

Abstract
β‐cell destruction in type 1 diabetes (T1D) results from the effect of inflammation and autoimmunity. In response to inflammatory signals, islet cells engage adaptive mechanisms to restore and maintain cellular homeostasis. Among these mechanisms, the unfolded protein response (UPR) leads to a reduction of the general protein translation rate, increased production of endoplasmic reticulum chaperones and the initiation of degradation by activation of the ER associated degradation pathway (ERAD) in which newly synthetized proteins are ubiquitinylated and processed through the proteasome. This adaptive phase is also believed to play a critical role in the development of autoimmunity by the generation of neoantigens. While we have previously investigated the effect of stress on transcription, translation and post‐translational events as possible source for neoantigens, the participation of the degradation machinery, yet crucial in the generation of antigenic peptides, remains to be investigated in the context of T1D pathology. In this review, we will describe the relation between the unfolded protein response and the Ubiquitin Proteasome System (UPS) and address the role of the cellular degradation machinery in the generation of antigens. Learning from tumour immunology, we propose how these processes may unmask β‐cells by triggering the generation of aberrant peptides recognized by the immune cells.
Keywords: autoimmunity, β‐cell, ER stress, neoantigen, proteasome
1. INTRODUCTION
Type 1 diabetes (T1D) is an autoimmune disease characterized by destruction of pancreatic β‐cells mediated by autoreactive T‐cells. During insulitis, proinflammatory cytokines released by infiltrating immune cells disturb the endoplasmic reticulum homeostasis leading to ER stress.1 In response to these environmental changes, β‐cells initiate an unfolded protein response (UPR)2, 3 by activation of ER sensors (i.e., IRE1a, PERK and ATF6) and induction of stress factors (ATF3, ATF4, XBP1 splicing). Activation of these pathways promotes cellular repair by reducing protein translation, promoting degradation of misfolded proteins and by activating chaperone protein synthesis to restore ER equilibrium.
This adaptive phase is believed to trigger the development of autoimmunity. Pioneer work from Eizirik et al has shown that the presence of inflammatory cytokines affects gene expression and leads to increased alternative mRNA splicing events providing evidence for β‐cell neo‐autoantigens generation during insulitis.4 The inflammatory milieu also impacts post‐translational modification (PTM) processes: the calcium‐dependent enzymes tissue transglutaminase 2 and peptidyl amine deaminase5 were shown to be activated in islets and dendritic cells leading to increase in deamidation and citrullination of islet‐autoantigens, improving their potency to bind HLA molecules and their immunogenic properties.6, 7, 8, 9, 10 Such modifications have been demonstrated to increase visibility of β‐cells to the immune system as seen by the increased T‐cell response observed after chemical induction of ER stress in β‐cells.11 Recently, we demonstrated that translation of the insulin mRNA was also influenced by environmental modifications and that ER stress induction led to recruitment of ribosomes at alternative translation initiation sites and to translation of a highly immunogenic insulin‐gene derived polypeptide targeted by T‐cell autoreactivity in T1D patients.12 Indeed, the rate of β‐cell destruction increased upon inflammatory stress, while expression of defective ribosomal products (DRIPs) of insulin was increased by the calcium‐dependent stressor thapsigargin, but not by calcium‐independendent tunicamycin. These translational errors, which are generated when converting genetic information into proteins as well as non‐functional proteins resulting from alternative RNA splicing or PTM modifications, are believed to undergo a rapid degradation after synthesis and to constitute a subset of peptides rapidly loaded on MHC class I molecules.13
The inflammatory milieu in type 1 diabetic islets triggers transcriptional, translational and post‐translational changes in β‐cells leading to the generation of neoepitopes and exposure to the immune system.
In this process, the ubiquitin proteasome system (UPS) plays a key role but little is known about the regulatory mechanisms involved, and how this degradation mechanism may participate in the development of autoimmunity. In this review, we describe the connection between the UPR and UPS in the generation of antigen class I peptides and its possible role in the generation of β‐cell neoantigens. Finally, we will propose possible strategies targeting the proteasome/immunoproteasome for the treatment of autoimmune diabetes.
2. DEGRADATION AND REGULATORY MECHANISMS
Insulin is synthesized by pancreatic β‐cells as preproinsulin that co‐translationally translocates into the ER lumen. After signal sequence cleavage and folding into its native conformation, proinsulin exits the ER and traffics via the Golgi system to secretory granules. The high demand in insulin, estimated to be up to a million molecules synthesized per minute and per cell14 (which is consistent with the high amount of insulin mRNA present in each β‐cell15), requires efficient folding of proinsulin molecules in the ER lumen of pancreatic β‐cells and targeting to the insulin secretory granules, while proper glucose regulation involves a severe quality control of the secretory pathway. Under stress, the ER associated degradation pathway (ERAD) triggers degradation of proteins that failed to achieve their functional conformation. Recognition of improper folded proteins involves chaperone proteins (BiP, HsP70) recognized by ERAD‐associated ligase gp78, HRD1, TEB4, TRC8 or TMEM129. In this process, proteins dislocation and retro‐translocation to the cytosol, rather than the secretory pathway, is facilitated by Derlin proteins and the dislocation complex is targeted to the proteasome by ATP dependent ubiquitin‐binding factors (Valosin‐Containing Protein p97). The close relation between the UPR and the Ubiquitin Proteasome System (UPS) in response to stress suggests that environmental changes may impair the degradation machinery. Dysregulation of this system and inappropriate degradation was shown to lead to the accumulation of cytoplasm aggregates and to the development of neurodegenerative diseases (eg, Huntington, Alzheimer or Parkinson disease16). Recently, accumulation of vesicles containing proinsulin in β‐cells from islet autoantibody positive pancreas donors, suggesting a defect in proinsulin conversion or an accumulation of immature vesicles caused by an increase in insulin demand and/or a dysfunction in vesicular trafficking.17 In a similar process, accumulation of islet amyloid polypeptide protein (IAPP) and the formation of amyloid fibrils has been associated with disease progression in type 1 diabetes as seen by the increased CD8 T‐cell response to ppIAPP in T1D patients.18, 19, 20, 21 Moreover, downregulation of derlin1, Hrd1 or p97 were shown to reduce proinsulin degradation,22 suggesting that these factors may participate to the generation of stress‐induced proinsulin‐derived peptides (Figure 1).
Figure 1.
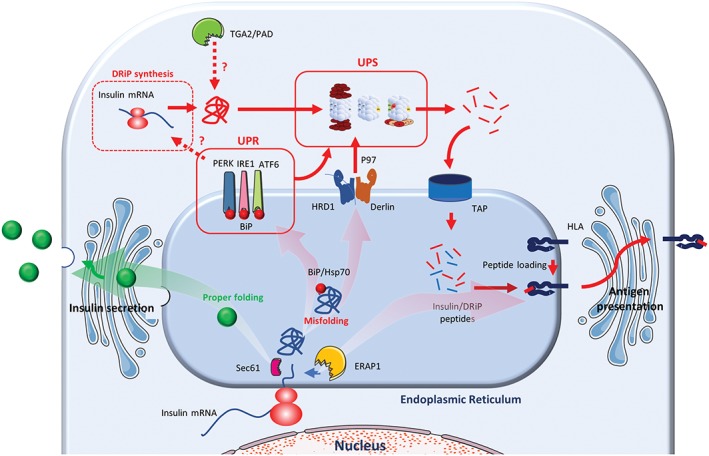
Schematic representation of preproinsulin processing. In normal environment, following cleavage of the signal peptide, proinsulin is targeted to the secretory pathway and further maturated in insulin and C‐peptide in secretory granules (green arrow). Under stress (red arrows), translation, post‐translational modifications, UPR and UPS contribute independently or synergistically to the generation of T1D autoantigenic peptides
In this degradation process, the proteasome plays a central role. This multiproteic complex, expressed in all cell types, is composed of a 20S proteolytic core and two 19S regulatory subunits acting as caps of the core and forming the final 26S holo‐structure. The crystal structures of the human 20S core and the 26S complex were unravelled at high resolution.23, 24 The proteolytic core forms a barrel of four stacked rings of seven subunits each. The two outer rings include the α subunits, and the two inner rings the β subunits. Amongst the β subunits, three different proteolytic activities are encountered. The β1, β2, β5 have caspase, trypsin, chymotrypsin‐like functions, respectively. The active residue relies on the N‐terminal threonine of the enzymatically active subunits exposed in the luminal part of the barrel. The regulatory 19S multi subunit complex acts as a check point for the entrance of proteins in the 20S barrel, by recognizing the ubiquitinated substrate, removing the ubiquitin chain and delivering the unfolded protein into the proteolytic core. Structurally, it can be stratified into lid and base. The base consists of six ATPases of the AAA family, responsible for unfolding and linearizing the substrate, at ATP expense, and three ubiquitin receptors in charge of substrate recognition. The lid comprises of nine subunits, Rpn3,5 to 9,11,12 and 15, some with undefined roles. Rpn11 is known for its ability to remove and promote recycling of ubiquitin chains. Evidence is provided that Rpn6 supports the stabilization of the complex through interactions with the α subunits. The degradation ability of the proteolytic subunits combined with the regulative role of the 19S complex is important for the maintenance of cellular proteostasis and for the definition of the MHC class I ligandome.
Similarly to transcription and translation, protein degradation is not a static process and can be regulated in response to extracellular signals to ensure homeostasis, survival or apoptosis. Upon cytokine treatment or after induction of oxidative stress, the β catalytic subunits of the proteasome are substituted by their induced counterparts β1i/LMP2, β2i/MECL‐1, β5i/LMP7, to generate the immunoproteasome (Figure 2). Strikingly, while immature dendritic cells present an equal proteasome/immunoproteasome ratio, significantly low amounts of standard proteasome are detected upon maturation,25 suggesting the importance of the immunoproteasome in the antigen presentation machinery.
Figure 2.
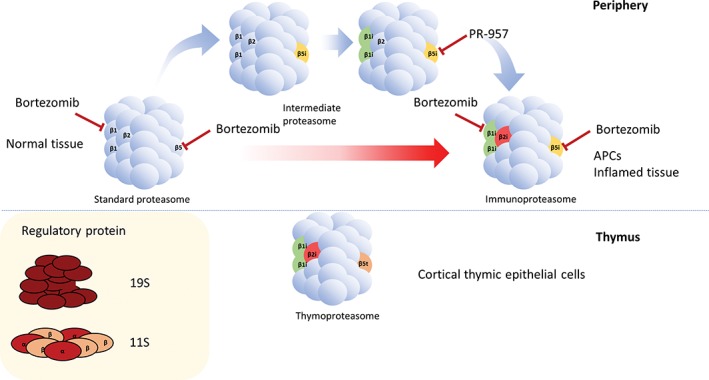
Scheme of the different types of 20S proteasomes. The β subunits of the standard proteasome (β1, β2 and β5) are expressed in all cells. Upon stress, some or all β subunits are substituted by their induced counterparts (β1i, β2i and β5i), composing the intermediate and immunoproteasome. APCs express immunoproteasomes constantly. Cortical thymic epithelial cells express a unique type of β5 subunit, indicated as β5t. The 20S cores can assemble, on 1 or both ends, with a variety of regulatory proteins, among them 19S and 11S were implicated in the antigen presentation process. Targets of the proteasome inhibitors Bortezomib and PR‐957 are indicated
The constitution of the proteasome and immunoproteasome is variable, highly regulated and cell‐specific. Therefore, different subunits can be incorporated in the same 20S core to generate intermediate proteasome forms. APCs can, for example, express β1‐β2‐β5i and β1i‐β2‐β5i influencing the nature of peptides generated, similar variants have also been described in liver, kidney, colon and heart tissues as well as in a variety of tumours, including melanoma, non‐small cell lung carcinoma (NSLC) and myeloma.26 Cortical thymic epithelial cells (cTEC), that participate to the expression of self‐antigens during T‐lymphocyte development and to the establishment of the T‐cell repertoire, express another class of proteasome,27 the thymoproteasome (Figure 2). In this conformation, the β5i subunit of the immunoproteasome is substituted by the β5t and is complexed with β1i and β2i in the 20S core, resulting in a poor chymotrypsin‐like activity.26 This diversity illustrates the complexity of the mechanisms underlying central and peripheral tolerance and the critical role played by the proteasome in the development of autoimmunity.
Moreover, the half‐life of the immunoproteasome was found to be approximately 5 times lower (27 h) than of the constitutive one (133 h), while its speed of assembly was 4 times higher (21 min vs. 82 min).28, 29 These values reflect the adaptive role held by the immunoproteasome. Inflammation is a common denominator in autoimmune diseases and the proteasomal composition has been shown to be regulated by inflammatory cytokines and IFN‐γ mediated oxidative stress.30 Inflammatory cytokines (IFNγ) can phosphorylate α subunits and affect 19S capped proteasomes assembly31, 32 and impact proteolytic properties. Similarly, IFNγ has been shown to induce the expression of the α and β subunits of the PA28 cap. Three α and four β subunits interact to form the PA28αβ heptameric ring. PA28αβ (11S) is mainly associated with 20S core containing the inducible catalytic subunits, via the outer α subunits of the barrel. PA28αβ acts in an ATP independent manner and is described to influence the generation of some peptides. Hybrid proteasomes composed of one 19S and one 11S were also described. Alternatively, also the presence of free 20S cores was described when coping with oxidative stress.33 In oxidative conditions, uncapped proteasomes were proven to be more resistant than the 26S holo‐complexes.28
Stress can also dictate the formation of the 20S core of the immunoproteasome in 3 different levels: during transcription, assembly and post‐translation. Increased transcription of the βi subunits upon IFNγ exposure is attributed to the action of IRF1 and STAT1 transcription factors.34, 35 Finally, the amino acids coating the substrate‐binding regions of the β1, β5 and β1i, β5i, respectively, were proposed to be responsible for the distinct catalytic properties. Also, incorporation of β5i was shown to enhance the activity of β1i and β2i via structural interactions.26 As a result, the peptide pool generated by the standard, the immunoproteasome and the intermediate forms was proposed to be quantitatively and qualitatively different. While the participation of the proteasome component still remains to be investigated in diabetes, IFNγ was shown to trigger an increase expression of the βi in human islets,36, 37 demonstrating the potential generation of inflammation‐specific epitopes and a possible subsequent CTL response.
In addition, during disease progression, the local increase of nitric oxide and endoplasmic reticulum (ER) Ca2+ depletion in response to proinflammatory cytokines secreted by infiltrated autoreactive immune cells, are believed to be the main causes for ER stress and β‐cell failure. This oxidative stress triggers expression of βi subunits via the activation of the cAMP responsive element protein (CREB) by nitric oxide, suggesting that such mechanisms may occur during T1D pathology.34
In diabetic conditions, the glycaemic environment was also shown to affect the proteasomal composition and activity of a cell but the results remain controversial. In human islets kept on high glucose for 24 hours an increase in trypsin activity was observed.37 Controversially, in another study, the rat β‐cell line INS‐1E exposed to high glucose and the pancreatic islets from the rat diabetic model GK, showed a decrease in all three catalytic activities of the proteasome and in increase in ER stress markers and apoptosis. Similar results were obtained in human islets grown in high glucose for 14 days.38 These discrepancies may rely on the technical variations (differences in timing and glucose levels), but further investigations are required to elucidate the role of glucose in proteasome and immunoproteasome function, since these proteolytic complexes are determinative for antigen presentation. These studies may give indications of the autoantigen origin in type 1 diabetes.
Fundamental knowledge on the mechanisms underlying the β‐cell adaptive and subsequent dysfunctional phases is critical to identify efficient novel therapeutic targets to decrease β‐cell stress, improve viability/function and reduce β‐cell visibility to the immune system.
3. PROTEASOME/IMMUNOPROTEASOME AND CELL LIGANDOME
The cell ligandome generated by the proteasome or immunoproteasome has been studied in detail in the case of viral infection and cancer. In tumour immunology, several MAGE antigenic peptides were shown to be derived from the immunoproteasome and the intermediate conformations and not by constitutive proteasome.26, 39 Among these peptides, MAGE‐A3(271‐279) is uniquely cleaved by the β1‐β2‐β5i form, while MAGE‐A10(254‐262) and MAGE ‐C2(191‐200) is cleaved by the β1i‐β2‐β5i form.39, 40
However, the different cleavage specificities between the constitutive and the induced catalytic subunits41 were challenged by a study by Mishto et al42 showing that peptidome differences relied more on peptide quantity rather than quality. Yet, these results based on digestions performed by isolated proteasomes and not on experiments related to antigen presentation and CTL activation illustrate the complexity of the antigen presentation pathway in vivo. Interestingly, while some peptides can be produced by both proteasomes, studies conducted on E1B(192‐200) of Listeria monocytogenes demonstrate that proteolytic capacities of the immuno‐ and standard proteasomes may differ in speed, affecting also the overall antigen presentation kinetics and CTL response.43 Moreover, the cellular localization of the immunoproteasome may also play a role. β1i and β5i were found to be in close proximity to the ER while the standard proteasomes were described to be homogenously distributed in both nucleus and cytoplasm of the IFNγ‐treated embryonic lung tissue cell line L132.44 Considering that the ER contains all the components needed for antigen presentation after the action of the proteasome, the determined localization of the immunoproteasome may explain the increased levels of antigen presentation noticed, but further investigation is needed.
Like in tumours, the immunoproteasome may have a role in autoimmune disease. Indeed, we previously reported differences in processing preproinsulin by immuno‐ vs. constitutive proteasomes.45, 46 Islet autoimmunity and β‐cell destruction could be demonstrated by CD8 T‐cells reactive with such proteasomal products, but also non‐proteosomally derived preproinsulin epitopes. Interestingly, destruction of β‐cells increased under high glucose conditions, pointing to hyperglycaemia and metabolic stress as environmental factors contributing to the demise of β‐cells in T1D.45, 47 Whether and how these factors influence proteasomal composition and antigen presentation of pancreatic β‐cells is still unknown. In multiple sclerosis, where CD8 cytotoxic T‐cells target epitopes of the self‐derived protein myelin, PA28αβ and immunoproteasome subunits were higher in brain tissue of MS patients compared to healthy donors. Association of PA28αβ with 20S immunoproteasome resulted in higher production of the myelin antigenic epitope MBP111–119.48, 49
While ultimately, HLA polymorphism define the ligandome, the variety of peptides generated is very large and even more amplified by more unconventional events occurring during degradation. In fact, examples of hybrid peptides, generated from fragments of two peptides from the same protein (cis‐splicing) or different proteins (trans‐splicing), have been described for tumour antigenic epitopes. Thus, peptides presented in class I HLA may exhibit different sequence than the parental unprocessed sequence as seen for peptides derived from gp100, tyrosinase, FGF‐5 or Sp100 in melanomas. Two different mechanisms for peptide splicing were described: transpeptidation and condensation. In the former, an acyl‐enzyme intermediate is formed between the proteasome and the first peptide via an ester bond, this bond receives a nucleophilic attack from the N‐terminal site of the second peptide. Condensation, describes the formation of peptide bond de novo. 50 While theoretically, both the constitutive and induced β subunits may perform transpeptidation or condensation, some SP100 derivatives are more pronounced in the presence of the immunoproteasome,51 and spliced gp100(47‐52)‐(40‐42) shown to occur by condensation by the action of β5i subunit.50
In autoimmune disease these processes are poorly investigated. Yet, DeLong et al illustrated the complexity of degradation and the diversity of the ligandome by describing immunoreactivity against hybrid peptides generated by a process of transpeptidation.52 CD4 T‐cells isolated from insulitic lesions of type 1 diabetic patients were shown to react with such hybrid peptides, suggesting the potential relevance to islet autoimmunity and β‐cell destruction in human disease.53 Prediction and identification of these hybrid peptides is challenging but these may constitute an important component of the peptide ligandome. Initially, this identification was exclusively dependent on the isolation of the corresponding patients CTLs, usually present in low amounts. Recently, a novel tool for spliced peptide characterization, combining both experimental and computational data, was developed by Liepe et al. In brief, theoretical m/z ratios from both spliced and linear peptides are compared with mass spectrometry data of in vitro digestions by the 20S proteasome. The occurrence of a specific peptide is then assured by tandem mass spectrometry.54 Another limiting factor is the representation of only linear epitopes in the different protein databases. In a study aiming at investigating the prevalence of such peptides, a database containing numerous spliced peptides that may occur by cis splicing of epitopes separated by a maximum of 25 amino acids was developed. According to this in silico analysis, approximately one third of the HLA class I ligandome is qualitatively produced by proteasomal‐catalyzed peptide splicing and these peptides comprise circa the 25% of the surface immunopeptidome. Importantly, spliced peptides are not produced by a random junction of epitopes, and their occurrence is highly dependent on protein sequence. However, these results are questioned by another study, supporting that only the 2% to 4% of the HLA class I ligands derive from spliced peptides. These ambiguous findings, highlight the necessity of a widely accepted methodology for spliced peptide identification.55 Nevertheless, the relevance of spliced peptides in antigen presentation is also highlighted by their high affinity to MHC class I molecules and by their ability to activate CTL responses as extensively as linear antigenic peptides.50, 56, 57
4. CONCLUSION
The high degradation rate of the immunoproteasome when compared to the standard proteasome suggest that it is an important element of the cellular adaptive mechanism to inflammation but also possibly in the generation of aberrant polypeptide derived epitopes. Therefore, targeting the immunoproteasome to prevent autoimmunity may represent an interesting alternative to current therapy. In mouse models for inflammatory bowel disease, inhibition of the chymotrypsin‐like activity of the β5‐subunit by Bortezomib was shown to reduce colitis.58 Similarly, the selective inhibition of LMP7 by PR‐957 (ONX0914) reduces inflammatory infiltration in a mouse model for rheumatoid arthritis.59 In a recent clinical trial, Bortezomib administration reduces the numbers of peripheral blood and bone marrow plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus.60 Yet, these beneficial effects are mainly mediated by a reduction of the Iκb degradation and the inhibition of NfκB regulated inflammatory mediators on effector cells (IFNγ, IL1β, TNFα)34, 59, 61 and not on the prevention of the generation of neoantigen by the target cells. Worse still, the few studies investigating the role of the proteasome inhibitors on β‐cell described an increase ER stress and an impaired islet function38, 62 but a better understanding of these degradation processes appears essential for the identification of stress induced antigens and ultimately for the identification of new biomarkers of disease progression.
Identification of new biomarkers of disease progression and establishment of a “diabetes immune signature” will provide a guide towards personalized medicine, defining the preferred immune modulation strategy in individual patients.
ACKNOWLEDGEMENTS
This work was supported by JDRF and by the Diabetes Fonds and the DON foundation. B.O.R. is supported by the Wanek Family Project for Type 1 Diabetes. Servier kindly provided medical art used in Figure 1.
Conflict of interest
No potential conflicts of interest relative to this article are reported.
Thomaidou S, Zaldumbide A, Roep BO. Islet stress, degradation and autoimmunity. Diabetes Obes Metab. 2018;20(Suppl. 2):88–94. 10.1111/dom.13387
Arnaud Zaldumbide and Bart O. Roep contributed equally to this work
Funding information DON foundation stichting DON (Diabetes Onderzoek Nederland); Diabetes Fonds; JDRF
REFERENCES
- 1. Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta‐cell loss in type 1 diabetes. Nat Rev Endocrinol. 2009;5(4):219‐226. [DOI] [PubMed] [Google Scholar]
- 2. Eizirik DL, Cardozo AK, Cnop M. The role for endoplasmic reticulum stress in diabetes mellitus. Endocr Rev. 2008;29(1):42‐61. [DOI] [PubMed] [Google Scholar]
- 3. Eizirik DL, Miani M, Cardozo AK. Signalling danger: endoplasmic reticulum stress and the unfolded protein response in pancreatic islet inflammation. Diabetologia. 2013;56(2):234‐241. [DOI] [PubMed] [Google Scholar]
- 4. Eizirik DL, Sammeth M, Bouckenooghe T, et al. The human pancreatic islet transcriptome: expression of candidate genes for type 1 diabetes and the impact of pro‐inflammatory cytokines. PLoS Genet. 2012;8(3):e1002552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McLaughlin RJ, de Haan A, Zaldumbide A, et al. Human islets and dendritic cells generate post‐translationally modified islet autoantigens. Clin Exp Immunol. 2016;185(2):133‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Khan MW, Sherwani S, Khan WA, Ali R. Characterization of hydroxyl radical modified GAD65: a potential autoantigen in type 1 diabetes. Autoimmunity. 2009;42(2):150‐158. [DOI] [PubMed] [Google Scholar]
- 7. Mannering SI, Harrison LC, Williamson NA, et al. The insulin A‐chain epitope recognized by human T cells is posttranslationally modified. J Exp Med. 2005;202(9):1191‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGinty JW, Chow IT, Greenbaum C, Odegard J, Kwok WW, James EA. Recognition of posttranslationally modified GAD65 epitopes in subjects with type 1 diabetes. Diabetes. 2014;63(9):3033‐3040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Rondas D, Crevecoeur I, D'Hertog W, et al. Citrullinated glucose‐regulated protein 78 is an autoantigen in type 1 diabetes. Diabetes. 2015;64(2):573‐586. [DOI] [PubMed] [Google Scholar]
- 10. van Lummel M, Duinkerken G, van Veelen PA, et al. Posttranslational modification of HLA‐DQ binding islet autoantigens in type 1 diabetes. Diabetes. 2014;63(1):237‐247. [DOI] [PubMed] [Google Scholar]
- 11. Marre ML, Profozich JL, Coneybeer JT, et al. Inherent ER stress in pancreatic islet beta cells causes self‐recognition by autoreactive T cells in type 1 diabetes. J Autoimmun. 2016;72:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kracht MJ, van Lummel M, Nikolic T, et al. Autoimmunity against a defective ribosomal insulin gene product in type 1 diabetes. Nat Med. 2017;23(4):501‐507. [DOI] [PubMed] [Google Scholar]
- 13. Eisenlohr LC, Huang L, Golovina TN. Rethinking peptide supply to MHC class I molecules. Nat Rev Immunol. 2007;7(5):403‐410. [DOI] [PubMed] [Google Scholar]
- 14. Scheuner D, Kaufman RJ. The unfolded protein response: a pathway that links insulin demand with beta‐cell failure and diabetes. Endocr Rev. 2008;29(3):317‐333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Van Lommel L, Janssens K, Quintens R, et al. Probe‐independent and direct quantification of insulin mRNA and growth hormone mRNA in enriched cell preparations. Diabetes. 2006;55(12):3214‐3220. [DOI] [PubMed] [Google Scholar]
- 16. Frake RA, Ricketts T, Menzies FM, Rubinsztein DC. Autophagy and neurodegeneration. J Clin Invest. 2015;125(1):65‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rodriguez‐Calvo T, Zapardiel‐Gonzalo J, Amirian N, et al. Increase in pancreatic proinsulin and preservation of beta‐cell mass in autoantibody‐positive donors prior to type 1 diabetes onset. Diabetes. 2017;66(5):1334‐1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Courtade JA, Klimek‐Abercrombie AM, Chen YC, et al. Measurement of pro‐islet amyloid polypeptide (1‐48) in diabetes and islet transplants. J Clin Endocrinol Metab. 2017;102(7):2595‐2603. [DOI] [PubMed] [Google Scholar]
- 19. Delong T, Baker RL, Reisdorph N, et al. Islet amyloid polypeptide is a target antigen for diabetogenic CD4+ T cells. Diabetes. 2011;60(9):2325‐2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ouyang Q, Standifer NE, Qin H, et al. Recognition of HLA class I‐restricted beta‐cell epitopes in type 1 diabetes. Diabetes. 2006;55(11):3068‐3074. [DOI] [PubMed] [Google Scholar]
- 21. Velthuis JH, Unger WW, Abreu JR, et al. Simultaneous detection of circulating autoreactive CD8+ T‐cells specific for different islet cell‐associated epitopes using combinatorial MHC multimers. Diabetes. 2010;59(7):1721‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hoelen H, Zaldumbide A, van Leeuwen WF, et al. Proteasomal degradation of proinsulin requires Derlin‐2, HRD1 and p97. PLoS One. 2015;10(6):e0128206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schrader J, Henneberg F, Mata RA, et al. The inhibition mechanism of human 20S proteasomes enables next‐generation inhibitor design. Science. 2016;353(6299):594‐598. [DOI] [PubMed] [Google Scholar]
- 24. Schweitzer A, Aufderheide A, Rudack T, et al. Structure of the human 26S proteasome at a resolution of 3.9 a. Proc Natl Acad Sci U S A. 2016;113(28):7816‐7821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Macagno A, Gilliet M, Sallusto F, Lanzavecchia A, Nestle FO, Groettrup M. Dendritic cells up‐regulate immunoproteasomes and the proteasome regulator PA28 during maturation. Eur J Immunol. 1999;29(12):4037‐4042. [DOI] [PubMed] [Google Scholar]
- 26. Vigneron N, Van den Eynde BJ. Proteasome subtypes and regulators in the processing of antigenic peptides presented by class I molecules of the major histocompatibility complex. Biomolecules. 2014;4(4):994‐1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nitta T, Murata S, Sasaki K, et al. Thymoproteasome shapes immunocompetent repertoire of CD8+ T cells. Immunity. 2010;32(1):29‐40. [DOI] [PubMed] [Google Scholar]
- 28. Heink S, Ludwig D, Kloetzel PM, Kruger E. IFN‐gamma‐induced immune adaptation of the proteasome system is an accelerated and transient response. Proc Natl Acad Sci U S A. 2005;102(26):9241‐9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yewdell JW. Immunoproteasomes: regulating the regulator. Proc Natl Acad Sci U S A. 2005;102(26):9089‐9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. van Deventer S, Neefjes J. The immunoproteasome cleans up after inflammation. Cell. 2010;142(4):517‐518. [DOI] [PubMed] [Google Scholar]
- 31. Bose S, Brooks P, Mason GG, Rivett AJ. Gamma‐interferon decreases the level of 26 S proteasomes and changes the pattern of phosphorylation. Biochem J. 2001;353(Pt 2):291‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schmidt M, Finley D. Regulation of proteasome activity in health and disease. Biochim Biophys Acta. 2014;1843(1):13‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Aiken CT, Kaake RM, Wang X, Huang L. Oxidative stress‐mediated regulation of proteasome complexes. Mol Cell Proteomics. 2011;10(5):R110.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Angeles A, Fung G, Luo H. Immune and non‐immune functions of the immunoproteasome. Front Biosci (Landmark Ed). 2012;17:1904‐1916. [DOI] [PubMed] [Google Scholar]
- 35. Namiki S, Nakamura T, Oshima S, et al. IRF‐1 mediates upregulation of LMP7 by IFN‐gamma and concerted expression of immunosubunits of the proteasome. FEBS Lett. 2005;579(13):2781‐2787. [DOI] [PubMed] [Google Scholar]
- 36. Freudenburg W, Gautam M, Chakraborty P, et al. Reduction in ATP levels triggers immunoproteasome activation by the 11S (PA28) regulator during early antiviral response mediated by IFNbeta in mouse pancreatic beta‐cells. PLoS One. 2013;8(2):e52408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lundh M, Bugliani M, Dahlby T, et al. The immunoproteasome is induced by cytokines and regulates apoptosis in human islets. J Endocrinol. 2017;233(3):369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Broca C, Varin E, Armanet M, et al. Proteasome dysfunction mediates high glucose‐induced apoptosis in rodent beta cells and human islets. PLoS One. 2014;9(3):e92066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guillaume B, Stroobant V, Bousquet‐Dubouch MP, et al. Analysis of the processing of seven human tumor antigens by intermediate proteasomes. J Immunol. 2012;189(7):3538‐3547. [DOI] [PubMed] [Google Scholar]
- 40. Guillaume B, Chapiro J, Stroobant V, et al. Two abundant proteasome subtypes that uniquely process some antigens presented by HLA class I molecules. Proc Natl Acad Sci U S A. 2010;107(43):18599‐18604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ferrington DA, Gregerson DS. Immunoproteasomes: structure, function and antigen presentation. Prog Mol Biol Transl Sci. 2012;109:75‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mishto M, Liepe J, Textoris‐Taube K, et al. Proteasome isoforms exhibit only quantitative differences in cleavage and epitope generation. Eur J Immunol. 2014;44(12):3508‐3521. [DOI] [PubMed] [Google Scholar]
- 43. Deol P, Zaiss DM, Monaco JJ, Sijts AJ. Rates of processing determine the immunogenicity of immunoproteasome‐generated epitopes. J Immunol. 2007;178(12):7557‐7562. [DOI] [PubMed] [Google Scholar]
- 44. Brooks P, Murray RZ, Mason GG, Hendil KB, Rivett AJ. Association of immunoproteasomes with the endoplasmic reticulum. Biochem J. 2000;352(Pt 3):611‐615. [PMC free article] [PubMed] [Google Scholar]
- 45. Skowera A, Ellis RJ, Varela‐Calvino R, et al. CTLs are targeted to kill beta cells in patients with type 1 diabetes through recognition of a glucose‐regulated preproinsulin epitope. J Clin Invest. 2008;118(10):3390‐3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Unger WW, Velthuis J, Abreu JR, et al. Discovery of low‐affinity preproinsulin epitopes and detection of autoreactive CD8 T‐cells using combinatorial MHC multimers. J Autoimmun. 2011;37(3):151‐159. [DOI] [PubMed] [Google Scholar]
- 47. Kronenberg‐Versteeg D, Eichmann M, Russell MA, et al. Molecular pathways for immune recognition of preproinsulin signal peptide in type 1 diabetes. Diabetes. 2018;67(4):687‐696. [DOI] [PubMed] [Google Scholar]
- 48. Bellavista E, Santoro A, Galimberti D, Comi C, Luciani F, Mishto M. Current understanding on the role of standard and immunoproteasomes in inflammatory/immunological pathways of multiple sclerosis. Autoimmune Dis. 2014;2014:739705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mishto M, Bellavista E, Ligorio C, et al. Immunoproteasome LMP2 60HH variant alters MBP epitope generation and reduces the risk to develop multiple sclerosis in Italian female population. PLoS One. 2010;5(2):e9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ebstein F, Textoris‐Taube K, Keller C, et al. Proteasomes generate spliced epitopes by two different mechanisms and as efficiently as non‐spliced epitopes. Sci Rep. 2016;6:24032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vigneron N, Ferrari V, Stroobant V, Abi Habib J, Van den Eynde BJ. Peptide splicing by the proteasome. J Biol Chem. 2017;292(51):21170‐21179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Delong T, Wiles TA, Baker RL, et al. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science. 2016;351(6274):711‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Babon JA, DeNicola ME, Blodgett DM, et al. Analysis of self‐antigen specificity of islet‐infiltrating T cells from human donors with type 1 diabetes. Nat Med. 2016;22(12):1482‐1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liepe J, Mishto M, Textoris‐Taube K, et al. The 20S proteasome splicing activity discovered by SpliceMet. PLoS Comput Biol. 2010;6(6):e1000830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mylonas R, Beer I, Iseli C, et al. Estimating the contribution of proteasomal spliced peptides to the HLA‐I ligandome. bioRxiv. 2018. [DOI] [PMC free article] [PubMed]
- 56. Berkers CR, de Jong A, Schuurman KG, et al. Peptide splicing in the proteasome creates a novel type of antigen with an Isopeptide linkage. J Immunol. 2015;195(9):4075‐4084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Vigneron N, Abi Habib J, Van den Eynde BJ. Learning from the proteasome how to fine‐tune cancer immunotherapy. Trends Cancer. 2017;3(10):726‐741. [DOI] [PubMed] [Google Scholar]
- 58. Schmidt N, Gonzalez E, Visekruna A, et al. Targeting the proteasome: partial inhibition of the proteasome by bortezomib or deletion of the immunosubunit LMP7 attenuates experimental colitis. Gut. 2010;59(7):896‐906. [DOI] [PubMed] [Google Scholar]
- 59. Muchamuel T, Basler M, Aujay MA, et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat Med. 2009;15(7):781‐787. [DOI] [PubMed] [Google Scholar]
- 60. Alexander T, Sarfert R, Klotsche J, et al. The proteasome inhibitior bortezomib depletes plasma cells and ameliorates clinical manifestations of refractory systemic lupus erythematosus. Ann Rheum Dis. 2015;74(7):1474‐1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Basler M, Mundt S, Bitzer A, Schmidt C, Groettrup M. The immunoproteasome: a novel drug target for autoimmune diseases. Clin Exp Rheumatol. 2015;33(4 Suppl. 92):S74‐S79. [PubMed] [Google Scholar]
- 62. Kitiphongspattana K, Khan TA, Ishii‐Schrade K, Roe MW, Philipson LH, Gaskins HR. Protective role for nitric oxide during the endoplasmic reticulum stress response in pancreatic beta‐cells. Am J Physiol Endocrinol Metab. 2007;292(6):E1543‐E1554. [DOI] [PubMed] [Google Scholar]