

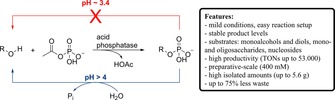
Abstract
Undesired product hydrolysis along with large amounts of waste in form of inorganic monophosphate by‐product are the main obstacles associated with the use of pyrophosphate in the phosphatase‐catalyzed synthesis of phosphate monoesters on large scale. In order to overcome both limitations, we screened a broad range of natural and synthetic organic phosphate donors with several enzymes on a broad variety of hydroxyl‐compounds. Among them, acetyl phosphate delivered stable product levels and high phospho‐transfer efficiency at the lower functional pH‐limit, which translated into excellent productivity. The protocol is generally applicable to acid phosphatases and compatible with a range of diverse substrates. Preparative‐scale transformations using acetyl phosphate synthesized from cheap starting materials yielded multiple grams of various sugar phosphates with up to 433 g L−1 h−1 space‐time yield and 75% reduction of barium phosphate waste.
Keywords: Enzymatic phosphorylation, phosphatase, phosphate donor, acetyl phosphate
Introduction
Synthesis of valuable phosphate (mono)esters, e. g. sugar phosphates,1, 2, 3 nucleotides,4,5 metabolites6,7 and prodrugs8, 9, 10 can be achieved by laborious chemical11, 12, 13, 14, 15, 16, 17, 18 or mild enzymatic routes.19, 20, 21 The latter usually employ kinases and ATP in conjunction with a second enzyme for the recycling of the phosphate donor. Although applied on preparative‐scale,22, 23, 24, 25 kinases are rather substrate‐specific and efficient cofactor recycling systems are still in an emerging phase.26
The ability of phosphatases – naturally active in the hydrolysis mode – to catalyze the (reverse) transphosphorylation reaction employing cheap high‐energy phosphate donors (P‐donors), e. g. pyrophosphate (PPi), was recognized decades ago.27,28 The avoidance of ATP recycling and the relaxed substrate spectrum of phosphatases rendered these enzymes attractive alternatives for the synthesis of a broad range of phosphate monoesters.29 However, on large scale, this method suffers from two major limitations: (i) the enzymatic hydrolysis of the formed phosphate ester leads to product depletion and unpredictable (optimum) reaction times, and (ii) generates large quantities of inorganic monophosphate (Pi) as by‐product. The first problem could be overcome by applying high substrate concentration,30, 31, 32 by protein engineering33, 34, 35 or by continuous flow technology.31,36,37 On the other hand, the formation of inorganic monophosphate as by‐product is a result of the nature of the P‐donor. Though being cheap, PPi is associated with excessive release of Pi, owing to liberation of a stoichiometric quantity of Pi during phosphate transfer and competing hydrolysis of the P‐donor. Application of triphosphate (PPPi) or other oligo‐ and polyphosphates would result in higher product levels, however, the efficiency of the phosphate transfer does not correlate with the number of transferable high‐energy phosphate moieties.30,31,37 The generation of high amounts of inorganic monophosphate also leads to technical difficulties in downstream processing. A straightforward and generally applicable way to separate Pi from the product phosphate monoester consists in fractional crystallization, whereby Pi as well as the unreacted P‐donor are sequentially precipitated as barium salts, thereby creating vast amounts of barium waste.31,35,37, 38, 39, 40, 41 Decreasing the latter would render the process overall more benign. As alternative to crystallization, ion‐exchange chromatography might be applied, however, due to similar pKa values of Pi and product phosphate monoesters, large amounts of eluent are required.30,42,43
Herein, we compare various organic P‐donors as alternatives to inorganic oligophosphates for the synthesis of phosphate monoesters by phosphatases. Organic P‐donors have been used with phosphatases in the hydrolysis‐ and transphosphorylation‐mode, however, product isolation has scarcely been reported.27,44,45 Aryl phosphates (e. g. phenyl phosphate, p‐nitrophenyl phosphate, phenolphthalein phosphate) and various metabolites (e. g. nucleoside phosphates, sugar‐phosphates) have been commonly used in biochemical characterization of novel enzymes due to their availability and features allowing facile spectrophotometric or NMR analysis. For reason of comparison, literature reports on enzyme‐catalyzed transphosphorylation reactions performed with organic P‐donors are collected in Table S1 (see the Electronic Supporting Information).
During the course of our study, we identified a specific pH range in which the phosphotransferase activity of the enzymes remained intact, while the phospho‐hydrolase activity was reduced. This proved to be a general feature among the tested acid phosphatases. The combination of pH control with use of acetyl phosphate as donor enabled rapid product synthesis on preparative scale, and hence formation of less barium phosphate waste.
Results and Discussion
P‐Donor Screening
Preliminary experiments were conducted at pH 4.2 with a range of acid phosphatases and an alkaline phosphatase (Table S2) using 1,4‐butanediol (1a) as model substrate (500 mM) and natural P‐donors, such as acetyl phosphate (AcP), phosphoenol pyruvate (PEP), carbamoyl phosphate (CP) or phosphocreatine (PC) (100 mM; Scheme 1). Importantly, at this pH value, AcP, CP and PC hydrolyze spontaneously with a half‐life of ∼7 h, ∼6 h and ∼3 h, respectively, while PPi and PEP are stable over at least 8 h (Figure S1). The results were compared with those obtained with PPi. All donors were accepted by the tested acid phosphatases, i. e. PhoN−Sf from Shigella flexneri, PhoN−Se from Salmonella typhimurium LT2, PiACP from Prevotella intermedia, Lw from Leptotrichia wadei, PhoC−Mm from Morganella morganii variant G92D/I171T, NSAP−Eb from Escherichia blattae 11‐variant and AphA−St from Salmonella typhimurium LT2 (Figure S2). The latter is a Mg‐dependent enzyme which shows marginal transphosphorylation activity using inorganic oligophosphates due to chelation of the metal causing inactivation. Magnesium supplementation is insufficient to recover its activity.31 However, using alternative donors, AphA−St showed good activity comparable to that of other acid phosphatases, highlighting an important benefit of non‐chelating phosphate donors. PhoK from Sphingomonas sp. BSAR‐1 at pH 9.0 and phytase from Aspergillus niger at pH 2.5 and 4.2 furnished only traces of product and were not further investigated. With less active variants of PhoC−Mm and NSAP−Eb,35 spontaneous hydrolysis of labile P‐donors (AcP, CP and PC) competed with transphosphorylation, leading to lower maximal product levels.
Scheme 1.
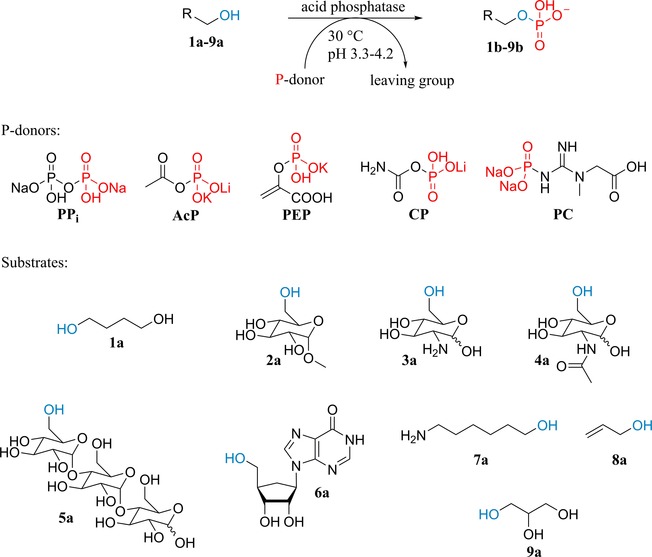
Substrates and phosphate donors tested in this study. PPi: disodium dihydrogenpyrophosphate; AcP: lithium potassium acetyl phosphate; PEP: potassium phosphoenolpyruvate; CP: lithium carbamoylphosphate; PC: phosphocreatine disodium salt.
Overall, AcP and PEP delivered the highest product concentrations, similar to those obtained with PPi (Figure S2 and Table S3). In the case of PEP, pyruvic acid (pKa 2.5) released upon phosphate transfer led to gradual decrease of the pH (to ∼3.2) and concomitant enzyme deactivation, which resulted in stable product concentrations but incomplete P‐donor consumption. Upon pH re‐adjustment, however, enzyme deactivation was reversible (Figure S3), indicating the existence of a pH threshold below which the enzymes reversibly turned into an inactive ‘stand‐by’ mode.
pH‐Controlled Product Hydrolysis
The dependence of catalytic activity on pH was tested with all enzymes. Since the use of AcP barely influences the pH during the course of the reaction (data not shown), the wild‐type enzymes were screened at various pH values with this donor. We found that lowering the initial pH led to dramatically reduced product hydrolysis. In contrast, the transphosphorylation activity was affected to a much lesser extent, which ultimately allows selective control of one activity over the other. Below a certain threshold, the enzymes turned inactive in the synthesis mode as well (Figure S4). This narrow pH range (3.3–3.8), in which the transphosphorylation activity was conserved, while the hydrolytic activity was strongly diminished, was identified for PhoN−Sf, PhoN−Se, PiACP, Lw and AphA−St (Figure 1). Overall, PiACP and Lw proved to be the most promising candidates for exploiting this property. The product concentrations were in the same range as those obtained at pH 4.2 and could be maintained over at least 1 d. Under these conditions, AcP was completely consumed when the product plateau was reached and no bis‐phosphorylated product was observed. Similar observations were made with acid phosphatases and PPi as donor,31,33,35,37 however, high product levels could not generally be maintained, if any, at the cost of large Pi side‐product formation. A difference between the pH optimum of phosphotransferase and phosphohydrolase activities has been also observed with calf intestine alkaline phosphatase.30
Figure 1.
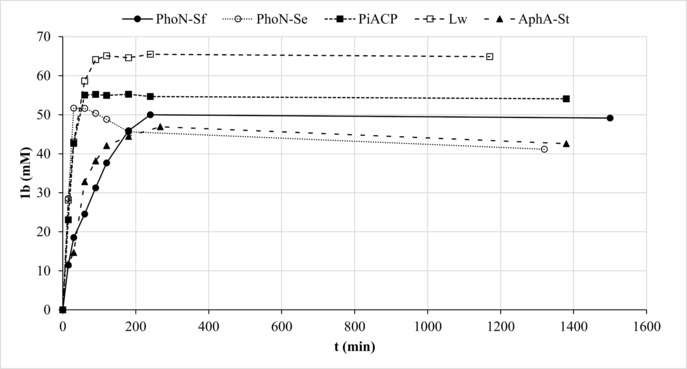
Phosphorylation of 1 a using AcP at optimum pH. Reaction conditions: 1 U mL−1 (0.4–1.0 μM) enzyme in 100 mM AcP at pH 3.8 (for PhoN−Sf), pH 3.5 (for PiACP and Lw), pH 3.3 (for PhoN−Se) or pH 2.9 (for AphA−St), 500 mM 1 a.
With PPi as donor at the optimum pH (Figure S5), the enzymes exhibited greater hydrolytic activity compared to that observed with AcP, which can be associated with concomittant pH‐increase over 4 h (ΔpH ∼+0.5 unit).
Next, various synthetic P‐donors, i. e. 2,2,2‐trifluoroethyl hydrogenphosphate monocyclohexylammonium salt (TFEP), 2,2,2‐trichloroethyl dihydrogen phosphate (TClEP), 2,5‐dioxopyrrolidin‐1‐yl hydrogen phosphate monocyclohexylammonium salt (NPS) were screened starting at the optimum pH (Figure S6–S8; for synthesis of donors, see ESI). Remarkably, most non‐natural P‐donors delivered product concentrations and product/Pi ratios similar to those obtained with AcP. However, as with PPi, hydrolytic activities were more pronounced due to increasing pH over time. In summary, AcP proved to be the ideal alternative to PPi, associated with rapid product formation, high phosphotransfer efficiency and low Pi waste.
P‐Donor, Enzyme and Substrate Concentration Test
Since Lw and PiACP delivered generally highest product concentrations at optimum pH (3.3–3.5), we investigated these enzymes in scale‐up reactions with 1 a. First, the concentration of AcP was enhanced (up to 400 mM) at constant enzyme concentration (1 U mL−1) and pH 3.4. Maximal product level and starting velocities decreased at above approx. 100 mM AcP (Figure S9), which indicates potential inhibition by AcP. Increasing the enzyme concentration (up to 10 U mL−1) improved product levels drastically (up to ∼270 mM; Figure S10). Finally, the concentration of 1 a was raised to 1.0 M and 1.5 M, however, both enzymes seemed to be sensitive to higher substrate concentrations (Figure S11).
Synthesis of Acetyl Phosphate (AcP)
So far, commercially available (expensive) acetyl phosphate was employed, setting serious limitations to large‐scale applications. However, this disadvantage along with the unstable character of AcP can be compensated by facile synthesis from cheap starting materials. AcP was synthesized by a modified protocol of Crans and Whitesides starting from phosphoric acid and acetic anhydride, followed by aqueous extraction and pH adjustment, to afford a mixture of 1.01 M AcP, containing only 47 mM Ac2P (diacetyl phosphate), 86 mM Pi and 12 mM PPi (Scheme S1, Figure S17).46 When stored at pH 7 at −20 °C, this crude solution exhibited only ∼5% degradation within 3 months without opening of the container and ∼10% degradation by regular usage.
Substrate Scope
The crude AcP preparation was diluted to ∼400 mM concentration and used for the phosphorylation of a range of substrates (1 a–9 a) with PiACP and Lw at previously identified optimum pH (∼3.4). In order to compensate for slight differences between batches of synthesized AcP, increased enzyme concentration was employed (15 U mL−1 corresponding to 6 μM PiACP and 9 μM Lw). In general, both enzymes could maintain stable product levels over at least 24 h (Figure 2 and Figure S12). PiACP yielded ∼260 mM 4‐hydroxybutyl phosphate (1 b), which is approx. twice as much as the amount formed by Lw (∼130 mM). The ratio of mono‐ versus bis‐phosphorylated product was ∼80:20 (PiACP) and ∼90:10 (Lw). In all other cases, however, PiACP delivered product concentrations lower than those obtained by Lw (Figure S12). Because of the sensitivity of this enzyme towards high substrate loadings, other substrates 3 a–5 a, 7 a and 8 a were used at 300 mM. In contrast, Lw had remarkable activity on 500 mM methyl α‐d‐glucopyranoside (2 a), d‐glucosamine (3 a) and maltotriose (5 a) resulting in ∼420 mM 2 b, ∼360 mM 3 b and ∼350 mM 5 b (Figure 2). These concentration levels correspond to >99%, 86% and 83% conversion (with respect to AcP), respectively. Furthermore, approx. 170 mM N‐acetyl‐d‐glucosamine‐6‐phosphate (4 b) was obtained (conv. ∼40%). Inosine (6 a) and 6‐amino‐1‐hexanol (7 a) proved to be poor substrates (product concentration <30 mM), while allyl alcohol (8 a) and glycerol (9 a) gave moderate results (∼50 mM and ∼100 mM, resp.). The amount of 4 b increased from 170 mM to 200 mM, while that of 8 b from 40 mM to 120 mM by employing elevated Lw loadings (from 15 to 20 and 25 U mL−1) (Figure S13–S14). The formation of 7 b was boosted by employing wild‐type PhoC−Mm from Morganella morganii,35 finally reaching 140 mM with 25 U mL−1 enzyme (Figure S15). The phosphorylation took place on the alcohol moiety as observed previously.35 40 mM 6 b could be also obtained by PhoC−Mm (Figure S16).
Figure 2.
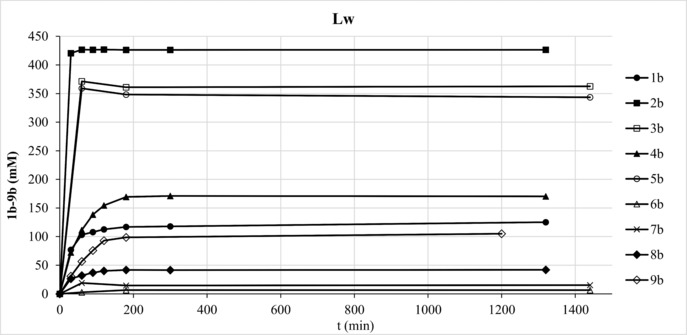
Substrate screening of Lw using crude AcP. Reaction conditions: 15 U mL−1 (9 μM) Lw in ∼400 mM AcP at pH 3.4, 500 mM 1 a–5 a, 7 a–9 a, 80 mM 6 a.
Preparative‐Scale Synthesis
Finally, scale‐up of conversions of 1 a–5 a (500 mM) was performed in 20–50 mL volume using ∼400 mM crude AcP preparation (Table 1, Figure 3) to avoid potential AcP inhibition. In the cases of 2 a, 3 a and 5 a, conversions (based on consumption of the limiting component AcP) reached >90%, highlighting a nearly perfect P‐transfer efficiency accompanied by remarkable space‐time yields (433, 197 and 425 g L−1 h−1, respectively). Turnover numbers (TONs) up to ∼50,000 highlight the robustness of the enzymes. The product phosphate esters were isolated as Ba salts.
Table 1.
Preparative‐scale synthesis of 1 b–5 b.
| P‐donor | Enzyme (U, μg mL−1) |
Conv. (%) |
Yield[g]
(%) |
STY[a]
(g L−1 h−1) |
TON | |
|---|---|---|---|---|---|---|
| 1b [b] | AcP | PiACP (750, 159) |
73[e] | 38[f]
(2.3 g) |
99 | 53,434 |
| PPi | PiACP (750, 159) |
71[e] | 46[f]
(2.8 g) |
64 | 51,970 | |
| 2b [b] | AcP | Lw (750, 263) |
99 | 68 (5.6 g) |
433 | 41,126 |
| 3b [b] | AcP | Lw (750, 263) |
95 | 78 (5.1 g) |
197 | 39,465 |
| 4b [c] | AcP | Lw (600, 350) |
51 | 33 (1.4 g) |
61 | 9,534 |
| 5b [d] | AcP | Lw (300, 263) |
91 | 62 (3.56 g) |
425 | 37,803 |
[a] Space‐time yield with respect to conversion (measured as depletion of substrate), monobasic form of phosphate product and reaction time needed to reach maximal product level.
[b] 50 mL reaction volume.
[c] 30 mL reaction volume.
[d] 20 mL reaction volume.
[e] mono‐/bis‐phosphorylated products ∼80:20.
[f] mono‐phosphorylated product.
[g] products isolated as Ba salt.
Reaction conditions: 15–20 U mL−1 (5.4–12.8 μM) enzyme in ∼400 mM AcP or PPi at pH 3.4, 500 mM 1 a–5 a, stirring at 30 °C.
Figure 3.
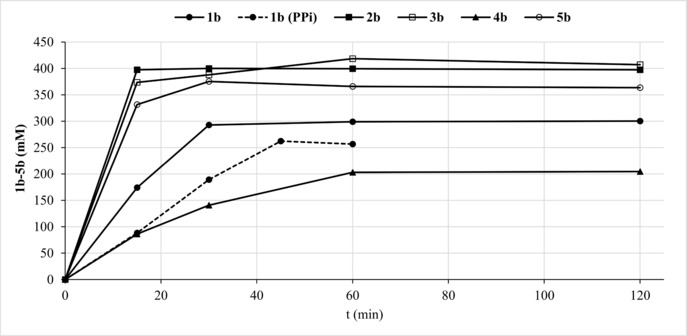
Preparative‐scale transformations of 1 a–5 a with ACP (1a‐5a) and PPi (1a).
In order to compare the AcP protocol with the one employing PPi as donor, the phosphorylation of 1 a was performed using 400 mM PPi (Figure 3, dashed line). Similar conversion, isolated yield and somewhat reduced reaction rate were obtained, however, the amount of BaHPO4 waste was significantly higher with PPi (8.0 g vs. 2.1 g). Phosphorylation of 2 a–5 a took place on the C6−OH position as proven by NMR (ESI).
Conclusion
A range of natural and synthetic high‐energy phosphate donors were evaluated to replace inorganic oligophosphates in phosphatase‐catalyzed transphosphorylation to reduce formation of inorganic monophosphate by‐product. Acetyl phosphate was identified as the most suitable alternative. Furthermore, a defined pH at which the hydrolytic activity of the enzymes could be disentangled from that of the phosphotransferase was identified, which turned highly practical to prevent product depletion. This, combined with the use of an easily obtained crude acetyl phosphate mixture, allowed gram‐scale synthesis of valuable phosphorylated sugars with high space‐time yields, and drastically reduced barium phosphate waste. The method is generally applicable to acid phosphatases and their substrates.
Experimental Section
General Remarks
All chemicals were purchased from commercial suppliers and used as received. Disodium pyrophosphate (PPi; purity ≥99%), lithium potassium acetyl phosphate (AcP), dilithium (carbamoyloxy)phosphonate hydrate (CP) and disodium N‐methyl‐N‐(phosphonocarbamimidoyl)glycinate hydrate (phosphocreatine disodium salt hydrate, PC) were purchased from Sigma, potassium 2‐(phosphonooxy)acrylate (phosphoenolpyruvate monopotassium salt, PEP), 4‐nitrophenyl phosphate disodium salt hexahydrate (p‐NPP) and dimethyl methylphosphonate were from Alfa Aesar. Ni−NTA column for His‐tag purification was from GE Healthcare Life Sciences. NMR spectra were recorded on a Bruker Avance III 300 MHz NMR spectrometer. Chemical shifts (δ) are given in parts per million (ppm) relative to TMS or H3PO4 as a reference. HPLC analysis was carried out on a Dionex Ultimate 3000 system equipped with Shodex RI‐101 refractory index detector (HPLC−RI; Alltech IOA‐2000 Organic Acids column, eluent: 8 mM H2SO4, flow rate: 0.4 mL min−1, 50 °C, injection volume: 20 μL).
Products were identified on an Agilent 1260 Infinity system equipped with Agilent Q6120 quadrupole mass spectrometer using electrospray ionization (HPLC−MS; Zorbax 300‐SCX cation exchanger column, eluent: 0.1% (v/v) formic acid, flow rate: 1 mL min−1, 40 °C, injection volume: 10 μL) and via co‐injection with reference materials. The ratio of mono‐ versus bis‐phosphorylated products in the phosphorylation of 1 a was determined as reported.31
PiACP from Prevotella intermedia,31 AphA‐St31 and PhoN‐Se35 from Salmonella typhimurium LT2, PhoN−Sf from Shigella flexneri,35 PhoK from Sphingomonas sp. BSAR‐1,47 PhoC−Mm wild‐type and G92D/I171T from Morganella morganii 35 and NSAP−Eb 11‐variant from Escherichia blattae 35 were expressed as reported. Phytase from Aspergillus niger was from BASF. The gene of acid phosphatase Lw from Leptotrichia wadei F0279 (signal peptide removed; UniProt Accession Number: U2QAK5) was purchased from IDT and subcloned into pET28a vector using standard molecular biology protocols. The enzyme was overexpressed in E. coli (ESI).
General Screening Conditions
A standard reaction mixture contained substrate and P‐donor in H2O at a concentration and pH indicated in the footnotes of tables and captions of figures and 1% (v/v) DMSO as internal standard in 1 mL final volume. No additional buffering agent was added. The reaction was initiated by adding the indicated amount of enzyme. The mixture was shaken in 1 mL glass vials at 30 °C and 600 rpm in an Eppendorf thermoshaker. Samples of 25 μL volume were taken at intervals, diluted with 475 μL of 8 mM aq. H2SO4 and analyzed on HPLC−RI. Experiments were performed in duplicate. Product levels were determined by consumption of substrate.
In the case of d‐glucosamine (3 a), maltotriose (5 a), inosine (6 a) and 6‐amino‐1‐hexanol (7 a), a 100 μL sample was taken at intervals and added to a mixture of 490 μL H2O, 100 μL 350 mM dimethyl methylphosphonate (internal standard, final concentration: 50 mM) in D2O and 10 μL 1 M HCl (for quenching). Then, a 31P−NMR spectrum was recorded using inverse gated decoupling (ns 32, d1=30 s, pw=11 μs).
Preparative‐Scale Synthesis of 1 b‐5 b
Preparative‐scale transformations were performed in 50 mL (1 a–3 a), 30 mL (4 a) or 20 mL (5 a) reaction volume in a round‐bottom flask containing 500 mM 1 a–5 a and 400 mM crude AcP or PPi. The mixture was adjusted to pH 3.4, the reactions were initiated by addition of enzyme [1 a: 15 U mL−1 (159 μg mL−1) PiACP; 2 a, 3 a, 5 a: 15 U mL−1 (263 μg mL−1) Lw; 4 a: 20 U mL−1 (350 μg mL−1) Lw] and were stirred at 30 °C. The progress of the reaction was monitored via HPLC−RI (1 a, 2 a and 4 a) or 31P−NMR (3 a and 5 a). When the product level reached a maximum, the mixture was ultrafiltered for enzyme removal. 450 mM Ba(OH)2×8H2O (for AcP reactions) or 800 mM Ba(OAc)2 (for PPi reaction) was added, the pH was set to 9–10 and the mixture was stirred for 15 min (AcP reactions) or 1 h (PPi reaction) at room temperature followed by filtration. To the filtrate was added EtOH (80 vol% final concentration) and the mixture was allowed to stand at 4 °C overnight to precipitate the barium salt of the product. In the case of 3 a and 4 a, the pH of the filtrate was set to 4 before the addition of EtOH. The solids were filtered and the products were dried at room temperature. The products were characterized without further purification (purity 1 b: ∼90%; 2 b: ∼93%; 3 b: ∼95%; 4 b: ∼88%; 5 b: ∼90%) by NMR (ESI).
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Funding by the Austrian BMWFW, BMVIT, SFG, Standortagentur Tirol, Government of Lower Austria and ZIT through the Austrian FFG‐COMET‐Funding Program is gratefully acknowledged.
G. Tasnádi, W. Jud, M. Hall, K. Baldenius, K. Ditrich, K. Faber, Adv. Synth. Catal. 2018, 360, 2394.
Contributor Information
Kai Baldenius, Phone: +43‐316‐380‐5332, FAX: +43‐316‐380‐9840.
Kurt Faber, Email: Kurt.Faber@Uni-Graz.at, Phone: +43‐316‐380‐5332, FAX: +43‐316‐380‐9840.
References
- 1. de Groeve M. R. M., Desmet T., Soetaert W., J. Biotechnol. 2011, 156, 253–260. [DOI] [PubMed] [Google Scholar]
- 2. Nakai H., Kitaoka M., Svensson B., Ohtsubo K., Curr. Opin. Chem. Biol. 2013, 17, 301–309. [DOI] [PubMed] [Google Scholar]
- 3. Liu Y., Nishimoto M., Kitaoka M., Carbohydr. Res. 2015, 401, 1–4. [DOI] [PubMed] [Google Scholar]
- 4. Suzuki E., Ishikawa K., Mihara Y., Shimba N., Asano Y., Bull. Chem. Soc. Jpn. 2007, 80, 276–286. [Google Scholar]
- 5. Sherstyuk Y. V., Abramova T. V., ChemBioChem 2015, 16, 2562–2570. [DOI] [PubMed] [Google Scholar]
- 6. Nobeli I., Ponstingl H., Krissinel E. B., Thornton J. M., J. Mol. Biol. 2003, 334, 697–719. [DOI] [PubMed] [Google Scholar]
- 7. Gauss D., Schönenberger B., Molla G. S., Kinfu B. M., Chow J., Liese A., Streit W. R., Wohlgemuth R., in Applied Biocatalysis, From Fundamental Science to Industrial Application (Eds.: L. Hilterhaus, A. Liese, U. Kettling, G. Antranikian), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2016, pp. 147–177. [Google Scholar]
- 8. Rautio J., Kumpulainen H., Heimbach T., Oliyai R., Oh D., Jarvinen T., Savolainen J., Nat. Rev. Drug Discovery 2008, 7, 255–270. [DOI] [PubMed] [Google Scholar]
- 9. Schultz C., Bioorg. Med. Chem. 2003, 11, 885–898. [DOI] [PubMed] [Google Scholar]
- 10. Zawilska J. B., Wojcieszak J., Olejniczak A. B., Pharmacol. Rep. 2013, 65, 1–14. [DOI] [PubMed] [Google Scholar]
- 11. Murray J. I., Woscholski R., Spivey A. C., Chem. Commun. 2014, 50, 13608–13611. [DOI] [PubMed] [Google Scholar]
- 12. Murray J. I., Woscholski R., Spivey A. C., Synlett 2015, 26, 985–990. [Google Scholar]
- 13. Coppola K. A., Testa J. W., Allen E. E., Sculimbrene B. R., Tetrahedron Lett. 2014, 55, 4203–4206. [Google Scholar]
- 14. Joseph A. A., Chang C.-W., Wang C.-C., Chem. Commun. 2013, 49, 11497–11499. [DOI] [PubMed] [Google Scholar]
- 15. Modro A. M., Modro T. A., Org. Prep. Proced. Int. 1992, 24, 57–60. [Google Scholar]
- 16. Lira L. M., Vasilev D., Pilli R. A., Wessjohann L. A., Tetrahedron Lett. 2013, 54, 1690–1692. [Google Scholar]
- 17. Bala S., Liao J.-Y., Mei H., Chaput J. C., J. Org. Chem. 2017, 82, 5910–5916. [DOI] [PubMed] [Google Scholar]
- 18. Jessen H. J., Ahmed N., Hofer A., Org. Biomol. Chem. 2014, 12, 3526–3530. [DOI] [PubMed] [Google Scholar]
- 19. Wever R., van Herk T., Hydrolysis and formation of P-O bonds, in Enzyme Catalysis in Organic Synthesis (Eds.: K. Drauz, H. Gröger, O. May), Wiley-VCH, 2012, pp. 1001–1033. [Google Scholar]
- 20. Wever R., Babich L., Hartog A. F., Transphosphorylation, in Science of Synthesis: Biocatalysis in Organic Synthesis (Eds.: K. Faber, W.-D. Fessner, N. Turner), Georg Thieme Verlag, 2015, vol. 1, pp. 223–254. [Google Scholar]
- 21. Wohlgemuth R., Liese A., Streit W., Trends Biotechnol. 2017, 35, 452–465. [DOI] [PubMed] [Google Scholar]
- 22. Chenault H. K., Mandes R. F., Hornberger K. R., J. Org. Chem. 1997, 62, 331–336. [DOI] [PubMed] [Google Scholar]
- 23. Crans D. C., Whitesides G. M., J. Am. Chem. Soc. 1985, 107, 7008–7018. [Google Scholar]
- 24. Crans D. C., Whitesides G. M., J. Am. Chem. Soc. 1985, 107, 7019–7027. [Google Scholar]
- 25. Drueckhammer D. G., Wong C. H., J. Org. Chem. 1985, 50, 5912–5913. [Google Scholar]
- 26. Andexer J. N., Richter M., ChemBioChem 2015, 16, 380–386. [DOI] [PubMed] [Google Scholar]
- 27. Axelrod B., J. Biol. Chem. 1948, 172, 1–13. [PubMed] [Google Scholar]
- 28. Appleyard J., Biochem. J. 1948, 42, 590–596. [PubMed] [Google Scholar]
- 29. Faber K., in Biotransformations in Organic Chemistry, 7th edn., Springer Verlag, Berlin Heidelberg, 2018, pp. 104–115. [Google Scholar]
- 30. Pradines A., Klaebe A., Perie J., Paul F., Monsan P., Tetrahedron 1988, 44, 6373–6386. [Google Scholar]
- 31. Tasnádi G., Lukesch M., Zechner M., Jud W., Hall M., Ditrich K., Baldenius K., Hartog A. F., Wever R., Faber K., Eur. J. Org. Chem. 2016, 45–50. [Google Scholar]
- 32. Schoevaart R., van Rantwijk F., Sheldon R. A., J. Org. Chem. 2000, 65, 6940–6943. [DOI] [PubMed] [Google Scholar]
- 33. Mihara Y., Utagawa T., Yamada H., Asano Y., Appl. Environ. Microbiol. 2000, 66, 2811–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mihara Y., Ishikawa K., Suzuki E., Asano Y., Biosci. Biotechnol. Biochem. 2004, 68, 1046–1050. [DOI] [PubMed] [Google Scholar]
- 35. Tasnádi G., Zechner M., Hall M., Baldenius K., Ditrich K., Faber K., Biotechnol. Bioeng. 2017, 114, 2187–2195. [DOI] [PubMed] [Google Scholar]
- 36. Babich L., Hartog A. F., Horst M. A. van der, Wever R., Chem. Eur. J. 2012, 18, 6604–6609. [DOI] [PubMed] [Google Scholar]
- 37. Tasnádi G., Hall M., Baldenius K., Ditrich K., Faber K., J. Biotechnol. 2016, 233, 219–227. [DOI] [PubMed] [Google Scholar]
- 38. Pollak A., Baughn R. L., Whitesides G. M., J. Am. Chem. Soc. 1977, 99, 2366–2367.193884 [Google Scholar]
- 39. Wong C.-H., Whitesides G. M., J. Am. Chem. Soc. 1981, 103, 4890–4899. [Google Scholar]
- 40. van Herk T., Hartog A., van der Burg A. M., Wever R., Adv. Synth. Catal. 2005, 347, 1155–1162. [Google Scholar]
- 41. Médici R., Garaycoechea J., Valino A., Pereira C., Lewkowicz E., Iribarren A., Appl. Microbiol. Biotechnol. 2014, 98, 3013–3022. [DOI] [PubMed] [Google Scholar]
- 42. Asano Y., Mihara Y., Yamada H., J. Mol. Catal. B Enzym. 1999, 6, 271–277. [Google Scholar]
- 43. Hokse B. H., Starch/Staerke 1983, 35, 101–102. [Google Scholar]
- 44. Fujimoto A., Smith R. A., Biochim. Biophys. Acta 1962, 56, 501–511. [DOI] [PubMed] [Google Scholar]
- 45. Meyerhof O., Green H., J. Biol. Chem. 1949, 178, 655–667. [PubMed] [Google Scholar]
- 46. Crans D. C., Whitesides G. M., J. Org. Chem. 1983, 48, 3130–3132. [Google Scholar]
- 47. Nilgiriwala K. S., Alahari A., Rao A. S., Apte S. K., Appl. Environ. Microbiol. 2008, 74, 5516–5523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary