

Abstract
Daclatasvir is a nonstructural protein 5A replication complex inhibitor, and asunaprevir is a nonstructural protein 3 protease inhibitor for hepatitis C virus (HCV). In 2014, the combination therapy of daclatasvir and asunaprevir received the first global approval in Japan as the first nonribavirin, all‐oral therapy for HCV treatment. The population pharmacokinetics (popPK) of daclatasvir and asunaprevir were characterized by nonlinear mixed‐effects modeling using 3801 and 2626 concentration data from 336 and 265 Japanese HCV subjects, respectively. The plasma pharmacokinetic profiles of daclatasvir and asunaprevir were described by a 1‐compartment model. Parameter estimates (interindividual variability) of daclatasvir apparent clearance (CL/F) and apparent volume of the central compartment (V/F) were 5.29 L/h (39.4%) and 64.2 L (38.1%). The effects of all statistically significant covariates on daclatasvir PK parameters were within or overlapped the 80% to 125% boundaries, suggesting a lack of clinical relevance. Parameter estimates (interindividual variability) of asunaprevir CL/F and V/F were 52.1 L/h (41.5%) and 75.1 L (93.4%), respectively. Baseline and time‐varying aspartate aminotransferase (AST) and cirrhosis on CL/F and formulation (soft‐gel capsule or tablet) on F were included as significant covariates in the asunaprevir popPK model. The effects of all covariates exceeded the 80% to 125% boundaries, indicating that the asunaprevir soft‐gel capsule had higher bioavailability than the tablet and that asunaprevir exposure increased with cirrhosis and increasing baseline and time‐varying AST values. The popPK models adequately described the PK profiles of daclatasvir and asunaprevir in Japanese HCV subjects.
Keywords: asunaprevir, clinical pharmacology, daclatasvir, hepatitis C virus, Japanese, pharmacometrics, population pharmacokinetics
Approximately 80‐185 million individuals are infected with hepatitis C virus (HCV) worldwide, and the number of HCV‐infected patients is estimated to be approximately 2 million in Japan.1, 2, 3 It is estimated that 20% of patients with chronic HCV infection will develop cirrhosis.4 In recent years, HCV treatment has evolved rapidly from peginterferon (pegIFN) plus ribavirin (RBV) to all‐oral combinations of direct‐acting antivirals.5, 6 Daclatasvir (Daklinza tablets) is a nonstructural protein 5A replication complex inhibitor with exceptional in vitro potency against HCV genotypes 1‐6.7 Asunaprevir (Sunvepra capsules) is an orally available small‐molecule inhibitor of the HCV nonstructural protein 3 protease and subsequent viral ribonucleic acid replication.8 The daclatasvir and asunaprevir combination (DUAL) regimen has demonstrated robust antiviral activity (including in patients with cirrhosis) with a low frequency of adverse events.9 The DUAL regimen received the first global approval as the first all‐oral pegIFN/RBV free HCV treatment in Japan. The approved dose of daclatasvir and asunaprevir was 60 mg administered orally once daily as a tablet and 100 mg administered orally twice daily as a soft‐gel capsule. After the first approval in Japan, the combination regimen has been approved in multiple countries.
Daclatasvir pharmacokinetics (PK) were evaluated in healthy Japanese volunteers.10 The dose‐exposure relationship for daclatasvir was generally linear within the dose range studied. Daclatasvir was readily absorbed, with median time at peak concentration (Tmax) ranging from 1.0 to 2.0 hours. Steady state was generally achieved between days 4 and 5.
Daclatasvir is a substrate and inhibitor of the P‐glycoprotein transporter and a substrate and weak inducer of, cytochrome P450 (CYP) 3A4 with minimal effects on the levels of the sensitive CYP3A probe midazolam in plasma.11 Daclatasvir is excreted primarily (∼88%) via feces in an unchanged form, with renal elimination accounting for a minor pathway for daclatasvir (∼7% of dose).12 There was no obvious association between exposure and degree of hepatic impairment or biochemical/serological markers of liver dysfunction.13
Asunaprevir was also readily absorbed, with median Tmax ranging from 2.0 to 4.0 hours. Asunaprevir exposure generally increased dose‐proportionally within the dose range studied. Steady state was generally achieved between days 3 and 5.10
Asunaprevir is eliminated primarily via CYP 3A4‐mediated hepatic metabolism.14, 15 Asunaprevir is a weak inducer and sensitive substrate of CYP3A4, a moderate inhibitor of CYP2D6, a weak inhibitor and sensitive substrate of organic anion transporting polypeptide(OATP)‐mediated uptake transport and a weak inhibitor of p‐glycoprotein.16
Significant food effects have been observed with the asunaprevir tablet formulation used in phase 1/2 studies.17 A soft‐gel capsule was developed as a food‐effect mitigating formulation and provided higher exposures (∼2‐fold for AUC) with or without food than the tablet formulation given with food.17 Subsequently, the soft‐gel capsule was selected in phase 3 studies and also for the commercial formulation.
Hepatic function impairment had been shown to significantly impact the steady‐state PK of asunaprevir. Asunaprevir steady‐state exposures were approximately 5‐ to 10‐fold and 20‐ to 30‐fold higher in subjects with moderate and severe hepatic impairment (Child‐Pugh class B and C cirrhosis, respectively), not changed in subjects with mild impairment (Child‐Pugh class A cirrhosis).18
The population pharmacokinetic (popPK) analyses were the first model development to characterize the PK of daclatasvir and asunaprevir integrating multiple studies including phase 3 data. The objectives of the popPK analyses were to help explain the source of variability in drug exposure by investigating the potential relationships between covariates and the PK of daclatasvir and asunaprevir in the target patient population. Also, the popPK model will be used to estimate individual patient exposures, which will be used for separate exposure‐response analyses.
Methods
Clinical Studies and Patient Population
Daclatasvir and asunaprevir plasma concentration‐time data were obtained from 4 clinical studies (AI444021, AI444022, AI447017, and AI447026)9, 19, 20, 21 and 2 clinical studies (AI447017 and AI447026), respectively (Supplementary Table 1). All clinical studies included in the popPK analyses were conducted in accordance with the ethical principles that have their origin in the Declaration of Helsinki. The study protocols were approval by the institutional review board prior to initiation of studies and obtaining written informed consent from each subject prior to study participation.
Daclatasvir was administered either as part of the DUAL regimen with asunaprevir or in combination with pegIFN/RBV. Daclatasvir doses investigated were 10 and 60 mg given once daily, and asunaprevir doses that were investigated included 200 and 600 mg twice daily as a tablet formulation and 100 mg twice daily as a soft‐gel capsule formulation. Eligible subjects were Japanese men or women ≥ 20 years of age, with chronic HCV genotype 1 infections. Subjects with compensated cirrhosis were included.
Assay Methodology
Daclatasvir and asunaprevir plasma concentrations were measured by a validated liquid chromatography‐tandem mass spectrometry method with a quantitation range of 0.500 to 500 ng/mL for AI444021 and AI444022, 1 to 1000 ng/mL for AI447017, and 2.0 to 1000 ng/mL and 2.0 to 2000 ng /mL for AI447026. Samples below the lower limit of quantification (LLOQ) were excluded from the analysis, and missing actual PK sampling time was imputed from nominal sampling time captured from the case report forms.
Analysis Platform
The popPK analysis was performed by nonlinear mixed‐effects modeling using the NONMEM computer program (version 7.2; Icon Development Solutions), compiled using GNU FORTRAN version 77, and installed on a Linux platform. Diagnostic graphics, exploratory analyses, and postprocessing of NONMEM output were performed using S‐Plus software (version 8.1 for Linux; Insightful, Seattle, Washington) on a Linux platform.
Population PK Model Development
The popPK methods were based on the US Food and Drug Administration Guidance for Industry Population Pharmacokinetics.22 The popPK of daclatasvir and asunaprevir was characterized by a nonlinear mixed‐effects model.
The pop PK model development comprised establishing a base and a full and final model. A base model was developed to represent the best description of the data without considering the effect of covariates. The structural, interindividual variability, and residual‐error models were determined. Linear compartmental models were tested as a base structural model. Selection of the base model was based on the Akaike information criterion, minimization success, diagnostic plots, and general observation of concentration‐time profiles.
Interindividual variability was described using a log‐normal distribution, as given below:
where is the parameter for the ith subject, is the typical value of the population, is an independent random interindividual variability with a mean of 0 and variance of ω2.
The residual error model for a log‐transform‐both‐sides approach was used. Residual variability was assumed to be log‐normally distributed, as follows:
where and represent the jth observed and predicted concentration, respectively, for the ith subject and ε is the residual intrasubject random error with a standard deviation of .
Supplementary Table 2 provides a list of prespecified covariates and their relationships to the PK parameters. The selection of the prespecified covariates and associated PK parameters was based on clinical interest and pharmacological plausibility. All prespecified covariates were included in the full model without considering significance level. If correlation was observed between covariates, the most significant term was kept in the full model.
The final popPK model was started from the full model and obtained by removing each covariate one at a time. The covariate with the smallest change in minimum objective function was removed from the model until all remaining covariates were significant (P < .001). The 95% confidence intervals (CIs) of estimated parameters were calculated by the bootstrap method.
The impact of significant covariates on the PK parameters was illustrated using forest plots. Pharmacokinetic parameters at the 5th and 95th percentiles of the population values of the continuous covariates or at different levels of the categorical covariates were compared with typical PK estimates. Effects of covariates at extreme values and associated 95%CIs, when wholly contained within the 80% to 125% boundaries of the typical PK estimates, may suggest a lack of clinical relevance.
Model Evaluation
The diagnostic graphs include the population predicted (PRED) and individual predicted (IPRED) mean concentrations versus observed concentrations and the conditional weighted residuals (CWRES) versus PRED or IPRED and time. This set of diagnostic graphs shows whether the predicted concentrations match the observed concentrations.
Prediction‐corrected visual predictive check (pcVPC) was created to show the time course of the predicted mean and spread of concentrations (5th to 95th percentiles) versus the observed data.23 A total of 1000 trial replicates was simulated using the observed covariates and dose regimens for each subject, the final model parameter estimates, and simulated subject‐specific random effects and residual errors. The pcVPC showed the overall model fit of all PK data with different dose regimens.
Results
Population PK Model Development
Daclatasvir
A total of 336 subjects were enrolled in the study, and demographic and baseline characteristics are summarized in Table 1. A total of 3808 pharmacokinetic records were obtained, and 7 pharmacokinetic records were excluded because the concentrations were below the LLOQ. Therefore, a total of 3801 pharmacokinetic records were included for model development.
Table 1.
Summary of Demographic Characteristics
| Daclatasvir | |
|---|---|
| Covariate (n = 336) | Value |
| Age, median (min, max), years | 61 (21, 75) |
| Sex | |
| Male, n (%) | 122 (36.3) |
| Female, n (%) | 214 (63.7) |
| Body weight, median (min, max), kg | 56 (36, 93) |
| Baseline AST, median (min, max), U/L | 51.0 (13, 377) |
| Baseline ALT, median (min, max), U/L | 48.0 (13, 595) |
| Baseline creatinine clearance, median (min, max), mL/min | 86.5 (39.56, 185.96) |
| Cirrhosis | |
| Yes, n (%) | 22 (6.5) |
| No, n (%) | 314 (93.5) |
| Patient type | |
| Nonresponder, null responder, or partial responder, n (%) | 143 (42.6) |
| pegIFN/RBV ineligible naive or intolerant subjects, n (%) | 157 (46.7) |
| Treatment naive | 36 (10.7) |
| Treatment description | |
| daclatasvir + PegIFN/RBV, n (%) | 71 (21.1) |
| daclatasvir + asunaprevir (DUAL), n (%) | 265 (78.9) |
| Genotype | |
| 1a, n (%) | 2 (0.6) |
| 1b, n (%) | 334 (99.4) |
| Asunaprevir | |
|---|---|
| Covariate (n = 256) | Value |
| Age, median (min, max), years | 62 (24, 75) |
| Sex | |
| Male, n (%) | 91 (34.3) |
| Female, n (%) | 174 (65.7) |
| Baseline body weight, median (min, max), kg | 55 (36, 93) |
| Cirrhosis | |
| Yes, n (%) | 22 (8.3) |
| No, n (%) | 243 (91.7) |
| Formulation | |
| Phase 2 formulation, n (%) | 43 (16.2) |
| Phase 3 formulation, n (%) | 222 (83.8) |
| Patient type | |
| Nonresponder, null responder, or partial responder, n (%) | 108 (40.8) |
| SOC‐ineligible naive or intolerant subjects, n (%) | 157 (59.2) |
| Baseline AST, median (min, max), U/L | 52 (13, 595) |
| Baseline ALT, median (min, max), U/L | 55 (13, 377) |
| Baseline creatinine clearance, median (min, max), mL/min | 84.6 (39.56, 171.95) |
| OATP haplotype | |
| *1B/*1B, n (%) | 51 (19.3) |
| *1B/*1A, n (%) | 86 (32.5) |
| *1A/*1A, n (%) | 28 (10.6) |
| Other, n (%) | 64 (24.2) |
| Missing, n (%) | 36 (13.6) |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; OATP, organic anion transporting polypeptide; SOC, standard of care.
A 1‐compartment PK structure model with first‐order absorption was identified as an optimal model both. Interindividual variability was estimated in apparent clearance (CL/F), apparent volume of the central compartment (V/F), and absorption rate constant (Ka), with correlation between CL/F and V/F. The residual error model was additive in log‐transformed daclatasvir plasma concentrations. Parameter estimates for the daclatasvir base model are presented in Supplementary Table 3.
To develop the full model, prespecified covariates and those correlations were investigated. Because the correlation between age, baseline body weight (BBWT), and baseline creatinine clearance (BCRCL), and the correlation between baseline aspartate aminotransferase (BAST) and baseline ALT (BALT) were moderate or high, the relationships between these covariates and CL/F were tested separately.
As a result, baseline creatinine clearance and baseline ALT on CL/F showed the greatest objective function change (OFV) among other covariates and were retained in the full model. All other prespecified covariates were included into the full model. Minimization and convergence were successful. The parameters of the full model are provided in Supplementary Table 4.
The final popPK model was obtained by removing nonsignificant covariates from the full model during backward elimination. The final model parameters are presented in Table 2.
Table 2.
Final Model Parameter Estimates
| Daclatasvir | ||||
|---|---|---|---|---|
| Parametera | Symbol | Estimateb | Standard Error (RSE%)c | 95% Confidence Intervald |
| Fixed effects | ||||
| CL/F (L/h) | θ1 | 5.29 | 0.161 (3.04) | 4.98‐5.59 |
| V/F (L) | θ2 | 64.2 | 2.04 (3.18) | 60.1‐68.2 |
| Ka (h‐1) | θ3 | 0.865 | 0.0516 (5.97) | 0.753‐0.974 |
| CL/F ∼SEX | θ6 | ‐0.110 | 0.0274 (24.9) | ‐0.197 to ‐0.053 |
| CL/F ∼TX | θ8 | ‐0.122 | 0.0336 (27.5) | ‐0.189‐0.132 |
| CL/F ∼BCRCL | θ10 | 0.235 | 0.0462 (19.7) | 0.142‐0.333 |
| V/F ∼BBWT | θ14 | 0.605 | 0.0989 (16.3) | 0.405‐0.974 |
| Random effects | ||||
| CL/F | ω1,1 | 0.155 (0.394) | 0.0157 (10.1) | 0.0176‐0.182 |
| V/F | ω2,2 | 0.145 (0.381) | 0.0216 (14.9) | 0.0168‐0.186 |
| Ka | ω3,3 | 0.756 (0.869) | 0.105 (13.9) | 0.590‐0.968 |
| CL/F: V/F | ω1,2 | 0.141 (0.941) | 0.0161 (11.4) | 0.00099‐0.171 |
| σ | ω4,4 | 0.107 (0.327) | 0.0193 (18.0) | 0.072‐0.148 |
| Residual error | ||||
| σ | θ4 | 0.375 | 0.0102 (2.72) | 0.358‐0.408 |
| Asunaprevir | ||||
|---|---|---|---|---|
| Parametera | Symbol | Estimateb | Standard Error (RSE%)c | 95% Confidence Intervald |
| Fixed effects | ||||
| CL/F (L/h) | θ1 | 52.1 | 7.35 (14.1) | 42.1‐66.8 |
| V/F (L) | θ2 | 75.1 | 13.5 (18.0) | 56.7‐101.0 |
| Ka (h‐1) | θ3 | 0.228 | 0.00395 (1.73) | 0.221‐0.234 |
| CL/F ∼BAST | θ7 | ‐0.598 | 0.0565 (9.45) | ‐0.707 to ‐0.486 |
| CL/F ∼AST | θ8 | ‐0.382 | 0.0443 (11.6) | ‐0.458 to ‐0.303 |
| F ∼FORM | θ10 | ‐0.314 | 0.144 (45.9) | ‐0.551 to ‐0.0922 |
| CL/F ∼CIRRHOSIS | θ11 | ‐0.428 | 0.140 (32.7) | ‐0.753 to ‐0.146 |
| Random effects | ||||
| CL/F | ω1,1 | 0.172 (0.415) | 0.0232 (13.5) | 0.127‐0.213 |
| V/F | ω2,2 | 0.872 (0.934) | 0.139 (15.9) | 0.663‐1.10 |
| σ | ω4,4 | 0.0672 (0.259) | 0.0176 (26.2) | 0.0364‐0.0974 |
| Residual error | ||||
| σ | θ4 | 0.68 | 0.0193 (2.84) | 0.648‐0.713 |
AST, aspartate aminotransferase at each time; BAST, baseline aspartate aminotransferase; BBWT, baseline body weight; BCRCL, baseline creatinine clearance; CIRRHOSIS, cirrhosis (yes vs no); CL/F, apparent clearance; FORM, formulation (phase 2 film‐coated tablet vs phase 3 soft‐gel capsule); Ka, absorption rate constant; TX, treatment description; V/F, apparent volume of distribution of central compartment; σ, parameters for additive residual error.
Random effects and residual error parameter estimates are shown as variance (standard deviation) for diagonal elements (ωi,i or ωi,i) and covariance (correlation) for off‐diagonal elements (ωi,j or ωi,j).
RSE, relative standard error (standard error as a percentage of estimate).
All confidence intervals are from a 500 bootstrap run.
For a typical HCV‐infected subject, the CL/F of daclatasvir was 5.29 L/h (3.04 relative standard error [RSE] %), V/F was 64.2 L (3.18 RSE%), and Ka was 0.865 h‐1 (5.97 RSE%). Interindividual variablility was estimated for CL/F (39.4%), V/F (38.1%), and Ka (86.9%). Significant covariates (P < .001) remaining in the final model included sex, treatment description, baseline creatinine clearance, on CL/F, and baseline body weight on V/F, as described in the equations below.
where TV,ref is the typical value at the reference or baseline values for appropriate covariates, BBWTref is 56 kg, BCRCLref is 86.48 mL/min, SEXref is male, and TXref is asunaprevir. The condition number of the final model was 224.8, indicating well‐conditioned because of being well below the condition number threshold of 1000. Interindividual variability of CL/F and V/F of the final model were reduced from that of the base model (39.7% to 39.4% and 40.7% to 38.1%, respectively). The residual errors were similar in the base model and the final model.
The shrinkage in CL/F, V/F, and Ka of subjects was 4.3%, 11.8%, and 19.2%, respectively, implying there is sufficient PK information from individual subjects to provide reliable individual parameter estimates for the majority of the patients.
Asunaprevir
A total of 265 subjects were enrolled in the study, and demographic and baseline characteristics are summarized in Table 1. A total of 2676 pharmacokinetic records were obtained, and 50 pharmacokinetic records were excluded because the concentrations were below the LLOQ. Therefore, a total of 2626 pharmacokinetic records were included for model development.
Same as the daclatasvir, a 1‐compartment PK structure model with first‐order absorption was identified as an optimal model. Interindividual variability was estimated in apparent clearance (CL/F), and apparent volume of the central compartment (V/F). The residual error model was additive in log‐transformed asunaprevir plasma concentrations. Parameters for the asunaprevir base model are presented in Supplementary Table 3. The random effect of Ka was fixed as zero because the sampling points around the peak concentrations were not enough for all subjects.
Also in the asunaprevir data set, because the correlation between age, baseline body weight, and baseline creatinine clearance and the correlation between baseline aspartate aminotransferase and baseline ALT were moderate or high, the relationships between these covariates and CL/F were separately tested. Age on CL/F showed the larger decrease in OFV compared with that of baseline body weight or baseline creatinine clearance on CL/F. In addition, the time‐varying effect was assessed for AST and ALT on CL/F and was highly significant compared with incorporating only baseline AST and ALT values, with a decrease of ‐139.192 and ‐108.05, respectively. Both AST and ALT resulted in a significant reduction in objective function value, but AST reduced the objective function value the greatest following incorporation into the full model. As a result, AST (baseline aspartate aminotransferase and time‐varying AST) and age were retained in the full model of ASV. All other prespecified covariates were included in the full model. Minimization of the full model in NONMEM was successful with covariance step completed. The parameters of the full model are provided in Supplementary Table 4.
The final popPK model was obtained by removing nonsignificant covariates from the full model during backward elimination. The final model parameters are provided in Table 2. For an HCV‐infected subject with typical covariate values (age 62 years old, 55 kg, female, with baseline AST of 52 U/L, no cirrhosis), CL/F was 52.1 L/h (14.1 RSE%), and V/F was 75.1 (18.0 RSE%). The first‐order absorption rate constant was 0.23 h‐1 (1.7% RSE%). Significant covariates (P < .001) remaining in the final model included baseline aspartate aminotransferase, time‐varying AST, cirrhosis status, formulation on F, as described in the equations below.
where TV,ref is the typical value at the reference or baseline values for appropriate covariates (BASTref, 52 U/L; FORMref, phase 2 tablet formulation; CIRRHOSISref, NO). The condition number of the final model was 172.4, indicating well‐conditioned because of being well below the condition number threshold of 1000. Interindividual variability of CL/F and V/F of the final model was reduced from that of the base model (51.5% to 41.5% and 97.4% to 93.4%, respectively). The residual errors were similar in the base model and the final model. Shrinkage of the final model parameters was 10.0% for CL/F and 18.2% for V/F.
Model Evaluations
Daclatasvir
Goodness‐of‐fit plots of the final model are presented in Figure 1. Overall, the plots reasonably supported the final model and demonstrated no bias over model‐predicted value and time. The predictive performance of the developed final PPK model was assessed using pcVPC. Figure 2 shows the pcVPC plot after 14 days at steady sate. The plot showed that the model adequately described the central tendency and the spread of the observed PK at steady state.
Figure 1.
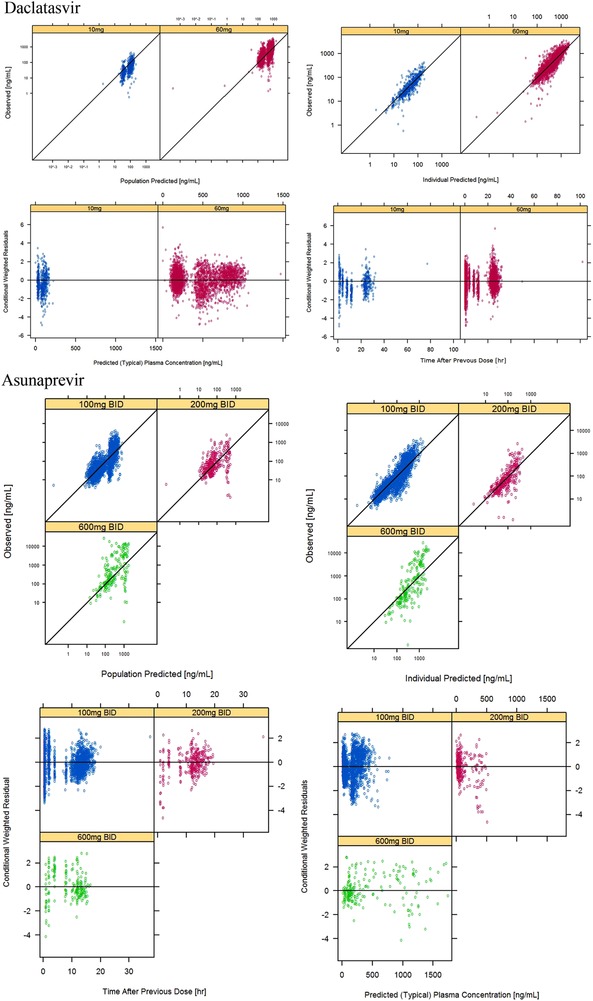
Goodness‐of‐fit plots of the final covariate population pharmacokinetic model (upper, daclatasvir; lower, asunaprevir).
Figure 2.
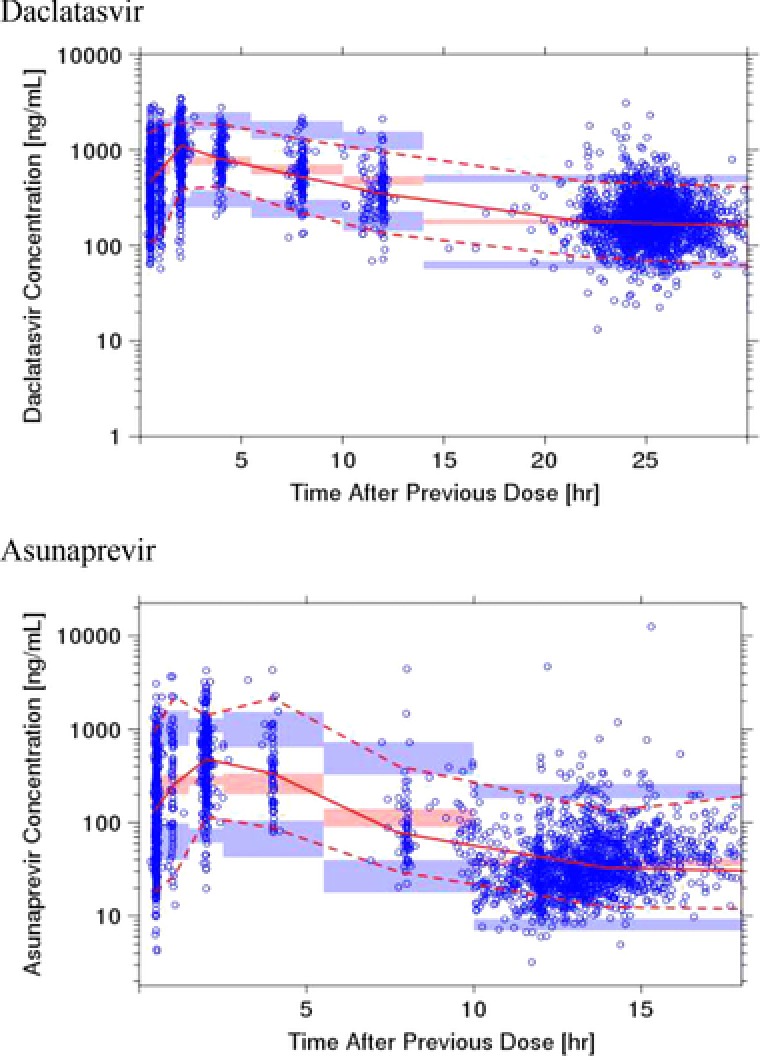
Prediction corrected visual predictive checks (upper, daclatasvir; lower, asunaprevir). Circles are observed asunaprevir plasma concentrations, solid red line represents the median observed value, and dotted red lines represent 5th and 95th percentiles of the observed values. Red‐shaded areas represent the spread of the median predicted values (5th to 95th percentiles), and blue‐shaded areas represent the spread (5th and 95th percentiles) of the 5th and 95th predicted percentile concentrations. Each bin was set to cover the sampling points.
Asunaprevir
Goodness‐of‐fit plots of the base model and final model are represented in Figure 1. Diagnostic plots of the final model showed good agreement between the predictions and the observations and CWRESs that were unbiased and with absolute value less than 5 and minimal correlations between PK parameters for HCV‐infected Japanese subjects. Only PK models of 600 mg twice daily have a tendency to underpredict because of the small number of sampling points.
Figure 2 shows the pcVPC plot after 14 days at steady state. Overall, the pcVPC plot demonstrates that the model adequately described the central tendency and the spread of the observed PK at steady state.
Effect of Covariates on Population PK Parameters
Daclatasvir
Impact of covariates on the daclatasvir PK parameters estimated from the final popPK model is represented in a forest plot (Figure 3). The impact of baseline body weight on V/F on the final popPK model was overwrapped with the 80% to 125% boundary. All other covariates effects were within the 80% to 125% range. The effect of baseline body weight on V/F suggests that for subjects with the extremes of baseline body weight (5th and 95th percentiles of baseline body weight were 42.8 and 78 kg, respectively), daclatasvir V/F would be ∼15% lower and ∼22% higher, respectively, than the median (reference) body weight of 56 kg. For the effects of treatment and sex, the daclatasvir CL/F was reduced by ∼10% for subjects receiving pegIFN/RBV compared with those receiving asunaprevir combination treatment, and for women relative to men. For subjects with baseline creatinine clearance at the 5th or 95th percentile (51.36 or 144.42 mL/min, respectively) relative to the typical subject was reduced or increased by approximately 10%.
Figure 3.
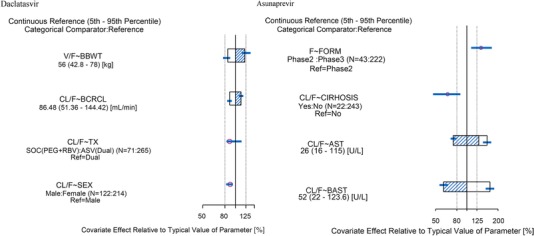
Impact of covariates on pharmacokinetic parameters of the final model (left, daclatasvir; right, asunaprevir). For daclatasvir, typical PK parameters were estimated for male, baseline body weight of 56 kg, baseline creatinine clearance of 88.48 mL/min using treatment description with administered dual (daclatasvir + asunaprevir). For asunaprevir, typical PK parameters were estimated for a 62‐year‐old, 55‐kg woman using the phase 2 formulation with baseline AST of 52 U/L, time‐varying AST of 26 U/L, and no cirrhosis. Relative bioavailability was computed from CL/F (phase 3 formulation, soft‐gel capsule)/CL/F (phase 2 formulation, film‐coated tablet), or V/F (phase 3 formulation)/V/F (phase 2 formulation). Categorical covariate effects (95%CI) are represented by open symbols (horizontal lines). Continuous covariate effects (95%CI) at the 5th/95th percentiles of the covariate are represented by the end of horizontal boxes (horizontal lines). Open/shaded area of boxes represents the range of covariate effects from the median to the 5th/95th percentiles of the covariate. AST, alanine aminotransferase; BAST, baseline alanine aminotransferase; BBWT, baseline body weight; BCRCL, baseline creatinine clearance; CI, confidence interval; CIRHOSIS, cirrhosis (yes or no); CL/F, apparent clearance of orally administered doses; F, relative bioavailability; FORM, formulation (phase 2 film‐coated tablet vs phase 3 soft‐gel capsule); TX, treatment description (standard of care or dual regimen); V/F, apparent volume of distribution.
Asunaprevir
The impact of significant covariates on the asunaprevir PK parameters estimated from the final popPK model is illustrated in a forest plot (Figure 3). The point estimates of the formulation on bioavailability and cirrhosis on CL/F exceeded the 80% to 125% boundary. The bioavailability of the phase 3 soft‐gel capsule (100‐mg twice‐daily) formulation was 1.37‐fold higher than the tablet formulation used in phase 2 (200 or 600 mg twice daily). ASV CL/F for cirrhotic subjects is expected to be 0.65‐fold lower relative to subjects without cirrhosis. The effect of baseline AST and time‐varying AST at the 5th and 95th percentiles on CL/F exceeds the 80%‐125% range. The effect of baseline AST suggests that for subjects with the extremes of baseline AST (5th and 95th percentiles of baseline AST were 22 and 123.6 U/L, respectively), ASV CL/F would be 1.67‐fold higher and 0.60‐fold lower, respectively, than the median baseline AST of 52 U/L. Similarly, the effect of the time‐varying effect of AST suggests that for subjects with extremes of AST at the last PK sampling point (5th and 95th percentiles of AST at the last sampling time were 16 and 115 U/L, respectively), ASV CL/F would be 1.57‐fold higher and 0.74‐fold lower, respectively, compared with the median baseline AST.
Discussion
Daclatasvir
The popPK of daclatasvir was characterized by a 1‐compartment model with first‐order absorption and linear clearance. In the final model, baseline creatinine clearance sex and treatment regimen on CL/F and BBWT on V/F were identified as statistically significant covariates.
The final model and covariate plot indicated that daclatasvir CL/F decreased with lower baseline creatinine clearance. These results are consistent with the results of the daclatasvir renal impairment study, in which, based on a regression analysis, daclatasvir AUC increased with decreasing baseline creatinine clearance, and the covariate effect was hypothesized to be a result of alteration of nonrenal clearance, given that renal excretion is not a major elimination pathway for daclatasvir.13
In terms of hepatic function, cirrhosis status and ALT/AST levels were not identified as significant covariates of daclatasvir PK parameters on CL/F; therefore, daclatasvir PK was not expected to be dependent on hepatic function in the subject population. These results are consistent with the results of the hepatic impairment study.13
Overall, the effects of all significant covariates included in the final model are within or overlapped the 80% to 125% boundaries, suggesting a lack of clinical relevance. This is also supported by the overall interindividual variability for CL/F being similar (from 39.7% and 39.4%) and for V/F being reduced only by approximately 2.5% (from 40.7% to 38.1%). This addition of the covariates to the base model suggests that the covariate effects do not contribute to interindividual variability of daclatasvir PK.
The daclatasvir popPK model has been further developed by adding the data from other clinical studies including non‐Japanese HCV patients after the current daclatasvir popPK analysis for Japanese HCV patients was performed.24 The effects of significant covariates included in the subsequent daclatasvir popPK analysis are also not considered clinically relevant, which is consistent with the result from the Japanese daclatasvir popPK analysis.
Asunaprevir
The popPK of asunaprevir was characterized by a 1‐compartment model with first‐order absorption and linear clearance. OATP1B1 haplotypes were evaluated for covariates, because differences in asunaprevir exposures between Japanese and non‐Japanese subjects have been investigated previously, and genetic variability in OATP1B1‐mediated transport of asunaprevir has been hypothesized to be a potential factor for the differences in exposure.25 Specifically, some single‐nucleotide polymorphisms with reduced function appear to be more common in the Asian population. The impact of different OATP1B1 haplotypes on asunaprevir CL/F was examined to evaluate the effect of OATP1B1 genetic variability on asunaprevir exposure. Consequently, the 4 categories of OATP1B1 haplotypes did not show statistical significance, suggesting that in the Japanese population, genetic variability of OATP1B1 has no impact on asunaprevir exposure. Other investigations had been evaluated by including non‐Japanese data, and the analyses did not reveal a relationship between OATP and asunaprevir exposure.17
In the final model, formulation on F and baseline aspartate aminotransferase and time‐varying AST and cirrhosis status on CL/F were identified as significant covariates for the PK of asunaprevir. The effect of formulation on bioavailability of asunaprevir, which was included in the base structural model, suggests that the bioavailability of the phase 3 soft‐gel capsule (100‐mg twice‐daily) formulation was 1.37‐fold higher than the tablet formulation used in phase 2 (200 or 600 mg twice daily). The result was similar to the findings in AI447017 (the phase 2 study used a 200‐mg tablet formulation) and AI447026 (the phase 3 study used the 100‐mg soft‐gel capsule). The observed asunaprevir AUC in AI447017 and AI447026 was 2950 and 2155 ng·h/mL, respectively.17 In addition, the trend was consistent with the result of the relative bioavailability study.
The other key covariates on asunaprevir CL/F were AST and cirrhosis status, suggesting that asunaprevir CL/F is a marker of hepatic function. Subjects with cirrhosis had higher baseline aspartate aminotransferase/time‐varying AST, resulting in a decrease in asunaprevir CL/F and therefore increased exposure to asunaprevir. The effect of cirrhosis on the regulation and expression of enzymes has been documented in the literature.26 The effect of AST on CL/F has been previously reported for sildenafil and tacrolimus, which are extensively metabolized by the liver.27, 28 Note that ALT and AST were highly correlated and were both tested as covariates, and AST was included in the final model because it had a slightly greater impact, although the magnitude of the effects was very similar in these covariates. Results of the current analysis are consistent with the metabolic profile of asunaprevir (asunaprevir is extensively metabolized and eliminated primarily through the feces) and with the results of the hepatic impairment study (subject with moderate and severe hepatic impairment had greater asunaprevir exposure). The presence of cirrhosis reduces blood flow, protein binding, and drug‐metabolizing enzymes, which could result in increasing asunaprevir distribution in the liver.29 Collectively, the presence of cirrhosis and increased AST, which reflect worsening hepatic function, is expected to increase exposure of asunaprevir.
Overall, cirrhosis status and AST were the significant covariates on asunaprevir CL/F. Asunaprevir CL/F decreases with cirrhosis and increasing baseline and time‐varying AST, indicating an association between hepatic function and asunaprevir CL/F.
The asunaprevir popPK model has been further developed by adding the data from other clinical studies including non‐Japanese HCV patients after the current asunaprevir popPK analysis for Japanese HCV patients was performed. Race was identified as a significant covariate on asunaprevir CL/F. Other than race, the significant covariates included in the subsequent asunaprevir popPK model and its effect on asunaprevir exposure were generally consistent with those in this asunaprevir popPK analysis for Japanese HCV patients.30
In summary, we developed population pharmacokinetic models for daclatasvir and asunaprevir characterized by nonlinear mixed‐effects models. None of the significant covariates included in the daclatasvir popPK model were considered clinically relevant. The asunaprevir soft‐gel capsule had higher bioavailability compared with the tablet, and asunaprevir exposure increased with cirrhosis and increasing baseline and time‐varying AST values. The significant covariates identified in the asunaprevir popPK model help to explain the source of variability in asunaprevir exposure.
Supporting information
Supporting Information
Acknowledgments
The authors thank Phyllis Chan, Timothy Eley, and Malaz Abu Tarif for their scientific contributions to the study concept, data analysis plan, and data interpretation for the study report and Megan Wind‐Rotolo and Rick Bertz for their scientific contributions to data interpretation for the study report.
Data Sharing
The individual deidentified participant data and related documents will not be shared.
Declaration of Conflicting Interests
Mayu Osawa, Takayo Ueno, Hiroki Ishikawa, Yasuhiko Imai, and Tushar Gallimera are employees of Bristol‐Myers Squibb K.K./Bristol‐Myers Squibb and/or stockholders.
Funding
The studies described in this report were funded by Bristol‐Myers Squibb.
References
- 1. Mohd Hanafiah K, Groeger J, Flaxman AD, Wiersma ST. Global epidemiology of hepatitis C virus infection: new estimates of age‐specific antibody to HCV seroprevalence. Hepatology. 2013;57(4):1333–1342. [DOI] [PubMed] [Google Scholar]
- 2. Gower E, Estes C, Blach S, Razavi‐Shearer K, Razavi H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J Hepatol. 2014;61(1 suppl):S45–S57. [DOI] [PubMed] [Google Scholar]
- 3. Chung H, Ueda T, Kudo M. Changing trends in hepatitis C infection over the past 50 years in Japan. Intervirology. 2010;53(1):39–43. [DOI] [PubMed] [Google Scholar]
- 4. Di Bisceglie AM. Natural history of hepatitis C: its impact on clinical management. Hepatology. 2000;31(4):1014–1018. [DOI] [PubMed] [Google Scholar]
- 5. EASL Recommendations on treatment of hepatitis C 2016. J Hepatol. 2017;66(1):153–194. [DOI] [PubMed] [Google Scholar]
- 6. Pawlotsky JM, Feld JJ, Zeuzem S, Hoofnagle JH. From non‐A, non‐B hepatitis to hepatitis C virus cure. J Hepatol. 2015;62(1 suppl):S87–S99. [DOI] [PubMed] [Google Scholar]
- 7. Gao M. Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr Opin Virol. 2013;3(5):514–520. [DOI] [PubMed] [Google Scholar]
- 8. McPhee F, Sheaffer AK, Friborg J, et al. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS‐650032). Antimicrob Agents Chemother. 2012;56(10):5387–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kumada H, Suzuki Y, Ikeda K, et al. Daclatasvir plus asunaprevir for chronic HCV genotype 1b infection. Hepatology. 2014;59(6):2083–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Shiozaki T, Ueno T, Nagashima H, et al. Single‐ and multiple‐ascending dose studies to evaluate the safety, tolerability, and pharmacokinetics of daclatasvir and asunaprevir in healthy male Japanese subjects. Int J Clin Pharmacol Ther. 2015;53(4):292–302. [DOI] [PubMed] [Google Scholar]
- 11. Bifano M SH, Stonier M, Jiang H, Bertz RJ. Daclatasvir, an HCV NS5A replication complex inhibitor, has minimal effect on pharmacokinetics of midazolam, a sensitive probe for cytochrome P450 3A4. Presented at: 8th International Workshop on Clinical Pharmacology of Hepatitis Therapy; June 26‐27, 2013; Cambridge, MA.
- 12. EMA . Daklinza product information. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/003768/WC500172848.pdf. 2015.
- 13. Gandhi Y, Eley T, Fura A, Li W, Bertz RJ, Garimella T. Daclatasvir: a review of preclinical and clinical pharmacokinetics. Clin Pharmacokinet [published online ahead of print 2018]. [DOI] [PubMed] [Google Scholar]
- 14. Gong J, Eley T, He B, et al. Characterization of ADME properties of [(14)C]asunaprevir (BMS‐650032) in humans. Xenobiotica. 2016;46(1):52–64. [DOI] [PubMed] [Google Scholar]
- 15. Eley T, Gardiner D, Persson A, et al. Evaluation of drug interaction potential of the HCV protease inhibitor asunaprevir (ASV; BMS‐650032) at 200 mg twice daily (BID) in metabolic cocktail and P‐glycoprotein (P‐gp) probe studies in healthy volunteers. Presented at: 62nd Annual Meeting of the American Association for the Study of Liver Diseases; November 6‐9, 2011; San Francisco, CA.
- 16. Eley T, Han YH, Huang SP, et al. Organic anion transporting polypeptide‐mediated transport of, and inhibition by, asunaprevir, an inhibitor of hepatitis C virus NS3 protease. Clin Pharmacol Ther. 2015;97(2):159–166. [DOI] [PubMed] [Google Scholar]
- 17. Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: a review of preclinical and clinical pharmacokinetics and drug‐drug interactions. Clin Pharmacokinet. 2015;54(12):1205–1222. [DOI] [PubMed] [Google Scholar]
- 18. Eley T, He B, Chang I, et al. The effect of hepatic impairment on the pharmacokinetics of asunaprevir, an HCV NS3 protease inhibitor. Antivir Ther. 2015;20(1):29–37. [DOI] [PubMed] [Google Scholar]
- 19. Izumi N, Yokosuka O, Kawada N, et al. Daclatasvir combined with peginterferon alfa‐2a and ribavirin in Japanese patients infected with hepatitis C genotype 1. Antivir Ther. 2014;19(5):501–510. [DOI] [PubMed] [Google Scholar]
- 20. Suzuki F, Toyota J, Ikeda K, et al. A randomized trial of daclatasvir with peginterferon alfa‐2b and ribavirin for HCV genotype 1 infection. Antivir Ther. 2014;19(5):491–499. [DOI] [PubMed] [Google Scholar]
- 21. Suzuki Y, Ikeda K, Suzuki F, et al. Dual oral therapy with daclatasvir and asunaprevir for patients with HCV genotype 1b infection and limited treatment options. J Hepatol. 2013;58(4):655–662. [DOI] [PubMed] [Google Scholar]
- 22. Guidance for Industry Population Pharmacokinetics. U.S. Department of Health and Human Services. Food and Drug Administration; 1999. [Google Scholar]
- 23. Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chan P, Li H, Zhu L, et al. Population pharmacokinetic analysis of daclatasvir in subjects with chronic hepatitis C virus infection. Clin Pharmacokinet. 2017;56(10):1173–1183. [DOI] [PubMed] [Google Scholar]
- 25. Eley T, Garimella T, Li W, Bertz RJ. Asunaprevir: an HCV protease inhibitor with preferential liver distribution. Clin Pharmacol Drug Dev. 2017;6(2):195–200. [DOI] [PubMed] [Google Scholar]
- 26. Elbekai RH, Korashy HM, El‐Kadi AO. The effect of liver cirrhosis on the regulation and expression of drug metabolizing enzymes. Curr Drug Metab. 2004;5(2):157–167. [DOI] [PubMed] [Google Scholar]
- 27. Milligan PA, Marshall SF, Karlsson MO. A population pharmacokinetic analysis of sildenafil citrate in patients with erectile dysfunction. Br J Clin Pharmacol. 2002;53(suppl 1):45s–52s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oteo I, Lukas JC, Leal N, et al. Tacrolimus pharmacokinetics in the early post‐liver transplantation period and clinical applicability via Bayesian prediction. Eur J Clin Pharmacol. 2013;69(1):65–74. [DOI] [PubMed] [Google Scholar]
- 29. Smolders EJ, de Kanter CT, van Hoek B, Arends JE, Drenth JP, Burger DM. Pharmacokinetics, efficacy, and safety of hepatitis C virus drugs in patients with liver and/or renal impairment. Drug Saf. 2016;39(7):589–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu L, Li H, Chan P, et al. Population pharmacokinetic analysis of asunaprevir in subjects with hepatitis C virus infection [published online ahead of print 2018]. Infect Dis Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information