

Abstract
Thiol‐ene click reaction was successfully employed for chemical modification of graphene oxide (GO) by one‐step synthesis. Herein, 2,2‐azobis(2‐methylpropionitrile) (AIBN) was used as thermal catalyst and cysteamine hydrochloride (HS−(CH2)2−NH2HCl) was used as thiol‐containing compound, which is incorporated to GO surface upon reaction with the C=C bonds. The hydrochloride acts as protecting group for the amine, which is finally eliminated by adding sodium hydroxide. The modified GO contains both S‐ and N‐containing groups (NS‐GO). We found that NS‐GO sheets form good dispersion in water, ethanol, and ethylene glycol. These graphene dispersions can be processed into functionalized graphene film. Besides, it was demonstrated that NS‐GO was proved to be an excellent host matrix for platinum nanoparticles. The developed method paves a new way for graphene modification and its functional nanocomposites.
Keywords: chemical modification, graphene, nanomaterials, platinum, thiol-ene reactions
Graphene is an atom‐thick crystal of sp2‐bonded carbon atoms arranged in a hexagonal lattice, which was reported for its existence the first time in 2004.1 Since then, many extraordinary properties of graphene have been established and reported, such as high thermal conductivity (≈5000 W mK−1),2 fast charged carrier mobility (≈200 000 cm2 V−1 s−1),3, 4 high Young's modulus (≈1 TPa),5 and huge surface area (2630 m2 g−1).6 Graphene has been widely considered as one of the most researched materials in the last decade owing to its exceptional properties as was mentioned above.6, 7 However, due to its high inertness, graphene needs to be chemically modified/functionalized for many applications, especially energy storages, such as electrodes in supercapacitors and batteries, catalyst supporters in fuel cells, and reinforcements in functional composites.8, 9, 10 The functionalization of graphene is the key to achieve stable graphene dispersions in various solvents leading to better processibility of these functional materials. In addition, it can be used in combination/incorporation with other materials to produce high performance multifunctional materials. Special attentions have been given to the modifications of graphene oxide by the oxygen functionalities;11 however, the effectiveness of modifications is limited due to low density/chemical activity of these oxygen‐containing groups. The chemical modifications of graphene sp2 backbone have been done to date including nucleophilic addition,12 cycloaddition,13 free radical addition,14 substitution,15 and rearrangement reactions.16
Click chemistry have been successfully applied in materials synthesis and modification owing to its advantages, such as highly efficient and simple to carry out under benign conditions with a vast range of chemical species.17 Recently, there have been few examples of using click reactions for graphene modification for polymer composites to increase electrically conductivity and mechanical strength. An alkyne–azide coupling by using CuI as catalyst used for graphene functionalization was reported.18 For this, alkyne–azide reaction, low molecular weight SH‐terminated polyethylene (PE) was used to covalently bond to the graphene sheets. This approach required another additional functionalization, in which graphene needs to be modified to have alkyne or azide functional groups and utilized toxic CuI catalyst. Thiol‐ene and thiol‐yne reactions have also been carried out for preparation of polymer/graphene composites.18 However, the reactivity of thiol group is known to decrease with increasing the size of thiol‐containing compound. Thus, yield of reaction between HS‐terminated PE and graphene would not be high enough for many applications, and it may be difficult to control.
Different from all above mentioned methods, herein, we have successfully employed thiol‐ene click reaction to functionalize graphene oxide (see Scheme 1 and the Experimental Section) by addition directly to C=C bond of graphene network in a reasonably mild reaction condition. The thiol‐ene click reactions offer many advantages including, for example, high regioselectivity, mild reaction conditions, and high conversion. By this chemistry, both sulfur and nitrogen atoms are able to be doped on graphene surface in one reaction, for example, by using cysteamine hydrochloride (HS−(CH2)2−NH2HCl) as the reagent in the reaction. It should be emphasized that in the click reaction, the thiol compounds can be added to every double bond in carbon network leading to extremely high functional groups on graphene surface, which are difficult to obtain otherwise. This developed method could be further applied to many other functional groups as long as the reagents containing thiol moieties. Different functionalities and their levels can be controlled by changing thiol agents and reaction parameters.
Scheme 1.
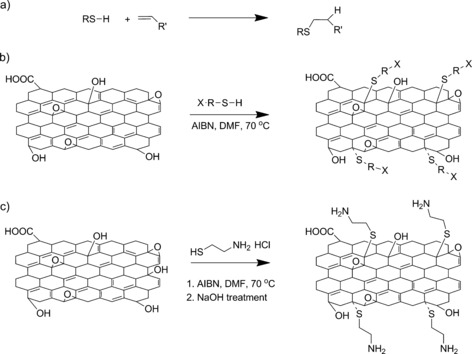
Preparation of functional graphene oxide by thiol‐ene click chemistry: Thiol‐ene reaction, which is hydrothiolation of a C=C bond with anti‐Markovnikov regioselectivity orientation (a); synthetic route for graphene oxide modification via thiol‐ene click reaction (b); and an example of the thiol‐ene approach by using GO and cysteamine hydrochloride (c).
It was noticed that cysteamine hydrochloride was used instead of cysteamine. Hydrochloride was used to protect the amine groups from reacting with the carboxylic functionalities of the GO surfaces, which forms amide functionalities. The hydrochloride was then eliminated from product by adding an excess amount of sodium hydroxide by acid/base reaction. X‐ray photoelectron spectroscopy (XPS) high‐resolution regional analysis confirmed the successful incorporation of cysteamine onto graphene oxide surfaces (Figure 1). The peaks at 284.9, 532.4, 400.0, and 163.8 eV correspond to C1s, O1s, N1s, and S2p, respectively. The overall spectrum is shown in Figure S1 in the Supporting Information. XPS quantitative data showed that the presence of S and N in chemically well‐defined states and in equal amounts (1.5±0.2 atom %) exhibit the successful functionalization via thiol‐ene reaction.
Figure 1.
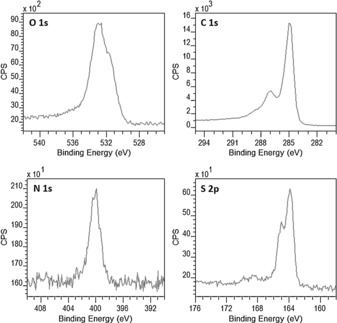
XPS spectra showing the presence of both S and N in equal amounts exhibiting the successful functionalization by thiol‐ene reaction.
Depending on the functional groups that are covalently bonded to the graphene network, the graphene solubility in both organic and inorganic media could be achieved. We found that the obtained NS‐GO flakes were dispersed well in water or ethanol upon mild sonication and the dispersion were stable for at least 24 h. We fabricated NS‐GO paper by vacuum filtration the graphene dispersion (Figure S2 in the Supporting Information). It was found that the paper was flexible and mechanically strong.
The presence of nitrogen and sulfur atoms on graphene flake in NS‐GO can play as anchoring sites to absorb and stabilize the nanoparticles on the graphene surface. Thus, the functional graphene can be a good supporter for nanoparticle catalysts, such as platinum, palladium, copper.19 We demonstrated herein that the NS‐GO is a good support for platinum nanoparticle deposition, as illustrated in Scheme 2 and described in the Experimental Section.
Scheme 2.
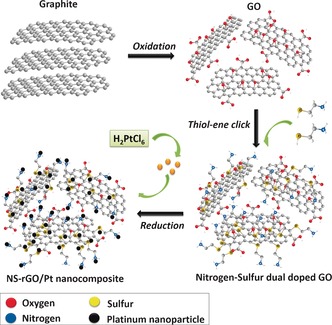
Preparation route for functional graphene by thiol‐ene click chemistry and preparation of electrically conductive graphene/Pt nanocomposites.
Size and size distribution of Pt nanoparticles in NS‐rGO/Pt nanocomposite were studied by using transmission electron microscopy (TEM, JEOL JEM‐2200FS). TEM images in Figure 2 show that Pt particles are uniformly coated NS‐rGO surfaces. The Pt sizes are estimated to be around five nanometers. The small size and uniform distribution of Pt nanoparticles on the graphene surfaces could be attributed to the present of several functional groups on the graphene surface, especially amine groups and the remaining oxygen functionalities. Morphology of the nanocomposite is additionally studied by a field emission scanning electron microscope (FE‐SEM, Jeol‐7500F). A SEM image showed that the whole graphene flake is completely covered by Pt nanoparticles, which supports TEM observation (Figures 3 and S3 in the Supporting Information).
Figure 2.
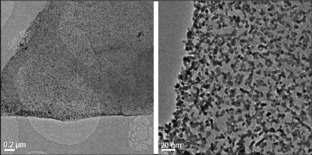
TEM images of NS‐rGO/Pt nanocomposites containing 38 wt % of Pt nanoparticles at two different magnifications revealing the homogeneous distribution of Pt nanoparticles with an average diameter of around 5 nm.
Figure 3.
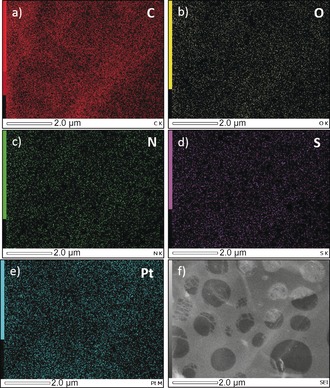
EDS mapping of NS‐rGO/Pt nanocomposite containing 38 wt % of Pt nanoparticles showing the homogeneous coating of C, O, N, S elements on the GO flake (a–d). Pt nanoparticles are uniformly distributed on GO (e). The mapping was taken from area shown in the SEM image (f). The sample for SEM observation was prepared by drop‐cast at very low concentration of NS‐rGO/Pt dispersion (in ethanol) on a TEM copper grid.
In another example, we mixed double carbon nanotube (DWCNT, Nanocyl NC2100) with GO, weight ratio of 50/50 by weight, and did the thiol‐ene modification of this mixture in the same way for NS‐GO. The obtained NS‐GO/DWCNT was then used to prepare NS‐rGO/DWCNT/Pt composite in the same way for NS‐rGO/Pt composite. The TEM demonstrates that Pt nanoparticles do not bind to the DWCNT surfaces but only to the functionalized GO flakes (Figure S4 in the Supporting Information). This phenomenon implies that the DWCNT surfaces were remained intact under thiol‐ene reaction. The result may indicate the presence of oxygen functionalities on GO surface could play an important role in thiol‐ene reaction of GO as activation of the C=C bonds. This mechanism should be studied in more detail and reported in the future.
Introduction of heteroatoms into graphene structure is an effective way to tailor the chemical, electrical, and catalyst properties of the materials.20 For example, graphene oxide has been doped with S by using benzyl disulfide,21 or mixture of SO2, H2S, and CS2.22 These S‐doped graphenes have been used as metal‐free catalyst for oxygen‐reduction reactions in fuel cells. However, it should be noted that these approaches used high temperature reactions and/or toxic chemical, which may limit the practical applications. Very recently, a work by Pumera's group has shown that doping with N, B, Al, S, Cl, and Mn can be achieved when graphite is oxidized to graphite oxide and graphite oxide is reduced to chemically functionalized graphene oxide.23 This study could provide us an alternative way of doping these elements and/or other elements into graphene structure by oxidation/reduction. Thus, more work should be dedicated to this approach.
In summary, a new thiol‐ene click approach has been employed successfully for graphene oxide by using cysteamine hydrochloride as an example. Varying the cysteamine hydrochloride amount and also the carbon double bond network of carbon in the GO may control the functionalization level. The thiol‐ene click reaction approach developed herein provides a powerful chemical tool for preparation of functional graphene materials.
Experimental Section
Chemical modification of GO through one‐step thiol‐ene click reaction (NS‐GO)
Graphite oxide was prepared by a modified Hummers’ method (see the Supporting Information).24 The graphite oxide was exfoliated in water by using tip sonication to obtain GO dispersion with a solid content of 5 mg mL−1. GO dispersion was freeze dried and subsequently vacuum dried to obtain GO powder. An amount of 100 mg of GO powder was then dispersed in 50 mL of N,N‐dimethylformamide (DMF) solvent by using ultrasonication for 30 min and was subsequently filled in three‐necked round bottom flask reactor equipped with a magnetic stirrer. Nitrogen bubbling was carried for 30 min to introduce inert environment. The solution of 2,2‐azobis(2‐methylpropionitrile) (AIBN, thermal catalyst) and cysteamine hydrochloride in 5 mL of DMF was injected to the reaction mixture. Nitrogen bubbling was continued for 30 min. The reaction mixture was heated to 70 °C using oil bath and kept for 12 h. The reaction was cooled to RT, and a solution of NaOH (1 m) in ethanol/water (15:5 mL) was added to the mixture while stirring. The mixture was washed by vacuum filtration to eliminate impurities with ethanol (2 times) and water (3 times). The product was freeze dried and subsequently vacuum dried at 60 °C to remove water. The color of NS‐GO is black compared to dark yellow of GO.
Preparation of NS‐rGO/Pt nanocomposite
An amount of NS‐GO (100 mg) was dispersed in ethylene glycol (EG) with a concentration of 1.5 mg mL−1. This mixture was treated with ultrasonic for 30 min to introduce good dispersion of NS‐GO sheets in the solvent. The mixture was supplied into a three‐necked round bottom flask equipped with a magnetic stirring. Nitrogen bubbling was carried out for 30 min. After that, an amount of H2PtCl6, which was pre‐dissolved in 5 mL EG, was injected to the solution. The amount of the salt was calculated with the Pt content of 38 wt % compared to that of the graphene. After 30 min of nitrogen bubbling, the solution was heated to 140 °C for 4 h to reduce Pt ions to Pt metallic nanoparticles. The solution was cooled to RT, and hydrazine (100 μL) was injected to the solution. The mixture was heated to 95 °C and kept for 1 h for further reduction. The reaction mixture was then cooled to RT and precipitated in DI water (200 mL). The precipitate was collected by centrifugation and washed with DI water five times. It was then freeze dried for 48 h and vacuum dried at 60 °C for 24 h.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Finnish Academy Funding 12137759 and 13272725 are acknowledged for the financial support. Dr. Hua Jang at Aalto University is thanked for the TEM measurement. This work made use of the Aalto University Nanomicroscopy Center (Aalto‐NMC) premises.
N. D. Luong, L. H. Sinh, L.-S. Johansson, J. Campell, J. Seppälä, Chem. Eur. J. 2015, 21, 3183.
References
- 1. Novoselov K. S., Geim A. K., Morozov S. V., Jiang D., Zhang Y., Dubonos S. V., Grigorieva I. V., Firsov A. A., Science 2004, 306, 666–669. [DOI] [PubMed] [Google Scholar]
- 2. Balandin A. A., Ghosh S., Bao W., Calizo I., Teweldebrhan D., Miao F., Lau C. N., Nano Lett. 2008, 8, 902–907. [DOI] [PubMed] [Google Scholar]
- 3. Bolotin K. I., Sikes K. J., Jiang Z., Klima M., Fudenberg G., Hone J., Kim P., Stormer H. L., Solid State Commun. 2008, 146, 351–355. [Google Scholar]
- 4. Du X., Skachko I., Barker A., Andrei E. Y., Nat. Nanotechnol. 2008, 3, 491–495. [DOI] [PubMed] [Google Scholar]
- 5. Lee C., Wei X., Kysar J. W., Hone J., Science 2008, 321, 385–388. [DOI] [PubMed] [Google Scholar]
- 6. Geim A. K., Novoselov K. S., Nat. Mater. 2007, 6, 183–191. [DOI] [PubMed] [Google Scholar]
- 7. Rodríguez-Pérez L., Herranz M. A., Martín N., Chem. Commun. 2013, 49, 3721–3735. [DOI] [PubMed] [Google Scholar]
- 8. Chua C. K., Pumera M., Chem. Soc. Rev. 2013, 42, 3222–3233. [DOI] [PubMed] [Google Scholar]
- 9. Hou J., Shao Y., Ellis M. W., Moore R. B., Yi B., Phys. Chem. Chem. Phys. 2011, 13, 15384–15402. [DOI] [PubMed] [Google Scholar]
- 10. Mahmood N., Zhang C., Yin H., Hou Y., J. Mater. Chem. A 2014, 2, 15–32. [Google Scholar]
- 11. Dreyer D. R., Park S., Bielawski C. W., Ruoff R. S., Chem. Soc. Rev. 2010, 39, 228–240. [DOI] [PubMed] [Google Scholar]
- 12. Economopoulos S. P., Rotas G., Miyata Y., Shinohara H., Tagmatarchis N., ACS Nano 2010, 4, 7499–7507. [DOI] [PubMed] [Google Scholar]
- 13. Chua C. K., Ambrosi A., Pumera M., Chem. Commun. 2012, 48, 5376–5378. [DOI] [PubMed] [Google Scholar]
- 14. Lomeda J. R., Doyle C. D., Kosynkin D. V., Hwang W. F., Tour J. M., J. Am. Chem. Soc. 2008, 130, 16201–16206. [DOI] [PubMed] [Google Scholar]
- 15. Chua C. K., Pumera M., Chem. Asian J. 2012, 7, 1009–1012. [DOI] [PubMed] [Google Scholar]
- 16. Collins W. R., Lewandowski W., Schmois E., Walish J., Swager T. M., Angew. Chem. Int. Ed. 2011, 50, 8848–8852; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9010–9014. [Google Scholar]
- 17. Hoyle C. E., Lowe A. B., Bowman C. N., Chem. Soc. Rev. 2010, 39, 1355–1387. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Castelaín M., Martínez G., Marco C., Ellis G., Salavagione H. J., Macromolecules 2013, 46, 8980–8987; [Google Scholar]
- 18b. Castelaín M., Martínez G., Ellis G., Salavagione H. J., Chem. Com. 2013, 49, 8967–8969; [DOI] [PubMed] [Google Scholar]
- 18c. Iskin B., Yilmaz G., Yagci Y., Chem. Eur. J. 2012, 18, 10254–10257. [DOI] [PubMed] [Google Scholar]
- 19. Yang Z., Nie H., Chen X., Chen X., Huang S., J. Power Sources 2013, 236, 238–249. [Google Scholar]
- 20. Wang H., Maiyalagan T., Wang X., ACS Catal. 2012, 2, 781–794. [Google Scholar]
- 21. Poh H. L., Simek P., Sofer Z., Pumera M., ACS Nano 2013, 7, 5262–5272. [DOI] [PubMed] [Google Scholar]
- 22. Yang Z., Yao Z., Li G., Fang G., Nie H., Liu Z., Zhou Z. X., Chen X., Huang S., ACS Nano 2012, 6, 205–211. [DOI] [PubMed] [Google Scholar]
- 23.C. K. Chua, A. Ambrosi, Z. Sofer, A. Mackova, V. Havranek, I. Tomandl, M. Pumera, Chem. Eur. J 2014, in press. [DOI] [PubMed]
- 24. Patel M. U. M., Luong N. D., Seppälä J., Tchernychova E., Dominko R., J. Power Sources 2014, 254, 55–61. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary