

Abstract
A metal‐free and direct alkene C−H cyanation is described. Directing groups are not required and the mechanism involves electrophilic activation of the alkene by a cyano iodine(III) species generated in situ from a [bis(trifluoroacetoxy)iodo]arene and trimethylsilyl cyanide as the cyanide source. This C−H functionalization can be conducted on gram scale, and for noncyclic 1,1‐ and 1,2‐disubstuted alkenes high stereoselectivity is achieved, thus rendering the method highly valuable.
Keywords: alkenes, C−H functionalization, cyanation, hypervalent compounds, synthetic methods
The nitrile functionality is an important structural moiety that can be found in various natural products, drugs, agrochemicals, and in polymers.1 Moreover, the cyano group can be used as a synthetic equivalent for primary amines, tetrazoles, aldehydes/ketones and their related functionalities.2
Aryl nitriles are generally prepared by cyanation of halides, pseudohalides, diazonium salts, and organometallic reagents.3 Step‐ and atom‐economy in the synthesis of cyanated arenes and alkenes can be improved following a direct C−H cyanation strategy. Significant advances in directing group (DG) assisted arene C−H cyanation have been achieved using transition‐metal catalysis (e.g., Pd, Cu, and Rh).4 Along these lines, direct C−H cyanation of indoles has also been reported.5 Recently, Wang and co‐workers disclosed an iron‐catalyzed electrophilic cyanation of arenes and heteroarenes using aryl(cyano)iodonium triflates,6a and the group of Nicewicz reported aromatic C−H cyanation with trimethylsilyl cyanide using photoredox catalysis.6b
Established approaches for the synthesis of acrylonitriles comprise carbocyanation,7 heterocyanation,8 and hydrocyanation9 of alkynes with X−CN‐type reagents (X=C, Si, B, Sn, Ge, S, O, Br, H). However, as compared to arene C−H cyanation, direct alkene C−H cyanation has not been well investigated. Anbarasan10a and Fu10b reported rhodium‐catalyzed direct C(sp2)−H cyanation of alkenes, bearing directing groups, with NCTS (N‐cyano‐N‐phenyl‐p‐methylbenzenesulfonamide) as the cyanating reagent (Scheme 1). However, nondirected alkene C−H cyanation has not yet been described. We report herein transition metal free alkene cyanation using an aryl(biscyano)iodine(III) reagent as an alkene activator and cyanide source.
Scheme 1.
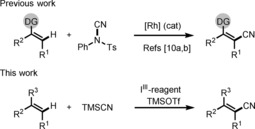
Direct C−H cyanation of alkenes. DG=directing group, Tf=trifluoromethanesulfonyl, TMS=trimethylsilyl.
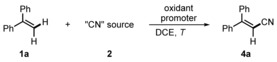
α‐Phenylstyrene (1 a) was chosen as a model substrate and various electrophilic cyanation reagents were screened in combination with or without an additive in 1,2‐dichloroethane (DCE) at 70 °C for 15 hours (Table 1, Figure 1).3g, 4b,4c,4i, 5d, 11 C−H cyanation of 1 a with the reagents 2 a–d, 2 i and 2 j failed (entries 1–6). Traces of the nitrile 4 a were detected when the benziodoxole 2 e was employed in combination with catalytic amounts of either CuCl or Fe(OAc)2 (entries 7 and 8). No improvement was noted by using 2 f (entries 9 and 10). TMSCN paired with oxone, (NH4)2S2O8, or tBuOOH did not provide 1 a (entries 11–13). The product 4 a was obtained in 7 % yield using 2 e in combination with TMSCN (entry 14). Replacing 2 e by 2 f led to a significant improvement of the yield (entry 15). The electronic nature of the substituents at the aryl moiety in the IIII reagent is important: the activity of 2 h was superior to that of 2 f and 2 g (entries 15–17). Pleasingly, direct C−H cyanation using the more stable and easily accessible [bis(trifluoroacetoxy)iodo]arene 3 d, which reacts in situ by sequential ligand exchange with TMSOTf and TMSCN to 2 h,12 gave a comparable result (entry 18). The best yield (90 %) was achieved upon lowering the temperature to 40 °C (entries 19 and 20). With 2 equivalents of TMSCN, a lower yield resulted, but using 3 and 4 equivalents provided good results (entries 22–24). The benziodoxole 3 e as an oxidant provided a worse result (entry 21). Other cyanide sources such as NaCN, KCN, and Bu4NCN did not lead to 4 a (entries 25–27).
