

Abstract
Giant cell arteritis (GCA) is an autoimmune vasculitis affecting large and medium‐sized arteries. Ample evidence indicates that GCA is a heterogeneous disease in terms of symptoms, immune pathology, and response to treatment. In the current review, we discuss the evidence for disease subsets in GCA. We describe clinical and immunologic characteristics that may impact the risk of cranial ischemic symptoms, relapse rates, and long‐term glucocorticoid requirements in patients with GCA. In addition, we discuss both proven and putative immunologic targets for therapy in patients with GCA who have an unfavorable prognosis. Finally, we provide recommendations for further research on disease subsets in GCA.
Introduction
Giant cell arteritis (GCA) is the most prevalent form of autoimmune vasculitis in elderly individuals. GCA is characterized by inflammation of medium‐sized cranial arteries and large systemic arteries. Cranial ischemic symptoms are well‐known complications of GCA and may include sight loss and stroke. In addition, many patients experience symptoms of systemic inflammation, such as fatigue, low‐grade fever, and weight loss. Inflammation marker levels are typically elevated in the blood of patients with GCA 1. Glucocorticoids are the cornerstone of treatment. Early initiation of high‐dose glucocorticoids led to a substantial decrease in visual symptoms among GCA patients over the last decades 2. However, side effects are frequently encountered with long‐term glucocorticoid treatment in elderly patients with GCA 3. Recently, novel targeted treatments have emerged as potent alternatives for maintenance of glucocorticoid‐free disease remission in patients with GCA 4, 5, 6.
Accumulating evidence indicates that GCA is a heterogeneous disease. The extent of the local and systemic inflammatory response may differ among GCA patients 1. Moreover, distinct immune cells and cytokines may predominate at the site of vascular inflammation in individual patients 7. Various clinical and immunologic factors have been linked to the risk of cranial ischemic symptoms, relapse rates, and overall glucocorticoid requirements in patients with GCA. Immunologic heterogeneity in GCA is further suggested by outcomes of recent trials with anti–interleukin‐6 receptor (anti–IL‐6R) and CTLA‐4Ig therapy, because these targeted treatments are not effective in all GCA patients 4, 5, 6. Taken together, these findings indicate that there may be distinct categories of GCA patients. Recognition of distinct GCA subsets is important, because it may eventually help to implement precision medicine for GCA.
In this review, we provide an overview of current evidence for disease subsets in GCA. We describe the prognostic relevance of clinical disease characteristics in patients with GCA, i.e., the systemic inflammatory response, coexistent polymyalgia rheumatica (PMR), and involvement of large systemic arteries in the disease. In addition, we discuss current insights into the immune pathology of GCA and highlight immune cells and cytokines that are associated with clinical outcomes in GCA. Finally, we evaluate open questions and research priorities that need to be solved before precision medicine for GCA patients can become a reality.
Evidence for distinct GCA subsets based on clinical features
Systemic inflammation. Systemic inflammation is present in the vast majority of patients with GCA 8. Symptoms resulting from systemic inflammation may include general malaise, weight loss, night sweats, and low‐grade fever. Laboratory findings suggestive of systemic inflammation include elevation of the erythrocyte sedimentation rate (ESR), C‐reactive protein (CRP) level, and thrombocyte count. In addition, anemia due to chronic inflammation is frequently observed in patients with GCA.
With the exception of 3 studies 9, 10, 11, a vast number of studies have shown that GCA patients with a strong systemic inflammatory response have a lower risk of cranial ischemic symptoms compared with patients with a weak systemic inflammatory response (Table 1) 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25. For instance, pretreatment ESRs and CRP levels are inversely correlated with the risk of visual symptoms in GCA (14, 17, 18, 19, 21, 22). The presence of fever is also associated with a lower risk of cranial ischemia in GCA patients 16, 23.
Table 1.
Characteristics predicting cranial ischemia or long‐term prognosis in GCA patients*
| Characteristic, study | Study design | Cranial ischemia risk | Relapse rate | Glucocorticoid requirement |
|---|---|---|---|---|
| Strong systemic inflammatory response | ||||
| Cid et al, 1998 12 | Retro | Decreased | NA | NA |
| Gonzalez‐Gay et al, 1998 13 | Retro | Decreased | NA | NA |
| Liozon et al, 2001 14 | Prosp | Decreased | NA | NA |
| Hernandez‐Rodriguez et al, 2002 15 | Retro | Decreased | Increased | Increased |
| Gonzalez‐Gay et al, 2004 16 | Retro | Decreased | NA | NA |
| Salvarani et al, 2005 17 | Retro | Decreased | NA | NA |
| Gonzalez‐Gay et al, 2005 18 | Retro | Decreased | NA | NA |
| Lopez‐Diaz et al, 2008 19 | Retro | Decreased | NA | NA |
| Nesher et al, 2008 9 | Retro | No effect | Increased | Increased |
| Chatelain et al, 2009 10 | Prosp | No effect | NA | NA |
| Gonzalez‐Gay et al, 2009 20 | Retro | Decreased | NA | NA |
| Salvarani et al, 2009 21 | Retro | Decreased | NA | NA |
| Martinez‐Lado et al, 2011 29 | Retro | NA | Increased | NA |
| Muratore et al, 2016 22 | Retro | Decreased | NA | NA |
| Liozon et al, 2016 23 | Prosp | Decreased | NA | NA |
| Grossman et al, 2017 24 | Retro | Decreased | NA | NA |
| Restuccia et al, 2017 30 | Retro | NA | NA | Increased |
| De Boysson et al, 2017 25 | Retro | Decreased | NA | NA |
| Yates et al, 2017 11 | Prosp | No effect | NA | NA |
| Presence of vasculitis of large systemic arteries | ||||
| Schmidt et al, 2008 37 | Retro | NA | NA | No effect |
| Schmidt et al, 2009 32 | Retro | Decreased | NA | NA |
| Prieto‐Gonzalez et al, 2012 31 | Prosp | Decreased | NA | NA |
| Espitia et al, 2012 36 | Retro | Increased | Increased | Increased |
| Czihal et al, 2012 33 | Retro | Decreased | NA | NA |
| Muratore et al, 2015 34 | Retro | Decreased | Increased | Increased |
| Czihal et al, 2015 38 | Retro | NA | No effect | NA |
| De Boysson et al, 2017 35 | Retro | Decreased | No effect | No effect |
| Presence of polymyalgia rheumatica | ||||
| Myklebust et al, 2001 44 | Prosp | NA | NA | Increased |
| Liozon et al, 2001 14 | Prosp | Decreased | NA | NA |
| Gonzalez‐Gay et al, 2005 18 | Retro | No effect | NA | NA |
| Gonzalez‐Gay et al, 2009 20 | Retro | No effect | NA | NA |
| Liozon et al, 2016 23 | Prosp | Decreased | NA | NA |
| Restuccia et al, 2017 30 | Retro | NA | NA | Increased |
| De Boysson et al, 2017 25 | Retro | No effect | NA | NA |
Studies published between January 1, 1997 and January 1, 2018 were included. A strong systemic inflammatory response is defined as an elevated erythrocyte sedimentation rate, elevated C‐reactive protein level, decreased hemoglobin level, and/or fever. GCA = giant cell arteritis; Prosp = prospective study; Retro = retrospective study; NA = not assessed.
A possible explanation is that patients with cranial ischemic symptoms present earlier in the disease course and therefore have not yet developed a strong systemic inflammatory response 12, 18, 23. Although 1 study documented that ESR levels are higher in GCA patients with a longer symptom duration 19, 3 studies showed no clear relationship between the magnitude of the systemic inflammatory response and symptom duration 12, 15, 26. As an alternative explanation, other investigators have reported that a strong systemic inflammatory response may prevent ischemic events by directly promoting neoangiogenesis in ischemic tissue 27, 28.
With respect to long‐term disease outcomes, 4 studies suggested that a strong systemic inflammatory response might be associated with higher relapse rates and glucocorticoid requirements in patients with GCA 9, 15, 29, 30.
Thus, current evidence indicates that the magnitude of the systemic inflammatory response may identify GCA patients with distinct disease outcomes. However, clearly defined criteria for “strong” and “weak” systemic inflammation are lacking. It has been proposed that “strong” systemic inflammation might be defined as the presence of ≥3 of the following 4 parameters: fever, weight loss, an ESR of >85 mm/hour, and a hemoglobin level of <11 gm/dl or <6.8 mmoles/liter 9, 12, 15. Clearly, this definition and its application in stratifying GCA patients need to be validated in protocolized study cohorts with long‐term follow‐up.
Vasculitis of large systemic arteries. Imaging studies have shown that inflammation of large systemic arteries (e.g., aorta, subclavian arteries, and axillary arteries) is frequently present in patients with GCA 31. In fact, some GCA patients present with vasculitis in these large systemic arteries in the absence of cranial vasculitis. Few studies have investigated the impact of large systemic artery involvement on disease outcomes in GCA. One prospective study and 4 retrospective studies demonstrated a lower risk of cranial ischemic symptoms in GCA patients with large systemic artery involvement, even in patients with positive temporal artery biopsy findings (Table 1) 31, 32, 33, 34, 35. In contrast, 1 retrospective study showed a higher risk of cranial ischemia in GCA patients with large systemic artery involvement 30. In the prospective study, large systemic artery involvement had no effect on laboratory markers of systemic inflammation 31. Two retrospective studies suggested that large systemic artery involvement is associated with higher relapse rates and increased requirements for glucocorticoids in GCA 34, 36, although this association was not observed in 3 other retrospective studies 35, 37, 38. It is important to note that in some of the retrospective studies, inflammation of large systemic arteries was not routinely investigated in all of the patients with GCA. Therefore, some patients might have been misclassified as not having large systemic artery involvement.
A general limitation in this context is the lack of a gold standard test for inflammation of large systemic arteries. Different imaging modalities have been used, including ultrasonography, 18F‐fluorodeoxyglucose positron emission tomography/computed tomography (18F‐FDG PET/CT), CT angiography, and magnetic resonance angiography 31, 32, 34, 35. Aortic inflammation may eventually lead to the development of aortic aneurysms and dissection in patients with GCA 39, 40. Little is known about the factors predictive of these late complications in GCA, although a recent study indicated that subclavian artery dilatation at diagnosis is associated with a higher risk of aortic aneurysms in GCA patients 41.
Limited data suggest that the presence of large systemic artery inflammation is associated with a lower risk of cranial ischemia, although it is unclear whether the long‐term outcomes differ between GCA patients with and those without large systemic artery involvement. Protocolized cohort studies are required in which all GCA patients are systematically assessed for both cranial and large systemic vasculitis. Importantly, recommendations for the use of imaging in GCA have recently been published 42. Further insight into the development and management of aortic aneurysms is also needed.
Polymyalgia rheumatica (PMR). Patients with GCA may present with clinical signs and symptoms consistent with PMR, a rheumatic syndrome characterized by symmetric pain and stiffness in both the shoulders and hips 1. 18F‐FDG PET/CT scans in these GCA patients often show combined vessel and (peri)articular inflammation 43. Few studies have investigated the impact of PMR on disease outcomes in patients with GCA. Two studies suggested that coexistent PMR identifies a subset of GCA patients with a low risk of cranial ischemic symptoms 14, 23. However, 3 other studies demonstrated no such effect 18, 20, 25. In addition, 2 retrospective studies showed higher glucocorticoid requirements in GCA patients with concomittant PMR when compared with GCA patients without PMR 30, 44. Clearly, current evidence is too contradictory to allow conclusions to be drawn regarding the impact of PMR on cranial ischemic symptoms, whereas limited data suggest that the presence of PMR is associated with a poor long‐term outcome.
Evidence for distinct GCA subsets based on immunologic features
In recent years, insight into the immune pathology of GCA has increased considerably. Different histologic patterns have been observed in the temporal arteries of GCA patients. Moreover, complex networks of immune cells and cytokines have been identified in the temporal arteries and blood of GCA patients. The predominance of particular immune cells and cytokines has been linked to disease outcomes in patients with GCA. Here, we discuss current insights into the immune pathology of GCA. Furthermore, we highlight the immune cells and cytokines that have been linked to poor disease outcomes and that may represent current or possibly future targets for treatment in GCA (Figure 1).
Figure 1.
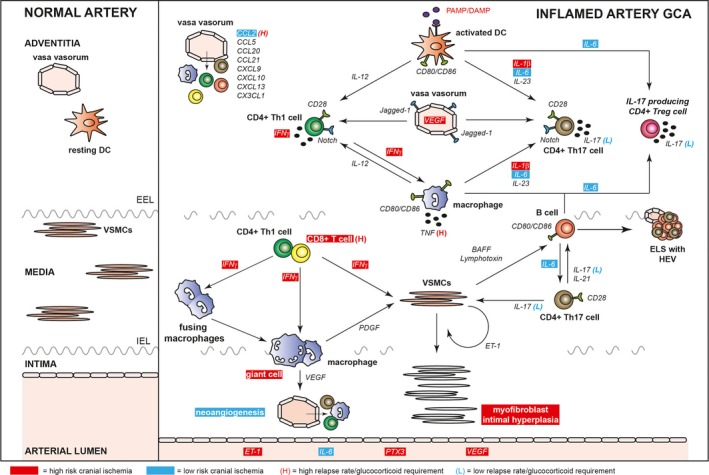
Overview of immune pathology of giant cell arteritis (GCA). Immune cells and cytokines involved in the arterial inflammatory process of GCA and the relationship of these cells and cytokines with disease outcomes in GCA are shown. PAMP = pathogen‐associated molecular pattern; DAMP = damage‐associated molecular pattern; DC = dendritic cell; IL‐12 = interleukin‐12; VEGF = vascular endothelial growth factor; IFNγ = interferon‐γ; EEL = external elastic lamina; VSMCs = vascular smooth muscle cells; TNF = tumor necrosis factor; ELS = ectopic lymphoid structure; HEV = high endothelial venule; IEL = internal elastic lamina; PDGF = platelet‐derived growth factor; ET‐1 = endothelin 1; PTX3 = pentraxin 3.
Histologic patterns in temporal artery biops y specimens. Temporal artery biopsy specimens are frequently obtained during the diagnostic workup of patients with GCA. Distinct histologic patterns of vascular inflammation have been observed in these temporal arteries. Typically, transmural inflammation in all 3 layers of the arterial wall, i.e., the adventitia, media, and intima, is observed 45, 46. In recent years, 3 alternative histologic patterns have been observed in temporal artery biopsy specimens: small vessel vasculitis (SVV) limited to periadventitial vessels surrounding a normal temporal artery, vasa vasorum vasculitis (VVV), and inflammation limited to the adventitia. Three studies showed no differences in cranial ischemic symptoms between GCA patients with inflammation limited to the adventitia versus those with transmural inflammation 45, 46, 47. In addition, various reports indicate that GCA patients with SVV or VVV show similar rates of visual symptoms when compared with GCA patients with transmural inflammation 45, 48, 49, 50. However, SVV, VVV, and inflammation limited to the adventitia have also been observed in the absence of a convincing diagnosis of GCA, i.e., in patients with PMR, cancer, or infections 47, 50, 51, 52, 53. Because multiple histologic patterns may coexist within a single temporal artery biopsy specimen from a GCA patient 46, 54, it might be possible that SVV, VVV, and inflammation limited to the adventitia are early and nonspecific histologic patterns that may progress to full‐blown transmural inflammation in GCA patients. Although it has been suggested that the predominance of these early histologic patterns in a temporal artery biopsy specimen identifies GCA patients with low glucocorticoid requirements 49, 55, the clinical relevance of these histologic patterns remains controversial.
Dendritic cells (DCs) and T lymphocytes in the inflamed artery. Vascular DCs reside in the proximity of the vasa vasorum in large and medium‐sized arteries 7. Activation of these cells by pathogen‐associated molecular patterns or damage‐associated molecular patterns, including ligands for Toll‐like receptors, may trigger the initial inflammatory response, resulting in chemokine‐mediated migration of immune cells via the vasa vasorum 7. Although it has been suggested that varicella zoster virus might be directly involved in the development of GCA 56, a growing number of studies strongly dispute this notion 57, 58, 59, 60. DCs stimulate T cells with their cognate peptides bound to class II major histocompatibility molecules, costimulatory molecules (i.e., CD80/CD86), and cytokines. Among these cytokines, interleukin‐12 (IL‐12) promotes the CD4+ Th1 cell response, whereas IL‐1β, IL‐6, and IL‐23 favor the expansion of CD4+ Th17 cells 7. In addition, decreased expression of the checkpoint molecule programmed death ligand 1 on DCs allows ongoing T cell activation in the arterial wall of GCA patients 61, 62, 63. In established GCA, macrophages around the external elastic lamina may further stimulate Th1 and Th17 cells by providing costimulation and Th1‐ and Th2‐skewing cytokines. Endothelial cells of the vasa vasorum may further augment Th1 and Th17 cell responses by activating the Notch receptor on these T cells via Jagged‐1 64. Endothelial cells up‐regulate Jagged‐1 upon exposure to high systemic levels of vascular endothelial growth factor (VEGF) 64.
Th1 and Th17 cells show proinflammatory effects on vascular smooth muscle cells (VSMCs) via interferon‐γ (IFNγ) and IL‐17, respectively 65, 66. Th1 cells also activate macrophages through IFNγ. IFNγ‐producing CD8+ T cells, small numbers of which have been detected in the inflamed arteries of GCA patients, might exert effects similar to those of Th1 cells in the vessel wall 67. CD8+ T cells may also promote vascular damage through secretion of granzyme B 67. Th17 cells are closely entwined with B cells in the inflamed arteries of GCA patients 66.
Cytokines involved in the Th1 and Th17 cell response have been linked to disease outcome in patients with GCA (Table 2). A previous study showed that high expression of IFNγ in temporal arteries is associated with an increased risk of cranial ischemic symptoms 68. Polymorphisms of the IFNγ gene have been associated with risk of cranial ischemia as well 69. In contrast, another study demonstrated that high local expression of IL‐17 is associated with low glucocorticoid requirements in patients with GCA and a trend for lower relapse rates 70. Strong local expression of the Th17 cell–polarizing cytokine IL‐6 has been associated with a low risk of cranial ischemia while showing no effect on long‐term glucocorticoid requirements 28, 71. In contrast, strong expression of IL‐1β in inflamed temporal arteries was linked to an increased risk of cranial ischemia 68. Thus far, the numbers of Th1 or Th17 cells in inflamed temporal arteries have not been linked to disease outcomes in GCA patients. However, a recent study demonstrated that a relatively high number of arterial CD8+ T cells (i.e., >6% of infiltrating cells) is associated with an increased risk of cranial ischemia and greater long‐term glucocorticoid requirements in GCA patients 67. All of these findings, however, still await validation in protocolized cohort studies.
Table 2.
Temporal artery biopsy findings associated with the risk of cranial ischemia, the relapse rate, and/or the glucocorticoid requirement in GCA*
| Temporal artery biopsy finding, study | Cranial ischemia risk | Relapse rate | Glucocorticoid requirement |
|---|---|---|---|
| Giant cell presence | |||
| Armstrong et al, 2008 72 | No effect | No effect | No effect |
| Chatelain et al, 2009 10 | Increased | NA | NA |
| Muratore et al, 2016 22 | Increased | NA | NA |
| CD8+ T cells† | |||
| Samson et al, 2016 67 | Increased | NA | Increased |
| Intimal hyperplasia | |||
| Kaiser et al, 1998 73 | Increased | NA | NA |
| Makkuni et al, 2008 77 | Increased | NA | NA |
| Neoangiogenesis | |||
| Cid et al, 2002 27 | Decreased | NA | NA |
| CCL2† | |||
| Cid et al, 2006 83 | Decreased | Increased | Increased |
| ET‐1† | |||
| Lozano et al, 2010 85 | No effect | NA | NA |
| IFNㆠ| |||
| Weyand et al, 1997 68 | Increased | NA | NA |
| IL‐1β† | |||
| Weyand et al, 1997 68 | Increased | NA | NA |
| Hernandez‐Rodriguez et al, 2004 71 | NA | NA | No effect |
| IL‐6† | |||
| Hernandez‐Rodriguez et al, 2003 28 | Decreased | NA | NA |
| Hernandez‐Rodriguez et al, 2004 71 | NA | NA | No effect |
| IL‐17† | |||
| Espigol‐Frigole et al, 2013 70 | NA | No effect | Decreased |
| TNF† | |||
| Hernandez‐Rodriguez et al, 2004 71 | NA | NA | Increased |
Studies published between January 1, 1997 and January 1, 2018 were included. GCA = giant cell arteritis; NA = not assessed; ET‐1 = endothelin 1; IFNγ = interferon‐γ; IL‐1β = interleukin‐1β; TNF = tumor necrosis factor.
High number of cells or protein expression level.
Macrophages in the inflamed artery. Whereas macrophages around the external elastic membrane are potent producers of Th1‐ and Th17‐polarizing cytokines (Figure 1), macrophages around the internal elastic membrane are involved in vascular damage and remodeling 7. Macrophages produce tumor necrosis factor (TNF) in the inflamed arteries of patients with GCA, and this was linked to high relapse rates in a previous study 71. Macrophages around the internal elastic membrane are activated by Th1 cells and CD8+ T cells and develop into multinucleated giant cells under the influence of IFNγ 68. The presence of giant cells was associated with an increased risk of cranial ischemia in 2 studies 10, 22, and also tended to correlate with more cranial ischemic symptoms in a third study 72. Macrophages and giant cells near the internal elastic membrane may secrete platelet‐derived growth factor (PDGF) and VEGF, which activate VSMCs 73, 74. VEGF also promotes local formation of neovessels 75. These neovessels allow for direct entry of immune cells into the deeper layers of the arterial wall. Nevertheless, one study showed a lower incidence of cranial ischemic symptoms in patients with neovessels in their temporal artery biopsy specimens 27.
VSMCs in the inflamed artery. VSMCs are central players in the immune pathology of GCA. Activated VSMCs migrate toward the intima and differentiate into myofibroblasts 74, 76. The latter cells may proliferate extensively, thereby resulting in intimal hyperplasia and luminal narrowing 73. Intimal hyperplasia has been linked to secretion of PDGF by macrophages and is strongly associated with the occurrence of cranial ischemic symptoms 73, 77. A recent study demonstrated that activated VSMCs may secrete BAFF and lymphotoxin, which promote the development of ectopic lymphoid structures in the inflamed arterial wall 66.
B cells and ectopic lymphoid structures in the inflamed artery. B cells are present predominantly in the adventitia of GCA patients and may form ectopic lymphoid structures together with T cells, follicular DCs, and high endothelial venules 66, 78, 79. Ectopic lymphoid structures are known to facilitate chronic inflammation by providing a framework for continuous B cell and T cell activation. Costimulatory molecules and cytokines such as IL‐21 and IL‐6 are likely to be involved in this process 66, 78. Ectopic lymphoid structures are more frequently observed in GCA patients with strong systemic inflammation, but show no relationship with the occurrence of cranial ischemic symptoms 66.
Chemokines in the inflamed artery. Immune cells and VSMCs secrete a wide variety of chemokines that attract different types of immune cells to the arterial wall of GCA patients 65, 66. Th1 and CD8+ T cells are attracted by CXCL9 and CXCL10 67, and Th17 cells are attracted by CCL20 80. Monocytes enter the arterial wall under the influence of CCL2 and CX3CL1 81, after which these cells differentiate into proinflammatory macrophages. B cells may migrate toward CCL20 and CXCL13 gradients in the arterial wall 66, 80. CCL5 promotes migration of both T cells and monocytes toward inflamed arteries 82. Little is known about the impact of local chemokine expression on clinical outcomes in GCA. Only one study showed that strong up‐regulation of CCL2 in temporal arteries is associated with a low risk of cranial ischemic symptoms, but higher relapse rates and glucocorticoid requirements 83.
Systemic cytokines. The levels of many proinflammatory cytokines are increased in the peripheral blood of GCA patients 84. Circulating levels of several cytokines have been identified as potential predictors of cranial ischemic symptoms in GCA (Table 3). In a previous study, high levels of systemic IL‐6 were observed in GCA patients with a low incidence of cranial ischemic symptoms 28. This finding is consistent with the lower risk of cranial ischemia in patients with a strong systemic inflammatory response (Table 1), because IL‐6 also drives the acute‐phase reactant CRP 15. It has been suggested that systemic IL‐6 may directly promote angiogenesis in the ischemic end organs of GCA patients 28. In contrast, high systemic levels of endothelin 1 (ET‐1), pentraxin 3, and VEGF have been linked to an increased risk of cranial ischemia 85, 86. Genetic variants of the VEGF gene have also been associated with the risk of cranial ischemic symptoms 87.
Table 3.
Serum or plasma proteins associated (when elevated) with the risk of cranial ischemia, the relapse rate, and/or the glucocorticoid requirement in GCA*
| Serum/plasma protein, study | Cranial ischemia risk | Relapse rate | Glucocorticoid requirement |
|---|---|---|---|
| ET‐1 | |||
| Lozano et al, 2010 85 | Increased | NA | NA |
| IL‐6 | |||
| Hernandez‐Rodriguez et al, 2003 28 | Decreased | NA | NA |
| IL‐1β | |||
| Hernandez‐Rodriguez et al, 2003 28 | No effect | NA | NA |
| PTX3 | |||
| Baldini et al, 2012 86 | Increased | NA | NA |
| TNF | |||
| Hernandez‐Rodriguez et al, 2003 28 | No effect | NA | NA |
| VEGF | |||
| Baldini et al, 2012 86 | Increased | NA | NA |
Studies published between January 1, 1997 and January 1, 2018 were included. GCA = giant cell arteritis; ET‐1 = endothelin 1; NA = not assessed; IL‐6 = interleukin‐6; PTX3 = pentraxin 3; TNF = tumor necrosis factor; VEGF = vascular endothelial growth factor.
Perspectives
Even though further studies are required to establish immunologic subsets of GCA patients, and correlations are not proof of causality between immunologic changes and disease outcomes in GCA, identification of immunologic subsets might provide a rationale for targeted treatments.
Targeting factors associated with cranial ischemic symptoms. Current evidence indicates that vascular predominance of IFNγ, IL‐1β, VEGF, and CD8+ T cells, as well as the presence of multinucleated giant cells and intimal hyperplasia, identify GCA patients with a high risk of cranial ischemic symptoms (Figure 1 and Table 2). Therefore, treatments directly or indirectly targeting these cytokines or immune cells are interesting to study.
Anti‐IFNγ treatment was shown to ameliorate vascular inflammation in an experimental model of GCA 65. In addition, anti–IL‐12/23 therapy was demonstrated to inhibit the induction of IFNγ‐producing Th1 cells 88. Anti–IL‐12/23 therapy showed promising therapeutic effects in a case series of GCA patients 89. Consequently, a randomized controlled trial with anti–IL‐12/23 therapy was recently initiated (ClinicalTrials.gov identifier: NCT02955147). In addition, targeting of the IFNγ response will likely ameliorate the effects of CD8+ T cells while also limiting formation of multinucleated giant cells. Furthermore, CTLA‐4Ig and JAK inhibitors will also limit activation of Th1 and CD8+ T cell responses in patients with GCA. A recent randomized controlled trial demonstrated the efficacy of CTLA‐4Ig for the maintenance of remission in patients with GCA 6. Currently, a single‐center trial of the JAK inhibitor baricitinib in GCA is ongoing (ClinicalTrials.gov identifier: NCT03026504). Another JAK inhibitor, tofacitinib, also ameliorated vascular inflammation in an experimental model of GCA 90. The same holds true for stress‐associated endoplasmic reticulum protein 1, a myxoma virus–derived serpin 91. The IL‐1R antagonist anakinra has shown promising results in a case series of GCA patients 92 and will be investigated in a randomized controlled trial (ClinicalTrials.gov identifier: NCT02902731). Although high systemic levels of VEGF have been associated with cranial ischemic symptoms in GCA patients, anti‐VEGF treatment might potentially be dangerous, because this treatment may inhibit angiogenesis in the ischemic tissue of GCA patients (e.g., the eyes). Intimal hyperplasia develops under the influence of PDGF and ET‐1. Anti‐PDGF treatment was shown to effectively inhibit intimal hyperplasia in a temporal artery explant model 74. Blocking ET‐1 reduced the outgrowth of VSMCs in another experimental study 76. Therefore, various immune cells and cytokines associated with cranial ischemic events are potential targets for therapy in patients with GCA.
Targeting factors associated with relapse and high glucocorticoid requirement. A strong systemic inflammatory response as well as high local expression of CCL2 and TNF have been associated with the development of relapse and/or increased glucocorticoid requirements in GCA patients (Tables 1 and 2). Although the prognostic value of systemic levels of IL‐6 in long‐term outcomes in GCA patients has not been formally studied, it is likely that GCA patients with a strong systemic inflammatory response also demonstrate the highest levels of systemic IL‐6. Therefore, GCA patients with disease relapses and an increased glucocorticoid requirement could potentially benefit from IL‐6–targeting treatment. Indeed, randomized controlled trials showed that anti–IL‐6R therapy effectively maintained glucocorticoid‐free remission in a significant proportion of patients with GCA 4, 5. Anti‐CCL2 treatment has not yet been tested in GCA patients or experimental models of GCA. Randomized controlled trials have demonstrated that anti‐TNF therapy lacks therapeutic efficacy in GCA 93, 94.
Open questions and research priorities
Evidence for distinct subsets of GCA patients is based mostly on retrospective cohort studies. Important questions regarding disease subsets in GCA are summarized in Table 4. Clearly, current evidence for distinct disease subsets of GCA patients requires validation in protocolized cohort studies with well‐characterized GCA patients. Evidence for disease subsets might be further obtained by post hoc analyses of the large randomized controlled trials in patients with newly diagnosed GCA 4, 6, 93, 94, 95, 96, 97. Although data on large vessel involvement have not been routinely obtained in these trials, baseline data on inflammation markers and the presence of PMR have been recorded. Eventually, dedicated randomized controlled trials are required to evaluate the effects of targeted treatments in distinct subsets of GCA patients, as was recently proposed for patients with small vessel vasculitis 98.
Table 4.
Open questions regarding disease subsets in GCA*
| 1. | What criteria can be used to identify GCA patients at high risk of severe cranial ischemic events? |
| 2. | What criteria should be used to identify GCA patients with a strong systemic inflammatory response that appears to be associated with a decreased risk of cranial ischemic events? |
| 3. | What are the optimal laboratory methods, or perhaps imaging methods, to measure immunologic markers in the tissue or blood of GCA patients, and which are the optimal prognostic cutoff levels for these markers? |
| 4. | In order to personalize the long‐term management of GCA, what criteria identify patients who are prone to develop future relapses, ischemic events, or aortic aneurysms? |
| 5. | Which immune cells or cytokines associated with poor long‐term disease outcomes should be targeted by treatment in GCA patients? |
| 6. | Are measurements of immunologic cells or cytokines predictive of the response to treatments targeting these cells or cytokines? |
GCA = giant cell arteritis.
Conclusions
Ample evidence indicates that GCA is a clinically and immunologically heterogeneous autoimmune disease. However, patients with GCA are currently treated according to standardized regimens. Retrospective studies have identified several clinical and immunologic characteristics associated with cranial ischemia and long‐term disease outcomes in GCA. Future studies should validate prognostic factors and the presence of disease subsets in GCA. Eventually, recognition of distinct GCA disease subsets may be helpful for implementing precision medicine for GCA patients.
Author Contributions
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published.
The opinions expressed herein are those of the authors and do not purport to reflect the official position or views of the funding agencies.
Supported by the European Union's Horizon 2020 Research and Innovation Programme (grant 668036 to Drs. Abdulahad, Heeringa, and Brouwer). Dr. van der Geest's work was supported by the Dutch Society for Rheumatology (Rheumatology grant 2017) and the Mandema Stipend. Dr. Sander's work was supported by the Dutch Kidney Foundation (grant 13OKJ39) and the Netherlands Organization for Scientific Research (grant 907‐14‐542).
Dr. Kallenberg has received consulting fees, speaking fees, and/or honoraria from GlaxoSmithKline and ImmuPharma (less than $10,000 each). Dr. Boots has received consulting fees from Gruenenthal (less than $10,000). Dr. Brouwer has received consulting fees from Roche (less than $10,000) and unrestricted grants from MSD, Pfizer, and Abbott.
References
- 1. Salvarani C, Cantini F, Hunder GG. Polymyalgia rheumatica and giant‐cell arteritis. Lancet 2008;372:234–45. [DOI] [PubMed] [Google Scholar]
- 2. Singh AG, Kermani TA, Crowson CS, Weyand CM, Matteson EL, Warrington KJ. Visual manifestations in giant cell arteritis: trend over 5 decades in a population‐based cohort. J Rheumatol 2015;42:309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broder MS, Sarsour K, Chang E, Collinson N, Tuckwell K, Napalkov P, et al. Corticosteroid‐related adverse events in patients with giant cell arteritis: a claims‐based analysis. Semin Arthritis Rheum 2016;46:246–52. [DOI] [PubMed] [Google Scholar]
- 4. Stone JH, Tuckwell K, Dimonaco S, Klearman M, Aringer M, Blockmans D, et al. Trial of tocilizumab in giant‐cell arteritis. N Engl J Med 2017;377:317–28. [DOI] [PubMed] [Google Scholar]
- 5. Villiger PM, Adler S, Kuchen S, Wermelinger F, Dan D, Fiege V, et al. Tocilizumab for induction and maintenance of remission in giant cell arteritis: a phase 2, randomised, double‐blind, placebo‐controlled trial. Lancet 2016;387:1921–7. [DOI] [PubMed] [Google Scholar]
- 6. Langford CA, Cuthbertson D, Ytterberg SR, Khalidi N, Monach PA, Carette S, et al. A randomized, double‐blind trial of abatacept (CTLA‐4Ig) for the treatment of Takayasu arteritis. Arthritis Rheumatol 2017;69:846–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Weyand CM, Goronzy JJ. Immune mechanisms in medium and large‐vessel vasculitis. Nat Rev Rheumatol 2013;9:731–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salvarani C, Hunder GG. Giant cell arteritis with low erythrocyte sedimentation rate: frequency of occurrence in a population‐based study. Arthritis Rheum 2001;45:140–5. [DOI] [PubMed] [Google Scholar]
- 9. Nesher G, Nesher R, Mates M, Sonnenblick M, Breuer GS. Giant cell arteritis: intensity of the initial systemic inflammatory response and the course of the disease. Clin Exp Rheumatol 2008;26 Suppl 49:S30–4. [PubMed] [Google Scholar]
- 10. Chatelain D, Duhaut P, Schmidt J, Loire R, Bosshard S, Guernou M, et al. Pathological features of temporal arteries in patients with giant cell arteritis presenting with permanent visual loss. Ann Rheum Dis 2009;68:84–8. [DOI] [PubMed] [Google Scholar]
- 11. Yates M, MacGregor AJ, Robson J, Craven A, Merkel PA, Luqmani RA, et al. The association of vascular risk factors with visual loss in giant cell arteritis. Rheumatology (Oxford) 2017;56:524–8. [DOI] [PubMed] [Google Scholar]
- 12. Cid MC, Font C, Oristrell J, de la Sierra A, Coll‐Vinent B, Lopez‐Soto A, et al. Association between strong inflammatory response and low risk of developing visual loss and other cranial ischemic complications in giant cell (temporal) arteritis. Arthritis Rheum 1998;41:26–32. [DOI] [PubMed] [Google Scholar]
- 13. Gonzalez‐Gay MA, Blanco R, Rodriguez‐Valverde V, Martinez‐Taboada VM, Delgado‐Rodriguez M, Figueroa M, et al. Permanent visual loss and cerebrovascular accidents in giant cell arteritis: predictors and response to treatment. Arthritis Rheum 1998;41:1497–504. [DOI] [PubMed] [Google Scholar]
- 14. Liozon E, Herrmann F, Ly K, Robert PY, Loustaud V, Soria P, et al. Risk factors for visual loss in giant cell (temporal) arteritis: a prospective study of 174 patients. Am J Med 2001;111:211–7. [DOI] [PubMed] [Google Scholar]
- 15. Hernandez‐Rodriguez J, Garcia‐Martinez A, Casademont J, Filella X, Esteban MJ, Lopez‐Soto A, et al. A strong initial systemic inflammatory response is associated with higher corticosteroid requirements and longer duration of therapy in patients with giant‐cell arteritis. Arthritis Rheum 2002;47:29–35. [DOI] [PubMed] [Google Scholar]
- 16. Gonzalez‐Gay MA, Garcia‐Porrua C, Amor‐Dorado JC, Llorca J. Fever in biopsy‐proven giant cell arteritis: clinical implications in a defined population. Arthritis Rheum 2004;51:652–5. [DOI] [PubMed] [Google Scholar]
- 17. Salvarani C, Cimino L, Macchioni P, Consonni D, Cantini F, Bajocchi G, et al. Risk factors for visual loss in an Italian population‐based cohort of patients with giant cell arteritis. Arthritis Rheum 2005;53:293–7. [DOI] [PubMed] [Google Scholar]
- 18. Gonzalez‐Gay MA, Barros S, Lopez‐Diaz MJ, Garcia‐Porrua C, Sanchez‐Andrade A, Llorca J. Giant cell arteritis: disease patterns of clinical presentation in a series of 240 patients. Medicine (Baltimore) 2005;84:269–76. [DOI] [PubMed] [Google Scholar]
- 19. Lopez‐Diaz MJ, Llorca J, Gonzalez‐Juanatey C, Pena‐Sagredo JL, Martin J, Gonzalez‐Gay MA. The erythrocyte sedimentation rate is associated with the development of visual complications in biopsy‐proven giant cell arteritis. Semin Arthritis Rheum 2008;38:116–23. [DOI] [PubMed] [Google Scholar]
- 20. Gonzalez‐Gay MA, Vazquez‐Rodriguez TR, Gomez‐Acebo I, Pego‐Reigosa R, Lopez‐Diaz MJ, Vazquez‐Trinanes MC, et al. Strokes at time of disease diagnosis in a series of 287 patients with biopsy‐proven giant cell arteritis. Medicine (Baltimore) 2009;88:227–35. [DOI] [PubMed] [Google Scholar]
- 21. Salvarani C, Della Bella C, Cimino L, Macchioni P, Formisano D, Bajocchi G, et al. Risk factors for severe cranial ischaemic events in an Italian population‐based cohort of patients with giant cell arteritis. Rheumatology (Oxford) 2009;48:250–3. [DOI] [PubMed] [Google Scholar]
- 22. Muratore F, Boiardi L, Cavazza A, Aldigeri R, Pipitone N, Restuccia G, et al. Correlations between histopathological findings and clinical manifestations in biopsy‐proven giant cell arteritis. J Autoimmun 2016;69:94–101. [DOI] [PubMed] [Google Scholar]
- 23. Liozon E, Dalmay F, Lalloue F, Gondran G, Bezanahary H, Fauchais AL, et al. Risk factors for permanent visual loss in biopsy‐proven giant cell arteritis: a study of 339 patients. J Rheumatol 2016;43:1393–9. [DOI] [PubMed] [Google Scholar]
- 24. Grossman C, Barshack I, Koren‐Morag N, Ben‐Zvi I, Bornstein G. Risk factors for severe cranial ischaemic events in patients with giant cell arteritis. Clin Exp Rheumatol 2017;35 Suppl 103:88–93. [PubMed] [Google Scholar]
- 25. De Boysson H, Liozon E, Lariviere D, Samson M, Parienti JJ, Boutemy J, et al. Giant cell arteritis‐related stroke: a retrospective multicenter case‐control study. J Rheumatol 2017;44:297–303. [DOI] [PubMed] [Google Scholar]
- 26. Gonzalez‐Gay MA, Lopez‐Diaz MJ, Barros S, Garcia‐Porrua C, Sanchez‐Andrade A, Paz‐Carreira J, et al. Giant cell arteritis: laboratory tests at the time of diagnosis in a series of 240 patients. Medicine (Baltimore) 2005;84:277–90. [DOI] [PubMed] [Google Scholar]
- 27. Cid MC, Hernandez‐Rodriguez J, Esteban MJ, Cebrian M, Gho YS, Font C, et al. Tissue and serum angiogenic activity is associated with low prevalence of ischemic complications in patients with giant‐cell arteritis. Circulation 2002;106:1664–71. [DOI] [PubMed] [Google Scholar]
- 28. Hernandez‐Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia‐Martinez A, Esteban MJ, et al. Elevated production of interleukin‐6 is associated with a lower incidence of disease‐related ischemic events in patients with giant‐cell arteritis: angiogenic activity of interleukin‐6 as a potential protective mechanism. Circulation 2003;107:2428–34. [DOI] [PubMed] [Google Scholar]
- 29. Martinez‐Lado L, Calvino‐Diaz C, Pineiro A, Dierssen T, Vazquez‐Rodriguez TR, Miranda‐Filloy JA, et al. Relapses and recurrences in giant cell arteritis: a population‐based study of patients with biopsy‐proven disease from northwestern Spain. Medicine (Baltimore) 2011;90:186–93. [DOI] [PubMed] [Google Scholar]
- 30. Restuccia G, Boiardi L, Cavazza A, Catanoso M, Macchioni P, Muratore F, et al. Long‐term remission in biopsy proven giant cell arteritis: a retrospective cohort study. J Autoimmun 2017;77:39–44. [DOI] [PubMed] [Google Scholar]
- 31. Prieto‐Gonzalez S, Arguis P, Garcia‐Martinez A, Espigol‐Frigole G, Tavera‐Bahillo I, Butjosa M, et al. Large vessel involvement in biopsy‐proven giant cell arteritis: prospective study in 40 newly diagnosed patients using CT angiography. Ann Rheum Dis 2012;71:1170–6. [DOI] [PubMed] [Google Scholar]
- 32. Schmidt WA, Krause A, Schicke B, Kuchenbecker J, Gromnica‐Ihle E. Do temporal artery duplex ultrasound findings correlate with ophthalmic complications in giant cell arteritis? Rheumatology (Oxford) 2009;48:383–5. [DOI] [PubMed] [Google Scholar]
- 33. Czihal M, Zanker S, Rademacher A, Tato F, Kuhlencordt PJ, Schulze‐Koops H, et al. Sonographic and clinical pattern of extracranial and cranial giant cell arteritis. Scand J Rheumatol 2012;41:231–6. [DOI] [PubMed] [Google Scholar]
- 34. Muratore F, Kermani TA, Crowson CS, Green AB, Salvarani C, Matteson EL, et al. Large‐vessel giant cell arteritis: a cohort study. Rheumatology (Oxford) 2015;54:463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. De Boysson H, Liozon E, Lambert M, Dumont A, Boutemy J, Maigne G, et al. Giant‐cell arteritis: do we treat patients with large‐vessel involvement differently? Am J Med 2017;130:992–5. [DOI] [PubMed] [Google Scholar]
- 36. Espitia O, Neel A, Leux C, Connault J, Espitia‐Thibault A, Ponge T, et al. Giant cell arteritis with or without aortitis at diagnosis: a retrospective study of 22 patients with longterm followup. J Rheumatol 2012;39:2157–62. [DOI] [PubMed] [Google Scholar]
- 37. Schmidt WA, Moll A, Seifert A, Schicke B, Gromnica‐Ihle E, Krause A. Prognosis of large‐vessel giant cell arteritis. Rheumatology (Oxford) 2008;47:1406–8. [DOI] [PubMed] [Google Scholar]
- 38. Czihal M, Piller A, Schroettle A, Kuhlencordt P, Bernau C, Schulze‐Koops H, et al. Impact of cranial and axillary/subclavian artery involvement by color duplex sonography on response to treatment in giant cell arteritis. J Vasc Surg 2015;61:1285–91. [DOI] [PubMed] [Google Scholar]
- 39. Kermani TA, Warrington KJ, Crowson CS, Hunder GG, Ytterberg SR, Gabriel SE, et al. Predictors of dissection in aortic aneurysms from giant cell arteritis. J Clin Rheumatol 2016;22:184–7. [DOI] [PubMed] [Google Scholar]
- 40. Kebed DT, Bois JP, Connolly HM, Scott CG, Bowen JM, Warrington KJ, et al. Spectrum of aortic disease in the giant cell arteritis population. Am J Cardiol 2018;121:501–8. [DOI] [PubMed] [Google Scholar]
- 41. Muratore F, Kermani TA, Crowson CS, Koster MJ, Matteson EL, Salvarani C, et al. Large vessel dilatation in giant cell arteritis: a different subset of disease? Arthritis Care Res (Hoboken) 2017. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 42. Dejaco C, Ramiro S, Duftner C, Besson FL, Bley TA, Blockmans D, et al. EULAR recommendations for the use of imaging in large vessel vasculitis in clinical practice. Ann Rheum Dis 2018. E‐pub ahead of print. [DOI] [PubMed] [Google Scholar]
- 43. Puppo C, Massollo M, Paparo F, Camellino D, Piccardo A, Shoushtari Zadeh Naseri M, et al. Giant cell arteritis: a systematic review of the qualitative and semiquantitative methods to assess vasculitis with 18F‐fluorodeoxyglucose positron emission tomography. Biomed Res Int 2014;2014:574248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Myklebust G, Gran JT. Prednisolone maintenance dose in relation to starting dose in the treatment of polymyalgia rheumatica and temporal arteritis: a prospective two‐year study in 273 patients. Scand J Rheumatol 2001;30:260–7. [DOI] [PubMed] [Google Scholar]
- 45. Cavazza A, Muratore F, Boiardi L, Restuccia G, Pipitone N, Pazzola G, et al. Inflamed temporal artery: histologic findings in 354 biopsies, with clinical correlations. Am J Surg Pathol 2014;38:1360–70. [DOI] [PubMed] [Google Scholar]
- 46. Hernandez‐Rodriguez J, Murgia G, Villar I, Campo E, Mackie SL, Chakrabarty A, et al. Description and validation of histological patterns and proposal of a dynamic model of inflammatory infiltration in giant‐cell arteritis. Medicine (Baltimore) 2016;95:e2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jia L, Couce M, Barnholtz‐Sloan JS, Cohen ML. Is all inflammation within temporal artery biopsies temporal arteritis? Hum Pathol 2016;57:17–21. [DOI] [PubMed] [Google Scholar]
- 48. Belilos E, Maddox J, Kowalewski RM, Kowalewska J, Turi GK, Nochomovitz LE, et al. Temporal small‐vessel inflammation in patients with giant cell arteritis: clinical course and preliminary immunohistopathologic characterization. J Rheumatol 2011;38:331–8. [DOI] [PubMed] [Google Scholar]
- 49. Restuccia G, Cavazza A, Boiardi L, Pipitone N, Macchioni P, Bajocchi G, et al. Small‐vessel vasculitis surrounding an uninflamed temporal artery and isolated vasa vasorum vasculitis of the temporal artery: two subsets of giant cell arteritis. Arthritis Rheum 2012;64:549–56. [DOI] [PubMed] [Google Scholar]
- 50. Esteban MJ, Font C, Hernández‐Rodriguez J, Valls‐Solé J, Sanmartí R, Cardellach F, et al. Small‐vessel vasculitis surrounding a spared temporal artery: clinical and pathological findings in a series of twenty‐eight patients. Arthritis Rheum 2001;44:1387–95. [DOI] [PubMed] [Google Scholar]
- 51. Chatelain D, Duhaut P, Loire R, Bosshard S, Pellet H, Piette JC, et al. Small‐vessel vasculitis surrounding an uninflamed temporal artery: a new diagnostic criterion for polymyalgia rheumatica? Arthritis Rheum 2008;58:2565–73. [DOI] [PubMed] [Google Scholar]
- 52. Le Pendu C, Meignin V, Gonzalez‐Chiappe S, Hij A, Galateau‐Salle F, Mahr A. Poor predictive value of isolated adventitial and periadventitial infiltrates in temporal artery biopsies for diagnosis of giant cell arteritis. J Rheumatol 2017;44:1039–43. [DOI] [PubMed] [Google Scholar]
- 53. Corcoran GM, Prayson RA, Herzog KM. The significance of perivascular inflammation in the absence of arteritis in temporal artery biopsy specimens. Am J Clin Pathol 2001;115:342–7. [DOI] [PubMed] [Google Scholar]
- 54. Chakrabarty A, Franks AJ. Temporal artery biopsy: is there any value in examining biopsies at multiple levels? J Clin Pathol 2000;53:131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Breuer GS, Nesher R, Reinus K, Nesher G. Association between histological features in temporal artery biopsies and clinical features of patients with giant cell arteritis. Isr Med Assoc J 2013;15:271–4. [PubMed] [Google Scholar]
- 56. Gilden D, Nagel MA. Varicella zoster virus triggers the immunopathology of giant cell arteritis. Curr Opin Rheumatol 2016;28:376–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bigler MB, Hirsiger JR, Recher M, Mehling M, Daikeler T, Berger CT. Varicella zoster virus‐specific T cell responses in untreated giant cell arteritis: comment on the article by England et al. Arthritis Rheumatol 2018;70:318–20. [DOI] [PubMed] [Google Scholar]
- 58. Muratore F, Croci S, Tamagnini I, Zerbini A, Bellafiore S, Belloni L, et al. No detection of varicella‐zoster virus in temporal arteries of patients with giant cell arteritis. Semin Arthritis Rheum 2017;47:235–40. [DOI] [PubMed] [Google Scholar]
- 59. Rondaan C, van der Geest KS, Eelsing E, Boots AM, Bos NA, Westra J, et al. Decreased immunity to varicella zoster virus in giant cell arteritis. Front Immunol 2017;8:1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Procop GW, Eng C, Clifford A, Villa‐Forte A, Calabrese LH, Roselli E, et al. Varicella zoster virus and large vessel vasculitis, the absence of an association. Pathog Immun 2017;2:228–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Zhang H, Watanabe R, Berry GJ, Vaglio A, Liao YJ, Warrington KJ, et al. Immunoinhibitory checkpoint deficiency in medium and large vessel vasculitis. Proc Natl Acad Sci U S A 2017;114:E970–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Watanabe R, Zhang H, Berry G, Goronzy JJ, Weyand CM. Immune checkpoint dysfunction in large and medium vessel vasculitis. Am J Physiol Heart Circ Physiol 2017;312:H1052–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weyand CM, Berry GJ, Goronzy JJ. The immunoinhibitory PD‐1/PD‐L1 pathway in inflammatory blood vessel disease. J Leukoc Biol 2018;103:565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wen Z, Shen Y, Berry G, Shahram F, Li Y, Watanabe R, et al. The microvascular niche instructs T cells in large vessel vasculitis via the VEGF‐Jagged1‐Notch pathway. Sci Transl Med 2017;9:eaal3322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Corbera‐Bellalta M, Planas‐Rigol E, Lozano E, Terrades‐Garcia N, Alba MA, Prieto‐Gonzalez S, et al. Blocking interferon γ reduces expression of chemokines CXCL9, CXCL10 and CXCL11 and decreases macrophage infiltration in ex vivo cultured arteries from patients with giant cell arteritis. Ann Rheum Dis 2016;75:1177–86. [DOI] [PubMed] [Google Scholar]
- 66. Ciccia F, Rizzo A, Maugeri R, Alessandro R, Croci S, Guggino G, et al. Ectopic expression of CXCL13, BAFF, APRIL and LT‐β is associated with artery tertiary lymphoid organs in giant cell arteritis. Ann Rheum Dis 2017;76:235–43. [DOI] [PubMed] [Google Scholar]
- 67. Samson M, Ly KH, Tournier B, Janikashvili N, Trad M, Ciudad M, et al. Involvement and prognosis value of CD8(+) T cells in giant cell arteritis. J Autoimmun 2016;72:73–83. [DOI] [PubMed] [Google Scholar]
- 68. Weyand CM, Tetzlaff N, Bjornsson J, Brack A, Younge B, Goronzy JJ. Disease patterns and tissue cytokine profiles in giant cell arteritis. Arthritis Rheum 1997;40:19–26. [DOI] [PubMed] [Google Scholar]
- 69. Gonzalez‐Gay MA, Hajeer AH, Dababneh A, Garcia‐Porrua C, Amoli MM, Llorca J, et al. Interferon‐γ gene microsatellite polymorphisms in patients with biopsy‐proven giant cell arteritis and isolated polymyalgia rheumatica. Clin Exp Rheumatol 2004;22 Suppl 36:S18–20. [PubMed] [Google Scholar]
- 70. Espigol‐Frigole G, Corbera‐Bellalta M, Planas‐Rigol E, Lozano E, Segarra M, Garcia‐Martinez A, et al. Increased IL‐17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant‐cell arteritis. Ann Rheum Dis 2013;72:1481–7. [DOI] [PubMed] [Google Scholar]
- 71. Hernandez‐Rodriguez J, Segarra M, Vilardell C, Sanchez M, Garcia‐Martinez A, Esteban MJ, et al. Tissue production of pro‐inflammatory cytokines (IL‐1β, TNFα and IL‐6) correlates with the intensity of the systemic inflammatory response and with corticosteroid requirements in giant‐cell arteritis. Rheumatology (Oxford) 2004;43:294–301. [DOI] [PubMed] [Google Scholar]
- 72. Armstrong AT, Tyler WB, Wood GC, Harrington TM. Clinical importance of the presence of giant cells in temporal arteritis. J Clin Pathol 2008;61:669–71. [DOI] [PubMed] [Google Scholar]
- 73. Kaiser M, Weyand CM, Bjornsson J, Goronzy JJ. Platelet‐derived growth factor, intimal hyperplasia, and ischemic complications in giant cell arteritis. Arthritis Rheum 1998;41:623–33. [DOI] [PubMed] [Google Scholar]
- 74. Lozano E, Segarra M, Garcia‐Martinez A, Hernandez‐Rodriguez J, Cid MC. Imatinib mesylate inhibits in vitro and ex vivo biological responses related to vascular occlusion in giant cell arteritis. Ann Rheum Dis 2008;67:1581–8. [DOI] [PubMed] [Google Scholar]
- 75. Kaiser M, Younge B, Bjornsson J, Goronzy JJ, Weyand CM. Formation of new vasa vasorum in vasculitis: production of angiogenic cytokines by multinucleated giant cells. Am J Pathol 1999;155:765–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Planas‐Rigol E, Terrades‐Garcia N, Corbera‐Bellalta M, Lozano E, Alba MA, Segarra M, et al. Endothelin‐1 promotes vascular smooth muscle cell migration across the artery wall: a mechanism contributing to vascular remodelling and intimal hyperplasia in giant‐cell arteritis. Ann Rheum Dis 2017;76:1624–34. [DOI] [PubMed] [Google Scholar]
- 77. Makkuni D, Bharadwaj A, Payne S, Hutchings A, Dasgupta B. Is intimal hyperplasia a marker of neuro‐ophthalmic complications of giant cell arteritis? Rheumatology (Oxford) 2008;47:488–90. [DOI] [PubMed] [Google Scholar]
- 78. Van der Geest KS, Abdulahad WH, Chalan P, Rutgers A, Horst G, Huitema MG, et al. Disturbed B cell homeostasis in newly diagnosed giant cell arteritis and polymyalgia rheumatica. Arthritis Rheumatol 2014;66:1927–38. [DOI] [PubMed] [Google Scholar]
- 79. Graver JC, Sandovici M, Diepstra A, Boots AM, Brouwer E. Artery tertiary lymphoid organs in giant cell arteritis are not exclusively located in the media of temporal arteries. Ann Rheum Dis 2017;76:235–43. [DOI] [PubMed] [Google Scholar]
- 80. Terrier B, Geri G, Chaara W, Allenbach Y, Rosenzwajg M, Costedoat‐Chalumeau N, et al. Interleukin‐21 modulates Th1 and Th17 responses in giant cell arteritis. Arthritis Rheum 2012;64:2001–11. [DOI] [PubMed] [Google Scholar]
- 81. Van Sleen Y, Wang Q, van der Geest KS, Westra J, Abdulahad WH, Heeringa P, et al. Involvement of monocyte subsets in the immunopathology of giant cell arteritis. Sci Rep 2017;7:6553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bruhl H, Vielhauer V, Weiss M, Mack M, Schlondorff D, Segerer S. Expression of DARC, CXCR3 and CCR5 in giant cell arteritis. Rheumatology (Oxford) 2005;44:309–13. [DOI] [PubMed] [Google Scholar]
- 83. Cid MC, Hoffman MP, Hernandez‐Rodriguez J, Segarra M, Elkin M, Sanchez M, et al. Association between increased CCL2 (MCP‐1) expression in lesions and persistence of disease activity in giant‐cell arteritis. Rheumatology (Oxford) 2006;45:1356–63. [DOI] [PubMed] [Google Scholar]
- 84. Van der Geest KS, Abdulahad WH, Rutgers A, Horst G, Bijzet J, Arends S, et al. Serum markers associated with disease activity in giant cell arteritis and polymyalgia rheumatica. Rheumatology (Oxford) 2015;54:1397–402. [DOI] [PubMed] [Google Scholar]
- 85. Lozano E, Segarra M, Corbera‐Bellalta M, Garcia‐Martinez A, Espigol‐Frigole G, Pla‐Campo A, et al. Increased expression of the endothelin system in arterial lesions from patients with giant‐cell arteritis: association between elevated plasma endothelin levels and the development of ischaemic events. Ann Rheum Dis 2010;69:434–42. [DOI] [PubMed] [Google Scholar]
- 86. Baldini M, Maugeri N, Ramirez GA, Giacomassi C, Castiglioni A, Prieto‐González S, et al. Selective up‐regulation of the soluble pattern‐recognition receptor pentraxin 3 and of vascular endothelial growth factor in giant cell arteritis: relevance for recent optic nerve ischemia. Arthritis Rheum 2012;64:854–65. [DOI] [PubMed] [Google Scholar]
- 87. Rueda B, Lopez‐Nevot MA, Lopez‐Diaz MJ, Garcia‐Porrua C, Martin J, Gonzalez‐Gay MA. A functional variant of vascular endothelial growth factor is associated with severe ischemic complications in giant cell arteritis. J Rheumatol 2005;32:1737–41. [PubMed] [Google Scholar]
- 88. Samson M, Ghesquiere T, Berthier S, Bonnotte B. Ustekinumab inhibits Th1 and Th17 polarisation in a patient with giant cell arteritis. Ann Rheum Dis 2018;77:e6. [DOI] [PubMed] [Google Scholar]
- 89. Conway R, O'Neill L, O'Flynn E, Gallagher P, McCarthy GM, Murphy CC, et al. Ustekinumab for the treatment of refractory giant cell arteritis. Ann Rheum Dis 2016;75:1578–9. [DOI] [PubMed] [Google Scholar]
- 90. Zhang H, Watanabe R, Berry GJ, Tian L, Goronzy JJ, Weyand C. Inhibition of JAK‐STAT signaling suppresses pathogenic immune responses in medium and large vessel vasculitis. Circulation 2017. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chen H, Zheng D, Ambadapadi S, Davids J, Ryden S, Samy H, et al. Serpin treatment suppresses inflammatory vascular lesions in temporal artery implants (TAI) from patients with giant cell arteritis. PLoS One 2015;10:e0115482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ly KH, Stirnemann J, Liozon E, Michel M, Fain O, Fauchais AL. Interleukin‐1 blockade in refractory giant cell arteritis. Joint Bone Spine 2014;81:76–8. [DOI] [PubMed] [Google Scholar]
- 93. Hoffman GS, Cid MC, Rendt‐Zagar KE, Merkel PA, Weyand CM, Stone JH, et al. Infliximab for maintenance of glucocorticosteroid‐induced remission of giant cell arteritis: a randomized trial. Ann Intern Med 2007;146:621–30. [DOI] [PubMed] [Google Scholar]
- 94. Seror R, Baron G, Hachulla E, Debandt M, Larroche C, Puechal X, et al. Adalimumab for steroid sparing in patients with giant‐cell arteritis: results of a multicentre randomised controlled trial. Ann Rheum Dis 2014;73:2074–81. [DOI] [PubMed] [Google Scholar]
- 95. Hoffman GS, Cid MC, Hellmann DB, Guillevin L, Stone JH, Schousboe J, et al. A multicenter, randomized, double‐blind, placebo‐controlled trial of adjuvant methotrexate treatment for giant cell arteritis. Arthritis Rheum 2002;46:1309–18. [DOI] [PubMed] [Google Scholar]
- 96. Jover JA, Hernandez‐Garcia C, Morado IC, Vargas E, Banares A, Fernandez‐Gutierrez B. Combined treatment of giant‐cell arteritis with methotrexate and prednisone: a randomized, double‐blind, placebo‐controlled trial. Ann Intern Med 2001;134:106–14. [DOI] [PubMed] [Google Scholar]
- 97. Chevalet P, Barrier JH, Pottier P, Magadur‐Joly G, Pottier MA, Hamidou M, et al. A randomized, multicenter, controlled trial using intravenous pulses of methylprednisolone in the initial treatment of simple forms of giant cell arteritis: a one year followup study of 164 patients. J Rheumatol 2000;27:1484–91. [PubMed] [Google Scholar]
- 98. Van der Geest KS, Brouwer E, Sanders JS, Sandovici M, Bos NA, Boots AM, et al. Toward precision medicine in ANCA‐associated vasculitis. Rheumatology (Oxford) 2017. [DOI] [PubMed] [Google Scholar]