in the image are used to create the Polynucleated Cell Mask. (Aii) Individually masked nuclei created using the Component mask highlighted in different colors (component 1 = purple, component 2 = red, component 3 = green, component 4 = yellow). Component mask 1 allows for identification of MONO cells while Component masks 2–4 allow for identification of TRI and QUAD cells. (B) The Spot Count feature created using the Polynucleated Cell Mask identifies cells with one, three or four distinct nuclei. (C–E) The Aspect Ratio (AR) and Circularity features were created using Component masks 1–4. Using the 1N population from panel B, a bivariate plot of these two features was created (C) to select for cells containing nuclei with normal morphology. MONO cells containing nuclei with high AR and Circularity values can then be separated from cells with nuclei possessing irregular nuclear morphology. (D, E) Using the 3N or 4N population from panel B, a bivariate plot of the Average AR and Average Circularity features was created to select for cells containing three (D) or four (E) nuclei with normal morphology. TRI and QUAD cells containing nuclei with high Average AR and high Average Circularity values can then be separated from cells with nuclei possessing irregular nuclear morphology. The percentages shown in each panel are taken from control samples. The values in panels C–E and will vary significantly from samples that have been exposed to chemical agents.
To identify and automatically quantify MONO cells, Component Mask 1 was used. This first component of the POLY cell mask selects, by default, the single nucleus in a MONO cell and therefore was used to identify these cells. The Aspect Ratio and Circularity features were created using Component Mask 1 and plotted against one another on a bivariate plot (Fig. 3C). The Circularity feature measures the degree of the mask's deviation from a circle; the higher the circularity value is, the more circular the masked object is. The Aspect Ratio values of all individual nuclei in MONO or POLY cells that possess normal morphology should be very close to one, and the Circularity values of these nuclei should be much higher than nuclei with irregular morphology. MONO cells containing a nucleus with normal morphology had Aspect Ratio values >0.85 and Circularity values >20. Using the POLY Cell Mask, tri‐ and quadranucleated cells were also automatically identified and scored. Based on the exposure and recovery schedules used, cells with five or more nuclei were never observed and were therefore were not scored. Tri‐ and quadranucleated cells were analyzed separately by applying the Aspect Ratio and Circularity features to each of the main nuclei using individual Component Masks 1–3 and 1–4, respectively. The average values of both Aspect Ratio and Circularity for each nucleus in tri‐ and quadranucleated cells were calculated and plotted against one another (Figs. 3D and 3E). Candidate cells for both tri‐ and quadranucleated cells contained multiple nuclei with normal morphology with average Aspect Ratio values >0.85 and average Circularity values >13. The number of tri‐ and quadranucleated cells were then summed to obtain the total number of POLY cells in each sample. Truth populations were used to determine the region boundaries and Table 2 shows the percentage of each truth population remaining following the application of each region in the template for mono‐, tri‐ and quadranucleated cells as well as the dimensions and parameters of all regions used in Figure 3. Supporting Information Table S2 contains the details of how to create the POLY Cell Mask.
Table 2.
Demonstration of the effectiveness of the gating strategy to retain positive events while removing negative events during data analysis in IDEAS®. In addition, the features, region boundaries and purpose for using each feature in the data analysis template are shown for each cell type. (A) The percentage of events remaining from the data acquisition positive and negative truth populations for mono‐, bi‐, tri‐, and quadranucleated cells following sequential application of each region. Region boundaries were adjusted to minimize the inclusion of negative events and to ensure inclusion of at least 85% of positive events. (A) The percentage of events remaining from the data analysis positive and negative truth populations following sequential application of the IDEAS data analysis regions for the mono‐, tri‐ and quadranucleated cell populations. Region boundaries were adjusted such that at least 85% of positive events remained. (B) Summary of features used in the IDEAS data analysis template to select for mono‐, bi‐ and quadranucleated cells. (C) Summary of features used in the IDEAS data analysis template to select for MN‐containing mononucleated and binucleated cells. In B and C, the purpose for the use of each feature is also shown. The percentages of the total events shown are taken from control samples and will vary from samples that have been exposed to chemical agents.
| (A) | ||
|---|---|---|
| Region | % Positive tagged events remaining following application of each region | % Negative tagged events remaining following application of each region |
| Trinucleated cells | ||
| Nonapoptotic | 100 | 100 |
| 3 nuclear spots | 100 | 84 |
| Trinucleated cells | 88 | 4 |
| Quadranucleated cells | ||
| Nonapoptotic | 100 | 100 |
| 4 nuclear spots | 100 | 88 |
| Quadranucleated cells | 96 | 4 |
| Mononucleated cells with Cyt‐B | ||
| Nonapoptotic | 100 | 84 |
| 1 nuclear spot | 100 | 54 |
| Mononucleated cells | 100 | 4 |
| Mononucleated cells without Cyt‐B | ||
| Nonapoptotic | 100 | 94 |
| 1 nuclear spot | 100 | 88 |
| Mononucleated cells | 96 | 2 |
| (B) | ||||
|---|---|---|---|---|
| Figure panel | Features | Region boundaries (min/max) | Purpose of feature/region | % of total events |
| S1B* |
Hoechst Area BF Contrast |
50/300 0/15 |
Removes apoptotic events | 92.2 |
| Mononucleated cells | ||||
| 3B | Hoechst spot count | 0.5/1.5 | Selects for events with one distinct nucleus identified by the polynucleated cell mask | 44.6 |
| 3C |
Hoechst Aspect Ratio Hoechst Circularity |
0.85/1 20/50 |
Selects for events that have one distinct nucleus with high Aspect Ratio and Circularity values | 38.5 |
| Trinucleated cells | ||||
| 3B | Hoechst Spot Count | 2.5/3.5 | Selects for events with three distinct nuclei identified by the polynucleated cell mask | 5.3 |
| 3D |
Average Hoechst Aspect Ratio (Components 1,2,3) Average Hoechst Circularity (Components 1,2,3) |
0.85/1 13/35 |
Selects for events that have three distinct nuclei with high Aspect Ratio and Circularity values | 2.5 |
| Quadranucleated cells | ||||
| 3B | Hoechst Spot Count | 3.5/4.5 | Selects for events with four distinct nuclei identified by the polynucleated cell mask | 3.0 |
| 3E |
Average Hoechst Aspect Ratio (Components 1,2,3,4) Average Hoechst Circularity (Components 1,2,3,4) |
0.85/1 13/35 |
Selects for events that have four distinct nuclei with high Aspect Ratio and Circularity values | 1.8 |
| *The gate in Figure S1A was used as an initial filter to remove apoptotic events from analysis. | ||||
| (C) | |||
|---|---|---|---|
| Figure panel | Features | Region boundaries (min/max) | Purpose of feature/region |
| 4A | MN mask spot count | 0.5/4 | Selects cells with spots identified by the MN spot mask |
| 4B | MN spot distance minimum | 0.2/25[Link]
0.2/18 b |
Filters cells where MN reside outside the cytoplasm |
| 4C |
MN aspect ratio Median pixel ratio(BN cell/MN) |
0.6/1 1/3 |
Selects for cells with circular MN that have a similar staining intensity as that of the main nuclei |
aIn BN cells.
bIn mononucleated cells.
Micronucleus Masking and Gating Strategy
In a previous publication, details for creating the MN mask are specified 43. For completeness, Supporting Information Figure S2 and Supporting Information Table S2 contains the full details regarding how to create the MN mask used in this work. As previously mentioned, the DNA stain used here was changed from DRAQ5 to Hoechst and data was collected using the 60× magnification lens. Therefore, slight modifications were made to the MN mask and gating strategy. The MN mask is a combination of the Spot, Dilate and Morphology masks that identify Hoechst‐stained spots separate from the main nuclei. Two spot identification masks were created; the first identifies spots with input radii between 2 and 6 pixels while the second identifies spots with radii between 1 and 6 pixels. At 60× magnification, one pixel has a width of 0.33 μm. In both spot identification masks, the spot‐to‐cell background value was set to 2. LevelSet and Morphology masks were then created to highlight the main nuclei and were subtracted from Spot masks 1 and 2. This process is described in more detail in previous work 43. Finally, the Range mask was created using the MN mask to allow proper identification of MN between 8 and 120 pixels (0.89 and 13.3 μm2). This range is slightly different than that proposed by published scoring criteria which indicates that MN should have areas that vary between 1/256th and 1/9th of the area of the main nuclei 9, 46. Normally this would correspond to a MN range between 0.40 and 11.1 μm2 since the mean area of the main nuclei was approximately 100 μm2. However, the increased lower area limit was necessary to avoid the incorrect masking of small image artifacts that can occur when attempting to mask spots below 1 μm2 and the increased upper area limit was necessary to ensure that larger legitimate MN were not missed. Supporting Information Table S2 contains the details of how to create the MN mask.
To quantify MN in both MONO and BN cells, the Spot Count feature, which highlights bright regions within an image, was created using the MN mask. This new MN Spot Count feature was then applied to the final MONO cell population (absence of Cyt‐B) or the final BN cell population (presence of Cyt‐B). A histogram of MN frequency was then generated to differentiate cells with zero MN from cells with at least one MN (Fig. 4A; Table 2C). In some cases, DNA positive debris that resides outside of the cytoplasm was masked by the MN mask and it was necessary to ensure that these events were not being scored as true MN. In addition, artifacts between the main nuclei of BN cells were sometimes also being incorrectly masked. In order to eliminate these events, the Spot Distance Minimum (SDM) feature was used. The SDM feature computes the minimum distance between the centers of two spots and was used to compute the distance between the MN and the main nucleus (Fig. 4B). In BN cells, this feature was used to compute the minimum distance between the MN and both main nuclei in all BN cells; both of these distances were then summed. A histogram of the SDM feature was then used to remove events in which masked DNA‐positive particles resided at distances outside of the cytoplasmic boundary (16 and 21 μm for MONO and BN cells, respectively) as well as artifacts that were masked between the main nuclei of BN cells (Fig. 4B; Table 2C). Finally, according to the scoring criteria developed by Fenech et al. MN should be morphologically similar to and usually have about the same staining intensity as the main nuclei 9, 46. The Aspect Ratio feature was created using the MN mask to identify circular MN. The Median Pixel feature, which computes the median of all background subtracted pixels in a mask, was created using both the BN cell and MN masks. The ratio of the Median Pixel values for both masks was then calculated to create a combined BN cell/MN Median Pixel feature. Figure 4C shows a bivariate plot of the MN mask Aspect Ratio and the BN Cell/MN Medial Pixel features. Positive MN had Aspect Ratio values higher than 0.6 and BN Cell/MN Median Pixel values between 1 and 3 (Table 2C). Events possessing an Aspect Ratio of <0.6 were largely found to be irregularly shaped artifacts. Furthermore, the Median Pixel ratio values between BN cells and MN vary between 1 and 3 simply because the ISX captures two dimensional images of three dimensional objects. As mentioned in previous work, some legitimate MN in an image may reside at a different depth of field than the main nuclei, and will therefore appear to be slightly less intense but should not be misinterpreted as false positive MN or artifacts 41, 43.
Figure 4.
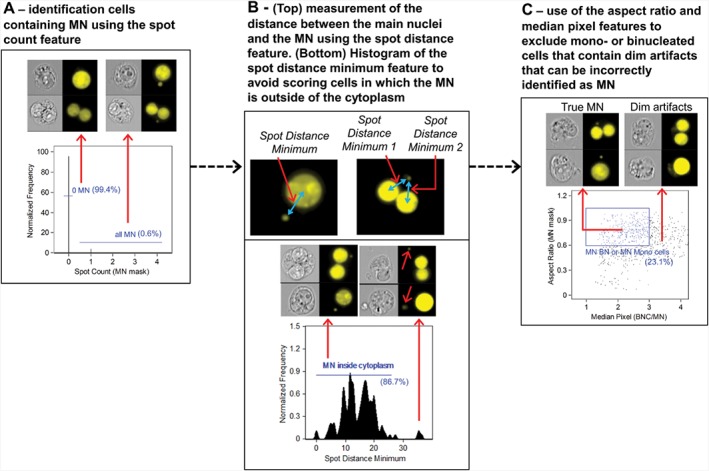
Gating strategy to identify micronuclei (MN) within mononucleated (MONO) and binucleated (BN) cells. (A) Histogram of the Spot Count function, which highlights bright regions within an image, created using either the final BN cell population (with cytochalasin B) or the final MONO cell population (without cytochalasin B) to identify all potential MN. (B—top) The Spot Distance Minimum (SDM) feature, which calculates the shortest distance in microns between two spots, is then used to measure the distance between the center of the main nucleus and the center of the MN (B—bottom) Histogram of the SDM feature illustrating how events with large SDM values contain DNA‐positive spots that are likely to reside outside of the cytoplasm (C) Bivariate plot of the Aspect Ratio feature created using the MN mask versus the Median Pixel ratio of the BN cell mask and the MN mask to identify true MN that are both circular and have a staining intensity comparable to that of the main nuclei. The percentages shown in each panel are taken from control samples. The values in panels A, C, and D will vary significantly from samples that have been exposed to chemical agents.
Experiments without Cytochalasin B
Results for experiments performed in the absence of Cyt‐B are shown in Figure 5 and in Supporting Information Table S3. In all cases except mannitol, statistically significant increases in the number of MN‐containing MONO cells were observed in all doses above and several doses below the 55 ± 5% cytotoxicity target 12 when compared with solvent controls. All cytotoxicity values discussed here are from the RPD parameter since the RICC parameter has been shown to overestimate cytotoxicity when compared with RPD 7, 48. For Colchicine (Fig. 5A) the 0.02 and 0.03 μg/mL doses were the only two below 55% cytotoxicity to produce a significant increases in MN frequencies, 0.28 and 0.62% respectively, corresponding to fold increases of 1.75 and 3.88 over the solvent control. For VS (Fig. 5B) all doses below the 55% cytotoxicity target induced statistically significant increases in MN frequency compared with the solvent control except for 0.005 μg/mL dose. The MN frequencies ranged from 0.4% at 0.01 μg/mL (a 2.5‐fold increase) to 1.6% at 0.02 μg/mL (a 10‐fold increase). In the case of the clastogens MMC and MMS (Figs. 5C and 5D) all doses below 55% cytotoxicity induced statistically significant MN frequencies when compared with solvent controls. For MMC, the MN frequencies ranged from 0.27 at 0.05 μg/mL (a 1.68‐fold increase) to 1.57% at 0.4 μg/mL (a 9.87‐fold increase) and for MMS, the MN frequencies ranged from 0.75 at 5 μg/mL (a 5‐fold increase) to 2.48% at 25 μg/mL (a 16.53‐fold increase). Finally, mannitol (Fig. 5E), a substance that has been shown previously to be non‐cytotoxic and non‐genotoxic 14, induced a cytotoxicity of 30.1% at the highest dose of 4,000 μg/ml, but did not induce statistically significant increases in the frequency of MN compared with the solvent control at any doses.
Figure 5.
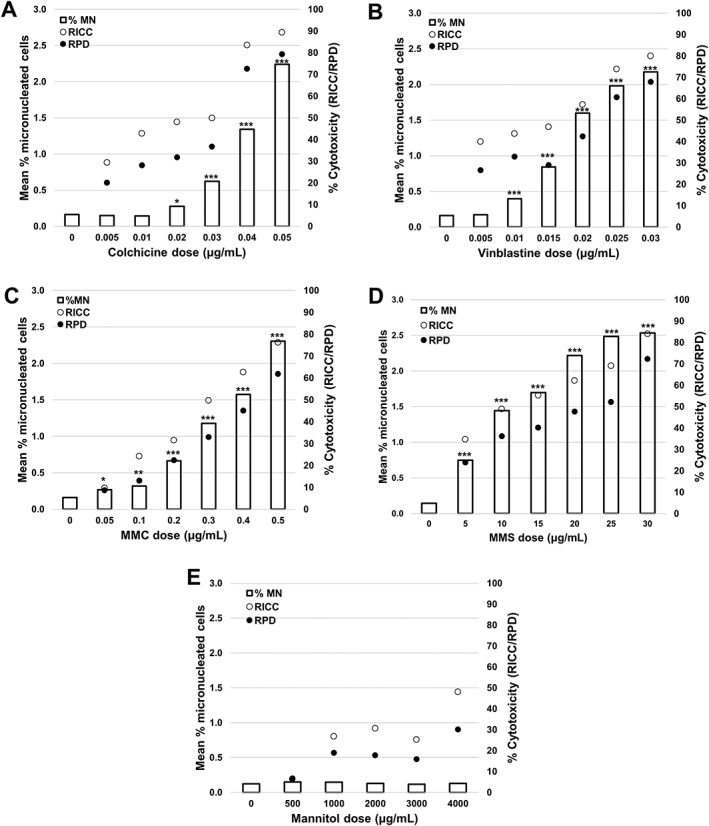
Genotoxicity measured by the percentage of MN (clear bars) and cytotoxicity measured by both RPD (black circles) or RICC (white circles) following a 3 h exposure and 24 h recovery in the absence of cytochalasin B for (A) colchicine, (B) vinblastine sulfate, (C) mitomycin C (MMC), (D) methyl methanesulfonate (MMS), and (E) mannitol. Statistically significant increases in MN frequency compared with solvent controls are indicated by stars (*P < 0.05; **P < 0.01; ***P < 0.001). All quantities are the average of two replicates at each dose point.
On average, a data file containing 20,000 total events took approximately 6 min to acquire and another 2–3 min to process in IDEAS depending on file size. Therefore, acquisition and analysis from 6 or 7 samples can be completed in about 1 h. Additionally, an average of 14,000 MONO cells per culture (28,000 cells per test concentration) were identified and scored in IDEAS.
Experiments with Cytochalasin B
Results for experiments performed in the presence of Cyt‐B are shown in Figure 6 and in Supporting Information Table S4. For all chemicals except mannitol, statistically significant increases in the number of BN cells containing MN were observed in all doses above and several doses below the 55 ± 5% cytotoxicity target 12 when compared with solvent controls. For Colchicine (Fig. 6A) the 0.02, 0.03, and 0.04 μg/mL doses were the only three below 55% cytotoxicity to produce a significant increases in MN frequencies, which ranged from 1.28 to 2.26% respectively, corresponding to fold increases that ranged from 3.56 to 6.28 over the solvent control. For VS (Fig. 6B) all doses induced statistically significant increases in MN frequency compared with the solvent control. The MN frequencies ranged from 0.58% at 0.005 μg/mL (a 2.9‐fold increase) to 2.29% at 0.03 μg/mL (an 11.45‐fold increase). In the case of the MMC (Fig. 6C) only the two top doses (0.4 and 0.5 μg/ml) induced statistically significant MN frequencies when compared with solvent controls. The MN frequencies were 0.93% at 0.4 μg/mL (a 2.58‐fold increase) and 1.02% at 0.5 μg/mL (a 2.83‐fold increase). Similarly for MMS, only the top two doses below the 55% cytotoxicity threshold (20 and 25 μg/ml) induced statistically significant increases in MN when compared with solvent controls. The MN frequencies were 0.61% at 20 μg/mL (a 6.10‐fold increase) and 0.53% at 25 μg/mL (a 5.30‐fold increase). Finally, for mannitol (Fig. 6E), the 3,000 μg/mL induced a cytotoxicity of 26.7% but no doses induced statistically significant increases in the frequency of MN when compared with solvent controls 14.
Figure 6.
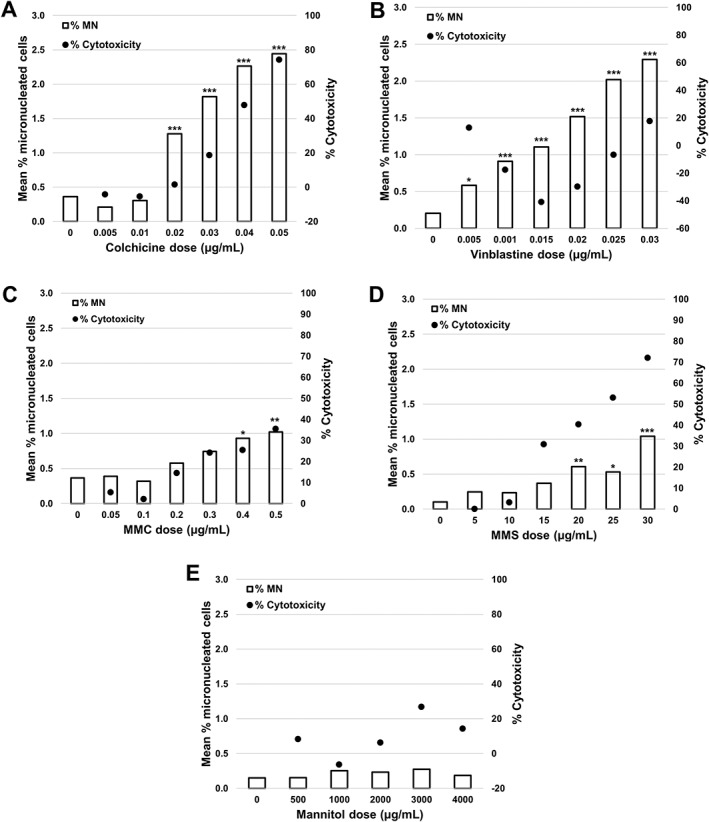
Genotoxicity measured by the percentage of MN (clear bars) and cytotoxicity measured by both RPD (black circles) or RICC (white circles) following a 3 h treatment and 24 h recovery in the presence of cytochalasin B for: (A) colchicine, (B) vinblastine sulfate, (C) mitomycin C (MMC), (D) methyl methanesulfonate (MMS), and (E) mannitol. Statistically significant increases in MN frequency compared with controls are indicated by stars (*P < 0.05; **P < 0.01; ***P < 0.001). All quantities are the average of two replicates at each dose point.
On average, a data file containing 20,000 total events took approximately 10 min to acquire and another 2–3 min to process in IDEAS depending on file size. Therefore, acquisition and analysis from 4 or 5 samples can be completed in about 1 h. The throughput of samples exposed to Cyt‐B is slightly lower than in the absence of Cyt‐B since highly focused BN cells are fewer in number than MONO cells and are therefore more difficult to capture on the ISX. Following processing of all data files in IDEAS, an average of 3,000 BN cells, 8,000 MONO cells and 400 POLY cells per culture (6,000, 16,000, and 800 cells per test concentration, respectively) were automatically identified and scored. In all samples except one, at least 1,000 BN cells were scored as per OECD scoring recommendations 12. The single exception was one replicate of one MMS sample exposed to 30 μg/mL where only 989 BN cells were scored and can likely be attributed to the high level of toxicity at this dose.
Discussion
This article demonstrates that the in vitro MN assay to assess cytotoxicity and genotoxicity can be adapted to an automated IFC method. In its current form, the sample preparation protocol, staining procedures and data collection are straightforward. However, the data analysis procedures are slightly more difficult, especially when considering that accurate identification of mono‐, bi‐, and polynucleated cells must be achieved. It is worth noting that if one examines (Fig. 2C), it may appear that these events may be able to be separated based purely on DNA intensity. In fact, this is not the case. Examination of the imagery in the peaks of the histogram reveals that each peak contains a mixture of cells with different numbers of nuclei. This is due to the fact that the uniformity in staining intensity can vary from one cell to the next. As such, separating populations based on DNA intensity alone does not work well, hence why the data analysis in this work is complex. Despite the difficulty of the image processing techniques, once an IDEAS template has been created, the batch processing option allows for data files from many samples to be rapidly analyzed automatically, removing any user bias or need for user intervention. All data files consisting of both BF and fluorescent images from all cellular images captured can be stored for re‐analysis if necessary. At the 60× magnification setting used in this work, the resolution of the ImageStream is 0.1 µm2. Therefore, no image enhancement has been performed and MN can be easily observed by eye in the images in the IDEAS analysis software, as well as by the masking algorithms used for automated enumeration. MN are also are fully visible in the image preview gallery during data acquisition on the ISX. The method presented here demonstrates several advantages over currently established techniques to perform the in vitro MN assay.
In this study, four chemicals (two aneugens and two clastogens) known to induce MN, as well as one negative control chemical, were evaluated for cytotoxicity and genotoxicity. In samples not exposed to Cyt‐B, increases in cytotoxicity were observed with increasing chemical dose as expected and all top doses induced a cytotoxicity of >55 ± 5%, similar to results presented in published literature 7, 48, 49, 50. In samples exposed to Cyt‐B however, cytotoxicity values at most doses from all chemicals tested tended to be lower than when Cyt‐B was not used. Similar results have been presented in the literature using several chemicals tested in a number of different cell lines, with varying exposure schedules 7, 51, 52. For VS and MMC, none of the tested doses induced a cytotoxicity of 55% when Cyt‐B was used; in fact in the case of VS, several negative values for cytotoxicity were observed with increasing dose. Similar trends have been observed by Massey and Hinchliffe who reported several negative cytotoxicity values when using VS in V79 cells and by Elhajouji who demonstrated cytotoxicity values below 15% up to doses of 1.0 μg/mL of MMC in TK6 cells 7, 52. Additionally, Hashimoto et al. 53 observed opposing cytotoxicity trends in the presence and absence of Cyt‐B using a number of clastogens and aneugens in TK6 cells, albeit with different exposure/recovery schedules. Exposure of cells to Mannitol showed an increase in cytotoxicity of <30% both in the presence and absence of Cyt‐B. This compares well to previous studies in which Mannitol was shown to be negative for cytotoxic effects up to 5,000 μg/mL 14. Overall, the cytotoxicity values obtained here for all tested chemicals compare well to published literature.
The results presented here demonstrate clear positive genotoxic responses in the form of MN induction from the four chemicals tested at most doses. Statistically significant increases in the frequency of MN when compared with controls were observed for doses of all test chemicals that induced a cytotoxicity of 55 ± 5% both in the absence and presence of Cyt‐B. In the absence of Cyt‐B, significant increases in MN frequencies were also observed at most doses that did not achieve 55% cytotoxicity with all tested chemicals when compared with solvent controls. Only the two lowest doses of Colchicine (0.005 and 0.01 μg/ml) and the lowest dose of VS (0.005 μg/ml) did not produce significant increases in MN. When using aneugens in the presence of Cyt‐B, only the lowest two doses of Colchicine (0.005 and 0.01 μg/ml) did not show significant increases in MN frequency. However, when using clastogens in the presence of Cyt‐B, only the top two doses of MMC (0.4 and 0.5 μg/ml) and the top three doses of MMS (20, 25, and 30 μg/ml) showed significant increases in MN frequency. For MMS in the absence of Cyt‐B, there is an apparent lack of correlation between cytotoxicity and MN induction between the 25 and 30 µg/mL doses (Fig. 5D). That is, the RPD increases from 52.5% (25 µg/ml) to 72.2% (30 µg/ml) while the corresponding MN frequencies are 2.48 and 2.53% for 25 and 30 µg/ml, respectively; a very small increase. This is likely due to two factors. The first is that manual hemocytometer counting performed on these samples to determine cytotoxicity has a high degree of uncertainty. The second is that the recovery time of 24 h is relatively short in comparison to the TK6 cell doubling time of 15 h (1.6 doubling times). This may be too short to allow MN to be fully expressed at higher doses. These results indicate that it may be worth increasing the culture time in future experimental work to more accurately quantify MN expression at higher doses.
When examining the literature for studies using TK6 cells exposed to the same test chemicals and similar exposure/recovery schedules, the fold increases in MN frequency compared well to the results obtained here in some cases, and in other cases they were slightly higher or lower. For example, with Colchicine, Elhajouji reported twofold and threefold increases MN frequency, with and without Cyt‐B, respectively at a dose level of 0.05 μg/mL 7. Here, at the same dose level, 7‐fold and 14‐fold increases in MN frequency were observed with and without Cyt‐B, respectively. In the case of VS, Lorge reported a 10‐fold increase in MN frequency over solvent controls in MONO cells at a dose level of 0.023 μg/mL 48. Here, at approximately the same dose, a 10‐fold increase in MN frequency when compared with solvent controls was also observed. Overall rates of MN induction using clastogens only slightly exceeded 1.0% at the top doses, an effect that has also been observed in the literature. For example, in the case of MMC, Elhajouji reported MN frequencies of 1.6% and 4.0% (1.7‐ and 2.4‐fold increases, respectively) with and without Cyt‐B, respectively, at a dose level of 0.4 μg/mL 7. Here, at the same dose level, MN frequencies of only 1.6% and 0.9% were observed with and without Cyt‐B, respectively. However, these correspond to 10‐fold and 3‐fold increases in MN frequency when compared with solvent controls. In general, the results presented here demonstrate that statistically significant increases in MN both in the presence and absence of Cyt‐B from four test chemicals are detectable above and below the 55 ± 5% cytotoxicity target using the ISX method to perform the in vitro MN assay. Caution must be taken when attempting to draw conclusions based on comparisons between the experimental results presented here and published literature. Laboratory conditions, sample preparation methods and exposure/recovery times all present variations in the methods as a whole. In addition, the large variations that exist between manual slide scoring and the mathematical algorithms used to identify MN in this ISX‐based method lead to difficulties when attempting to compare results.
Our group (MilliporeSigma, Seattle, WA and the Consumer and Clinical Radiation Protection Bureau, Health Canada, Ottawa, ON, Canada) has previously reported on several limitations with the ISX‐based methods to score MN; such as the observation of a lower MN frequency when compared with manual slide scoring for triage radiation biodosimetry 40, 41. When compared with automated slide‐scoring methods however, the MN frequency was more consistent with results in published literature 43. The explanation provided for such variations in MN frequency between the ISX method and manual scoring in the context of radiation biodosimetry likely applies in this work also. The discrepancy most probably arises from a combination of the strictness of scoring parameters implemented in automated methods and the depth of field of the ISX. Tight‐fitting nuclear masks can be created in IDEAS, but some MN that reside very close to or touch the main nuclei may still be incorrectly enveloped within the nuclear mask and can be missed. Additionally, some very small MN that would likely be scored through manual methods will be missed due to the need for an increased lower area limit on the MN mask to avoid the incorrect masking of small debris or artifacts. In a recent publication, we optimized the data analysis template created in IDEAS in an attempt to capture some of these missed MN, but only partial improvements in the way of masking modifications were possible 43. As described in that publication, we attempted to reduce the area of the BN cell mask to avoid enveloping a MN in close proximity to the main nuclei. This sometimes caused smaller, DNA‐positive debris to be incorrectly masked as a main nucleus. By the same token, reducing the area of the MN mask to score smaller MN remained an issue since this caused small image artifacts to be falsely counted as MN, especially at the periphery of the main nuclei. In addition to the complexities of image‐based data analysis, the ISX captures two dimensional images of three dimensional objects which may cause MN that are hidden behind a main nucleus to be missed. Also, some MN may reside at a different depth of field than the main nuclei which then makes identification and masking difficult due to their lower intensity. The higher MN frequencies in MONO cells when compared with BN cells observed here is likely because it is more difficult to capture highly focused BN cells containing in‐focus MN than it is to capture highly focused MONO cells containing in‐focus MN. The larger area of the BN cells may result in MN residing out of the focal plane, thus resulting in a lower rate of MN detection
In several unpublished experimental studies, the extended depth of field (EDF) option on the ISX was applied in the hopes of capturing MN lying at a different focal depths than the main nuclei. The EDF option on the ISX allows light from different focal planes to be imaged on the detector plane simultaneously. The EDF is a useful option that allows structures and probes, such as individual chromosomes in suspension 54, 55 or γH2AX foci 56, 57 to be focused onto a single two‐dimensional image. This improves the ability to count spots at different focus depths within the cell. In the case of MN assays however, the EDF has thus far been unsuccessful using both irradiated human lymphocytes in radiation biodosimetry studies and TK6 cells in toxicology studies. With the EDF option enabled, the main nuclei appear highly punctate and structures that may be candidate MN are significantly blurred. This makes resolving and masking these events nearly impossible in IDEAS. Therefore, the EDF option was not used in this work. The overall reduction in MN frequency observed here when compared with manual microscopy may simply be a general phenomenon of automated analysis methods and may not be possible to overcome with current technologies and image processing algorithms. Historically, the background frequency of MN has been shown to vary between 0.32% and 1.38% using flow cytometry and various microscopy‐based methods in genetic toxicology testing 58. In this work, the background MN frequency is 0.19% on average slightly lower than the historical lower limit. As such, caution should be observed when attempting to interpret statistically significant results at low dose levels. It is for these reasons that a separate, dedicated investigation must be performed to quantify the overall reduction in MN frequency obtained by the ISX method in comparison to most other established and validated methods including manual and automated microscopy as well as conventional flow cytometry. With respect to the conventional flow cytometry method, the lack of compatibility with Cyt‐B will make direct comparisons to other all methods that are compatible with Cyt‐B difficult or impossible. However, direct comparisons between all methods in the absence of Cyt‐B can be performed. Such a study will also provide insight into the lower limit of sensitivity of the ISX method as compared with other methods.
Despite these shortcomings, the ISX‐based method to perform the in vitro MN method offers many advantages over all other techniques established to date. Fenech et al. recently proposed several criteria that must be considered when developing automated image cytometry systems for MN assays 59. Some of these criteria include visualization of both the cytoplasm and nuclear material in MONO, BN, and POLY cells, MN assessment from cultures at varying degrees of cytotoxicity, sufficiently high magnification to ensure confidence in the quality of MN detected and the ability to quantitatively characterize nuclear morphology and MN positioning within the cytoplasm. The results demonstrated here illustrate that the ISX satisfies, or possesses the potential to satisfy, all of the proposed criteria. All samples are run on the ISX in suspension and therefore the need to create microscope slides has been eliminated. Furthermore, automated slide‐scoring methods typically do not allow visualization of the cytoplasm, which can make some positive events questionable (e.g., BN cells versus doublets) 21, 23. On the ISX, cytoplasmic imagery can be captured by the BF LED and all nuclear imagery by the fluorescent lasers, demonstrating that all cell types of interest, with and without MN, can be rapidly collected and resolved. Images containing cells with nuclei of normal morphology can then be automatically separated from other events and debris by using a series of masks and features. For example, apoptotic cells have been shown to be differentiable from healthy cells simply through the use of BF Contrast and DNA Area, without the requirement to use EMA or other viability dyes 43, 60. Healthy cells will have a lower, more uniform contrast in the brightfield images and the DNA imagery will be large in area. By comparison, images of apoptotic/necrotic cells will demonstrate dark spots and blebbing (characteristic of chromatin condensation) in Brightfield imagery and the nuclear imagery will be much smaller and more fragmented. This allows relatively simple differentiation between healthy and apoptotic/necrotic cells without the need for additional sample processing. Moreover, conventional flow cytometry methods still suffer from the lack of ability to confidently differentiate between legitimate MN and DNA‐positive debris or fragments produced during apoptosis or necrosis. This can lead to significantly overestimated MN frequencies at higher doses that induce toxicity in excess of 55% 32. Direct visualization of MN in all acquired images eliminates these concerns in the ISX‐based method. Furthermore, POLY cells and cells with multiple MN can be easily identified using manual microscopy but these events become difficult to score in the case of automated slide scoring and impossible to score in the case of conventional flow cytometry where lysis of all cells is required. In the ISX method, all cells remain intact as they flow through the cytometer and due to the spot counting features available during image processing, all of these events can be automatically identified and scored, as demonstrated in this work and in previous publications 40, 41, 42, 43, 44.
Additional advantages of the ISX method are ease of sample processing, rapid rate of data acquisition and the high numbers of cells that can be scored in a relatively short timeframe. While samples for microscope‐based methods can be prepared relatively quickly (<1 h) conventional flow methods currently require a minimum of 2.5 h to process a single sample, including multiple staining and cell lysing steps 30, 31, 35. The sample processing technique used in this work requires <2 h to process a single sample and includes only a cytoplasmic swelling step, two fixation steps and a single staining step; all of which are relatively easy to perform. The OECD Test Guideline 487 for the in vitro MN assay recommends that at least 2,000 BN cells be scored for the presence of MN per test concentration for samples treated in the absence of Cyt‐B 12. When Cyt‐B is used, it is recommended to score 2,000 BN cells per test concentration for the presence of MN (genotoxicity) along with at least 500 cells per test concentration to determine cytotoxicity. In this work, an average of 28,000 MONO cells per test concentration for samples tested in the absence of Cyt‐B were collected and scored by the ISX in about 10 min. For samples tested in the presence of Cyt‐B, an average of about 16,000 MONO, 6,000 BN and 800 POLY cells per test concentration were collected and scored in approximately 20 min. The high numbers of cells captured by the ISX illustrate the improvement in statistical robustness that can be achieved in comparison to microscope‐based versions of the assay. In addition, conventional flow cytometry methods that do not use Cyt‐B typically analyze between 20,000 and 40,000 lysed nuclei 30, 31, 61. The number of MONO cells scored from samples treated without Cyt‐B using the ISX method fall roughly in the middle of this range and have the added benefit of associated BF and fluorescent imagery for every cell collected. Furthermore, conventional flow cytometry methods do not possess the ability to distinguish between MONO, BN, and POLY cells 32, 50, whereas the data analysis template created in this work can do this relatively easily, allowing for cytotoxicity calculations to be performed. Finally, data files from all processed samples containing all required imagery (e.g., cytoplasm, nuclear material, etc.) can be stored indefinitely for re‐evaluation if necessary. This is not possible with conventional flow cytometry methods but can be performed using automated slide scanning however, slides can only be stored for a few months and must be re‐loaded to examine specific events of interest 23. In addition, in the majority of cases, the entire sample does not have to be passed through the ISX to acquire the required number of events. Any unused, fixed samples can be returned and stored for several weeks in the event that additional data collection is required. The advantages and limitations of all methods for performing the MN assay for toxicology discussed in this article are summarized in Table 3.
Table 3.
Summary of the advantages and disadvantages of visual microscopy, automated microscopy, conventional flow cytometry and imaging flow cytometry methods in the context of performing the in vitro micronucleus assay
| Scoring method | Advantages | Disadvantages |
|---|---|---|
| Visual microscopy |
Direct visualization of all required events (e.g., MONO, BN, and POLY cells and MN) Slides can be stored and re‐scored if required Validated sample preparation protocols and scoring procedures |
Laborious and suffers from low throughput Inter‐laboratory and scorer variations make MN scoring subjective High quality microscope slides are required and slide quality can vary according to laboratory conditions and technical expertise Limited number of cells can be scored; restricts statistical robustness |
| Automated microscopy |
Automatic scanning of slides allows identification of MN and MONO, BN, and POLY cells Images can be stored and slides can be re‐scanned if necessary Improved throughput when compared with visual microscopy (1,000–2,000 BN cells can be detected in about 2 min) |
High‐quality microscope slides are required and slide quality can vary according to laboratory conditions and technical expertise Lack of cytoplasmic visualization with many systems Visual verification of the image gallery required due to high rates of false positive BN cells and MN; may re‐introduce scorer bias and reduces throughput In many cases, slides must be scanned twice: first for BN cells and then again for MONO and POLY cells |
| Conventional flow cytometry |
Fully automated scoring of nuclei and MN High throughput data acquisition (several thousand events can be scored in just a few minutes) Viable nuclei can be easily differentiated from necrotic or apoptotic cells |
Cell lysis creates a solution that may contain debris; does not guarantee clear distinction between MN and DNA positive debris Absence of cytochalasin B does not permit the use of the cytokinesis‐blocked version of the MN assay; BN and POLY cells cannot be quantified |
| Imaging flow cytometry |
Direct visualization and potential for fully automated scoring of all required events (e.g., MONO, BN, and POLY cells and MN) Cytoplasm can be visualized through the use of brightfield imagery Multiple excitation lasers and image channels will allow for several cellular structures to be imaged simultaneously (e.g., nuclei, centromeres, telomeres) High throughput data acquisition (several thousand events can be scored in just a few minutes) All cellular imagery is stored in data files permitting re‐analysis if required Custom data analysis templates created using image processing algorithms allow scoring criteria for BN cells and MN to be implemented |
MN frequency obtained using IFC has been shown to be roughly 30% of that measured using manual microscopy in radiation biodosimetry Two‐dimensional projection image of a three‐dimensional cell likely causes some MN to be hidden behind the main nucleus or to reside at a different depth of focus than the main nuclei Mathematical algorithms used to perform image analysis lack the flexibility and judgment of the human eye; this may cause smaller MN or MN that reside very close to the main nuclei to be missed |
In a recent publication Verma et al. stated that “a test system that combines the high‐throughput, high‐content and multiplexing potential of flow cytometry, with the re‐validation and data storage benefits for image analysis, would be a major step forward in achieving a truly twenty‐first century approach” 50. The ISX‐based method developed here for performing the in vitro MN assay satisfies this statement and overcomes many of the challenges in slide‐based microscopy and conventional flow cytometry techniques. The work in this article presents the first step toward the development of a fully automated, ISX‐based method for performing the in vitro MN assay to assess cytotoxicity and genotoxicity. Further development, optimization and validation are required; however, these results indicate that this method may represent the next technological advancement for performing automated MN assays.
Future Directions
The methodology presented here to perform the in vitro MN assay using imaging flow cytometry can be improved upon and expanded to better identify and quantify MONO, BN, and POLY cells as well as MN. For example, since all true MN form their own nuclear envelope, markers that stain the nuclear envelope can be incorporated into sample preparation methods and imagery can be captured on the ISX. This will allow further confirmation that the identified MN are indeed genuine. Furthermore, since most MN appear relatively large at 60× magnification, it may be possible to incorporate centromere, telomere, and nuclear protein (e.g., histone) markers to better differentiate MN from nonspecific DNA positive events, and identify MN that originate from whole chromosomes versus those from chromosomal fragments. This may also allow for the development of a protocol to distinguish aneugenicity from clastogenicity. Additionally, as is done with conventional flow cytometry methods, apoptotic/necrotic markers can be added to confirm the effectiveness of the current methodology (e.g., BF contrast vs. Nuclear area) to separate these apoptotic or necrotic cells from viable cells. Due to the multi‐spectral nature of the ISX, and the wide range of masks and features available in IDEAS for data analysis, it is conceivable that many (or all) of these markers could be introduced at the same time to all samples, providing a broader picture of the cytotoxic and genotoxic processes taking place.
There are several working methods to perform the in vitro MN assay: visual microscopy, automated microscopy and conventional flow cytometry and studies in the literature have been performed to assess the comparability between these methods 50. In order to assess the accuracy of the ISX method described in this article, a dedicated investigation should performed that compares results to the existing three established methods. This will allow for the determination of how well the results obtained from the ISX method compares to other validated methods.
The current OECD guideline for performing the in vitro MN assay permits the use of several cell lines, both human and rodent as well as primary human or other mammalian peripheral blood lymphocytes 12. Several studies have been performed to test the in vitro MN assay using a number of different cell types including human peripheral blood lymphocytes 14, 49, Chinese Hamster Ovary cells 13, Chinese Hamster Lung cells 17, and mouse lymphoma L5178Y cells 16, 48, 62, demonstrating positive results for many test agents. Therefore, the assay should be performed on the ISX using additional cell types and chemical agents to develop applicable sample processing methods and to demonstrate the adaptability of the method.
Finally, the ISX possesses an autosampler option in which a 96 well plate can be loaded into the instrument allowing samples to be run with very little user intervention. A plate definition template can be defined that assigns specific instrument settings to particular wells, allowing customizable data collection for each sample. The use of the autosampler would greatly expand the number of samples that could be analyzed per experiment. This would allow laboratories using this technique to run experiments in triplicate or quadruplicate (or higher replicates) enabling robust measurements of typical background MN frequencies to be obtained. Additionally, the use of the autosampler would permit a larger number of samples to be run per experiment, allowing for a wider range of dose levels to be tested than may be possible using other methods and to examine the dynamic range of a number of cell types. All of this, combined with an increased number of replicates per dose point, may help to better interpret any chance statistically significant increases observed that may not be biologically meaningful.
All of these proposed future research directions would increase the capacity of the ISX to provide more robust and comprehensive results from the in vitro MN assay than are currently available from all other methods.
Conflict of Interest
The author is employed by MilliporeSigma, the maker of the ImageStream® imaging flow cytometer that was used in this work.
Supporting information
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Tabel 1
Supporting Information Tabel 2
Supporting Information Tabel 3
Supporting Information Table 4
Supporting Information MIFlowCyt Item Checklist
Supporting Information Figure legends
Supporting Information Table Titles
Acknowledgments
The author thanks Christine Probst (MilliporeSigma) for her efforts in developing previous forms of the IDEAS® data analysis template, as well as Dr. Haley Pugsley (MilliporeSigma), Dr. Phil Morrissey (MilliporeSigma) and Dr. Sherree Friend (MilliporeSigma) for reviewing and editing the manuscript.
Literature Cited
- 1. Bonassi S, Znaor A, Ceppi M, Lando C, Chang WP, Holland N, Kirsch‐Volders M, Zeiger E, Ban S, Barale R. An increased micronucleus frequency in peripheral blood lymphocytes predicts the risk of cancer in humans. Carcinogenesis 2006;28:625–631. [DOI] [PubMed] [Google Scholar]
- 2. Fenech M. The in vitro micronucleus technique. Mutat Res 2000;455:81–95. [DOI] [PubMed] [Google Scholar]
- 3. Fenech M. The lymphocyte cytokinesis‐block micronucleus cytome assay and its application in radiation biodosimetry. Health Phys 2010;98:234–243. [DOI] [PubMed] [Google Scholar]
- 4. Hintzsche H, Hemmann U, Poth A, Utesch D, Lott J, Stopper H. Fate of micronuclei and micronucleated cells. Mutat Res 2017;771:85–98. [DOI] [PubMed] [Google Scholar]
- 5. Fenech M. The advantages and disadvantages of the cytokinesis‐block micronucleus method. Mutat Res 1997;392:11–18. [DOI] [PubMed] [Google Scholar]
- 6. Elhajouji A, Cunha M, Kirsch‐Volders M. Spindle poisons can induce polyploidy by mitotic slippage and micronucleate mononucleates in the cytokinesis‐block assay. Mutagenesis 1998;13:193–198. [DOI] [PubMed] [Google Scholar]
- 7. Elhajouji A. Mitomycin C, 5‐fluoruracil, colchicine and etoposide tested in the in vitro mammalian cell micronucleus test (MNvit) in the human lymphoblastoid cell line TK6 at Novartis in support of OECD draft Test Guideline 487. Mutat Res 2010;702:157–162. [DOI] [PubMed] [Google Scholar]
- 8. Topham CH, Taylor SS. Mitosis and apoptosis: How is the balance set? Curr Opin Cell Biol 2013;25:780–785. [DOI] [PubMed] [Google Scholar]
- 9. Fenech M. Cytokinesis‐block micronucleus cytome assay. Nat Protoc 2007;2:1084–1104. [DOI] [PubMed] [Google Scholar]
- 10. Fenech M, Holland N, Chang WP, Zeiger E, Bonassi S. The HUman MicroNucleus Project—An international collaborative study on the use of the micronucleus technique for measuring DNA damage in humans. Mutat Res 1999;428:271–283. [DOI] [PubMed] [Google Scholar]
- 11. Kirsch‐Volders M, Sofuni T, Aardema M, Albertini S, Eastmond D, Fenech M, Ishidate M, Jr Kirchner S, Lorge E, Morita T, et al. Report from the in vitro micronucleus assay working group. Mutat Res 2003;540:153–163. [DOI] [PubMed] [Google Scholar]
- 12.OECD Library. Test No. 487: In vitro mammalian cell micronucleus test. OECD Guidelines for the Testing of Chemicals, Section 4 2016; 10.1787/20745788:ISSN 2074-5788. [DOI]
- 13. Aardema MJ, Snyder RD, Spicer C, Divi K, Morita T, Mauthe RJ, Gibson DP, Soelter S, Curry PT, Thybaud V, et al. SFTG international collaborative study on in vitro micronucleus test. III. Using CHO cells. Mutat Res 2006;607:61–87. [DOI] [PubMed] [Google Scholar]
- 14. Clare MG, Lorenzon G, Akhurst LC, Marzin D, van Delft J, Montero R, Botta A, Bertens A, Cinelli S, Thybaud V, et al. SFTG international collaborative study on in vitro micronucleus test. II. Using human lymphocytes. Mutat Res 2006;607:37–60. [DOI] [PubMed] [Google Scholar]
- 15. Lorge E, Thybaud V, Aardema MJ, Oliver J, Wakata A, Lorenzon G, Marzin D. SFTG international collaborative study on in vitro micronucleus test. I. General conditions and overall conclusions of the study. Mutat Res 2006;607:13–36. [DOI] [PubMed] [Google Scholar]
- 16. Oliver J, Meunier JR, Awogi T, Elhajouji A, Ouldelhkim MC, Bichet N, Thybaud V, Lorenzon G, Marzin D, Lorge E. SFTG international collaborative study on in vitro micronucleus test. V. Using L5178Y cells. Mutat Res 2006;607:125–152. [DOI] [PubMed] [Google Scholar]
- 17. Wakata A, Matsuoka A, Yamakage K, Yoshida J, Kubo K, Kobayashi K, Senjyu N, Itoh S, Miyajima H, Hamada S, et al. SFTG international collaborative study on in vitro micronucleus test. IV. Using CHL cells. Mutat Res 2006;607:88–124. [DOI] [PubMed] [Google Scholar]
- 18. Fenech M, Bonassi S, Turner J, Lando C, Ceppi M, Chang WP, Holland N, Kirsch‐Volders M, Zeiger E, Bigatti MP, et al. Intra‐ and inter‐laboratory variation in the scoring of micronuclei and nucleoplasmic bridges in binucleated human lymphocytes: Results of an international slide‐scoring exercise by the HUMN project. Mutat Res 2003;534:45–64. [DOI] [PubMed] [Google Scholar]
- 19. Decordier I, Papine A, Plas G, Roesems S, Vande Loock K, Moreno‐Palomo J, Cemeli E, Anderson D, Fucic A, Marcos R, et al. Automated image analysis of cytokinesis‐blocked micronuclei: an adapted protocol and a validated scoring procedure for biomonitoring. Mutagenesis 2008;24:85–93. [DOI] [PubMed] [Google Scholar]
- 20. Decordier I, Papine A, Vande Loock K, Plas G, Soussaline F, Kirsch‐Volders M. Automated image analysis of micronuclei by IMSTAR for biomonitoring. Mutagenesis 2011;26:163–168. [DOI] [PubMed] [Google Scholar]
- 21. Rossnerova A, Spatova M, Schunck C, Sram RJ. Automated scoring of lymphocyte micronuclei by the MetaSystems Metafer image cytometry system and its application in studies of human mutagen sensitivity and biodosimetry of genotoxin exposure. Mutagenesis 2011;26:169–175. [DOI] [PubMed] [Google Scholar]
- 22. Schunck C, Johannes T, Varga D, Lorch T, Plesch A. New developments in automated cytogenetic imaging: unattended scoring of dicentric chromosomes, micronuclei, single cell gel electrophoresis, and fluorescence signals. Cytogenet Genome Res 2004;104:383–389. [DOI] [PubMed] [Google Scholar]
- 23. Seager AL, Shah U‐K, Brüsehafer K, Wills J, Manshian B, Chapman KE, Thomas AD, Scott AD, Doherty AT, Doak SH, et al. Recommendations, evaluation and validation of a semi‐automated, fluorescent‐based scoring protocol for micronucleus testing in human cells. Mutagenesis 2014;29:155–164. [DOI] [PubMed] [Google Scholar]
- 24. Darzynkiewicz Z, Smolewski P, Holden E, Luther E, Henriksen M, Francois M, Leifert W, Fenech M. Laser scanning cytometry for automation of the micronucleus assay. Mutagenesis 2011;26:153–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Francois M, Hochstenbach K, Leifert W, Fenech MF. Automation of the cytokinesis‐block micronucleus cytome assay by laser scanning cytometry and its potential application in radiation biodosimetry. Biotechniques 2014;57:309–312. [DOI] [PubMed] [Google Scholar]
- 26. Smolewski P, Ruan Q, Vellon L, Darzynkiewicz Z. Micronuclei assay by laser scanning cytometry. Cytometry 2001;45:19–26. [DOI] [PubMed] [Google Scholar]
- 27. Nüsse M, Marx K. Flow cytometric analysis of micronuclei in cell cultures and human lymphocytes: Advantages and disadvantages. Mutat Res 1997;392:109–115. [DOI] [PubMed] [Google Scholar]
- 28. Roman D, Locher F, Suter W, Cordier A, Bobadilla M. Evaluation of a new procedure for the flow cytometric analysis of in vitro , chemically induced micronuclei in V79 cells. Environ Mol Mutagen 1998;32:387–396. [PubMed] [Google Scholar]
- 29. Wessels JM, Nüsse M. Flow cytometric detection of micronuclei by combined staining of DNA and membranes. Cytometry 1995;19:201–208. [DOI] [PubMed] [Google Scholar]
- 30. Avlasevich SL, Bryce SM, Cairns SE, Dertinger SD. in vitro micronucleus scoring by flow cytometry: Differential staining of micronuclei versus apoptotic and necrotic chromatin enhances assay reliability. Environ Mol Mutagen 2006;47:56–66. [DOI] [PubMed] [Google Scholar]
- 31. Bryce SM, Bemis JC, Avlasevich SL, Dertinger SD. in vitro micronucleus assay scored by flow cytometry provides a comprehensive evaluation of cytogenetic damage and cytotoxicity. Mutat Res 2007;630:78–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Avlasevich S, Bryce S, De Boeck M, Elhajouji A, Van Goethem F, Lynch A, Nicolette J, Shi J, Dertinger S. Flow cytometric analysis of micronuclei in mammalian cell cultures: Past, present and future. Mutagenesis 2011;26:147–152. [DOI] [PubMed] [Google Scholar]
- 33. Bryce SM, Avlasevich SL, Bemis JC, Lukamowicz M, Elhajouji A, Van Goethem F, De Boeck M, Beerens D, Aerts H, Van Gompel J, et al.. Interlaboratory evaluation of a flow cytometric, high content in vitro micronucleus assay. Mutat Res 2008;650:181–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bryce SM, Avlasevich SL, Bemis JC, Phonethepswath S, Dertinger SD. Miniaturized flow cytometric in vitro micronucleus assay represents an efficient tool for comprehensively characterizing genotoxicity dose–response relationships. Mutat Res 2010;703:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bryce SM, Avlasevich SL, Bemis JC, Tate M, Walmsley RM, Saad F, Van Dijck K, De Boeck M, Van Goethem F, Lukamowicz‐Rajska M, et al. Flow cytometric 96‐well microplate‐based in vitro micronucleus assay with human TK6 cells: Protocol optimization and transferability assessment. Environ Mol Mutagen 2013;54:180–194. [DOI] [PubMed] [Google Scholar]
- 36. Plamadeala C, Wojcik A, Creanga D. Micronuclei versus chromosomal aberrations induced by X‐ray in radiosensitive mammalian cells. Iran J Public Health 2015;44:325–331. [PMC free article] [PubMed] [Google Scholar]
- 37. Fenech M. Commentary on the SFTG international collaborative study on the in vitro micronucleus test: To Cyt‐B or not to Cyt‐B? Mutat Res 2006;607:9–12. [DOI] [PubMed] [Google Scholar]
- 38. Zuba‐Surma EK, Kucia M, Abdel‐Latif A, Lillard JW, Jr. , Ratajczak MZ. The ImageStream System: A key step to a new era in imaging. 2007;279–290. [PubMed]
- 39. Basiji DA. Principles of Amnis imaging flow cytometry. Methods Mol Biol 2016;1389:13–21. [DOI] [PubMed] [Google Scholar]
- 40. Rodrigues MA, Beaton‐Green LA, Kutzner BC, Wilkins RC. Automated analysis of the cytokinesis‐block micronucleus assay for radiation biodosimetry using imaging flow cytometry. Radiat Environ Biophys 2014;53:273–282. [DOI] [PubMed] [Google Scholar]
- 41. Rodrigues MA, Beaton‐Green LA, Kutzner BC, Wilkins RC. Multi‐parameter dose estimations in radiation biodosimetry using the automated cytokinesis‐block micronucleus assay with imaging flow cytometry. Cytom A 2014;85:883–893. [DOI] [PubMed] [Google Scholar]
- 42. Rodrigues MA, Beaton‐Green LA, Wilkins RC. Validation of the cytokinesis‐block micronucleus assay using imaging flow cytometry for high throughput radiation biodosimetry. Health Phys 2016;110:29–36. [DOI] [PubMed] [Google Scholar]
- 43. Rodrigues MA, Probst CE, Beaton‐Green LA, Wilkins RC. Optimized automated data analysis for the cytokinesis‐block micronucleus assay using imaging flow cytometry for high throughput radiation biodosimetry. Cytom A 2016;89:653–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rodrigues MA, Probst CE, Beaton‐Green LA, Wilkins RC. The effect of an optimized imaging flow cytometry analysis template on sample throughput in the reduced culture cytokinesis‐block micronucleus assay. Radiat Prot Dosim 2016;172:223–229. [DOI] [PubMed] [Google Scholar]
- 45. Wilkins RC, Rodrigues MA, Beaton‐Green LA. The application of imaging flow cytometry to high‐throughput biodosimetry. Genome Integr 2017;8:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fenech M, Chang WP, Kirsch‐Volders M, Holland N, Bonassi S, Zeiger E. HUMN project: Detailed description of the scoring criteria for the cytokinesis‐block micronucleus assay using isolated human lymphocyte cultures. Mutat Res 2003;534:65–75. [DOI] [PubMed] [Google Scholar]
- 47. Högstedt B, Karlsson A. The size of micronuclei in human lymphocytes varies according to inducing agent used. Mutat Res 1985;156:229–232. [DOI] [PubMed] [Google Scholar]
- 48. Lorge E. Comparison of different cytotoxicity measurements for the in vitro micronucleus assay using L5178Y and TK6 cells in support of OECD draft Test Guideline 487. Mutat Res 2010;702:199–207. [DOI] [PubMed] [Google Scholar]
- 49. Fowler P, Smith R, Smith K, Young J, Jeffrey L, Carmichael P, Kirkland D, Pfuhler S. Reduction of misleading (“false”) positive results in mammalian cell genotoxicity assays. III: Sensitivity of human cell types to known genotoxic agents. Mutat Res 2014;767:28–36. [DOI] [PubMed] [Google Scholar]
- 50. Verma JR, Rees BJ, Wilde EC, Thornton CA, Jenkins GJS, Doak SH, Johnson GE. Evaluation of the automated MicroFlow® and Metafer™ platforms for high‐throughput micronucleus scoring and dose response analysis in human lymphoblastoid TK6 cells. Arch Toxicol 2017;91:2689–2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Fellows MD, O'Donovan MR. Etoposide, cadmium chloride, benzo[a]pyrene, cyclophosphamide and colchicine tested in the in vitro mammalian cell micronucleus test (MNvit) in the presence and absence of cytokinesis block using L5178Y mouse lymphoma cells and 2‐aminoanthracene tested in MNvit in the absence of cytokinesis block using TK6 cells at AstraZeneca UK, in support of OECD draft Test Guideline 487. Mutat Res 2010;702:163–170. [DOI] [PubMed] [Google Scholar]
- 52. Massey ED, Hinchliffe S. 2‐Aminoanthracene, diethylstilboesterol and vinblastine tested in the in vitro mammalian cell micronucleus test at British American Tobacco UK in support of the OECD draft Test Guideline 487. Mutat Res 2010;702:208–211. [DOI] [PubMed] [Google Scholar]
- 53. Hashimoto K, Nakajima Y, Matsumura S, Chatani F. Comparison of four different treatment conditions of extended exposure in the in vitro micronucleus assay using TK6 lymphoblastoid cells. Regul Toxicol Pharmacol 2011;59:28–36. [DOI] [PubMed] [Google Scholar]
- 54. Beaton LA, Ferrarotto C, Kutzner BC, McNamee JP, Bellier PV, Wilkins RC. Analysis of chromosome damage for biodosimetry using imaging flow cytometry. Mutat Res 2013;756:192–195. [DOI] [PubMed] [Google Scholar]
- 55. Beaton‐Green LA, Rodrigues MA, Lachapelle S, Wilkins RC. Foundations of identifying individual chromosomes by imaging flow cytometry with applications in radiation biodosimetry. Methods 2017;112:18–24. [DOI] [PubMed] [Google Scholar]
- 56. Bourton EC, Plowman PN, Zahir SA, Senguloglu GU, Serrai H, Bottley G, Parris CN. Multispectral imaging flow cytometry reveals distinct frequencies of gamma‐H2AX foci induction in DNA double strand break repair defective human cell lines. Cytom A 2012;81:130–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Parris CN, Zahir SA, Al‐Ali H, Bourton EC, Plowman C, Plowman PN. Enhanced gamma‐H2AX DNA damage foci detection using multimagnification and extended depth of field in imaging flow cytometry. Cytom A 2015;87:717–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lovell DP, Fellows M, Marchetti F, Christiansen J, Elhajouji A, Hashimoto K, Kasamoto S, Li Y, Masayasu O, Moore MM, et al.. Analysis of negative historical control group data from the in vitro micronucleus assay using TK6 cells. Mutat Res 2018;825:40–50. [DOI] [PubMed] [Google Scholar]
- 59. Fenech M, Kirsch‐Volders M, Rossnerova A, Sram R, Romm H, Bolognesi C, Ramakumar A, Soussaline F, Schunck C, Elhajouji A, et al. HUMN project initiative and review of validation, quality control and prospects for further development of automated micronucleus assays using image cytometry systems. Int J Hyg Environ Health 2013;216:541–552. [DOI] [PubMed] [Google Scholar]
- 60. Zuba‐Surma EK, Kucia M, Abdel‐Latif A, Lillard JW Jr., Ratajczak MZ. The ImageStream System: A key step to a new era in imaging. Folia Histochem Cytobiol 2007;45:279–290. [PubMed] [Google Scholar]
- 61. Lukamowicz M, Woodward K, Kirsch‐Volders M, Suter W, Elhajouji A. A flow cytometry based in vitro micronucleus assay in TK6 cells—Validation using early stage pharmaceutical development compounds. Environ Mol Mutagen 2011;52:363–372. [DOI] [PubMed] [Google Scholar]
- 62. Kirchner S, Zeller A. Comparison of different cytotoxicity measures for the in vitro micronucleus test (MNVit) in L5178Y tk(+/−) cells: Summary of 4 compounds (Mitomycin C, Cyclophosphamide, Colchicine and Diethylstilboestrol) with clastogenic and aneugenic mode of action. Mutat Res 2010;702:193–198. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Figure 1
Supporting Information Figure 2
Supporting Information Tabel 1
Supporting Information Tabel 2
Supporting Information Tabel 3
Supporting Information Table 4
Supporting Information MIFlowCyt Item Checklist
Supporting Information Figure legends
Supporting Information Table Titles