

Abstract
The network hypothesis of depression proposes that mood disorders reflect problems in information processing within particular neural networks. Antidepressants (AD), including selective serotonin reuptake inhibitors (SSRI), function by gradually improving information processing within these networks. AD have been shown to induce a state of juvenile‐like plasticity comparable to that observed during developmental critical periods: Such critical‐period‐like plasticity allows brain networks to better adapt to extrinsic and intrinsic signals. We have coined this drug‐induced state of juvenile‐like plasticity ‘iPlasticity.’ A combination of iPlasticity induced by chronic SSRI treatment together with training, rehabilitation, or psychotherapy improves symptoms of neuropsychiatric disorders and issues underlying the developmentally or genetically malfunctioning networks. We have proposed that iPlasticity might be a critical component of AD action. We have demonstrated that iPlasticity occurs in the visual cortex, fear erasure network, extinction of aggression caused by social isolation, and spatial reversal memory in rodent models. Chronic SSRI treatment is known to promote neurogenesis and to cause dematuration of granule cells in the dentate gyrus and of interneurons, especially parvalbumin interneurons enwrapped by perineuronal nets in the prefrontal cortex, visual cortex, and amygdala. Brain‐derived neurotrophic factor (BDNF), via its receptor tropomyosin kinase receptor B, is involved in the processes of synaptic plasticity, including neurogenesis, neuronal differentiation, weight of synapses, and gene regulation of synaptic formation. BDNF can be activated by both chronic SSRI treatment and neuronal activity. Accordingly, the BDNF/tropomyosin kinase receptor B pathway is critical for iPlasticity, but further analyses will be needed to provide mechanical insight into the processes of iPlasticity.
Keywords: brain‐derived neurotrophic factor/tropomyosin kinase receptor B, dematuration, neurogenesis, neuronal plasticity, parvalbumin/perineuronal nets
Antidepressants (AD), such as tricyclic AD, selective serotonin reuptake inhibitors (SSRI), and monoamine oxidase inhibitors, increase the available monoamines in the synaptic cleft by, respectively, inhibiting the reuptake of serotonin and norepinephrine, selectively inhibiting the reuptake of serotonin by the presynaptic nerve terminal, and inhibiting their degradation of monoamines, thereby increasing the available monoamines in the synaptic cleft. This increase in monoamine signaling associated with AD effects has led to what is defined as the ‘monoamine hypothesis’ of depression,1 which proposes that serotonin and norepinephrine are involved in the etiology and treatment of depressive disorders and that there is a causal relation between the observed physiological effects of AD on monoamine metabolism and their behavioral outcomes.1 Recently, however, the network hypothesis of depression and AD action has offered an alternative to the ‘imbalance of monoamines’ theory.2, 3, 4 The network hypothesis proposes that mood disorders reflect problems in information processing within particular neural networks in the brain and that AD act by gradually improving information processing within these networks.
More recently, it has been demonstrated that AD treatment activates a state of plasticity in the adult brain that resembles plasticity during critical or juvenile periods in postnatal development. When AD‐induced plasticity is combined with training (learning a particular skill or behavior), rehabilitation (restoring to health or normal state mentally and/or physically through training and therapy), or psychotherapy that guides the plastic connectivity, networks miswired by abnormal early experiences can be repaired in adulthood. In this review, we introduce the concept of and evidence for the phenomena and discuss possible underlying biological mechanisms.
CRITICAL‐PERIOD PLASTICITY
During development, the brain adapts to the environment through activity‐dependent neuronal plasticity. In this process, neural networks that are originally manifested by genetic guidance are modified through experience and gradually adapt to the external and internal milieu.5, 6 Experience‐dependent modifications peak during the critical or sensitive periods of early life7, 8, 9 when neuronal networks are plastic and adjust to environmental conditions. A breakthrough in the study of critical‐period plasticity was the discovery of ocular dominance columns in the kitten visual cortex by Hubel and Wiesel.10 These columns, though not visible as columns by eye, are organized such that they are predominantly innervated by axons from the left or right eye. Plasticity studies have shown that activity‐dependent reorganization of these columns is highest during critical periods and very restricted in adulthood after closure of critical periods.11 Abnormal visual input during critical periods, including monocular deprivation, leads to abnormal network structure within the visual cortex, including loss of innervation of a deprived eye and increased innervation of the fellow eye. These effects persist into adulthood if the pattern is not corrected during the critical period, a phenomenon known as amblyopia.12 Since then, the shift in the ocular dominance paradigm in the visual cortex has been used as a robust model to study plasticity. With advances in pharmacological and genetic manipulations, our understanding of the mechanisms underlying critical periods has been growing extensively. This plasticity has been considered to permanently close at the end of the critical periods, and plasticity in adults is thought to be more restricted and to mainly involve changes in synaptic strength.13, 14, 15, 16
This decade, however, new evidence has suggested that the adult brain is more plastic than previously expected. This is especially apparent through interventions that affect cognition and mood, such as learning, environmental enrichment, exercise, and chronic treatment with AD.17, 18, 19
AD INDUCE JUVENILE‐LIKE PLASTICITY (iPLASTICITY)
As discussed above, plasticity is a permissive state of neuronal networks that are affected by internal and environmental stimuli and ultimately display behavioral changes. Increasing evidence shows that AD can activate a plastic state in the adult brain that is comparable to that observed during developmental critical periods, and that this plastic state can be used to rewire the brain in an activity‐dependent manner by stimuli such as training or rehabilitation. Our group has coined this phenomenon induced juvenile‐like plasticity (iPlasticity; Fig. 1). Clinical studies have shown that combinations of AD treatment with psychotherapy or training are more effective than either treatment alone,20, 21, 22, 23 which is consistent with the iPlasticity principle. Pampallona et al. have shown in a clinical meta‐analysis that patients suffering from depression respond better to AD treatment combined with psychotherapy than AD treatment alone.23 The results of a clinical trial demonstrated that SSRI treatment combined with cognitive behavioral therapy leads to improved clinical response in SSRI‐resistant depressed adolescents.22 Branchi and coworkers recently showed that depressed patients living under more favorable living conditions showed a better response to citalopram than patients living under unfavorable conditions.24 In addition, Amin et al. showed higher efficacy of a combination of clomipramine (a tricyclic AD) and behavioral therapy in the treatment of obsessive–compulsive and phobic disorders.21
Figure 1.
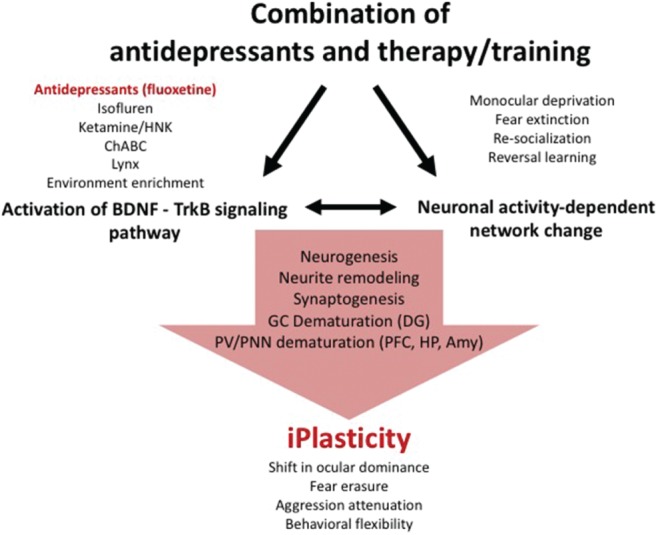
Concept of iPlasticity. Amy, amygdala; BDNF, brain‐derived neurotrophic factor; ChABC, chondroitinase ABC; DG, dentate gyrus; GC, granule cell; HNK, hydroxynorketamine; HP, hippocampus; PFC, prefrontal cortex; PV/PNN, parvalbumin/ perineuronal nets; TrkB, tropomyosin kinase receptor B.
PROOF‐OF‐CONCEPT EXPERIMENTS OF iPLASTICITY IN RODENT MODELS
Shift of ocular dominance by monocular deprivation together with chronic fluoxetine treatment
The visual cortex has become a popular model for studying brain iPlasticity as its measurements are robust, reliable, and conserved across species (including monkeys, cats, and rats).10, 12, 25 Inputs from the left and right eye segregate into eye‐specific ocular dominance columns in the visual cortex during critical periods. This plasticity declines with age.26 For the segregation to be successful, it is crucial to maintain a balance between excitation and inhibition.11 Even small alterations to inhibitory neurotransmission can result in profound effects on visual cortex plasticity. For example, in transgenic mice lacking the 65‐kDa isoform of the gamma‐aminobutyric acid (GABA)‐synthesizing enzyme GAD (GAD65), ocular dominance plasticity in response to monocular deprivation is deficient and can be rescued if GABAergic transmission is enhanced by the administration of benzodiazepines,27 indicating that the critical period is dependent on inhibitory transmission. Conversely, enhancing intracortical inhibition with benzodiazepines28, 29 or promoting the maturation of GABA neurons via transgenic overexpression of brain‐derived neurotrophic factor (BDNF)30 is associated with accelerated onset of critical periods. This close relation between neuronal activity, BDNF release, and GABAergic transmission is also shown in the effects of dark rearing. If animals are raised without visual input, BDNF levels and GABAergic transmission are reduced in the visual cortex, which delays the peak of plasticity into adulthood. Infusion with diazepam, a positive allosteric modulator of the GABAA receptor,29 or overexpression of BDNF,31 however, abolishes the extension of the critical period. These results suggest that the GABAergic intracortical networks mature in correspondence with the critical period.
Later studies, however, have provided evidence for reactivation of critical‐period‐like plasticity in the adult visual cortex induced by drugs currently in clinical use and belonging to a number of pharmacological classes.25, 32 The first groundbreaking study showed that chronic treatment with the AD fluoxetine can reopen visual cortex plasticity in the adult rat brain.25 The authors in this study combined chronic treatment with the AD fluoxetine (4 weeks) with monocular deprivation for a week in adult rats and used the following two classical assays: (i) the ocular dominance shift of visual cortical neurons in the adult brain; and (ii) the recovery of visual acuity in amblyopic adult rats whose ocular dominance had been shifted with monocular deprivation during the critical period. By recording visually evoked potentials in the binocular region of the primary visual cortex contralateral to the deprived eye, the responses of a population of neurons to patterned visual stimuli can be measured and used to evaluate visual acuity and binocularity alterations. Ocular dominance is then calculated by the contralateral‐to‐ipsilateral visually evoked potential ratio. While rodents naturally display contralateral eye dominance, the authors demonstrated that after chronic treatment with the AD fluoxetine, they could reinstate visual cortex plasticity in the adult rat, inducing a shift in ocular dominance towards the weaker, ipsilateral non‐deprived eye. Moreover, this treatment promoted the recovery of visual functions in adult amblyopic rats. These effects were accompanied by a reduction in intracortical inhibition (as assessed by in vivo microdialysis) and increased BDNF protein levels (quantified by enzyme‐linked immunosorbent assay). Finally, cortical diazepam administration prevented the effects induced by fluoxetine. These results indicate different roles of interneurons in the developing and mature visual cortex, or between the opening of the critical period and maintaining the state of the critical period.
Previously, Maffei et al.33 reported that enzymatic removal of perineuronal nets (PNN), which are extracellular matrix structures mainly enwrapping the maturing synaptic circuitry of GABAergic parvalbumin‐positive (PV+) inhibitory neurons, can reactivate critical period plasticity by decreasing inhibition33 (discussed in the ‘Cortical inhibition’ section below), thereby promoting the generation of gamma oscillations.34 Harauzov et al. pharmacologically reduced GABAergic action using GABA antagonists, picrotoxin, or 3‐mercaptopropionic acid and induced a shift in ocular dominance by monocular deprivation, which was inefficient in saline‐treated animals.35 The studies support the hypothesis by Takao Hensch, which posits that a reduction in intracortical inhibition promotes visual cortical plasticity in the adult brain, contrasting with a different role of GABAergic inhibition during and after the critical period. These findings are not only relevant for the clinical application of fluoxetine but also demonstrate the idea of iPlasticity and suggest new mechanisms of AD effects.36
Fear erasure by fear extinction training with chronic fluoxetine treatment
Pavlovian fear conditioning37 is another validated and popular paradigm in plasticity studies, especially in studying networks that are more complex. A variety of studies have shown that the fear and anxiety circuit is conserved across species and has been studied in both animal models and humans, and they can be modeled by the fear‐conditioning paradigm in mice. Consistent throughout animal models and human studies, the fear and anxiety circuit involves the prefrontal cortex, amygdala, and hippocampus, which are each responsible for different aspects of fear.38, 39, 40 The primary choices of treatment in such conditions are either extinction through exposure therapy41 or pharmacotherapy, mostly using SSRI AD.42 Exposure therapy extinguishes or suppresses fear responses by repeatedly exposing the subject to the fear‐inducing stimulus under a safe environment; however, the effect of extinction is transient and a spontaneous recovery typically appears. Additionally, clinical experience has shown that a combination of AD treatment and psychotherapy is more effective than either treatment alone.23 Our group has shown that chronic treatment with the AD fluoxetine can extinguish long‐term fear memory only when it is combined with extinction training, while fluoxetine alone is ineffective and extinction training alone leads to spontaneous recovery.43 In the study, we used the common cued and contextual fear conditioning protocol consisting of the fear conditioning/acquisition phase, extinction training in a chamber different from the one used in the conditioning/acquisition phase, spontaneous recovery (same chamber as extinction training), and fear renewal (the chamber used for conditioning) phases. Chronic fluoxetine treatment was started either 3 weeks before fear conditioning or immediately after conditioning, and was continued throughout the whole experiment. While control water‐treated mice recovered and renewed conditioned fear in context A and B, respectively, after extinction training fluoxetine‐treated mice showed attenuated fear recovery and renewal. Aiming to identify possible underlying mechanisms, we found that fluoxetine treatment reduced the percentage of PNN‐positive neurons expressing PV, but upregulated polysialylated neuronal cell‐adhesion molecule (PSA‐NCAM) and KCC2, which are increased during postnatal development. In addition, electrophysiological experiments showed that fluoxetine treatment increased synaptic plasticity in the lateral amygdala, as measured by increases in field excitatory postsynaptic potential responses and long‐term potentiation (LTP) induction. Furthermore, BDNF‐knockout mice and infection with a BDNF‐expressing lentivirus in the basolateral amygdala demonstrated that an absence of or overexpression of BDNF blocked or enhanced the effect of fluoxetine treatment on fear erasure, respectively. Together, the study suggested that fear erasure induced by a combination of fluoxetine treatment and extinction training relies on a shift of PV‐containing and PNN‐containing neurons towards a juvenile or immature state in the basolateral amygdala and CA1 region of the hippocampus through the activation of the BDNF/tropomyosin kinase receptor B (TrkB) pathway, which opens a window of plasticity in the adult brain.
Attenuation of aggression by resocialization together with chronic fluoxetine treatment
Rat models of early social neglect were previously established by post‐weaning social isolation.44, 45 This model can be used to investigate early adversity‐induced aggression and explore novel possibilities for intervention.46 The characteristic feature of social isolation is the increase in biting attacks that are preferentially aimed at vulnerable body parts of the opponent (head, throat, and belly) in a resident‐intruder test.44, 45 Resocialization of isolated rodents in the adult phase has been proposed as a laboratory model for behavioral therapy,47 but resocialization failed to decrease the social isolation‐induced escalation of aggression, which confirms the assumption that this paradigm induces long‐term changes in emotional processing.48, 49 We hypothesized that there is a critical period for suppression of social aggression that is lost during early isolation, and that resocialization is not effective in adult animals after the closure of this critical period. However, resocialization as an adaptive process to environmental factors might be enhanced by reopening the critical period by chronic fluoxetine treatment in adulthood. In fact, a combination of resocialization therapy and chronic fluoxetine treatment in isolated rats largely reversed escalated aggression, while neither treatment alone exerted significant effects.50 In resocialization‐resistant early‐isolation‐induced aggressive rats, BDNF expression is reduced in the amygdala and medial prefrontal cortex (mPFC). Only combined treatment with fluoxetine and resocialization was able to recover BDNF expression via epigenetic regulation (DNA methylation) in the mPFC. In addition, experiments using TrkB inhibitors demonstrated that behavioral improvement after combined treatment was dependent on TrkB activity. Furthermore, cholera toxin subunit B, a retrograde tracer combined with FosB immunostaining, showed that the input from the ventral hippocampus (vHP) to the mPFC was specifically strengthened by combined treatment. These results suggest that BDNF expression in the mPFC and vHP–mPFC pathway is an important mediator of the reduced aggression induced by resocialization combined with chronic fluoxetine treatment. Together, synergistic interactions between psychosocial therapy and temporarily increased plasticity by SSRI specifically ameliorate escalated aggression induced by socially adverse early experiences.
Increased behavioral flexibility by spatial reversal learning together with fluoxetine treatment
The effects of fluoxetine on spatial learning have been assessed in several studies. It has been shown that both one‐time (acute) and prolonged (chronic) fluoxetine treatment caused a significant improvement in reference spatial memory in rats.51 However, some studies show negative effects or no effects of fluoxetine treatment on spatial learning skills.52, 53, 54, 55 Thus, it is still unclear whether chronic fluoxetine treatment can improve spatial learning. Considering that typically used spatial learning tasks are designed to identify spatial learning deficits, we hypothesized that chronic fluoxetine treatment specifically affects more complex learning tasks, such as reversal learning and behavioral flexibility.56 In this case, an individual learned response to a single decision among multiple options was elicited during an initial learning or acquisition phase. After obtaining the desired level of performance, the task pattern was reversed or altered: A previously incorrect option was now correct and the previously correct option incorrect. To test this more complex spatial learning task, we used an Intellicage (NewBehavior AG, Zurich, Switzerland), in which transponder‐implanted female mice were group‐housed.57, 58 In the IntelliCage protocol, during the acquisition phase, water‐deprived mice are granted access to water in a certain corner chamber. Then, the next corner becomes available for drinking in a clockwise manner during the acquisition phase. If a mouse enters the correct corner, the door opens upon nose poke and water becomes available. Thus, animals have to patrol the corners to find the correct chamber that offers water access. After the mice have learnt the clockwise pattern for water access and display low‐error trials, the direction switches to the opposite direction, counterclockwise. Our studies indicate that chronic treatment with fluoxetine does not affect acquisition of spatial memory (clockwise pattern) but does improve ‘reversal’ learning (counterclockwise pattern) (Umemori and Castrén, manuscript in preparation). These results indicate that chronic fluoxetine enhances behavioral flexibility of the spatial leaning process, which may reflect the same underlying phenomenon of iPlasticity.
Effects of AD under adverse environmental stimuli
If AD treatment acts permissively to potentiate responses to environmental stimuli, then AD treatment under adverse stimuli might worsen the state of a patient. Recently, Branchi and coworkers provided experimental evidence suggesting that this, indeed, may be the case.59 They exposed mice to stress and then either switched the mice to an enriched environment (EE) or continued stress while treating them with an SSRI or a vehicle. While SSRI treatment counteracted an anhedonia‐like state in mice kept in an EE, fluoxetine treatment of stressed mice significantly elevated depression‐like behavior when compared to vehicle treatment.59 These data emphasize that plasticity does not have a direction, but rather that favorable stimuli promote adaptability while unfavorable stimuli are maladaptive.
AD OTHER THAN SSRI AND FACTORS MEDIATING iPLASTICITY
All AD drugs increase TrkB signaling in the brain60 (see below), and BDNF signaling through TrkB has been shown to be necessary and sufficient for fluoxetine‐induced iPlasticity, indicating that most if not all AD might induce plasticity. In agreement with this notion, preliminary findings show that tianeptine, an AD not acting through serotonin reuptake inhibition61 reactivates ocular dominance plasticity in the adult rat visual cortex (Maya‐Vetencourt, Cattaneo, and Castrén, unpublished).
Ketamine, an anesthetic compound and a non‐competitive NMDA receptor antagonist,62 has been shown to induce a fast and long‐lasting AD effect in treatment‐resistant patients with bipolar disorder and depression.63, 64, 65 Moreover, administration of ketamine also decreases suicidal ideation in treatment‐resistant depressed patients.64, 66, 67 One feature of ketamine is its fast‐acting production of AD effects, which can already be observed within 4 h after a single administration,68 whereas other conventional AD take several weeks to produce AD effects.69 In the fear‐conditioning paradigm, a combination of acute (single) administration of ketamine and fear extinction training promotes the erasure of conditioned fear70 similarly to that observed for chronic treatment with fluoxetine.43 Despite its fast and long‐lasting AD effects, the psychotomimetic effects of the compound are concerning. A possible way to overcome this problem may be to instead use specific intermediate compounds produced during the metabolism of ketamine, such as norketamine and hydroxynorketamine (HNK).71 In rodent models, HNK appears to provide the same AD‐like effects as ketamine, but without its NMDA‐binding capacity. Interestingly, this promotes the expression of BDNF through activation of α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid (AMPA)‐mediated currents.72 It will be important to use appropriate experimental models to test whether ketamine and HNK might induce plasticity in the same manner as fluoxetine.
Electroconvulsive stimulation (ECS) is a non‐drug‐based alternative approach in the treatment of psychiatric diseases.73 Previous studies have shown an involvement of neural plasticity in the therapeutic effects of ECS in human patients.74, 75, 76 In rodent models, it has been shown that ECS increases the expression of BDNF and its receptor TrkB73, 77, 78 in cortical and limbic areas and increases the duration of their expression, especially in the granular layer of the dentate gyrus.78 In addition, it has been observed that ECS increases neurogenesis79, 80 and synaptogenesis.81, 82, 83
Morishita et al. showed that Lynx, a bungarotoxin‐like endogenous pro‐toxin that binds to nicotinic acetylcholine receptors and decreases their sensitivity to acetylcholine, is expressed in the visual cortex only after the closure of the critical period, and that adult Lynx‐deficient mice show ocular dominance plasticity, indicating a role for Lynx in restricting visual cortex plasticity.84 The same paper also demonstrated that an inhibitor of acetylcholinesterase, physostigmine, restored the visual cortex of monocularly deprived adult mice,84 indicating that the ability to induce iPlasticity is not restricted to AD drugs.
Finally, Maffei and coworkers have demonstrated that housing rats in an EE, which provides high motor‐sensory stimulation, produces similar iPlasticity effects to fluoxetine in the adult visual cortex and restores visual acuity in a rat model of amblyopia.18, 85 In addition, it has been shown that an EE can improve other plasticity‐related features of the brain, such as memory and learning86 and neurogenesis in the hippocampus.87 These results provide a promising view regarding a non‐pharmacological way to reactivate plasticity in the adult brain.
In summary, iPlasticity is induced by several drugs acting on different neurotransmitter systems. Additional experiments are needed to test whether iPlasticity is a common phenomenon shared by all AD drugs88 such as monoamine oxidase inhibitors, serotonin and norepinephrine reuptake inhibitors1 and inhibitors of histone deacetylase.89
iPLASTICITY, NEUROGENESIS, AND DEMATURATION
In addition to iPlasticity, AD drug treatment has been shown to promote adult neurogenesis and dematuration in the hippocampal dentate gyrus. Below, we will introduce these plastic phenomena and discuss their potential relationship to iPlasticity.
Adult neurogenesis
Neurons are known to newly arise in both the dentate gyrus and the subventricular zone of adults, and comprise up to 10% of the entire granule cell (GC) population in the dentate gyrus (DG).90 Adult hippocampal neurogenesis is associated with pattern separation, which is the ability to differentiate overlapping contextual representations.91 Chronic SSRI treatment has been shown to stimulate all stages of adult neurogenesis, including the proliferation, differentiation, and survival of adult‐born granule cells (abGC)19, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101 (Fig. 2, 102, 103). Fluoxetine does not affect the division of stem‐like cells but increases symmetric divisions of early progenitor/precursor cells.104 Chronic SSRI treatment prompts young abGC to rapidly mature with heightened synaptic plasticity and to integrate into the DG.92, 93, 105, 106 Young abGC are highly active and have elevated plasticity for a few weeks,106, 107, 108 and eventually become functionally similar to the mature, developmentally born GC 8 weeks after neurogenesis.106, 108 In addition, chronic fluoxetine treatment has been shown to affect the dendritic arborization of immature neurons that express doublecortin and to enhance LTP in a neurogenesis‐dependent manner.109
Figure 2.
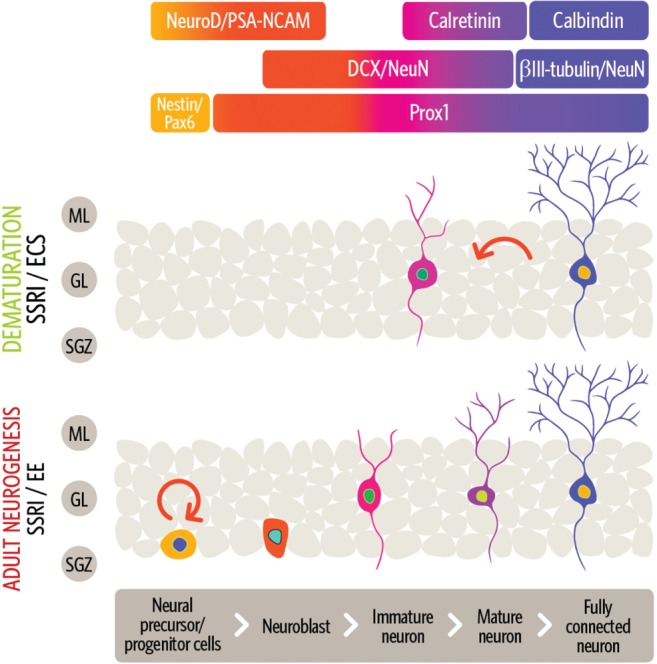
Model of dematuration and adult neurogenesis in the dentate gyrus (DG) by chronic selective serotonin reuptake inhibitors (SSRI) treatment. The upper section indicates expression markers of each neuron state.102, 103 In the middle section, a mature neuron change to an immature state by dematuration after chronic SSRI treatment or electroconvulsive stimulation (ECS). In the bottom section, chronic SSRI treatment or environmental enrichment (EE) promote the proliferation of progenitor granule cells in the DG and rapid maturation of young adult‐born cells and integration into the DG. Finally, the matured adult‐born cell is functionally similar to other mature cells. The picture is combined and modified from Zhang and Jiao102 and Segi‐Nishida.103 DCX, doublecortin; ML, molecular layer; GL, granule layer; PSA‐NCAM, polysialic acid neural cell adhesion molecule; SGZ, subgranular zone. Copyright © 2017 Segi‐Nishida. Copyright © 2015 Juan Zhang and Jianwei Jiao.
The 5‐HT1A receptors on mature DG GC are critical for the AD response, as absence of the serotonin 1A receptor (5HT1AR, a receptor required for the SSRI response) specifically abolishes from DG GC the effects of fluoxetine on behavior and on the hypothalamic–pituitary–adrenal axis.110 These observations suggest that the involvement of the serotonergic system in neurogenesis in the DG is most likely due to SSRI action itself.
Chronic corticosterone exposure (mimicking the effect of chronic stress)100, 111, 112 and an unpredictable chronic mild stress prompt a depressive‐like state and have decreased numbers of newly born cells in the DG of the adult hippocampus, which are completely recovered from after 2–3 weeks of fluoxetine treatment.96, 100 In addition, it has been reported that there are neurogenesis‐dependent and neurogenesis‐independent ameliorating effects of fluoxetine on anxiety, as well as on depression‐like behaviors caused by chronic corticosterone exposure or an unpredictable chronic mild stress paradigm.96, 113 However, periodic food restriction induces iPlasticity in the adult visual cortex and this effect was shown to be mimicked by a periodic increase in corticosteroid release,114 demonstrating that short‐lasting and chronic treatment with corticoids have opposite effects on plasticity. In addition, studies using focal radiation or genetic manipulation demonstrated that ablation or impairment of the neurogenic niche attenuate AD effects on depression‐related behaviors, indicating that hippocampal neurogenesis is required for some AD effects.93, 95, 96, 115 An EE has also been reported to increase adult neurogenesis in the DG of mice housed in an EE87 and in young and aged rats when housed in an EE.116, 117 In addition, ketamine has been shown to promote synaptogenesis in the rodent PFC118 and neurogenesis in the hippocampus.119
These observations indicate that a range of AD treatments exert effects on adult neurogenesis and ameliorate depression‐like behaviors. However, it is unknown whether neurogenesis is involved in iPlasticity, especially in the visual cortex and brain regions related to fear erasure, social behavior, and reversal spatial learning. Adult neurogenesis after chronic SSRI treatment has also been reported to occur in the amygdala,120 PFC,121 hypothalamus, and habenula,122 but not in the visual cortex. Thus, the neurogenesis and their maturation promoted by fluoxetine might contribute to the formation of new networks, although further studies are needed.
Dematuration of dentate gyrus by chronic fluoxetine treatment
As discussed above, chronic fluoxetine treatment is generally known to increase adult neurogenesis in the hippocampal DG, but some studies have reported that the behavioral effects of AD do not always coincide with increased neurogenesis.98, 99, 123, 124, 125 Thus, not only neurogenesis but also functional modifications of existing neurons would be necessary for AD action.126 Kobayashi et al. found that chronic fluoxetine treatment reverts the molecular and functional properties of GC to an immature state in the DG of the hippocampus126, 127 (Fig. 2), which is a substrate for both cognition and mood regulation.128 This ‘dematuration’ is characterized by downregulated expression of mature GC markers, such as calbindin, desmoplakin, tryptophan‐2,3‐dioxygenase, and interleukin‐1 receptor type I, as well as by upregulated immature‐like GC markers, including calretinin.121, 126, 129 The dematured GC induced by chronic fluoxetine treatment exhibited electrophysiological features of actual immature or developing GC, such as the following: (i) higher excitability upon somatic current injections and active membrane properties resembling those in young GC; (ii) enhanced long‐term depression at the perforant path synapse, but reduced LTP; and (iii) mossy fiber synaptic facilitation reduced to the juvenile level. Mossy fiber‐CA3 pyramidal cell synapses formed by immature‐like GC exhibit smaller frequency facilitation126 than 3‐week‐old young mice.130 Chronic fluoxetine treatment had no significant effects either on the expression level of calbindin or on frequency facilitation in 5‐HT4‐receptor‐deficient mice, suggesting that the 5‐HT4 receptor plays a critical role in the dematuration of GC126 and in adult neurogenesis in the DG, and that these phenomena are closely associated.129 Thus, the serotonergic system seems to be important probably due to SSRI action on the maturity of GC in the DG. Recently, it has been shown that a brief neuronal activation by ECS induces dematuration of the mature GC. Namely, in all GC of the DG, there was observed reduced expression of the mature GC markers and physiological properties of immature GC (such as elevated somatic intrinsic excitability and smaller frequency facilitation at the dentate‐to‐CA3 synapse) were observed.131 In addition, while a single ECS lasts a short time, repeated ECS causes long‐lasting (greater than 2 weeks) dematuration in an NMDA‐receptor‐dependent but not serotonergic‐dependent manner.131 These results suggest that altered functioning of the mature dentate GC is commonly involved in the cellular mechanisms of physical and chemical AD treatments.132 A dematuration‐like response to fluoxetine also occurred in interneurons in other brain regions, such as the PFC,121, 133 amygdala,43 and the visual cortex (Umemori, Winkel, and Castrén, unpublished observations). In these brain areas, chronic fluoxetine treatment caused a decrease in the number of PV‐positive or PV/PNN double‐positive interneurons,43, 134 which are negatively associated with plastic or immature states of PV interneurons, suggesting that dematuration of PV interneurons in these areas is important for iPlasticity (discussed in the ‘Cortical inhibition’ section). These findings strongly support the idea of iPlasticity, namely that AD produce an immature or plastic state and a heightened adaptability to neuronal activity in response to internal or external stimuli (or both)19, 32 during the maturation process. An immature DG is also observed in gene‐modified and drug‐induced mice showing psychiatric‐disease‐like phenotypes.127, 135, 136, 137, 138
In the human brain, dematuration of GC in the DG has been observed in postmortem brains of bipolar and schizophrenic patients, as well as in alcoholics.139, 140, 141 It has been suggested that dematuration in the DG might be an endophenotype of certain psychiatric diseases.127, 138 In addition, it has been shown that destabilized cage activity was accompanied by increased anxiety‐related behaviors after high doses and long‐term treatment with high‐dose fluoxetine.142 Moreover, maladaptive effects were observed after the combination of chronic fluoxetine treatment and stress as described above.59 Given the concept of iPlasticity, psychiatric‐disease‐like phenotypes or behavioral consequences might be produced by a combination of dematuration with maladaptive, unstable, or uncontrolled environmental factors (also discussed in the section above, ‘Effects of AD under adverse environmental stimuli’).
Taken together, iPlasticity, neurogenesis, and dematuration are all induced after chronic treatment with AD and increase plasticity in neuronal networks in several regions of the brain, which promotes the adaptation to environmental signals. Although these three plastic phenomena are induced by the same treatment and share at least some properties, it is premature to conclude that they are all reflections of a single underlying mechanism. It will be important in future studies to directly compare these phenomena to each other and investigate the biological mechanisms underlying them to reveal whether they are produced by the same mechanism and, if so, what this mechanism might be.
MECHANISTIC INSIGHTS INTO iPLASTICITY: NEURONAL AND SYNAPTIC CHANGES AFTER CHRONIC FLUOXETINE TREATMENT
The behavioral consequences of iPlasticity have been described in the context of several brain networks, but the actual biological mechanisms underlying iPlasticity are not fully understood. In this last section, we will discuss the observed biological phenomena and possible underlying mechanism of iPlasticity at the level of neuronal circuits, cells, and molecules.
Cortical inhibition
The establishment of an excitation and inhibition balance is crucial for a successful critical period and disturbances in this balance can lead to maladapted networks across different brain regions, including the visual and insular cortex.27, 143, 144 Critical orchestrators of inhibitory network activity are PV+ interneurons, whose activity is tightly regulated.36 PV interneurons are known for fast‐spiking capability and for projection onto the axosomatic region of principal neurons.145 Hence, PV interneurons are capable of strongly regulating principal neuron output (feedforward inhibition) by shaping oscillatory activity.146, 147 Previous research has shown that the maturation of the PV+ interneurons coincides with the production of PNN, whose functions are still largely unknown but should involve the regulation of synaptic plasticity and neuroprotection.33, 143, 148, 149, 150 Both maturation of PV+ interneurons and PNN production promote the closure of critical periods and restrict plasticity of the adult brain, hence being referred to as ‘molecular brakes’ whose inactivation and removal, respectively, is thought to reset the excitation–inhibition balance.11, 143 Recent research has focused on PV and PNN and the underlying mechanisms of critical periods and adult plasticity (Fig. 3, 151). Thus far, accumulated evidence suggests that resetting the PV network to an immature state and removing PNN from the network can induce critical‐period‐like plasticity or iPlasticity in distinct brain regions.33, 34, 134 For example, in the visual cortex, PNN removal results in a decrease in PV activity and subsequent promotion of gamma oscillations mediated through pyramidal cells, finally resulting in ocular dominance plasticity.34 Disruption of PNN in the amygdala has been shown to enhance the erasure of drug addiction with extinction training.152 Removal of PNN in the hippocampus resulted in disrupted contextual and trace fear memory and their removal from the medial prefrontal cortex impaired long‐term trace‐ and conditioned‐stimulus‐elicited fear memory in the trace fear conditioning task.153
Figure 3.
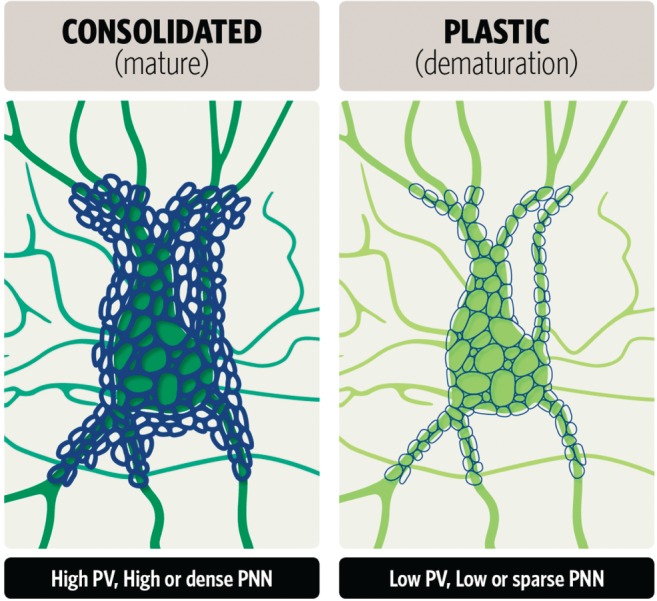
Dematuration of parvalbumin (PV)/perineuronal nets (PNN) interneurons. PNN surround synaptic boutons on neuronal soma and proximal dendrites and consist of chondroitin sulfate proteoglycans (CSPG) assembled on the hyaluronan scaffold of PV interneurons. The development of PNN is controlled by synaptic activity and their formation terminates the critical period of synaptic plasticity, consolidating the neuronal network. CSPG has a unique geometric structure of polygonal mesh shapes with the number of vertices varying from three to nine. Sizes of the mesh vary.151 A reduction in PV (green) and PNN (blue) expression is associated with a plastic and immature network observed in iPlasticity by chronic SSRI treatment.
Interesting work has been published by Donato et al., who proposed a new concept of PV network states that correlate with brain plasticity and consolidation.154 While fear conditioning that involves memory consolidation results in a network state mainly composed of PV cells expressing high levels of PV, an EE, an established way to induce plasticity in the brain, promotes a network state mainly composed of PV cells expressing low levels of PV154 (Fig. 3). A high‐PV/consolidated network is attained by enhanced excitation from local pyramidal cells or external input as memories are consolidated. In contrast, a low‐PV network is maintained by increased inhibitory input from vasoactive intestinal peptide neurons, which are readily engaged by neuromodulators under EE conditions. Therefore, low or high PV states are paralleled by increased inhibition or excitation, respectively, of PV cells themselves.154
Cellular regulators of this change in PV expression likely involve molecules maintaining their mature state through PNN, which interact and release factors such as neurotrophins (BDNF and GDNF) and homeoproteins (Otx2).155 Recently, the microstructure of PNN has been shown to be of a heterogeneous nature with uniform (nonpolar) and node‐enriched (polar) patterns of chondroitin sulfate distribution within a single mesh.151 This pattern might be related to the molecular composition of the PNN of the neuronal cell surface and might be functionally connected to the regulation of synaptic signaling processes, including synaptic plasticity.151 While the function of PNN is still incompletely understood, evidence points towards a role in neuronal protection from oxidative stress.156 Considering the fast‐spiking properties of PV interneurons, metabolic demand is high and mitochondrial density is increased, which specifically renders PV cells particularly sensitive to oxidative stress.156 Therefore, to ensure the control of feedforward inhibition and rhythmic neuronal synchrony, protection through PNN might maintain the fast‐spiking activity of PV cells in adulthood and limit their intrinsic vulnerability.
The BDNF/TrkB pathway
As described above, previous studies have indicated that the activation of BDNF signaling through TrkB is a key molecular pathway in iPlasticity in the adult brain19, 25, 43, 88, 157, 158 (Fig. 4, 159, 160, 161, 162, 163, 164, 165, 166, 167, 168, 169, 170, 171, 172, 173, 174, 175, 176, 177, 178, 179, 180, 181, 182, 183). One possible molecular mechanism of iPlasticity is a synergy of SSRI‐induced and activity‐dependent activation of the BDNF/TrkB pathway (Fig. 4).
Figure 4.

Brain‐derived neurotrophic factor (BDNF)/tropomyosin kinase receptor B (TrkB) pathway. BDNF is synthesized as a precursor protein (pro‐BDNF) that is proteolytically processed into mature BDNF by intracellular proteases and/or extracellular proteases.159, 160, 161 Synthesis, release, and action of BDNF are regulated by neuronal activity. BDNF mRNA increases after neuron depolarization162 and BDNF levels are mediated by calcium‐calmodulin‐dependent protein kinases (CaMK).163 TrkB is a tyrosine kinase membrane protein highly expressed in both presynaptic axons and postsynaptic densities.164, 165, 166 TrkB activation promotes spine formation, neuronal survival, and long‐term potentiation.167, 168, 169, 170, 171 TrkB consists of the following three main domains: an extracellular domain where the ligand (usually BDNF) binds, a transmembrane domain, and a cytosolic domain, which contains the catalytic sites that are responsible for a transphosphorylation reaction.172 Binding of BDNF to the recognition site increases its affinity for the TrkB monomer, stabilizing to form a TrkB dimer.173 This dimerization activates the catalytic sites, and each TrkB phosphorylates the other on specific tyrosine residues (Y706).174 Secondary messenger molecules (such as sarc homology containing [Shc] and phospholipase C gamma [PLCγ]) bind to the phospho‐tyrosines in positions 515 (Y515) and 816 (Y816) and activate the Ras/Erk and PIP3/Ca+2 dependent pathways, respectively. The latter pathway leads to activation of protein kinase C (PKC) and eventually to the phosphorylation of cyclic AMP response element binding protein (CREB).175 CREB in turn induces gene expression involved in neuroplasticity.176, 177, 178, 179, 180, 181, 182 Neuronal activation and high‐frequency stimulation facilitate the localization of TrkB from intracellular pools to the cell surface, requiring a Ca+2 influx.183 Akt, cellular homolog of murine thymoma virus akt8 oncogene; CBP, CREB‐binding protein; ERK, extracellular signal‐regulated kinase; P, phosphorylation.
Neurotrophins, including nerve grow factor, BDNF, neurotrophin‐3, and neurotrophin‐4, play a key role in supporting neurons and neuronal survival during development.184, 185 These family members bind to two types of receptors, namely the Trk‐family members (nerve grow factor to TrkA, BDNF and neurotrophin‐4 to TrkB, and neurotrophin‐3 to TrkC) and a common low‐affinity neurotrophin receptor (p75NTR).159, 186, 187, 188, 189, 190, 191, 192 Generally, AD treatment positively regulates BDNF levels both in animal models and humans. In rodents, BDNF mRNA and protein expression are increased in the hippocampus, cortex, and amygdala after long‐term AD therapies, such as electroconvulsive therapy or commonly used AD drugs.25, 43, 78, 193, 194, 195, 196, 197 Phosphorylation of TrkB and downstream signaling is also increased by AD. Specifically, AD act by phosphorylating the phospholipase C γ‐binding site (Y816) independently of BDNF.198 Interestingly, plasticity represented as LTP and learning seems to be mediated by signaling through the TrkB‐ phospholipase C γ pathway, as deletion of the Y816 residue disrupts LTP and results in deficient learning.175, 199
Hence, BDNF signaling through TrkB is necessary and sufficient for AD behavioral responses200 and for effects in iPlasticity induced by fluoxetine treatment.43 Clear examples of the BDNF–TrkB pathway implication for AD effects have been reported; BDNF knockout mice do not respond to AD treatment,201, 202, 203, 204, 205 whereas BDNF infusion into the DG of the rat hippocampus produces an AD‐like effect in learned helplessness in forced‐swim‐test paradigms.206 Further, the overexpression of a dominant‐negative TrkB leads to a loss of AD efficacy,201 whereas TrkB overexpression and increasing TrkB signaling results in AD‐like behavioral effects.207 In addition to TrkB and BDNF, other downstream molecules of the BDNF/TrkB pathway are also regulated by AD. For example, AD treatment upregulates the expression of CREB2, 208 and CREB overexpression in the rat hippocampus induces an AD‐like behavior.209 Evidence for increased BDNF in human patients after chronic AD treatment has also been reported; post‐mortem tissues had an increased level of BDNF in the hippocampi of depressed patients receiving long‐term AD therapy.210 Besides SSRI, ketamine at low doses seems to increase the expression of BDNF.210 Ketamine does not exhibit an AD response in an inducible BDNF knockout or in conditional TrkB knockout mice.211 Also, mice with the Met allele of the BDNF V66M mutation, in which the cleavage site in pro‐BDNF is mutated (Fig. 4), are resistant to ketamine.212 Finally, an EE has been associated with increased levels of BDNF in the rat cortex and hippocampus.18, 213 In addition, heterozygous BDNF knockout mice (BDNF+/−) have impaired response in exploratory behavior and impaired adult neurogenesis induced by an EE.214, 215 These results suggest that ketamine and EE affect BDNF expression similarly to SSRI.
The BDNF/TrkB pathway is also known to be involved in adult hippocampal neurogenesis stimulated by both chronic AD treatment and voluntary exercise.95, 216 AD specifically act through the BDNF/TrkB pathway, enhance neuronal turnover and promote long‐term survival of newborn neurons.95, 217 In addition, BDNF can promote differentiation and maturation of newborn cells by enhancing GABA release and is thought to at least partially mechanistically act through TrkB localized in PV‐expressing GABAergic interneurons.218
Synthesis, release, and action of BDNF are regulated by neuronal activity, experience, and environmental stimulation (Fig. 4). At the cellular level, BDNF mRNA increases after depolarization of hippocampal culture cells via non‐NMDA glutamate receptors, mainly by AMPA‐receptor pathways.162 In addition, changes in BDNF levels are mediated by calcium‐calmodulin‐dependent protein kinases.163 Recent studies have demonstrated that BDNF expression displays an all‐or‐nothing type of response that only takes place when neuronal activity reaches a certain threshold.219 In vivo, an activity‐dependent change in BDNF levels is also observed after environmental stimulation; limbic seizures increase BDNF mRNA expression in the rat forebrain220 and BDNF synthesis in the visual cortex is regulated by visual stimulation. While darkness decreases mRNA BDNF levels, subsequent light exposure rapidly restores normal mRNA BDNF levels.221 Localization of TrkB to the cell membrane is also regulated in a neuronal activity‐dependent manner. TrkB is rapidly recruited to the plasma membrane by translocation from intracellular stores after neuronal activation accompanied by depolarization and subsequent increase of cAMP levels.222 High‐frequency electric stimulation also facilitates the movement of TrkB from intracellular pools to the cell surface, which requires a Ca+2 influx.183 These results clearly show the activity‐dependent regulation of the BDNF/TrkB pathway.
Taken together, the BDNF/TrkB pathway is involved in all processes of synaptic plasticity, including neurogenesis, neuronal differentiation, synaptic strength, and gene regulation of synaptic formation, and is activated by both chronic SSRI treatment and neuronal activity. Thus, this pathway is considered a key factor of iPlasticity. However, further research is needed to understand at the molecular level the synergic relationship between increased synaptic plasticity and training/behavioral guidance via the SSRI‐induced and activity‐dependent BDNF/TrkB pathway.
CONCLUSIONS AND FUTURE DIRECTIONS
The concept of iPlasticity is a state in the adult brain that resembles plasticity during critical or juvenile periods in postnatal development. iPlasticity permits rewiring of neuronal networks that adjust to internal or external stimuli, such as training or rehabilitation in an activity‐dependent manner. The concept has thus far been demonstrated in rodent brains, such as in the visual cortex, amygdala, and prefrontal cortex, and is mediated by activation of the BDNF/TrkB pathway. However, its detailed mechanisms remain unknown. Further molecular and network analyses will be needed to provide deeper and mechanical insight into the processes of iPlasticity and open up possibilities for treatment of neuropsychiatric disorders, including amblyopia, post‐traumatic stress disorder, and social illness.
DISCLOSURE STATEMENT
The authors declare no competing financial interests.
AUTHOR CONTRIBUTIONS
J.U. and E.C. conceived of and designed the review. J.U., F.W., G.D., M.L., and E.C. wrote the draft. J.U. and M.L. drew or made drafts of the figures.
ACKNOWLEDGMENTS
The original research in our laboratory was supported by the ERC grant # 322742 – iPLASTICITY, the Sigrid Jusélius foundation, the EU Joint Programme – Neurodegenerative Disease Research (JPND) project # JPCOFUND_FP‐829‐007, HiLife Fellows program, and Academy of Finland grants # 294710, 303124, and 307416. We especially thank Miquel Llach for drawing unique and outstanding pictures and Mark Cowlishaw for excellent language revision.
Contributor Information
Juzoh Umemori, Email: juzoh.umemori@helsinki.fi.
Eero Castrén, Email: eero.castren@helsinki.fi.
REFERENCES
- 1. Hillhouse TM, Porter JH. A brief history of the development of antidepressant drugs: From monoamines to glutamate. Exp. Clin. Psychopharmacol. 2015; 23: 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch. Gen. Psychiatry 1997; 54: 597–606. [DOI] [PubMed] [Google Scholar]
- 3. Castrén E. Is mood chemistry? Nat. Rev. Neurosci. 2005; 6: 241–246. [DOI] [PubMed] [Google Scholar]
- 4. Castrén E. Neuronal network plasticity and recovery from depression. JAMA Psychiatry 2013; 70: 983–989. [DOI] [PubMed] [Google Scholar]
- 5. Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science 1996; 274: 1133–1138. [DOI] [PubMed] [Google Scholar]
- 6. Hua JY, Smith SJ. Neural activity and the dynamics of central nervous system development. Nat. Neurosci. 2004; 7: 327–332. [DOI] [PubMed] [Google Scholar]
- 7. Knudsen EI. Sensitive periods in the development of the brain and behavior. J. Cogn. Neurosci. 2004; 16: 1412–1425. [DOI] [PubMed] [Google Scholar]
- 8. Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr. Opin. Neurobiol. 2000; 10: 138–145. [DOI] [PubMed] [Google Scholar]
- 9. Hensch TK. Critical period regulation. Annu. Rev. Neurosci. 2004; 27: 549–579. [DOI] [PubMed] [Google Scholar]
- 10. Hubel DH, Wiesel TN. Shape and arrangement of columns in cat's striate cortex. J. Physiol. 1963; 165: 559–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morishita H, Hensch TK. Critical period revisited: Impact on vision. Curr. Opin. Neurobiol. 2008; 18: 101–107. [DOI] [PubMed] [Google Scholar]
- 12. Hubel DH, Wiesel TN, LeVay S. Plasticity of ocular dominance columns in monkey striate cortex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1977; 278: 377–409. [DOI] [PubMed] [Google Scholar]
- 13. Huang Y‐Y, Colino A, Selig DK, Malenka RC. The influence of prior synaptic activity on the induction of long‐term potentiation. Science 1992; 255: 730–733. [DOI] [PubMed] [Google Scholar]
- 14. Cummings JA, Mulkey RM, Nicoll RA, Malenka RC. Ca 2+ signaling requirements for long‐term depression in the hippocampus. Neuron 1996; 16: 825–833. [DOI] [PubMed] [Google Scholar]
- 15. Govindarajan A, Kelleher RJ, Tonegawa S. A clustered plasticity model of long‐term memory engrams. Nat. Rev. Neurosci. 2006; 7: 575–583. [DOI] [PubMed] [Google Scholar]
- 16. Kroes MCW, Fernández G. Dynamic neural systems enable adaptive, flexible memories. Neurosci. Biobehav. Rev. 2012; 36: 1646–1666. [DOI] [PubMed] [Google Scholar]
- 17. Sale A, Berardi N, Maffei L. Environment and brain plasticity: Towards an endogenous pharmacotherapy. Physiol. Rev. 2014; 94: 189–234. [DOI] [PubMed] [Google Scholar]
- 18. Sale A, Maya Vetencourt JF, Medini P et al Environmental enrichment in adulthood promotes amblyopia recovery through a reduction of intracortical inhibition. Nat. Neurosci. 2007; 10: 679–681. [DOI] [PubMed] [Google Scholar]
- 19. Castrén E, Hen R. Neuronal plasticity and antidepressant actions. Trends Neurosci. 2013; 36: 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chollet F, Tardy J, Albucher JF et al Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): A randomised placebo‐controlled trial. Lancet Neurol. 2011; 10: 123–130. [DOI] [PubMed] [Google Scholar]
- 21. Amin MM, Ban TA, Pecknold JC, Klingner A. Clomipramine (Anafranil) and behaviour therapy in obsessive‐compulsive and phobic disorders. J. Int. Med. Res. 1977; 5(Suppl 5): 33–37. [PubMed] [Google Scholar]
- 22. Brent D, Emslie G, Clarke G et al Switching to another SSRI or to venlafaxine with or without cognitive behavioral therapy for adolescents with SSRI‐resistant depression: The TORDIA randomized controlled trial. JAMA 2008; 299: 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pampallona S, Bollini P, Tibaldi G, Kupelnick B, Munizza C. Combined pharmacotherapy and psychological treatment for depression: A systematic review. Arch. Gen. Psychiatry 2004; 61: 714–719. [DOI] [PubMed] [Google Scholar]
- 24. Chiarotti F, Viglione A, Giuliani A, Branchi I. Citalopram amplifies the influence of living conditions on mood in depressed patients enrolled in the STAR*D study. Transl. Psychiatry 2017; 7: e1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vetencourt JFM, Sale A, Viegi A et al The antidepressant fluoxetine restores plasticity in the adult visual cortex. Science 2008; 320: 385–388. [DOI] [PubMed] [Google Scholar]
- 26. Berardi N, Pizzorusso T, Ratto GM, Maffei L. Molecular basis of plasticity in the visual cortex. Trends Neurosci. 2003; 26: 369–378. [DOI] [PubMed] [Google Scholar]
- 27. Hensch TK, Fagiolini M, Mataga N, Stryker MP, Baekkeskov S, Kash SF. Local GABA circuit control of experience‐dependent plasticity in developing visual cortex. Science 1998; 282: 1504–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fagiolini M, Fritschy J‐M, Löw K, Möhler H, Rudolph U, Hensch TK. Specific GABAA circuits for visual cortical plasticity. Science 2004; 303: 1681–1683. [DOI] [PubMed] [Google Scholar]
- 29. Iwai Y, Fagiolini M, Obata K, Hensch TK. Rapid critical period induction by tonic inhibition in visual cortex. J. Neurosci. 2003; 23: 6695–6702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanover JL, Huang ZJ, Tonegawa S, Stryker MP. Brain‐derived neurotrophic factor overexpression induces precocious critical period in mouse visual cortex. J. Neurosci. 1999; 19: RC40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gianfranceschi L, Siciliano R, Walls J et al Visual cortex is rescued from the effects of dark rearing by overexpression of BDNF. Proc. Natl. Acad. Sci. 2003; 100: 12486–12491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Castrén E, Rantamäki T. The role of BDNF and its receptors in depression and antidepressant drug action: Reactivation of developmental plasticity. Dev. Neurobiol. 2010; 70: 289–297. [DOI] [PubMed] [Google Scholar]
- 33. Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002; 298: 1248–1251. [DOI] [PubMed] [Google Scholar]
- 34. Lensjø KK, Lepperød ME, Dick G, Hafting T, Fyhn M. Removal of perineuronal nets unlocks juvenile plasticity through network mechanisms of decreased inhibition and increased gamma activity. J. Neurosci. 2017; 37: 1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harauzov A, Spolidoro M, DiCristo G et al Reducing intracortical inhibition in the adult visual cortex promotes ocular dominance plasticity. J. Neurosci. 2010; 30: 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hensch TK. Critical period plasticity in local cortical circuits. Nat. Rev. Neurosci. 2005; 6: 877–888. [DOI] [PubMed] [Google Scholar]
- 37. Pavlov IP. Conditioned reflex. Feldsher Akush. 1927; 11: 6–12. [PubMed] [Google Scholar]
- 38. LeDoux J. The emotional brain, fear and the amygdala. Cell. Mol. Neurobiol. 2003; 23: 727–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Milad MR, Quirk GJ. Neurons in medial prefrontal cortex signal memory for fear extinction. Nature 2002; 420: 70–74. [DOI] [PubMed] [Google Scholar]
- 40. Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav. Neurosci. 1992; 106: 274–285. [DOI] [PubMed] [Google Scholar]
- 41. Bisson J, Andrew M. Psychological treatment of post‐traumatic stress disorder (PTSD). Cochrane Database Syst. Rev. 2005; 2: CD003388. [DOI] [PubMed] [Google Scholar]
- 42. Stein DJ, Ipser J, McAnda N. Pharmacotherapy of posttraumatic stress disorder: A review of meta‐analyses and treatment guidelines. CNS Spectr. 2009; 14(Suppl 1): 25–31. [PubMed] [Google Scholar]
- 43. Karpova NN, Pickenhagen A, Lindholm J et al Fear erasure in mice requires synergy between antidepressant drugs and extinction training. Science 2011; 334: 1731–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tóth M, Halász J, Mikics É, Barsy B, Haller J. Early social deprivation induces disturbed social communication and violent aggression in adulthood. Behav. Neurosci. 2008; 122: 849–854. [DOI] [PubMed] [Google Scholar]
- 45. Toth M, Mikics E, Tulogdi A, Aliczki M, Haller J. Post‐weaning social isolation induces abnormal forms of aggression in conjunction with increased glucocorticoid and autonomic stress responses. Horm. Behav. 2011; 60: 28–36. [DOI] [PubMed] [Google Scholar]
- 46. Sandi C, Haller J. Stress and the social brain: Behavioural effects and neurobiological mechanisms. Nat. Rev. Neurosci. 2015; 16: 290–304. [DOI] [PubMed] [Google Scholar]
- 47. Miczek KA, De Boer SF, Haller J. Excessive aggression as model of violence: A critical evaluation of current preclinical methods. Psychopharmacology (Berl.) 2013; 226: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pascual R, Zamora‐León SP, Valero‐Cabré A. Effects of postweaning social isolation and re‐socialization on the expression of vasoactive intestinal peptide (VIP) and dendritic development in the medial prefrontal cortex of the rat. Acta Neurobiol. Exp. (Wars.) 2006; 66: 7–14. [DOI] [PubMed] [Google Scholar]
- 49. Wright IK, Upton N, Marsden CA. Resocialisation of isolation‐reared rats does not alter their anxiogenic profile on the elevated X‐maze model of anxiety. Physiol. Behav. 1991; 50: 1129–1132. [DOI] [PubMed] [Google Scholar]
- 50. Mikics É, Guirado R, Umemori J et al Social learning requires plasticity enhanced by fluoxetine through prefrontal Bdnf‐TrkB signaling to limit aggression induced by post‐weaning social isolation. Neuropsychopharmacology 2017; 43: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nowakowska E, Kus K, Chodera A, Rybakowski J. Behavioural effects of fluoxetine and tianeptine, two antidepressants with opposite action mechanisms, in rats. Arzneimittelforschung 2000; 50: 5–10. [DOI] [PubMed] [Google Scholar]
- 52. Majlessi N, Naghdi N. Impaired spatial learning in the Morris water maze induced by serotonin reuptake inhibitors in rats. Behav. Pharmacol. 2002; 13: 237–242. [DOI] [PubMed] [Google Scholar]
- 53. Yau JL, Hibberd C, Noble J, Seckl JR. The effect of chronic fluoxetine treatment on brain corticosteroid receptor mRNA expression and spatial memory in young and aged rats. Brain Res. Mol. Brain Res. 2002; 106: 117–123. [DOI] [PubMed] [Google Scholar]
- 54. Valluzzi JA, Chan K. Effects of fluoxetine on hippocampal‐dependent and hippocampal‐independent learning tasks. Behav. Pharmacol. 2007; 18: 507–513. [DOI] [PubMed] [Google Scholar]
- 55. Akers KG, Martinez‐Canabal A, Restivo L et al Hippocampal neurogenesis regulates forgetting during adulthood and infancy. Science 2014; 344: 598–602. [DOI] [PubMed] [Google Scholar]
- 56. Hamilton DA, Brigman JL. Behavioral flexibility in rats and mice: Contributions of distinct frontocortical regions. Genes Brain Behav. 2015; 14: 4–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Codita A, Mohammed AH, Willuweit A et al Effects of spatial and cognitive enrichment on activity pattern and learning performance in three strains of mice in the IntelliMaze. Behav. Genet. 2012; 42: 449–460. [DOI] [PubMed] [Google Scholar]
- 58. Endo T, Maekawa F, Võikar V et al Automated test of behavioral flexibility in mice using a behavioral sequencing task in IntelliCage. Behav. Brain Res. 2011; 221: 172–181. [DOI] [PubMed] [Google Scholar]
- 59. Alboni S, Van Dijk RM, Poggini S et al Fluoxetine effects on molecular, cellular and behavioral endophenotypes of depression are driven by the living environment. Mol. Psychiatry 2017; 22: 552–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Rantamäki T, Hendolin P, Kankaanpää A et al Pharmacologically diverse antidepressants rapidly activate brain‐derived neurotrophic factor receptor TrkB and induce phospholipase‐Cgamma signaling pathways in mouse brain. Neuropsychopharmacology 2007; 32: 2152–2162. [DOI] [PubMed] [Google Scholar]
- 61. McEwen BS, Chattarji S, Diamond DM et al The neurobiological properties of tianeptine (Stablon): From monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry 2010; 15: 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Abdallah CG, Sanacora G, Duman RS, Krystal JH. Ketamine and rapid‐acting antidepressants: A window into a new neurobiology for mood disorder therapeutics. Annu. Rev. Med. 2015; 66: 509–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ibrahim L, Diazgranados N, Luckenbaugh DA et al Rapid decrease in depressive symptoms with an N‐methyl‐d‐aspartate antagonist in ECT‐resistant major depression. Prog. Neuropsychopharmacol. Biol. Psychiatry 2011; 35: 1155–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. DiazGranados N, Ibrahim LA, Brutsche NE et al Rapid resolution of suicidal ideation after a single infusion of an N‐methyl‐D‐aspartate antagonist in patients with treatment‐resistant major depressive disorder. J. Clin. Psychiatry 2010; 71: 1605–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zarate CA, Machado‐Vieira R. Ketamine: Translating mechanistic discoveries into the next generation of glutamate modulators for mood disorders. Mol. Psychiatry 2017; 22: 324–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Larkin GL, Beautrais AL. A preliminary naturalistic study of low‐dose ketamine for depression and suicide ideation in the emergency department. Int. J. Neuropsychopharmacol. 2011; 14: 1127–1131. [DOI] [PubMed] [Google Scholar]
- 67. Thakurta RG, Das R, Bhattacharya AK et al Rapid response with ketamine on suicidal cognition in resistant depression. Indian J. Psychol. Med. 2012; 34: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Berman RM, Cappiello A, Anand A et al Antidepressant effects of ketamine in depressed patients. Biol. Psychiatry 2000; 47: 351–354. [DOI] [PubMed] [Google Scholar]
- 69. Nestler EJ. Antidepressant treatments in the 21st century. Biol. Psychiatry 1998; 44: 526–533. [DOI] [PubMed] [Google Scholar]
- 70. Ju LS, Yang JJ, Lei L et al The combination of long‐term ketamine and extinction training contributes to fear erasure by Bdnf methylation. Front. Cell. Neurosci. 2017; 11: 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Paul RK, Singh NS, Khadeer M et al (R,S)‐ketamine metabolites (R,S)‐norketamine and (2S,6S)‐hydroxynorketamine increase the mammalian target of rapamycin function. Anesthesiology 2014; 121: 149–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zanos P, Moaddel R, Morris PJ et al NMDAR inhibition‐independent antidepressant actions of ketamine metabolites. Nature 2016; 533: 481–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Duman RS, Vaidya VA. Molecular and cellular actions of chronic electroconvulsive seizures. J. ECT 1998; 14: 181–193. [PubMed] [Google Scholar]
- 74. Bouckaert F, Sienaert P, Obbels J et al ECT. J. ECT 2014; 30: 143–151. [DOI] [PubMed] [Google Scholar]
- 75. Abbott CC, Jones T, Lemke NT et al Hippocampal structural and functional changes associated with electroconvulsive therapy response. Transl. Psychiatry 2014; 4: e483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dukart J, Regen F, Kherif F et al Electroconvulsive therapy‐induced brain plasticity determines therapeutic outcome in mood disorders. Proc. Natl. Acad. Sci. U. S. A. 2014; 111: 1156–1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lindefors N, Brodin E, Metsis M. Spatiotemporal selective effects on brain‐derived neurotrophic factor and trkB messenger RNA in rat hippocampus by electroconvulsive shock. Neuroscience 1995; 65: 661–670. [DOI] [PubMed] [Google Scholar]
- 78. Nibuya M, Morinobu S, Duman RS. Regulation of BDNF and trkB mRNA in rat brain by chronic electroconvulsive seizure and antidepressant drug treatments. J. Neurosci. 1995; 15: 7539–7547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Scott BW, Wojtowicz JM, Burnham WM. Neurogenesis in the dentate gyrus of the rat following electroconvulsive shock seizures. Exp. Neurol. 2000; 165: 231–236. [DOI] [PubMed] [Google Scholar]
- 80. Madsen TM, Treschow A, Bengzon J, Bolwig TG, Lindvall O, Tingström A. Increased neurogenesis in a model of electroconvulsive therapy. Biol. Psychiatry 2000; 47: 1043–1049. [DOI] [PubMed] [Google Scholar]
- 81. Zhao C, Warner‐Schmidt J, Duman RS, Gage FH. Electroconvulsive seizure promotes spine maturation in newborn dentate granule cells in adult rat. Dev. Neurobiol. 2012; 72: 937–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Vaidya VA, Siuciak JA, Du F, Duman RS. Hippocampal mossy fiber sprouting induced by chronic electroconvulsive seizures. Neuroscience 1999; 89: 157–166. [DOI] [PubMed] [Google Scholar]
- 83. Chen F, Madsen TM, Wegener G, Nyengaard JR. Repeated electroconvulsive seizures increase the total number of synapses in adult male rat hippocampus. Eur. Neuropsychopharmacol. 2009; 19: 329–338. [DOI] [PubMed] [Google Scholar]
- 84. Morishita H, Miwa JM, Heintz N, Hensch TK. Lynx1, a cholinergic brake, limits plasticity in adult visual cortex. Science 2010; 330: 1238–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Baroncelli L, Sale A, Viegi A et al Experience‐dependent reactivation of ocular dominance plasticity in the adult visual cortex. Exp. Neurol. 2010; 226: 100–109. [DOI] [PubMed] [Google Scholar]
- 86. Eckert MJ, Abraham WC. Effects of environmental enrichment exposure on synaptic transmission and plasticity in the hippocampus. Curr. Top. Behav. Neurosci. 2013; 15: 165–187. [DOI] [PubMed] [Google Scholar]
- 87. Kempermann G, Kuhn HG, Gage FH. More hippocampal neurons in adult mice living in an enriched environment. Nature 1997; 386: 493–495. [DOI] [PubMed] [Google Scholar]
- 88. Castrén E, Antila H. Neuronal plasticity and neurotrophic factors in drug responses. Mol. Psychiatry 2017; 22: 1085–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Covington HE, Maze I, LaPlant QC et al Antidepressant actions of histone deacetylase inhibitors. J. Neurosci. 2009; 29: 11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Imayoshi I, Sakamoto M, Ohtsuka T et al Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat. Neurosci. 2008; 11: 1153–1161. [DOI] [PubMed] [Google Scholar]
- 91. Sahay A, Scobie KN, Hill AS et al Increasing adult hippocampal neurogenesis is sufficient to improve pattern separation. Nature 2011; 472: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Malberg JE, Eisch AJ, Nestler EJ, Duman RS. Chronic antidepressant treatment increases neurogenesis in adult rat hippocampus. J. Neurosci. 2000; 20: 9104–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Santarelli L, Saxe M, Gross CT et al Requirement of hippocampal neurogenesis for the behavioral effects of antidepressants. Science 2003; 301: 805–809. [DOI] [PubMed] [Google Scholar]
- 94. Airan RD, Meltzer LA, Roy M, Gong Y, Chen H, Deisseroth K. High‐speed imaging reveals neurophysiological links to behavior in an animal model of depression. Science 2007; 317: 819–823. [DOI] [PubMed] [Google Scholar]
- 95. Li Y, Luikart BW, Birnbaum S et al TrkB regulates hippocampal neurogenesis and governs sensitivity to antidepressive treatment. Neuron 2008; 59: 399–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. David DJ, Samuels BA, Rainer Q et al Neurogenesis‐dependent and ‐independent effects of fluoxetine in an animal model of anxiety/depression. Neuron 2009; 62: 479–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. David DJ, Wang J, Samuels BA et al Implications of the functional integration of adult‐born hippocampal neurons in anxiety‐depression disorders. Neuroscientist 2010; 16: 578–591. [DOI] [PubMed] [Google Scholar]
- 98. Holick KA, Lee DC, Hen R, Dulawa SC. Behavioral effects of chronic fluoxetine in BALB/cJ mice do not require adult hippocampal neurogenesis or the serotonin 1A receptor. Neuropsychopharmacology 2007; 33: 406–417. [DOI] [PubMed] [Google Scholar]
- 99. Huang G‐J, Bannerman D, Flint J. Chronic fluoxetine treatment alters behavior, but not adult hippocampal neurogenesis, in BALB/cJ mice. Mol. Psychiatry 2008; 13: 119–121. [DOI] [PubMed] [Google Scholar]
- 100. Murray F, Smith DW, Hutson PH. Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur. J. Pharmacol. 2008; 583: 115–127. [DOI] [PubMed] [Google Scholar]
- 101. Wu X, Castrén E. Co‐treatment with diazepam prevents the effects of fluoxetine on the proliferation and survival of hippocampal dentate granule cells. Biol. Psychiatry 2009; 66: 5–8. [DOI] [PubMed] [Google Scholar]
- 102. Zhang J, Jiao J. Molecular biomarkers for embryonic and adult neural stem cell and neurogenesis. Biomed. Res. Int. 2015; 2015: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Segi‐Nishida E. The effect of serotonin‐targeting antidepressants on neurogenesis and neuronal maturation of the hippocampus mediated via 5‐HT1A and 5‐HT4 receptors. Front. Cell. Neurosci. 2017; 11: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Encinas JM, Vaahtokari A, Enikolopov G. Fluoxetine targets early progenitor cells in the adult brain. Proc. Natl. Acad. Sci. 2006; 103: 8233–8238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Hajszan T, MacLusky NJ, Leranth C. Short‐term treatment with the antidepressant fluoxetine triggers pyramidal dendritic spine synapse formation in rat hippocampus. Eur. J. Neurosci. 2005; 21: 1299–1303. [DOI] [PubMed] [Google Scholar]
- 106. Ge S, Yang CH, Hsu KS, Ming GL, Song H. A critical period for enhanced synaptic plasticity in newly generated neurons of the adult brain. Neuron 2007; 54: 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Schmidt‐Hieber C, Jonas P, Bischofberger J. Enhanced synaptic plasticity in newly generated granule cells of the adult hippocampus. Nature 2004; 429: 184–187. [DOI] [PubMed] [Google Scholar]
- 108. Laplagne DA, Espósito MS, Piatti VC et al Functional convergence of neurons generated in the developing and adult hippocampus. PLoS Biol. 2006; 4: 2349–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Wang J‐W, David DJ, Monckton JE, Battaglia F, Hen R. Chronic fluoxetine stimulates maturation and synaptic plasticity of adult‐born hippocampal granule cells. J. Neurosci. 2008; 28: 1374–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Samuels BA, Anacker C, Hu A et al 5‐HT1A receptors on mature dentate gyrus granule cells are critical for the antidepressant response. Nat. Neurosci. 2015; 18: 1606–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Ardayfio P, Kim K‐S. Anxiogenic‐like effect of chronic corticosterone in the light‐dark emergence task in mice. Behav. Neurosci. 2006; 120: 249–256. [DOI] [PubMed] [Google Scholar]
- 112. Gourley SL, Kiraly DD, Howell JL, Olausson P, Taylor JR. Acute hippocampal brain‐derived neurotrophic factor restores motivational and forced swim performance after corticosterone. Biol. Psychiatry 2008; 64: 884–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Bessa JM. A trans‐dimensional approach to the behavioral aspects of depression. Front. Behav. Neurosci. 2009; 3: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Spolidoro M, Baroncelli L, Putignano E, Maya‐Vetencourt JF, Viegi A, Maffei L. Food restriction enhances visual cortex plasticity in adulthood. Nat. Commun. 2011; 2: 320. [DOI] [PubMed] [Google Scholar]
- 115. Sahay A, Hen R. Adult hippocampal neurogenesis in depression. Nat. Neurosci. 2007; 10: 1110–1115. [DOI] [PubMed] [Google Scholar]
- 116. Speisman RB, Kumar A, Rani A et al Environmental enrichment restores neurogenesis and rapid acquisition in aged rats. Neurobiol. Aging 2013; 34: 263–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Segovia G, Yagüe AG, García‐Verdugo JM, Mora F. Environmental enrichment promotes neurogenesis and changes the extracellular concentrations of glutamate and GABA in the hippocampus of aged rats. Brain Res. Bull. 2006; 70: 8–14. [DOI] [PubMed] [Google Scholar]
- 118. Li N, Lee B, Liu R‐J et al mTOR‐dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science 2010; 329: 959–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Keilhoff G, Bernstein H‐G, Becker A, Grecksch G, Wolf G. Increased neurogenesis in a rat ketamine model of schizophrenia. Biol. Psychiatry 2004; 56: 317–322. [DOI] [PubMed] [Google Scholar]
- 120. Castro JE, Varea E, Márquez C, Cordero MI, Poirier G, Sandi C. Role of the amygdala in antidepressant effects on hippocampal cell proliferation and survival and on depression‐like behavior in the rat. PLoS One 2010; 5: e8618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Ohira K, Takeuchi R, Shoji H, Miyakawa T. Fluoxetine‐induced cortical adult neurogenesis. Neuropsychopharmacology 2013; 38: 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Sachs BD, Caron MG. Chronic fluoxetine increases extra‐hippocampal neurogenesis in adult mice. Int. J. Neuropsychopharmacol. 2015; 18: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Surget A, Saxe M, Leman S et al Drug‐dependent requirement of hippocampal neurogenesis in a model of depression and of antidepressant reversal. Biol. Psychiatry 2008; 64: 293–301. [DOI] [PubMed] [Google Scholar]
- 124. Petersén Å, Wörtwein G, Gruber SHM, El‐Khoury A, Mathé AA. Nortriptyline mediates behavioral effects without affecting hippocampal cytogenesis in a genetic rat depression model. Neurosci. Lett. 2009; 451: 148–151. [DOI] [PubMed] [Google Scholar]
- 125. Bessa JM, Ferreira D, Melo I et al The mood‐improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol. Psychiatry 2009; 14: 764–773. [DOI] [PubMed] [Google Scholar]
- 126. Kobayashi K, Ikeda Y, Sakai A et al Reversal of hippocampal neuronal maturation by serotonergic antidepressants. Proc. Natl. Acad. Sci. U. S. A. 2010; 107: 8434–8439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Hagihara H, Takao K, Walton NM, Matsumoto M, Miyakawa T. Immature dentate gyrus: An endophenotype of neuropsychiatric disorders. Neural Plast. 2013; 2013: 318596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Doetsch F, Hen R. Young and excitable: The function of new neurons in the adult mammalian brain. Curr. Opin. Neurobiol. 2005; 15: 121–128. [DOI] [PubMed] [Google Scholar]
- 129. Imoto Y, Kira T, Sukeno M et al Role of the 5‐HT4 receptor in chronic fluoxetine treatment‐induced neurogenic activity and granule cell dematuration in the dentate gyrus. Mol. Brain 2015; 8: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: A potential role for kainate receptors. J. Physiol. 2004; 561: 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Imoto Y, Segi‐Nishida E, Suzuki H, Kobayashi K. Rapid and stable changes in maturation‐related phenotypes of the adult hippocampal neurons by electroconvulsive treatment. Mol. Brain 2017; 10: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Kobayashi K. Activity modifies adult brain maturity. Oncotarget 2017; 8: 46708–46709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Guirado R, Perez‐Rando M, Sanchez‐Matarredona D, Castrén E, Nacher J. Chronic fluoxetine treatment alters the structure, connectivity and plasticity of cortical interneurons. Int. J. Neuropsychopharmacol. 2014; 17: 1635–1646. [DOI] [PubMed] [Google Scholar]
- 134. Ohira K, Takeuchi R, Iwanaga T, Miyakawa T. Chronic fluoxetine treatment reduces parvalbumin expression and perineuronal nets in gamma‐aminobutyric acidergic interneurons of the frontal cortex in adult mice. Mol. Brain 2013; 6: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Yamasaki N, Maekawa M, Kobayashi K et al Alpha‐CaMKII deficiency causes immature dentate gyrus, a novel candidate endophenotype of psychiatric disorders. Mol. Brain 2008; 1: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Ohira K, Kobayashi K, Toyama K et al Synaptosomal‐associated protein 25 mutation induces immaturity of the dentate granule cells of adult mice. Mol. Brain 2013; 6: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Takao K, Kobayashi K, Hagihara H et al Deficiency of schnurri‐2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology 2013; 38: 1409–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Shin R, Kobayashi K, Hagihara H et al The immature dentate gyrus represents a shared phenotype of mouse models of epilepsy and psychiatric disease. Bipolar Disord. 2013; 15: 405–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Mertens J, Wang QW, Kim Y et al Differential responses to lithium in hyperexcitable neurons from patients with bipolar disorder. Nature 2015; 527: 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Hagihara H, Ohira K, Takao K, Miyakawa T. Transcriptomic evidence for immaturity of the prefrontal cortex in patients with schizophrenia. Mol. Brain 2014; 7: 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Murano T, Koshimizu H, Hagihara H, Miyakawa T. Transcriptomic immaturity of the hippocampus and prefrontal cortex in patients with alcoholism. Sci. Rep. 2017; 7: 44531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Kobayashi K, Ikeda Y, Suzuki H. Behavioral destabilization induced by the selective serotonin reuptake inhibitor fluoxetine. Mol. Brain 2011; 4: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Gogolla N, Caroni P, Lüthi A, Herry C. Perineuronal nets protect fear memories from erasure. Science 2009; 325: 1258–1261. [DOI] [PubMed] [Google Scholar]
- 144. Hensch TK, Fagiolini M. Excitatory‐inhibitory balance and critical period plasticity in developing visual cortex. Prog. Brain Res. 2004; 147: 115–124. [DOI] [PubMed] [Google Scholar]
- 145. Szabadics J, Varga C, Molnár G, Oláh S, Barzó P, Tamás G. Excitatory effect of GABAergic axo‐axonic cells in cortical microcircuits. Science 2006; 311: 233–235. [DOI] [PubMed] [Google Scholar]
- 146. Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature 2009; 459: 698–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Hu H, Gan J, Interneurons JP. Fast‐spiking, parvalbumin+ GABAergic interneurons: From cellular design to microcircuit function. Science 2014; 345: 1255263. [DOI] [PubMed] [Google Scholar]
- 148. Miyata S, Kitagawa H. Mechanisms for modulation of neural plasticity and axon regeneration by chondroitin sulphate. J. Biochem. 2015; 157: 13–22. [DOI] [PubMed] [Google Scholar]
- 149. Romberg C, Yang S, Melani R et al Depletion of perineuronal nets enhances recognition memory and long‐term depression in the perirhinal cortex. J. Neurosci. 2013; 33: 7057–7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Wang D, Fawcett J. The perineuronal net and the control of CNS plasticity. Cell Tissue Res. 2012; 349: 147–160. [DOI] [PubMed] [Google Scholar]
- 151. Arnst N, Kuznetsova S, Lipachev N et al Spatial patterns and cell surface clusters in perineuronal nets. Brain Res. 2016; 1648: 214–223. [DOI] [PubMed] [Google Scholar]
- 152. Xue Y‐X, Xue L‐F, Liu J‐F et al Depletion of perineuronal nets in the amygdala to enhance the erasure of drug memories. J. Neurosci. 2014; 34: 6647–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Hylin MJ, Orsi SA, Moore AN, Dash PK. Disruption of the perineuronal net in the hippocampus or medial prefrontal cortex impairs fear conditioning. Learn. Mem. 2013; 20: 267–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Donato F, Rompani SB, Caroni P. Parvalbumin‐expressing basket‐cell network plasticity induced by experience regulates adult learning. Nature 2013; 504: 272–276. [DOI] [PubMed] [Google Scholar]
- 155. Hensch TK. Bistable parvalbumin circuits pivotal for brain plasticity. Cell 2014; 156: 17–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Cabungcal J‐H, Steullet P, Kraftsik R, Cuenod M, Do KQ. Early‐life insults impair parvalbumin interneurons via oxidative stress: Reversal by N‐acetylcysteine. Biol. Psychiatry 2013; 73: 574–582. [DOI] [PubMed] [Google Scholar]
- 157. Mikics É, Guirado R, Umemori J et al Social learning requires plasticity enhanced by fluoxetine through prefrontal Bdnf‐TrkB signaling to limit aggression induced by post‐weaning social isolation. Neuropsychopharmacology 2017; 43: 235–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Castrén E, Kojima M. Brain‐derived neurotrophic factor in mood disorders and antidepressant treatments. Neurobiol. Dis. 2017; 97: 119–126. [DOI] [PubMed] [Google Scholar]
- 159. Deinhardt K, Chao MV. Shaping neurons: Long and short range effects of mature and proBDNF signalling upon neuronal structure. Handb. Exp. Pharmacol. 2014; 220: 103–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Seidaha NG, Benjannet S, Pareek S, Chrétien M, Murphy RA. Cellular processing of the neurotrophin precursors of NT3 and BDNF by the mammalian proprotein convertases. FEBS Lett. 1996; 379: 247–250. [DOI] [PubMed] [Google Scholar]
- 161. Wetsel WC, Rodriguiz RM, Guillemot J et al Disruption of the expression of the proprotein convertase PC7 reduces BDNF production and affects learning and memory in mice. Proc. Natl. Acad. Sci. 2013; 110: 17362–17367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zafra F, Hengerer B, Leibrock J, Thoenen H, Lindholm D. Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non‐NMDA glutamate receptors. EMBO J. 1990; 9: 3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Zafra F, Lindholm D, Castrén E, Hartikka J, Thoenen H. Regulation of brain‐derived neurotrophic factor and nerve growth factor mRNA in primary cultures of hippocampal neurons and astrocytes. J. Neurosci. 1992; 12: 4793–4799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wu K, Xu JL, Suen PC et al Functional trkB neurotrophin receptors are intrinsic components of the adult brain postsynaptic density. Mol. Brain Res. 1996; 43: 286–290. [DOI] [PubMed] [Google Scholar]
- 165. Helgager J, Liu G, McNamara JO. The cellular and synaptic location of activated TrkB in mouse hippocampus during limbic epileptogenesis. J. Comp. Neurol. 2013; 521: 499–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Xu B, Gottschalk W, Chow A et al The role of brain‐derived neurotrophic factor receptors in the mature hippocampus: Modulation of long‐term potentiation through a presynaptic mechanism involving TrkB. J. Neurosci. 2000; 20: 6888–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Teng HK. ProBDNF induces neuronal apoptosis via activation of a receptor complex of p75NTR and Sortilin. J. Neurosci. 2005; 25: 5455–5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Woo NH, Teng HK, Siao C‐J et al Activation of p75NTR by proBDNF facilitates hippocampal long‐term depression. Nat. Neurosci. 2005; 8: 1069–1077. [DOI] [PubMed] [Google Scholar]
- 169. Zagrebelsky M. The p75 neurotrophin receptor negatively modulates dendrite complexity and spine density in hippocampal neurons. J. Neurosci. 2005; 25: 9989–9999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Koshimizu H, Kiyosue K, Hara T et al Multiple functions of precursor BDNF to CNS neurons: Negative regulation of neurite growth, spine formation and cell survival. Mol. Brain 2009; 2: 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Deinhardt K, Kim T, Spellman DS et al Neuronal growth cone retraction relies on proneurotrophin receptor signaling through Rac. Sci. Signal. 2011; 4: ra82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Klein R, Conway D, Parada LF, Barbacid M. The trkB tyrosine protein kinase gene codes for a second neurogenic receptor that lacks the catalytic kinase domain. Cell 1990; 61: 647–656. [DOI] [PubMed] [Google Scholar]
- 173. Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell 1990; 61: 203–212. [DOI] [PubMed] [Google Scholar]
- 174. Segal RA, Bhattacharyya A, Rua LA et al Differential utilization of Trk autophosphorylation sites. J. Biol. Chem. 1996; 271: 20175–20181. [DOI] [PubMed] [Google Scholar]
- 175. Minichiello L, Calella AM, Medina DL, Bonhoeffer T, Klein R, Korte M. Mechanism of TrkB‐mediated hippocampal long‐term potentiation. Neuron 2002; 36: 121–137. [DOI] [PubMed] [Google Scholar]
- 176. Mabuchi T, Kitagawa K, Kuwabara K et al Phosphorylation of cAMP response element‐binding protein in hippocampal neurons as a protective response after exposure to glutamate in vitro and ischemia in vivo. J. Neurosci. 2001; 21: 9204–9213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Reppert SM, Weaver DR. Molecular analysis of mammalian circadian rhythms. Annu. Rev. Physiol. 2001; 63: 647–676. [DOI] [PubMed] [Google Scholar]
- 178. Kandel ER. The molecular biology of memory storage: A dialogue between genes and synapses. Science 2001; 294: 1030–1038. [DOI] [PubMed] [Google Scholar]
- 179. West AE, Griffith EC, Greenberg ME. Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 2002; 3: 921–931. [DOI] [PubMed] [Google Scholar]
- 180. Lonze BE, Ginty DD. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002; 35: 605–623. [DOI] [PubMed] [Google Scholar]
- 181. Kida S, Josselyn SA, Peña de Ortiz S et al CREB required for the stability of new and reactivated fear memories. Nat. Neurosci. 2002; 5: 348–355. [DOI] [PubMed] [Google Scholar]
- 182. Pittenger C, Huang YY, Paletzki RF et al Reversible inhibition of CREB/ATF transcription factors in region CA1 of the dorsal hippocampus disrupts hippocampus‐dependent spatial memory. Neuron 2002; 34: 447–462. [DOI] [PubMed] [Google Scholar]
- 183. Du J, Feng L, Yang F, Lu B. Activity‐ and Ca2+‐dependent modulation of surface expression of brain‐derived neurotrophic factor receptors in hippocampal neurons. J. Cell Biol. 2000; 150: 1423–1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Buchman VL, Davies AM. Different neurotrophins are expressed and act in a developmental sequence to promote the survival of embryonic sensory neurons. Development 1993; 118: 989–1001. [DOI] [PubMed] [Google Scholar]
- 185. Barde YA. Trophic factors and neuronal survival. Neuron 1989; 2: 1525–1534. [DOI] [PubMed] [Google Scholar]
- 186. Chao MV, Bothwell MA, Ross AH et al Gene transfer and molecular cloning of the human NGF receptor. Science 1986; 232: 518–521. [DOI] [PubMed] [Google Scholar]
- 187. Kaplan DR, Hempstead BL, Martin‐Zanca D, Chao MV, Parada LF. The trk proto‐oncogene product: A signal transducing receptor for nerve growth factor. Science 1991; 252: 554–558. [DOI] [PubMed] [Google Scholar]
- 188. Klein R, Nanduri V, Jing S et al The trkB tyrosine protein kinase is a receptor for brain‐derived neurotrophic factor and neurotrophin‐3. Cell 1991; 66: 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Huang EJ, Reichardt LF. Trk receptors: Role in neuronal signal transduction. Annu. Rev. Plant Biol. 2003; 72: 609–642. [DOI] [PubMed] [Google Scholar]
- 190. Ibáñez CF, Simi A. P75 neurotrophin receptor signaling in nervous system injury and degeneration: Paradox and opportunity. Trends Neurosci. 2012; 35: 431–440. [DOI] [PubMed] [Google Scholar]
- 191. Radeke MJ, Misko TP, Hsu C, Herzenberg LA, Shooter EM. Gene transfer and molecular cloning of the rat nerve growth factor receptor. Nature 1987; 325: 593–597. [DOI] [PubMed] [Google Scholar]
- 192. Kraemer BR, Yoon SO, Carter BD. The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb. Exp. Pharmacol. 2014; 220: 121–164. [DOI] [PubMed] [Google Scholar]
- 193. Balu DT, Hoshaw BA, Malberg JE, Rosenzweig‐Lipson S, Schechter LE, Lucki I. Differential regulation of central BDNF protein levels by antidepressant and non‐antidepressant drug treatments. Brain Res. 2008; 1211: 37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Dias BG, Banerjee SB, Duman RS, Vaidya VA. Differential regulation of brain derived neurotrophic factor transcripts by antidepressant treatments in the adult rat brain. Neuropharmacology 2003; 45: 553–563. [DOI] [PubMed] [Google Scholar]
- 195. Russo‐Neustadt A, Beard RC, Cotman CW. Exercise, antidepressant medications, and enhanced brain derived neurotrophic factor expression. Neuropsychopharmacology 1999; 21: 679–682. [DOI] [PubMed] [Google Scholar]
- 196. Tsankova NM, Berton O, Renthal W, Kumar A, Neve RL, Nestler EJ. Sustained hippocampal chromatin regulation in a mouse model of depression and antidepressant action. Nat. Neurosci. 2006; 9: 519–525. [DOI] [PubMed] [Google Scholar]
- 197. Altar CA, Whitehead RE, Chen R, Wörtwein G, Madsen TM. Effects of electroconvulsive seizures and antidepressant drugs on brain‐derived neurotrophic factor protein in rat brain. Biol. Psychiatry 2003; 54: 703–709. [DOI] [PubMed] [Google Scholar]
- 198. Rantamäki T, Vesa L, Antila H et al Antidepressant drugs transactivate trkb neurotrophin receptors in the adult rodent brain independently of Bdnf and monoamine transporter blockade. PLoS One 2011; 6: e20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Gruart A, Sciarretta C, Valenzuela‐Harrington M, Delgado‐Garcia JM, Minichiello L. Mutation at the TrkB PLC‐docking site affects hippocampal LTP and associative learning in conscious mice. Learn. Mem. 2007; 14: 54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Autry AE, Monteggia LM. Brain‐derived neurotrophic factor and neuropsychiatric disorders. Pharmacol. Rev. 2012; 64: 238–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Saarelainen T, Hendolin P, Lucas G et al Activation of the TrkB neurotrophin receptor is induced by antidepressant drugs and is required for antidepressant‐induced behavioral effects. J. Neurosci. 2003; 23: 349–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Monteggia LM, Barrot M, Powell CM et al Essential role of brain‐derived neurotrophic factor in adult hippocampal function. Proc. Natl. Acad. Sci. U. S. A. 2004; 101: 10827–10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Monteggia LM, Luikart B, Barrot M et al Brain‐derived neurotrophic factor conditional knockouts show gender differences in depression‐related behaviors. Biol. Psychiatry 2007; 61: 187–197. [DOI] [PubMed] [Google Scholar]
- 204. Adachi M, Barrot M, Autry AE, Theobald D, Monteggia LM. Selective loss of brain‐derived neurotrophic factor in the dentate gyrus attenuates antidepressant efficacy. Biol. Psychiatry 2008; 63: 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Lindholm JSO, Castrén E. Mice with altered BDNF signaling as models for mood disorders and antidepressant effects. Front. Behav. Neurosci. 2014; 8: 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Shirayama Y, Chen AC‐H, Nakagawa S, Russell DS, Duman RS. Brain‐derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 2002; 22: 3251–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207. Koponen E, Rantamäki T, Voikar V, Saarelainen T, MacDonald E, Castrén E. Enhanced BDNF signaling is associated with an antidepressant‐like behavioral response and changes in brain monoamines. Cell. Mol. Neurobiol. 2005; 25: 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol. Psychiatry 1999; 46: 1181–1191. [DOI] [PubMed] [Google Scholar]
- 209. Chen AC, Shirayama Y, Shin KH, Neve RL, Duman RS. Expression of the cAMP response element binding protein (CREB) in hippocampus produces an antidepressant effect. Biol. Psychiatry 2001; 49: 753–762. [DOI] [PubMed] [Google Scholar]
- 210. Chen B, Dowlatshahi D, MacQueen GM, Wang JF, Young LT. Increased hippocampal BDNF immunoreactivity in subjects treated with antidepressant medication. Biol. Psychiatry 2010; 50: 260–265. [DOI] [PubMed] [Google Scholar]
- 211. Autry AE, Adachi M, Nosyreva E et al NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature 2011; 475: 91–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Liu RJ, Lee FS, Li XY, Bambico F, Duman RS, Aghajanian GK. Brain‐derived neurotrophic factor Val66Met allele impairs basal and ketamine‐stimulated synaptogenesis in prefrontal cortex. Biol. Psychiatry 2012; 71: 996–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Falkenberg T, Mohammed AK, Henriksson B, Persson H, Winblad B, Lindefors N. Increased expression of brain‐derived neurotrophic factor mRNA in rat hippocampus is associated with improved spatial memory and enriched environment. Neurosci. Lett. 1992; 138: 153–156. [DOI] [PubMed] [Google Scholar]
- 214. Rossi C, Angelucci A, Costantin L et al Brain‐derived neurotrophic factor (BDNF) is required for the enhancement of hippocampal neurogenesis following environmental enrichment. Eur. J. Neurosci. 2006; 24: 1850–1856. [DOI] [PubMed] [Google Scholar]
- 215. Zhu SW, Codita A, Bogdanovic N et al Influence of environmental manipulation on exploratory behaviour in male BDNF knockout mice. Behav. Brain Res. 2009; 197: 339–346. [DOI] [PubMed] [Google Scholar]
- 216. Banasr M, Duman RS. Keeping “Trk” of antidepressant actions. Neuron 2008; 59: 349–351. [DOI] [PubMed] [Google Scholar]
- 217. Sairanen M, Lucas G, Ernfors P, Castrén M, Castrén E. Brain‐derived neurotrophic factor and antidepressant drugs have different but coordinated effects on neuronal turnover, proliferation, and survival in the adult dentate gyrus. J. Neurosci. 2005; 25: 1089–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Waterhouse EG, An JJ, Orefice LL et al BDNF promotes differentiation and maturation of adult‐born neurons through GABAergic transmission. J. Neurosci. 2012; 32: 14318–14330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Fukuchi M, Izumi H, Mori H et al Visualizing changes in brain‐derived neurotrophic factor (BDNF) expression using bioluminescence imaging in living mice. Sci. Rep. 2017; 7: 4949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: Temporal patterns of induction distinct from NGF. Neuron 1991; 6: 937–948. [DOI] [PubMed] [Google Scholar]
- 221. Castrén E, Zafra F, Thoenen H, Lindholm D. Light regulates expression of brain‐derived neurotrophic factor mRNA in rat visual cortex. Proc. Natl. Acad. Sci. U. S. A. 1992; 89: 9444–9448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Meyer‐Franke A, Wilkinson GA, Kruttgen A et al Depolarization and cAMP elevation rapidly recruit TrkB to the plasma membrane of CNS neurons. Neuron 1998; 21: 681–693. [DOI] [PMC free article] [PubMed] [Google Scholar]