

Abstract
Exposure to botanicals in dietary supplements is increasing across many geographies; with increased expectations from consumers, regulators, and industry stewards centered on quality and safety of these products. We present a tiered approach to assess the safety of botanicals, and an in silico decision tree to address toxicity data gaps. Tier 1 describes a Threshold of Toxicologic Concern (TTC) approach that can be used to assess the safety of conceptual levels of botanicals. Tier 2 is an approach to document a history of safe human use for botanical exposures higher than the TTC. An assessment of botanical‐drug interaction (BDI) may also be necessary at this stage. Tier 3 involves botanical chemical constituent identification and safety assessment and the in silico approach as needed. Our novel approaches to identify potential hazards and establish safe human use levels for botanicals is cost and time efficient and minimizes reliance on animal testing.
Exposure to natural ingredients of botanical nature, particularly through the use of dietary supplements and herbal medicines, continues to increase globally. In the United States for example, 2016 marked the 13th consecutive year of sales growth for herbal‐based supplements.1 In a 2017 survey conducted by the Council for Responsible Nutrition, growth of dietary supplement use over the last decade shows that 76% of Americans are taking dietary supplements, up from 64% in 2008.2 The Council for Responsible Nutrition survey also shows that 39% of the total dietary supplement use consists of herbals and botanicals, including green tea, cranberry, turmeric, garlic, ginseng, ginkgo biloba, milk thistle, and echinacea as the most popular. Furthermore, increases in supplement use is increasing across all age groups surveyed (18–55+ years of age), with the greatest increase in the 55 + age group (80% up from 76% in 2016). More recently, Agbabiaka et al.3 using a systematic review of the literature, found that herbal medicinal product use was common in older adults (≥65 years of age). Exposure to dietary supplements, including botanicals, is also increasing in children. According to the National Health and Nutrition Examination Survey data from 2007 to 2010, 1.7% of children used supplements containing botanicals, primarily to boost immunity and prevent colds.4 Most supplement use in children does not occur under the recommendation of a healthcare provider.5 Kantor et al.6 analyzed the National Health and Nutrition Examination Survey data from 1999–2012 and reported that only 23% of all supplement products were used at the recommendation of a health care provider.
The increased consumer exposure to botanical supplements has led to heightened scrutiny and compliance expectations by regulatory authorities, industry stewards, and consumers. Unfortunately, there is no global consensus on how to define dietary supplements, or regulatory expectations for quality, safety, and labeling across geographies. Even more challenging are the emotional and polarizing opinions on how to regulate this category; ranging from an approach that is similar to conventional drugs and foods, to a more tailored approach that relies on traditional or historical usages.7 As evidenced above, the growing popularity and lucrative nature of the category has led to an increase in industry participants marketing novel and innovative botanical products.
Although regulatory authorities and industry organizations are improving the safety expectations related to dietary supplements, there remain significant data gaps or conflicting data for critical toxicity end points. In many cases, safety data gaps may be attributed to an over‐reliance on limited historical information rather than empirical testing of these complex botanical mixtures. Botanicals may include the use of whole plants or a specific plant part (e.g., flower, stem, leaves, bark, root, and fruit), in a dietary supplement. Typically, plant material is subjected to some form of extraction by a solvent or solvent mixture (e.g., water and ethanol) or high‐pressure extraction (supercritical CO2) to create the botanical ingredients that may be used in products. These botanical ingredients are typically complex mixtures consisting of numerous individual phytochemical constituents and potential contaminants; as shown in Figure 1 with a representative chromatogram using high‐performance liquid chromatography photodiode array detection for a botanical ingredient. Thus, comparing published literature reports for botanicals is challenging due to highly variable material in the marketplace and poorly described and characterized test materials. Likewise, testing botanicals is equally challenging due to the lack of characterized material, natural variations in botanical composition, presence of contaminants, and a lack of knowledge on toxicologically active phytochemical constituent(s). Testing these complex mixtures in in vitro systems pose particularly unique challenges, such as determining appropriate testing concentrations, solubility, and inherent antibacterial or cytotoxicity properties of plant‐based constituents. Extrapolating findings from in vitro studies to typical human doses is also difficult without well characterized material and knowledge of dose form parameters (e.g., dissolution and disintegration) and bioavailability of phytochemical constituents.
Figure 1.
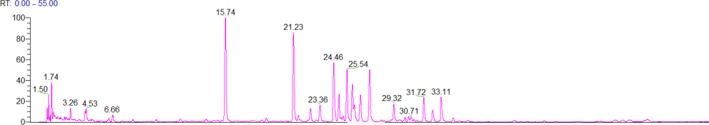
A representative chromatogram using high‐performance liquid chromatography photodiode array detection for a botanical extract.
Herein, we describe development of a tiered approach to support safety of botanical ingredients and a strategic way to fill certain toxicity end point gaps that allows for higher throughput, is more cost and time efficient, and may avoid the use of animal testing. The approach presented below provides guidance for the evaluation of safety of complex botanical mixtures for use in products marketed as herbal medicines, foods, or dietary supplements. Thus, it applies to botanical ingredients intended to deliver a product benefit to the consumer (e.g., health or function claim). In most geographies, a dietary supplement by definition is administered orally; however, our approach is applicable with other routes of administration as well as to consumer products (e.g., cosmetics).
Although identity quality of botanical raw materials is also a concern within the industry, and a significant effort is underway to make improvements in this arena, this topic will not be covered here. The approach outlined in this article assumes that the quality of the botanical ingredient(s) has been assured.
Tiered approach for assessing safety of botanical dietary supplements
Tier 1—Conceptual levels of botanicals
Establishing safe botanical exposure levels in the absence of carcinogenicity and genotoxicity data, which address the most sensitive end point, becomes a critical and often rate‐limiting step in botanical risk assessment. For chemical ingredients (including botanicals) that are used in products at low levels, a threshold of toxicological concern (TTC) approach can be used to rapidly assess safety when toxicological data are lacking.8 The TTC allows for a level of exposure for any chemical, even without chemical‐specific toxicity data, below which the assumption is that there would be no appreciable risk to human health.9 This approach uses conservative assumptions for systemic exposure (i.e., assumes 100% bioavailability). The TTC decision tree approach starts with the identification and evaluation of possible structural alerts for genotoxicity and high potency carcinogenicity.10 This step applies an exposure threshold of 0.15 μg/person/day.11, 12 Other authoritative bodies, including the European Food Safety Authority, have proposed the use of TTC to assess the safety of individual substances in botanical ingredients.13
Our laboratory has proposed to extend the TTC approach to botanicals, relying on this initial TTC exposure limit of 0.15 μg/day (0.0025 μg/kg bw/day) and adjusting it based on the concentration of phytochemical constituents of concern found in plants for genotoxicity and carcinogenicity end points.14 We evaluated over 50 genotoxic/DNA reactive and carcinogenic phytochemical constituents found in plants and compiled concentration data from several hundred plant species (over 2,300 observations). Phytochemical constituent concentration values ranged from 0.00015 to 136,000 ppm; with the vast majority of the concentrations residing in lower ppm levels, which were best fitted with a Weibull distribution model. The distribution of the data took into account single chemical occurrences; co‐occurrences remain to be done. The concentration probability at the 95th percentile for the concentration of phytochemical constituents of concern in plants can be used to adjust the TTC at the most conservative level for phytochemical constituents with genotoxic potential:
Thus, when classical toxicological data are lacking, we suggest using a health protective TTC exposure limit for the botanical mixtures and their simple extracts.14
Tier 2—Exposure > TTC with documented history of significant human use
When the botanical ingredient used in a dietary supplement exceeds the TTC, a logical starting place in the safety assessment is determining whether a documented significant human use (SHU) exists. An SHU may be considered as a surrogate measure for safety in the absence of relevant toxicological data. However, one must consider the context of SHU relative to traditional uses. A comparison of the two is presented in Table 1. As outlined in Table 1, an SHU considers relevant information about historical use, including route of administration (e.g., oral ingestion) and a comparison of intake levels and patterns (frequency of use). A variety of resources can help confirm food uses or components of food as well as food exposure data, including Research Institute for Fragrance Materials and Flavor Extract Manufacturers Association databases and Generally Recognized as Safe and Everything Added to Food in the United States for the US Food and Drug Administration approved food uses.15, 16, 17, 18 Additionally, a careful examination of the population that has traditionally used the botanical ingredient is also critical, including demographics, geographical location, method and purpose of use, and any associated side effects reported.19
Table 1.
Comparison between significant history of use and traditional use
| Significant history of use | Traditional use |
|---|---|
| A concept used to describe the qualified presumption of safety, where there is evidence for safety from compositional data and from experience as an ongoing part of the diet (and possibly from other relevant routes of exposure) for a number of generations in a large, genetically diverse population. | Is based upon knowledge and experience in a population/culture but may have limited scientific documentation. |
| Includes a scientific evaluation of the information, which should include conclusions about safe use. | Traditional use in this regard may provide information on acute toxicity but it is unlikely to provide information on chronic toxicity and those effects that are delayed and, thus, less likely to be detected, such as cancer, developmental toxicity (including teratogenicity) and reproductive toxicity. |
| A description of history of use covers the use in different defined geographic areas with information on intake levels, intake patterns, years of use, preparation, handling methods, and impact on human health as well as addressing any potential adverse effect issues. | Information from traditional use will be influenced by the general health of the particular population and the available health care and health monitoring facilities. |
A classic example of the importance of understanding traditional use of a botanical ingredient can be seen with Ephedra (ma‐huang). This Chinese shrub has been known in Traditional Chinese Medicine for at least 5,000 years.20 The historical use of Ephedra was as a tea, taken for the treatment of respiratory symptoms (e.g., cough and congestion), with minimal reported side effects. Beginning in the 1990s in the United States, Ephedra was frequently used in weight‐loss and energy‐enhancement products taken chronically. During this same time period, serious adverse effects associated with Ephedra‐containing dietary supplements, including heart attack, stroke, seizure, high blood pressure, and heart rhythm problems were increasingly reported. Due to the serious nature of the adverse events, including several deaths in otherwise healthy individuals, the US Food and Drug Administration banned the use of Ephedra in dietary supplements in 2004. This example highlights the potential change in side effect profile that can occur when a botanical ingredient is used in a different form, indication, pattern of use, and population than its traditional use. In other words, no SHU was established for the new usage paradigm for Ephedra.
There are a number of reputable sources and databases available to confirm documented use (of same species, plant part, method of preparation, and similar exposure levels) to support SHU (Table 2). When utilizing traditional medicine sources, it is important to confirm widespread availability of use, and not just via a learned intermediary and/or preparation of tailor‐made complex mixtures. To further confirm widespread availability, a review of pharmacovigilance data and adverse event databases should also be conducted to ensure adequate reporting systems are in place and that no serious health problems exist with the botanicals under investigation. It should be noted that similar adverse event data are typically unavailable for traditional uses because formal reporting systems are not used.
Table 2.
Sources to support significant history of use
| Source | Weblink or Reference |
|---|---|
| WHO Monographs on Selected Medicinal Plants14 | http://apps.who.int/medicinedocs/en/d/Js2200e/ |
| German Commission E Monographs15 | Translated from German and available online by the American Botanical Council, http://cms.herbalgram.org/commissione/intro/comm_e_int.html |
| European Medicines Agency Committee on Herbal Medicines16 | http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/herbal_search.jsp&mid=WC0b01ac058001fa1d |
| European Scientific Cooperative on Phytotherapy17 | http://escop.com/ |
| Natural Standard Monographs18 | https://naturalmedicines.therapeuticresearch.com/ J Med Libr Assoc 93, 4, (2005) |
Published literature, monographs, and assessment reports, examples of which are outlined in Table 2, may also provide data on safety end points, including nonclinical toxicity, clinical, and case report data to inform on an overall weight of evidence (WoE) conclusion for whether a safety concern exists regarding developmental and reproductive toxicity (DART), genotoxicity, and safety pharmacology parameters (cardiovascular, central nervous, and respiratory systems). These end points are particularly highlighted because, in the case of the latter, they are central to the function of primary organ systems or, in the case of genotoxicity/carcinogenicity and DART, these are end points that may be difficult to detect through human use alone. When applying a WoE approach, the relative robustness of published literature, including supporting references and scientific quality of reported studies, should be evaluated, as indicated in Table 3.
Table 3.
Considerations on relative robustness of available information on botanical of interest
| Data type | Key considerations | General strengths | General limitations |
|---|---|---|---|
| Epidemiological studies involving safety‐related endpoints |
|
|
|
| Clinical studies |
|
|
|
| Human use experience (outside the context of epidemiology studies and clinical trials) |
|
|
|
| Nonclinical animal studies |
|
|
|
| In vitro studies |
|
|
|
| Structure‐activity relationships | This capability is available for individual constituents of botanicals |
|
|
Epidemiology studies with safety‐related end points can be very useful when the botanical composition and exposure being studied are very similar to the proposed product use. Prospective epidemiology studies can be expensive and retrospective studies are subject to recall bias. Clinical studies typically have smaller populations compared to epidemiology studies but can be carefully controlled as to the subject population, product use, and duration and safety end points measured. The regimented use in clinical studies may not mimic real world use and there is limited ability to detect rare events. With a robust monitoring system in place, human use experience outside of epidemiology or clinical studies can provide useful data, although may times these reports do not come from medical professionals and causality is difficult to assess. Nonclinical animal studies have been the traditional approach to safety testing and can be used to address many toxicity end points over critical developmental/reproductive periods or up to lifetime exposure. However, no one animal model can completely mimic a human response and animal testing bans exist for ingredients used in some product types in certain countries.21 In vitro studies can be a fast, inexpensive way to obtain data on very specific end points but do not model the entire in vivo response. Structure‐activity relationships (SARs) can be a fast and inexpensive tool for the chemical constituents of botanicals to provide an estimate of toxicity.
One assumes (but should verify) objective reporting when studies are published in peer‐reviewed scientific journals; however, traditional uses may be reported in the form of anecdotal reports and, thus, potentially subjective reporting. An SHU should be based on fully vetted literature sources and/or well‐established databases. As the WoE approach to safety evaluation of botanicals includes a comprehensive review of a diverse compilation of information and data (as shown in Table 3 and discussed above), assistance from a number of experts may be required. Individuals with expertise in pharmacognosy and toxicology of botanicals, and healthcare practitioners with clinical experience in the use of herbal medicines, and natural products chemists can contribute to an overall WoE assessment.
Additionally, a consideration of interaction potential between the botanical ingredient and conventional medicines (botanical‐drug interaction (BDI)) should be considered as part of Tier 2. This assessment is also made with a consideration of SHU but with recognition that as an aging population there is the potential for concomitant use of a number of prescription medications.3, 22 The scientific literature is replete with nonclinical reports of botanical ingredients and/or phytochemical constituents as potent inhibitors of drug‐metabolizing enzymes.23, 24 However, without the use of robust analytical characterization and quantitation of phytochemical constituents, dose performance data, use of physiologically relevant in vitro metabolic systems, and follow‐up human clinical studies when necessary; extrapolating these preliminary reports to determine clinical relevance in humans is likely impossible. This lack of clear determination of risk hinders healthcare professionals and consumers from making informed decisions about the safety of taking dietary supplements with prescription medications. Various strategies for assessing BDI potential have been proposed, including more systematic approaches similar to studying drug‐drug interactions. For example, the integration of in vitro data with the pharmacokinetics of individual botanical constituents into physiologically based pharmacokinetic (PBPK) models can help prioritize and design follow‐up clinical studies.25, 26, 27, 28 Other researchers have suggested quite extensive in vitro workflows to study BDI that include an initial assessment of CYP450 interactions by individual botanical constituents. Botanical constituents are then tested individually or as an extract for absorption, efflux, and transporter interactions using a Caco‐2 permeability assay. Primary hepatocytes or cell lines, such as HepaRG cells, are proposed to study induction of CYP450 enzyme activity and mRNA expression.26
Important considerations that should be included in performing an overall BDI assessment are similar to those for other toxicity end points, including establishing an SHU, utilization of published literature data, and phytochemical constituent information based on careful analytical characterization (Table 4).27 However, there are some nuances associated with some of these considerations that are unique to assessing BDI. For example, in assessing SHU in the context of BDI, a general consideration of prescription medication use (polypharmacy) in the population for which the botanical supplement is targeted may be helpful. Understanding the profile of the consumer who might be attracted to a particular botanical supplement can provide perspective on potential underlying diseases/conditions, and associated comedications used by that consumer population. For example, one might consider that a botanical‐based dietary supplement aimed for inflammation and joint pain relief might be attractive as adjunctive therapy to individuals using nonsteroidal anti‐inflammatory drugs. If there are literature data indicating the potential for the botanical under consideration to inhibit the enzymes involved in the metabolism of many nonsteroidal anti‐inflammatory drugs, then perhaps follow‐up BDI studies should be considered to further delineate risk associated with their co‐administration.
Table 4.
Important considerations that warrant inclusion when assessing potential botanical‐drug interactions (BDIs)
History of safe use:
|
Literature data:
|
Incorporation of analytical characterization:
|
Dose performance:
|
©2015 American Botanical Council. Reprinted with permission.
A robust analytical characterization of phytochemical constituents in a given botanical can allow for further mining of the scientific literature for available absorption, distribution, metabolism, and excretion (ADME)‐related data, including BDI potential. In addition, identification of phytochemical constituents may allow for molecular modeling (docking studies) to determine potential binding to enzyme active sites and in silico prediction of enzyme‐inhibitor interactions.29 Last, dose performance data that include dissolution of the dose formulation and solubility of the botanical extract/phytochemicals in gastric and intestinal fluids is also critical for predicting absorption of key constituents.
A number of well‐established in vitro models exist for studying drug‐drug interactions; some of which may be useful for re‐application to studying BDI.30, 31 Findings from screening‐level studies in simplistic metabolic systems (e.g., liver microsomes and membrane vesicles) can be followed up in more physiologically relevant whole cell models, such as primary hepatocytes. Whole cell systems, particularly sandwich‐cultured hepatocytes, have an advantage in that they integrate multiple processes (transport, metabolism, and associated cellular regulatory pathways), provide more in vivo‐relevant intracellular concentrations, and allow for studying inhibition and induction simultaneously.32 Recently, sandwich‐cultured human hepatocytes have shown considerable promise in predicting clinically relevant BDI.33 Other in vitro models (e.g., gut‐liver co‐cultures) need to be investigated in order to improve predictions of BDI occurring at the level of the gut. Data from an overall assessment of BDI potential, including in vitro assays, PBPK modeling, and/or human clinical studies can be used to inform on product label, formulation‐adjusted or dose‐adjusted, and postmarket surveillance strategies (Figure 2).
Figure 2.
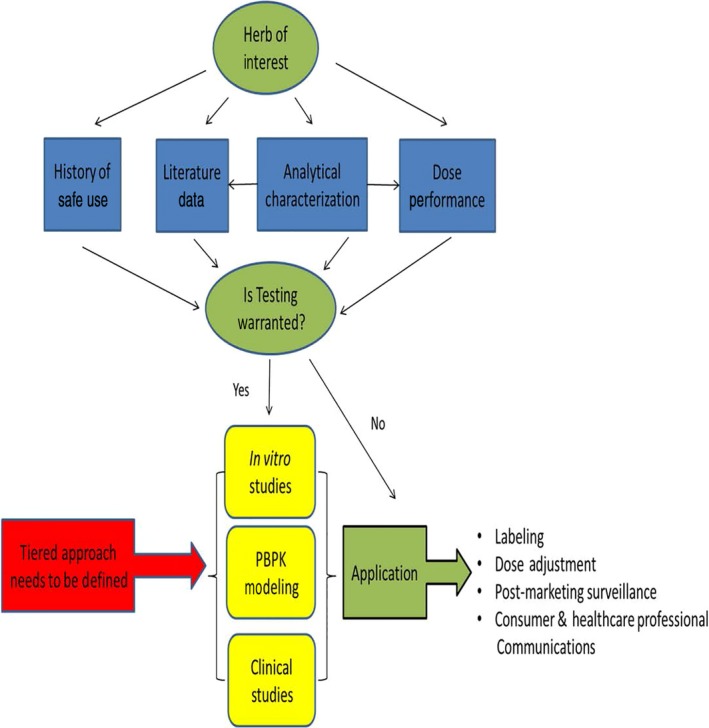
Key components of a framework for assessing botanical‐drug interactions; ©2015 American Botanical Council. Reprinted with permission.
Tier 3—Decision tree approach to address botanical hazard assessment data gaps
When safety data are insufficient (specific gaps identified) from the Tier 2 assessment, an additional level of assessment may be necessary to confirm the safe use of a botanical ingredient (Tier 3). A novel decision tree approach has been developed that utilizes both SHU information and phytochemical constituent‐based safety evaluations to resolve toxicity end points of concern (Figure 3).34 This approach requires state‐of‐the‐art analytical techniques to identify and quantify botanical constituents. Individual phytochemical constituents are then assessed according to both food intake levels and established in silico toxicology assessment tools to identify hazards. Combining this analysis with the appropriate dosing and phytochemical constituent co‐exposure considerations can be used to establish a risk characterization for the various constituents of the botanical preparation.
Figure 3.
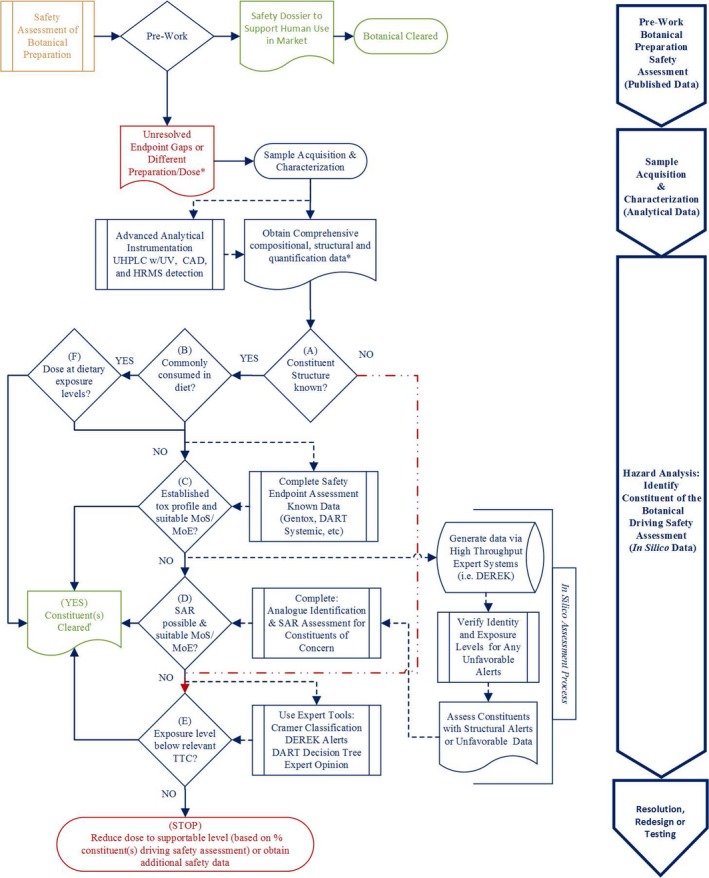
Decision tree for botanical constituent(s). CAD, Charged Aerosol Detector; HRMS, High Resolution Mass Spectrometry; MoE, Margin of Exposure; MoS, Margin of Safety; SAR, structure‐activity relationship; TTC, threshold of toxicological concern; UHPLC, ultra‐high‐performance liquid chromatography. Reprinted from Food and Chemical Toxicology, Vol 107, Little, J., Marsman, D., Baker, T. & Mahony, C., In silico approach to safety of botanical dietary supplement ingredients utilizing constituent‐level characterization. 418–429, (C) 2017, with permission from Elsevier.
Once a botanical ingredient of interest is fully characterized for individual phytochemical constituents and an estimate of human exposure identified, one can determine if the constituents are known structures (e.g., commonly consumed in the diet), and whether the proposed dose associated with the botanical supplement is comparable to dietary exposure levels. For many botanical constituents, this approach alone may be sufficient to support their safe use.
The decision tree approach can also be used to bridge safety between botanicals that are prepared by different methods. It has been well‐established that different methods of botanical preparation and extraction (e.g., water, solvent, and CO2) have potential to influence the phytochemical constituent profile. Situations can exist in which SHU, toxicology data, and clinical reports may exist for some preparations, whereas alternate methods of preparation of the same botanical may lack similar safety data. By making a comparison of both the similarities and unique chemical differences among two or more preparations, any qualitative or quantitative differences in the phytochemical constituents can then be addressed individually by using existing data or applying established in silico toxicology tools (e.g., TTC).
Last, this approach can be used to address toxicity end points for the identified phytochemical constituents, or as an early screen for toxicity alerts; particularly for less commonly known botanicals. As previously mentioned under Tier 2, toxicity data must address a number of different end points. When insufficient data are available for one or more of these end points, the typical approach is to conduct traditional in vitro and in vivo animal toxicity studies to generate safety data on the botanical preparation. When botanical safety has been sufficiently characterized at the level of its phytochemical constituents, these toxicity studies become more meaningful. Using the in silico approach, a safety assessor can assess hazards and establish exposure levels of low safety concern for each phytochemical constituent by conducting a risk assessment using data on a constituent itself, or by using well‐established toxicology assessment tools, such as TTC or SAR, for those constituents with insufficient toxicity data.35, 36 An SAR approach utilizes available toxicity data on structurally similar analogs of the phytochemical constituent in question. Additionally, phytochemical constituent structures with potential data gaps can be processed through rule‐based software programs, such as DEREK, to evaluate them for possible structural alerts for various toxicity end points or ontologies where available (e.g., DART).37 Alerts should be considered cautionary evidence and are not themselves definitive.
Special populations—pregnant/breastfeeding women and children
It is generally recognized that medicines and supplements should not be taken during pregnancy/breastfeeding unless the benefit to the mother outweighs any possible risk to the fetus or nursing infant. One of the major problems in drawing conclusions on the risk:benefit associated with an ingredient used during such periods is a lack of safety information (i.e., limited information during pregnancy or breastfeeding). Similarly, such data are often lacking in children. Furthermore, there is a subsection of products that are specifically designed to support pregnancy/nursing health and well‐being, as well as child growth and development.38 To ensure minimal risk to these special populations, ingredients used in products intended for these populations should be supported by appropriate toxicity data and/or a rationale for low toxicity risk based on SHU during pregnancy/period of nursing or infant/child development. Alternatively, if safety (or preclinical and/or clinical data) data are indicated for these populations, the ingredient may be used at sufficiently low levels in the formula to assure adequate margins of safety. Otherwise, in the absence of data to conclude safe use at a defined dose for the consumer, the mother or unborn/nursing infant or developing child, the ingredient should not be targeted toward pregnant/nursing women unless data gaps are satisfied through testing.
Summary, Challenges, and Future Directions
Continued interest in botanical ingredients, particularly as botanical dietary supplements, coupled with challenges of testing these complex mixtures of phytochemical constituents, and cost, time, and animal usage constraints, presents some unique challenges for this category of ingredients. For example, critical data end points that are often lacking may include raw material characterization, DART, genotoxicity/carcinogenicity testing, and/or ADME considerations (including botanical‐drug and botanical‐botanical interactions). Across the dietary supplement industry, there are various approaches for safety assessment of botanicals intended for use in food/dietary supplements, from heavy reliance on capturing adverse events via post‐marketing surveillance, to tiered approaches building an assessment based on existing data and new data generated specifically to meet safety assessment needs as presented here.
We present a tiered‐approach and proposed in silico decision tree methodology for botanical safety evaluation that provides a useful and pragmatic approach based on dietary intake levels and sufficient safe margins of exposure from existing safety information. We have also borrowed from traditional chemical‐based safety assessment methodologies to include TTC and SAR approaches for establishing safe exposure thresholds. Although more work remains to be done, our botanical safety assessment approach begins to evaluate the critical data end points mentioned previously using more recent and advanced toxicology methodologies that are intended to eliminate or reduce the need for animal testing.
We believe that emphasis should be put against in vitro testing approaches to address critical end points where safety data are lacking. For example, high throughput approaches are assisting in characterizing the toxicological mode of action of botanical mixtures by way of gene expression studies, which allows for identification of functional analogues through the Connectivity Mapping Approach,39 and receptor binding and enzyme activity data, which allows for identification of molecular targets.40 These data streams (and extensions thereof) seem promising for follow‐up on safety questions identified by the in silico decision tree methodology, either by informing on a lack of interaction with DART relevant targets or by enabling potency comparisons of in vitro targets, which can be linked to DART data for functional analogue(s) and used to judge whether human exposures would likely require follow‐up (unpublished data Vandermolen, K., Naciff, J., Daston, G., & Mahony, C.). In the area of assessing BDI, utility of more physiologically relevant in vitro models, including human hepatocytes that are fully functional for uptake and efflux transporters, metabolism, and requisite regulatory pathways are now being used.33 These advanced models, coupled with PBPK modeling techniques may be more useful for predicting clinical relevance of BDI, and/or aid in the design of follow‐on clinical studies.
Central to our approach is the use of an advanced analytical method, using multiple simultaneous detectors, to adequately characterize botanical constituents sufficiently to enable the application of these modeling tools to botanical ingredients. In fact, the need to further develop and validate analytical methods to enable complete chemical characterization of complex botanical mixtures has been identified as a critical need by other experts in the field of botanical safety assessment.41
Future focus should include the investigation of relative source contributions or exposure source allocation factors in the assessment, as well as a dedicated effort toward building a database of chemical constituents of food substance mixtures (important for both botanical dietary supplements and botanical‐containing consumer products). An emerging need is guidance on how to generate and interpret ADME‐related data on complex botanical mixtures in order to extrapolate from in vitro assays to in vivo animal toxicity studies and ultimately to human exposures. The question of which marker of the botanical constituent(s) to extrapolate with across these studies is challenging. A “Best Practices” for characterizing ADME of botanicals should address which and how many constituents should be followed, and the best method(s) of analysis to use (unpublished data Ryan, K. et al.). Addressing these additional challenges will further support positive assurances of safety in the use of botanical preparations across various consumer product categories; and may ultimately lead to more innovative botanical‐based products, including dietary supplements.
Funding
Any work referenced to the authors of this article was funded privately by The Procter & Gamble Company.
Conflict of Interest
Authors work for a company that manufactures and distributes dietary supplements.
Acknowledgments
Studies and analyses that have contributed to the strategies discussed in this article have been funded by The Procter & Gamble Company or its wholly owned subsidiary, New Chapter, Inc.
References
- 1. Smith, T. , Kawa, K. , Eckl, V. , Morton, C. & Stredney, R. Herbal supplement sales in US increase 7.7% in 2016. Herbalgram 115, 56–65 (2017). [Google Scholar]
- 2. Council for Responsible Nutrition . CRN 2017 Annual Survey on Dietary Supplements (Council for Responsible Nutrition, Washington, DC). < http://www.crnusa.org/survey >.
- 3. Agbabiaka, T. , Wider, B. , Watson, L. & Goodman, C. Concurrent use of prescription drugs and herbal medicinal products in older adults: a systematic review. Drug Aging 34, 891–905 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fryar, C. , Gu, Q. & Ogden, C. Anthropometric reference data for children and adults: United States, 2007–2010. Vital. Health Stat. 11, 1–48 (2012). [PubMed] [Google Scholar]
- 5. Bailey, R. , Gahche, J. , Thomas, P. & Dwyer, J. Why US children use dietary supplements. Pediatr. Res. 74, 737–741 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kantor, E.D. , Rehm, C.D. , Du, M.M. , White, E. & Giovannucci, E.L. Trends in dietary supplement use among US adults from 1999–2012. JAMA 316, 1464–1474 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dwyer, J. , Coates, P. & Smith, M. Dietary supplements: regulatory challenges and research resources. Nutrients 10, 1–24 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Blackburn, K. , Stickney, J. , Carlson‐Lynch, H. , McGinnis, P. , Chappell, L. & Felter, S. Application of the threshold of toxicological concern approach to ingredients in personal and household care products. Regul. Toxicol. Pharmacol. 43, 249–259 (2005). [DOI] [PubMed] [Google Scholar]
- 9. Kroes, R. , Kleiner, J. & Renwich, A. Structure‐based 2005. The threshold of toxicological concern concept in risk assessment. Toxicol. Sci. 86, 226–230 (2005). [DOI] [PubMed] [Google Scholar]
- 10. Munro, I.C. Safety assessment procedures for indirect food‐additives—an overview. Regul. Toxicol. Pharmacol. 12, 2–12 (1990). [DOI] [PubMed] [Google Scholar]
- 11. Munro, I. , Ford, R. , Kennepohl, E. & Sprenger, J. Correlation of structural class with no‐observed‐effect levels: a proposal for establishing a threshold of concern. Food Chem. Toxicol. 34, 829–867 (1996). [DOI] [PubMed] [Google Scholar]
- 12. Kroes, R. et al Structure‐based thresholds of toxicological concern (TTC): guidance for application to substances present at low levels in the diet. Food Chem. Toxicol. 42, 65–83 (2004). [DOI] [PubMed] [Google Scholar]
- 13. European Food Safety Authority (EFSA) . Guidance on safety assessment of botanicals and botanical preparations intended for use as ingredients in food supplements. EFSA J. 7, 1249 (2009). [Google Scholar]
- 14. McMillan, D. , Kosemund, K. , Mahony, C. , Huber, M. & Bowtell, P. Threshold of toxicological concern (TTC) for botanicals‐data analysis to substantiate and extend the TTC approach to botanicals. Toxicol. Suppl. Toxicol. Sci. 150, PS1831 (2017). [DOI] [PubMed] [Google Scholar]
- 15. Research Institute for Fragrance Material (RIFM) . The RIFM Database 2018. < http://www.rifm.org/rifm-science-database.php#.WvCEmvnwbDc >.
- 16. FEMA (2018). < https://www.femaflavor.org/ >.
- 17. U.S. Food and Drug Administration . GRAS Notices. < https://www.accessdata.fda.gov/scripts/fdcc/?set=GRASNotices >.
- 18. U.S. Food and Drug Administration . Everything Added to Food in the United States (EAFUS). < https://www.accessdata.fda.gov/scripts/fcn/fcnNavigation.cfm?rpt=eafusListing >.
- 19. Institute of Medicine and National Research Council . Dietary Supplements: A Framework for Evaluating Safety (The National Academies Press, Washington, DC, 2005). [PubMed] [Google Scholar]
- 20. Lee, M. The history of Ephedra (ma‐huang). J. R. Coll. Physicians Edinb. 41, 78–84 (2011). [DOI] [PubMed] [Google Scholar]
- 21. European Parliament . Directive 2003/15/EC of the European Parliament and of the Council of 27 February 2003 amending Council Directive 76/768/EEC on the approximation of the laws of the Member States relating to cosmetic products (Text with EEA relevance). In: Council of the European Union, editor (2003).
- 22. Farina, E.K. , Austin, K.G. & Lieberman, H.R. Concomitant dietary supplement and prescription medication use is prevalent among US adults with doctor‐informed medical conditions. J. Acad. Nutr. Diet. 114, 1784 (2014). [DOI] [PubMed] [Google Scholar]
- 23. Frank, A. & Unger, M. Analysis of frankincense from various Boswellia species with inhibitory activity on human drug metabolising cytochrome P450 enzymes using liquid chromatography mass spectrometry after automated on‐line extraction. J. Chromatogr. A 1112, 255–262 (2006). [DOI] [PubMed] [Google Scholar]
- 24. Iwata . Identification and characterization mass spectrometry after automated on‐line extraction.
- 25. Brantley, S. , Argikar, A. , Lin, Y. , Nagar, S. & Paine, M. Herb‐drug interactions: challenges and opportunities for improved predictions. Drug Metab. Dispos. 42, 301–317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sprouse, A. & van Breemen, R. Pharmacokinetic interactions between drugs and botanical dietary supplements. Drug Metab. Dispos. 44, 162–171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roe, A. Assessing potential herb‐drug interactions in the use of herbal dietary supplements: need for a common framework approach. Herbalgram 12, 40–43 (2015). [Google Scholar]
- 28. Roe, A. , Paine, M. , Gurley, B. , Brouwer, K. , Jordan, S. & Griffiths, J. Assessing natural product‐drug interactions: an end‐to‐end safety framework. Regul. Toxicol. Pharmacol. 76, 1–6 (2016). [DOI] [PubMed] [Google Scholar]
- 29. Barr, J. , Jones, J. , Oberlies, N. & Paine, M.F. Inhibition of human aldehyde oxidase activity by diet‐derived constituents: structural influence, enzyme‐ligand interactions, and clinical relevance. Drug Metab. Dispos. 43, 34–41 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. European Medicines Agency . Herbal medicines for human use. < http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/landing/herbal_search.jsp&mid=WC0b01ac058001fa1d >. Accessed 28 March 2018.
- 31. U.S. Food and Drug Administration . In vitro metabolism‐ and transporter‐mediated drug‐drug interaction studies: Guidance for industry. < https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM581965.pdf > (2017).
- 32. Korzekwa, K. , Nagar, S. , Tucker, J. , Weiskitcher, E. , Bhoopathy, S. & Hidalgo, I. Models to predict unbound intracellular drug concentrations in the presence of transporters. Drug Metab. Dispos. 40, 865–876 (2012). [DOI] [PubMed] [Google Scholar]
- 33. Jackson, J. et al Prediction of clinically relevant herb‐drug clearance interactions using sandwich‐cultured human hepatocytes: Schisandra spp. case study. Drug Metab. Dispos. 45, 1019–1026 (2017). [DOI] [PubMed] [Google Scholar]
- 34. Little, J. , Marsman, D. , Baker, T. & Mahony, C. In silico approach to safety of botanical dietary supplement ingredients utilizing constituent‐level characterization. Food Chem. Toxicol. 107, 418–429 (2017). [DOI] [PubMed] [Google Scholar]
- 35. Wu, S. , Blackburn, K. , Amburgey, J. , Jaworska, J. & Federle, T. A framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for SAR‐based toxicology assessments. Regul. Toxicol. Pharmacol. 56, 67–81 (2010). [DOI] [PubMed] [Google Scholar]
- 36. Blackburn, K. , Bjerke, D. , Daston, G. , Felter, S. , Mahony, C. & Naciff, J. Case studies to test: a framework for using structural, reactivity, metabolic and physicochemical similarity to evaluate the suitability of analogs for SAR‐based toxicological assessments. Regul. Toxicol. Pharmacol. 60, 120–135 (2011). [DOI] [PubMed] [Google Scholar]
- 37. Wu, S.D. et al Framework for identifying chemicals with structural features associated with the potential to act as developmental or reproductive toxicants. Chem. Res. Toxicol. 26, 1840–1861 (2013). [DOI] [PubMed] [Google Scholar]
- 38. Picciano, M. & Mcgurie, M. Use of dietary supplements by pregnant and lactating women in North America. Am. J. Clin. Nutr. 89, 663S–667S (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. De Abrew, K. et al Grouping 34 chemicals based on mode of action using connectivity mapping. Toxicol. Sci. 151, 447–461 (2016). [DOI] [PubMed] [Google Scholar]
- 40. Bowes, J. et al Reducing safety‐related drug attrition: the use of in vitro pharmacological profiling. Nat. Rev. Drug Discov. 11, 909–922 (2012). [DOI] [PubMed] [Google Scholar]
- 41. Rietjens, I. , Slob, W. , Galli, C. & Silano, V. Risk assessment of botanicals and botanical preparations intended for use in food and food supplements: emerging issues. Toxicol. Lett. 180, 131–136 (2008). [DOI] [PubMed] [Google Scholar]