Table 1.
Optimization studies.[a]
| Entry | “CN” 2
(equiv) |
Oxidant (equiv) | Additive (equiv) | Yield [%][b] |
|---|---|---|---|---|
| 1 | 2 a (2.0) | none | none | – |
| 2 | 2 b (2.0) | none | AuCl3 (0.05) | – |
| 3 | 2 c (2.0) | none | CuCl (0.1) | – |
| 4 | 2 d (2.0) | none | BF3⋅Et2O (2.0) | – |
| 5 | 2 i (2.0) | none | none | – |
| 6 | 2 j (2.0) | none | none | – |
| 7 | 2 e (2.0) | none | CuCl (0.1) | traces |
| 8 | 2 e (2.0) | none | Fe(OAc)2 (0.1) | traces |
| 9 | 2 f (2.0) | none | none | traces |
| 10 | 2 f (2.0) | none | Fe(OAc)2 (0.1) | traces |
| 11 | 2 k (2.0) | 3 a (2.0) | none | – |
| 12 | 2 k (2.0) | 3 b (2.0) | none | – |
| 13 | 2 k (2.0) | 3 c (2.0) | none | – |
| 14 | 2 k (4.0) | 2 e (1.5) | none | 7 |
| 15 | 2 k (4.0) | 2 f (1.5) | none | 32 |
| 16 | 2 k (4.0) | 2 g (1.5) | none | 44 |
| 17 | 2 k (4.0) | 2 h (1.5) | none | 87 |
| 18 | 2 k (5.5) | 3 d (1.5) | TMSOTf (1.5) | 88 |
| 19 [c] | 2 k (5.5) | 3 d (1.5) | TMSOTf (1.5) | 90 [d] |
| 20[e] | 2 k (5.5) | 3 d (1.5) | TMSOTf (1.5) | 14 |
| 21 | 2 k (5.5) | 3 e (1.5) | TMSOTf (1.5) | 34 |
| 22[c] | 2 k (2.0) | 3 d (1.5) | TMSOTf (1.5) | 27 |
| 23[c] | 2 k (3.0) | 3 d (1.5) | TMSOTf (1.5) | 81 |
| 24[c] | 2 k (4.0) | 3 d (1.5) | TMSOTf (1.5) | 85 |
| 25[c] | NaCN (5.5) | 3 d (1.5) | TMSOTf (1.5) | – |
| 26[c] | KCN (5.5) | 3 d (1.5) | TMSOTf (1.5) | – |
| 27[c] | Bu4NCN (5.5) | 3 d (1.5) | TMSOTf (1.5) | – |
[a] Reaction conditions: 1 a (0.20 mmol, 1.0 equiv), 2, oxidant, additive, DCE (2 mL), 70 °C, 15 h. [b] Yield determined by 1H NMR analysis using MeNO2 as an internal standard. [c] Conducted at 40 °C. [d] Isolated in 87 % yield. [e] Conducted at room temperature.
Figure 1.
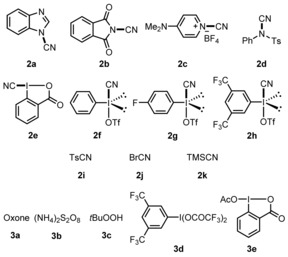
Various “CN” sources and oxidants tested. Ts=p‐tolylsulfonyl.
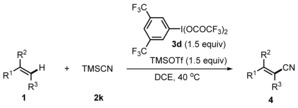
Under optimized reaction conditions, various alkenes were tested (Table 2). α‐Methylstyrenes possessing either electron‐withdrawing or electron‐donating groups at the para‐position of the arene ring afforded the acrylonitriles 4 b–l in moderate to high yields with good to excellent E selectivity. Generally, systems bearing electron‐withdrawing groups provided lower E selectivities in this series. An α‐methylstyrene bearing a meta‐substituent (4 m) worked well and the β‐naphthyl congener reacted with similar efficiency (4 n). Increasing the size of the α‐alkyl substituent in the styrene substrate leads to diminished selectivity, as shown by switching from α‐methylstyrene to the n‐propyl derivative (4 o), and reversed selectivity was obtained for the α‐cyclohexyl styrene (4 p).
Table 2.
Substrate scope.[a,b]
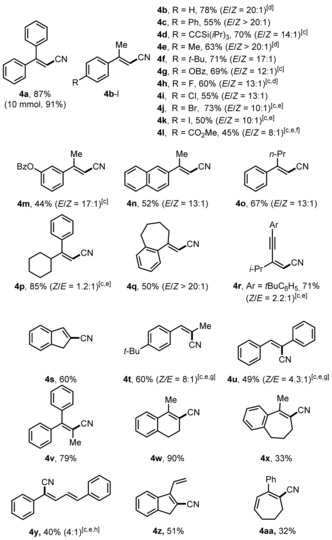
|
[a] Reaction conditions: 1 (0.20 mmol, 1.0 equiv), 2 k (1.1 mmol, 5.5 equiv), 3 d (0.30 mmol, 1.5 equiv), TMSOTf (0.30 mmol, 1.5 equiv), DCE (2 mL), 40 °C, 15 h. [b] Yields refer to the yield of the isolated major isomer if not otherwise noted. [c] Conducted at 70 °C. [d] These yields are based on 1H NMR analysis with MeNO2 as an internal standard. [e] Combined yield of both isomers. [f] 2 k (1.4 mmol, 7.0 equiv), 3 d (0.30 mmol, 1.5 equiv), TMSOTf (0.30 mmol, 1.5 equiv). [g] 2 k (0.30 mmol, 1.5 equiv), 2 h (0.30 mmol, 1.5 equiv). [h] The ratio of the major isomer to other isomers is given within parentheses.
A bicyclic styrene with an exocyclic double bond reacted with excellent selectivity (4 q), and 1,3‐enynes engage in this transformation, providing the Z product as major isomer (see 4 r; Table 2). The reaction works on β‐substituted styrenes and indene was converted into 4 s in a good yield. A trans‐β‐methylstyrene and trans‐stilbene reacted with complete regioselectivity and good stereoselectivity (4 t, 4 u). Both acyclic and cyclic trisubstituted styrenes could be converted (4 v–x) and conjugated dienes were also competent reaction partners, affording the monocyanated products with high regioselectivity (4 y–4 aa). To document the practicability, reaction of 1 a on gram scale gave 4 a in 91 % yield. Unfortunately, styrene did not react to cinnamonitrile.
Notably, Z selectivity of 4 p could be improved from 1.2:1 to 17:1 upon E→Z isomerization under UV‐irradiation (402 nm) following the Gilmour protocol in the presence of (−)‐riboflavin (Scheme 2).13 This method was also applied to isomerize (E)‐4 q to its Z derivative, showing that both isomers are accessible using our approach.
Scheme 2.
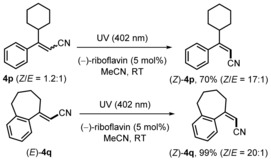
E→Z isomerization.
To elucidate the mechanism, control experiments were conducted. A radical pathway14 could be excluded based on the result obtained with the radical probe 1 ab, which reacted to give 4 ab without formation of any of the ring‐opening product 5 (Scheme 3 a). In the absence of Lewis acid, the cyanation did not occur, indicating that TMSOTf is essential for generating the active IIII species (Scheme 3 b, left). ArI(CN)OTf (2 h; 1.5 equiv) in combination with TMSCN (4 equiv) provided 4 a in comparable yield, showing that 2 h is a potentially active reagent that can be formed by reaction of 3 d with TMSCN and TMSOTf (Scheme 3 b, right). However, we found that 2 h is not an active IIII species, since cyanation of 1 a with 2 h did not provide 4 a, and 1 a decomposed (Scheme 3 c, left). Other nucleophilic CN reagents such as NaCN, KCN, and (nBu)4NCN did not work in combination with 2 h, showing the importance of the TMS moiety (Scheme 3 c, right). Replacing TMSCN by TMSCl provided the chlorination product 6 in 34 % yield, and 4 a was not formed (Scheme 3 d). The analogue Ph2IOTf was not a competent oxidant for cyanation of 1 a with TMSCN and the phenylated product 7 was also not identified (Scheme 3 e). These results revealed that the active IIII reagent is likely the highly electrophilic [bis(cyano)iodo]arene15 formed in situ from 2 h and TMSCN.
Scheme 3.
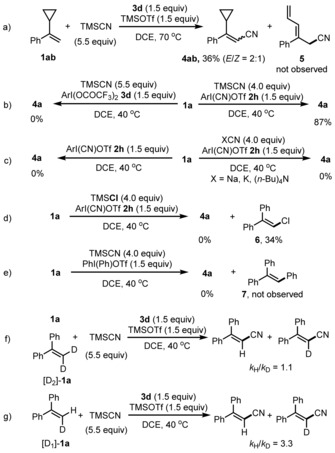
Mechanistic studies.
We also determined intermolecular and intramolecular kinetic isotope effects (KIEs). α‐Phenylstyrene (1 a) and [D2]‐1 a (1:1) were subjected to the reaction conditions and a k H/k D value of 1.1 was measured for the intermolecular KIE (Scheme 3 f). A significant intramolecular KIE of 3.3 was determined for cyanation of [D1]‐1 a (Scheme 3 g). These KIE experiments indicate that activation of the alkene by the IIII reagent is likely reversible and C−H bond cleavage is a slow step.16
Based on these investigations, the following mechanism is suggested (Scheme 4). The aryl(cyano)iodonium triflate 2 h, generated from 3 d by ligand exchange, undergoes renewed ligand exchange with TMSCN to give the active species [bis(cyano)iodo]arene A and TMSOTf. The electrophilic activation of alkene 1 with A, which is additionally supported by interaction with TMSOTf, then reversibly leads to the cyclic iodonium intermediate B.17 Regioselective ring‐opening of B by the cyanide anion provides the intermediate C and diastereoselective deprotonation of C in a conformation where the cyano group is positioned anti to the bulkier RL substituent, and eventually provides 4.
Scheme 4.
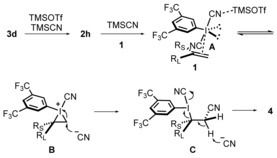
Suggested mechanism.
In summary, we have described a metal‐free C(sp2)−H cyanation of various alkenes using TMSCN in combination with the [bis(trifluoroacetoxy)iodo]arene 3 d. Reactions proceed under mild reaction conditions and show broad substrate scope: 1,1‐disubstituted, 1,2‐disubstituted, and trisubstituted alkenes are smoothly converted into the corresponding cyanated products in high yields and good to excellent diastereoselectivities.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the European Research Council (ERC Advanced Grant agreement No. 692640) for financial support.
X. Wang, A. Studer, Angew. Chem. Int. Ed. 2018, 57, 11792.
References
- 1.
- 1a. Larock R. C., Comprehensive Organic Transformations: A Guide to Functional Group Preparations, VCH, New York, 1989; [Google Scholar]
- 1b. Fleming F. F., Yao L., Ravikumar P. C., Funk L., Shook B. C., J. Med. Chem. 2010, 53, 7902–7917; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Kleemann A., Engel J., Kutscher B., Reichert D., Pharmaceutical Substances: Syntheses, Patents, Applications, 4th ed., Thieme, Stuttgart, 2001. [Google Scholar]
- 2.
- 2a. Rappoport Z., Chemistry of the Cyano Group, Wiley, London, 1970; [Google Scholar]
- 2b. Larock R. C., Comprehensive Organic Transformations: A Guide to Functional Group Preparations, Wiley, New York, 1999. [Google Scholar]
- 3.
- 3a. Sandmeyer T., Ber. Dtsch. Chem. Ges. 1884, 17, 1633–1635; [Google Scholar]
- 3b. Lindley J., Tetrahedron 1984, 40, 1433–1456; [Google Scholar]
- 3c. Galli C., Chem. Rev. 1988, 88, 765–792; For reviews: [Google Scholar]
- 3d. Anbarasan P., Schareina T., Beller M., Chem. Soc. Rev. 2011, 40, 5049–5067; [DOI] [PubMed] [Google Scholar]
- 3e. Ding S., Jiao N., Angew. Chem. Int. Ed. 2012, 51, 9226–9237; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 9360–9371; [Google Scholar]
- 3f. Kim J., Kim H. J., Chang S., Angew. Chem. Int. Ed. 2012, 51, 11948–11959; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 12114–12125; [Google Scholar]
- 3g. Yan G., Zhang Y., Wang J., Adv. Synth. Catal. 2017, 359, 4068–4105. Selected examples: [Google Scholar]
- 3h. Kaupp G., Schmeyers J., Boy J., Chem. Eur. J. 1998, 4, 2467–2474; [Google Scholar]
- 3i. Luo F.-H., Chu C., Cheng C.-H., Organometallics 1998, 17, 1025–1030; [Google Scholar]
- 3j. Sundermeier M., Mutyala S., Zapf A., Spannenberg A., Beller M., J. Organomet. Chem. 2003, 684, 50–55; [Google Scholar]
- 3k. Sato N., Yue Q., Tetrahedron 2003, 59, 5831–5836; [Google Scholar]
- 3l. Anbarasan P., Neumann H., Beller M., Angew. Chem. Int. Ed. 2011, 50, 519–522; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 539–542; [Google Scholar]
- 3m. Jiang Z., Huang Q., Chen S., Long L., Zhou X., Adv. Synth. Catal. 2012, 354, 589–592; [Google Scholar]
- 3n. Okamoto K., Sakata N., Ohe K., Org. Lett. 2015, 17, 4670–4673. [DOI] [PubMed] [Google Scholar]
- 4.DG-assisted arene C−H cyanation:
- 4a. Chen X., Hao X.-S., Goodhue C. E., Yu J.-Q., J. Am. Chem. Soc. 2006, 128, 6790–6791; [DOI] [PubMed] [Google Scholar]
- 4b. Jia X., Yang D., Zhang S., Cheng J., Org. Lett. 2009, 11, 4716–4719; [DOI] [PubMed] [Google Scholar]
- 4c. Kim J., Chang S., J. Am. Chem. Soc. 2010, 132, 10272–10274; [DOI] [PubMed] [Google Scholar]
- 4d. Chaitanya M., Yadagiri D., Anbarasan P., Org. Lett. 2013, 15, 4960–4963; [DOI] [PubMed] [Google Scholar]
- 4e. Gong T.-J., Xiao B., Cheng W.-M., Su W., Xu J., Liu Z.-J., Liu L., Fu Y., J. Am. Chem. Soc. 2013, 135, 10630–10633; [DOI] [PubMed] [Google Scholar]
- 4f. Liu W., Ackermann L., Chem. Commun. 2014, 50, 1878–1881; [DOI] [PubMed] [Google Scholar]
- 4g. Yu D. G., Gensch T., Azambuja F., Céspedes S. V., Glorius F., J. Am. Chem. Soc. 2014, 136, 17722–17725; [DOI] [PubMed] [Google Scholar]
- 4h. Pawar A. B., Chang S., Org. Lett. 2015, 17, 660–663; [DOI] [PubMed] [Google Scholar]
- 4i. Li J., Ackermann L., Angew. Chem. Int. Ed. 2015, 54, 3635–3638; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 3706–3709. [Google Scholar]
- 5.Indole C−H cyanation:
- 5a. Yan G., Kuang C., Zhang Y., Wang J., Org. Lett. 2010, 12, 1052–1055; [DOI] [PubMed] [Google Scholar]
- 5b. Yang Y., Zhang Y., Wang J., Org. Lett. 2011, 13, 5608–5611; [DOI] [PubMed] [Google Scholar]
- 5c. Ding S.-T., Jiao N., J. Am. Chem. Soc. 2011, 133, 12374–12377; [DOI] [PubMed] [Google Scholar]
- 5d. Kim J., Kim H., Chang S., Org. Lett. 2012, 14, 3924–3927; [DOI] [PubMed] [Google Scholar]
- 5e. Okamoto K., Watanabe M., Murai M., Hatano R., Ohe K., Chem. Commun. 2012, 48, 3127–3129. [DOI] [PubMed] [Google Scholar]
- 6.Direct C−H cyanation of arenes:
- 6a. Shu Z., Ji W., Wang X., Zhou Y., Zhang Y., Wang J., Angew. Chem. Int. Ed. 2014, 53, 2186–2189; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2218–2221; [Google Scholar]
- 6b. McManus J. B., Nicewicz D. A., J. Am. Chem. Soc. 2017, 139, 2880–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Transition-metal-catalyzed carbocyanations:
- 7a. Nakao Y., Oda S., Hiyama T., J. Am. Chem. Soc. 2004, 126, 13904–13905; [DOI] [PubMed] [Google Scholar]
- 7b. Nakao Y., Yukawa T., Hirata Y., Oda S., Satoh J., Hiyama T., J. Am. Chem. Soc. 2006, 128, 7116–7117; [DOI] [PubMed] [Google Scholar]
- 7c. Arai S., Sato T., Koike Y., Hayashi M., Nishida A., Angew. Chem. Int. Ed. 2009, 48, 4528–4531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 4598–4601; [Google Scholar]
- 7d. Hirata Y., Yada A., Morita E., Nakao Y., Hiyama T., Ohashi M., Ogoshi S., J. Am. Chem. Soc. 2010, 132, 10070–10077; [DOI] [PubMed] [Google Scholar]
- 7e. Nakao Y., Yada A., Ebata S., Hiyama T., J. Am. Chem. Soc. 2007, 129, 2428–2429; [DOI] [PubMed] [Google Scholar]
- 7f. Rondla N. R., Levi S. M., Ryss J. M., Vanden Berg R. A., Douglas C. J., Org. Lett. 2011, 13, 1940–1943; [DOI] [PubMed] [Google Scholar]
- 7g. Arai S., Amako Y., Yang X., Nishida A., Angew. Chem. Int. Ed. 2013, 52, 8147–8150; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8305–8308; [Google Scholar]
- 7h. Wang F., Wang D., Wan X., Wu L., Chen P., Liu G., J. Am. Chem. Soc. 2016, 138, 15547–15550. [DOI] [PubMed] [Google Scholar]
- 8.Transition-metal-catalyzed heterocyanations:
- 8a. Chatani N., Hanafusa T., J. Chem. Soc. Chem. Commun. 1985, 838–839; [Google Scholar]
- 8b. Chatani N., Horiuchi N., Hanafusa T., J. Org. Chem. 1990, 55, 3393–3395; [Google Scholar]
- 8c. Suginome M., Kinugasa H., Ito Y., Tetrahedron Lett. 1994, 35, 8635–8638; [Google Scholar]
- 8d. Obora Y., Baleta A. S., Tokunaga M., Tsuji Y., J. Organomet. Chem. 2002, 660, 173–177; [Google Scholar]
- 8e. Suginome M., Yamamoto A., Murakami M., J. Am. Chem. Soc. 2003, 125, 6358–6359; [DOI] [PubMed] [Google Scholar]
- 8f. Suginome M., Yamamoto A., Murakami M., Angew. Chem. Int. Ed. 2005, 44, 2380–2382; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 2432–2434; [Google Scholar]
- 8g. Murai M., Hatano R., Kitabata S., Ohe K., Chem. Commun. 2011, 47, 2375–2377; [DOI] [PubMed] [Google Scholar]
- 8h. Koester D. C., Kobayashi M., Werz D. B., Nakao Y., J. Am. Chem. Soc. 2012, 134, 6544–6547; [DOI] [PubMed] [Google Scholar]
- 8i. Wang X., Studer A., J. Am. Chem. Soc. 2016, 138, 2977–2980. [DOI] [PubMed] [Google Scholar]
- 9.Transition-metal-catalyzed hydrocyanation:
- 9a. Funabiki T., Yamazaki Y., Tarama K., J. Chem. Soc. Chem. Commun. 1978, 63–65; [Google Scholar]
- 9b. Jackson W. R., Lovel C. G., J. Chem. Soc. Chem. Commun. 1982, 1231–1232; [Google Scholar]
- 9c. Alonso P., Pardo P., Galván A., Fañanás F. J., Rodríguez F., Angew. Chem. Int. Ed. 2015, 54, 15506–15510; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 15726–15730; [Google Scholar]
- 9d. Ye F., Chen J., Ritter T., J. Am. Chem. Soc. 2017, 139, 7184–7187. [DOI] [PubMed] [Google Scholar]
- 10.Directing-group-assisted alkene C−H cyanation:
- 10a. Chaitanya M., Anbarasan P., Org. Lett. 2015, 17, 3766–3769; [DOI] [PubMed] [Google Scholar]
- 10b. Su W., Gong T.-J., Xiao B., Fu Y., Chem. Commun. 2015, 51, 11848–11851. [DOI] [PubMed] [Google Scholar]
- 11.Electrophilic cyanation:
- 11a. Dohi T., Morimoto K., Takenaga N., Goto A., Maruyama A., Kiyono Y., Tohma H., Kita Y., J. Org. Chem. 2007, 72, 109–116; [DOI] [PubMed] [Google Scholar]
- 11b. Anbarasan P., Neumann H., Beller M., Chem. Eur. J. 2010, 16, 4725–4728; [DOI] [PubMed] [Google Scholar]
- 11c. Brand J. P., González D. F., Nicolai S., Waser J., Chem. Commun. 2011, 47, 102–115; [DOI] [PubMed] [Google Scholar]
- 11d. Kamijo S., Hoshikawa T., Inoue M., Org. Lett. 2011, 13, 5928–5931; [DOI] [PubMed] [Google Scholar]
- 11e. Yang Y., Buchwald S. L., Angew. Chem. Int. Ed. 2014, 53, 8677–8681; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8821–8825; [Google Scholar]
- 11f. Frei R., Courant T., Wodrich M. D., Waser J., Chem. Eur. J. 2015, 21, 2662–2668; [DOI] [PubMed] [Google Scholar]
- 11g. Vita M. V., Caramenti P., Waser J., Org. Lett. 2015, 17, 5832–5835; [DOI] [PubMed] [Google Scholar]
- 11h. Talavera G., Peña J., Alcarazo M., J. Am. Chem. Soc. 2015, 137, 8704–8707; [DOI] [PubMed] [Google Scholar]
- 11i. Reeves J. T., Malapit C. A., Buono F. G., Sidhu K. P., Marsini M. A., Sader C. A., Fandrick K. R., Busacca C. A., Senanayake C. H., J. Am. Chem. Soc. 2015, 137, 9481–9488; [DOI] [PubMed] [Google Scholar]
- 11j. Zhao W., Montgomery J., J. Am. Chem. Soc. 2016, 138, 9763–9766. [DOI] [PubMed] [Google Scholar]
- 12. Zhdankin V. V., Scheuller M. C., Stang P. J., Tetrahedron Lett. 1993, 34, 6853–6856. [Google Scholar]
- 13. Metternich J. B., Artiukhin D. G., Holland M. C., von Bremen-Kühne M., Neugebauer J., Gilmour R., J. Org. Chem. 2017, 82, 9955–9977. [DOI] [PubMed] [Google Scholar]
- 14. Wang X., Studer A., Acc. Chem. Res. 2017, 50, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frohn H.-J., Hirschberg M. E., Boese R., Bläser D., Flörke U., Z. Anorg. Allg. Chem. 2008, 634, 2539–2550. [Google Scholar]
- 16. Simmons E. M., Hartwig J. F., Angew. Chem. Int. Ed. 2012, 51, 3066–3072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 3120–3126. [Google Scholar]
- 17.
- 17a. Dohi T., Ito M., Yamaoka N., Morimoto K., Fujioka H., Kita Y., Angew. Chem. Int. Ed. 2010, 49, 3334–3337; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 3406–3409; [Google Scholar]
- 17b. Wang F., Wang D., Mu X., Chen P., Liu G., J. Am. Chem. Soc. 2014, 136, 10202–10205. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary