

Abstract
Objective
To assess the safety and efficacy of 2 repeated intrathecal injections of autologous bone marrow–derived mesenchymal stem cells (BM‐MSCs) in amyotrophic lateral sclerosis (ALS).
Methods
In a phase 2 randomized controlled trial (NCT01363401), 64 participants with ALS were randomly assigned treatments (1:1) of riluzole alone (control group, n = 31) or combined with 2 BM‐MSC injections (MSC group, n = 33). Safety was assessed based on the occurrence of adverse events. The primary efficacy outcome was changes in Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS‐R) score from baseline to 4 and 6 months postinjection. Post hoc analysis includes investigation of cerebrospinal fluid biomarkers and long‐term survival analysis.
Results
Safety rating showed no groupwise difference with absence of serious treatment‐related adverse events. Mean changes in ALSFRS‐R scores from baseline to 4 and 6 months postinjection were reduced in the MSC group compared with the control group (4 months: 2.98, 95% confidence interval [CI] = 1.48–4.47, p < 0.001; 6 months: 3.38, 95% CI = 1.23–5.54, p = 0.003). The MSC group showed decreased proinflammatory and increased anti‐inflammatory cytokines. In good responders, transforming growth factor β1 significantly showed inverse correlation with monocyte chemoattractant protein‐1. There was no significant difference in long‐term survival between groups.
Interpretation
Repeated intrathecal injections of BM‐MSCs demonstrated a possible clinical benefit lasting at least 6 months, with safety, in ALS patients. A plausible action mechanism is that BM‐MSCs mediate switching from pro‐ to anti‐inflammatory conditions. A future randomized, double‐blind, large‐scale phase 3 clinical trial with additional BM‐MSC treatments is required to evaluate long‐term efficacy and safety. Ann Neurol 2018;84:361–373
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder characterized by selective and progressive loss of motor neurons. Disease progression leads to death within 2 to 4 years.1 The pathophysiological mechanisms of cell death (mostly of motor neurons) in ALS remain unclear, but recent studies using models of SOD1 mutant and ALS‐associated genes including C9orf72, FUS, TDP43, UBQLN2, and TBK1 revealed that diverse molecular mechanisms such as altered protein degradation, RNA dysregulation, oxidative stress, glutamate toxicity, mitochondrial dysfunction, altered immunoinflammation, and abnormal axonal transport are responsible for motor neuronal cell death.2, 3, 4 The concept of a non–cell‐autonomous mechanism resulting in motor neuronal cell death supports further study of immunoinflammatory modulation as a stratergy for clinical therapeutics. Previous ALS clinical trials based on single molecular targets suggest the importance of integration of multiple molecular targets in the overall therapeutic strategy. Mesenchymal stem cell (MSC) therapy is a desired approach for addressing this issue. MSCs exert diverse actions, such as stimulating intrinsic neurogenesis, releasing diverse neurotrophic factors, and modulating immunoinflammatory processes.5, 6
The wide range of MSC effects could be incorporated in an alternative treatment strategy for ALS. Moreover, autologous MSCs have multiple advantages in clinical practice, especially in relation to ethical concerns, lack of possible tumorigenesis, and graft rejections.7 MSCs regulate both innate and adaptive immune cells, through the release of soluble factors such as prostaglandin E2, indoleamine 2,3‐dioxygenase, and transforming growth factor (TGF)‐β, thereby switching the patient's environment from a proinflammatory and toxic one to an anti‐inflammatory and neuroprotective condition.8, 9 Recently, we reported that immunoregulatory mechanisms of MSCs, such as elevation of regulatory T cells (Tregs) and T helper‐2 cells, play important roles in the mediation of neuroprotective effects on motor neuronal cell death in ALS, in a manner similar to the secretion of neurotrophic factors that are crucial to the effectiveness of MSCs in ALS.8 In addition, MSCs can modulate the functional properties of microglia via TGF‐β secretion, switching them from a classically activated phenotype to an inflammation‐resolving phenotype. These effects of MSCs could be an important therapeutic strategy to inhibit toxic neuroinflammatory processes in the symptomatic stage of ALS.2, 8, 9
In our previous phase 1 trial, we reported the safety and feasibility of 2 repeated intrathecal injections of autologous bone marrow–derived (BM)‐MSCs over 12 months.10 Furthermore, we reported that factors such as TGF‐β, angiogenin (ANG), and vascular endothelial growth factor (VEGF), cytokines secreted by BM‐MSCs, play crucial roles in the response of an ALS patient to intrathecal autologous BM‐MSC injection.11 Based on the action mechanisms of MSCs and findings from an in vivo transgenic mouse study, we hypothesized that repeated intrathecal BM‐MSC administration could be a valuable therapeutic strategy for ALS.12
Herein, we conducted a phase 2 clinical trial in patients with ALS to evaluate the safety and efficacy of 2 repeated intrathecal injections of BM‐MSCs for up to 4 months. In addition to this protocol, we evaluated the safety and efficacy for an additional 2 months (total of 6 months), in accordance with Korean Ministry of Food and Drug Safety (KMFDS) recommendations. To further understand the mechanism underlying the effectiveness of BM‐MSC therapy in ALS patients, post hoc analyses of cytokines in cerebrospinal fluid (CSF) that reflect the immunomodulatory effects were conducted. In addition, we evaluated the long‐term safety and survival benefit of 2 repeated autologous BM‐MSC treatments for up to a maximum 75 months to determine optimal protocols in a planned phase 3 clinical trial.
Patients and Methods
Study Design
This study was a parallel‐group, randomized, and controlled phase 2 trial (Fig 1A and Supplementary Table 1) performed at Hanyang University Hospital (Seoul, Republic of Korea), a tertiary referral center for ALS. The study was conducted in 2 phases: first within a 3‐month lead‐in period, and the second within a 4‐month initial follow‐up period, with an additional 2‐month follow‐up, for a total follow‐up of 6‐months.
Figure 1.
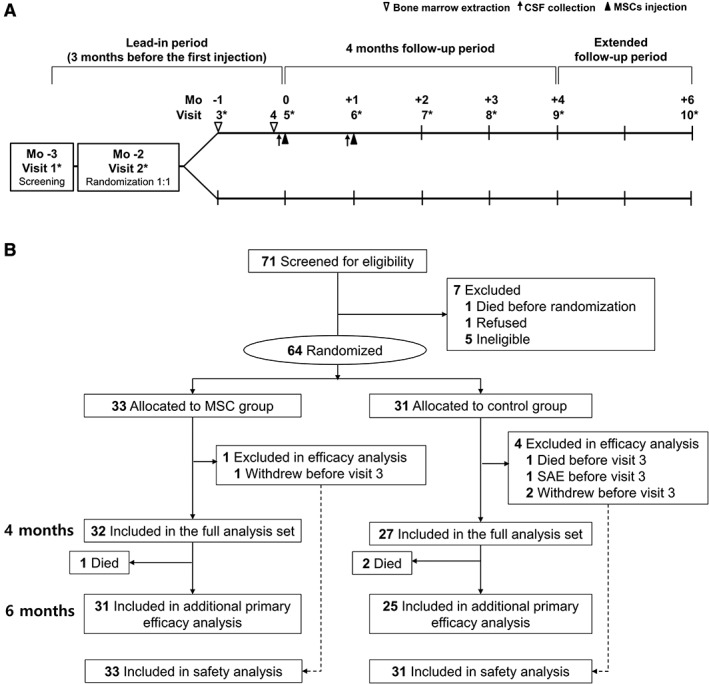
Study design and trial profile. (A) The study design. (B) Scheduling of screening, randomization, treatment, and follow‐up of the participants. The full analysis set was defined as all randomized participants with baseline data and at least 1 efficacy value. *Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised score was assessed for the primary efficacy outcome. CSF = cerebrospinal fluid; MSC = mesenchymal stem cell; SAE = serious adverse event.
Participants and Study Approval
Participants (age = 25–75 years) were diagnosed with clinically probable or definite ALS according to the revised El Escorial criteria.13 Other inclusion criteria were as follows: (1) Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS‐R) score between 31 and 46, (2) stable riluzole treatment (50mg, twice daily) for at least 3 months before screening, and (3) disease duration no longer than 5 years after the onset of the first symptom. Participants were excluded if they met any of the following criteria: (1) participation in other clinical trials within the past 12 months, (2) forced vital capacity (FVC) of <40% of the predicted value, (3) presence of any comorbidity that might interfere with the outcome, (4) tracheostomy or noninvasive ventilation, (5) any hemorrhagic tendency, and (6) administration of any drug that could affect the bone marrow. Participants were enrolled between December 2011 and November 2012, and follow‐up of patients was conducted until July 2013.
The clinical trial protocol was approved and monitored by the local institutional review board (HYUH IRB 2010‐C‐70) and the KMFDS (MFDS‐2413). This study was registered at http://ClinicalTrials.gov (NCT01363401). The overall trial‐related activities and documents were monitored by the clinical research organization (Dream Clinical Investigation Services, Seoul, Korea). An external trial monitor was enlisted to protect the rights and well‐being of the participants, verify the accuracy of the trial data, and guarantee compliance with Good Clinical Practice guidelines. Written informed consent was obtained from all participants before screening.
Randomization and Masking
All eligible participants were randomized (1:1) into 2 groups using the interactive Web response system: (1) MSC group (receiving 2 injections of BM‐MSCs at a 26‐day interval, 1 × 106 cells/kg) and (2) control group. Patients from both groups received continuous riluzole treatment (100mg/day), and symptomatic treatments were also allowed in both groups. In the control group, the sham procedures related with stem cell therapy including BM aspiration, CSF collection for BM‐MSC suspension, and lumbar puncture were not performed due to ethical considerations. Assessment of ALSFRS‐R score, Appel ALS Rating Scale (AALS), and FVC were performed by neurologists and evaluators who were blinded to treatment assignments.
Procedures
The procedures comprised 2 bone marrow extractions (BMEs), at Visit 3 (−1 month) and Visit 4 (2 days prior to Visit 5), and 2 intrathecal injections, at Visit 5 (0 months) and Visit 6 (+1 month; see Fig 1A). To allow sufficient time for ex vivo MSC expansion, each BME was performed 28 days prior to each BM‐MSC injection.
BM‐MSCs were isolated, expanded, and analyzed under Good Manufacturing Practice (GMP) conditions at CORESTEM Inc. (Seoul, Korea), based on the International Society of Cellular Therapy guidelines.14
BM mononuclear cells were isolated using Ficoll (Ficoll‐Paque Premium; GE Healthcare Bio‐Sciences, Uppsala, Sweden) density gradient centrifugation. The mononuclear cells (2 × 105 cells) were placed in a 175cm2 flask (Thermo Scientific Nunc, Roskilde, Denmark) and cultured in CSBM‐A06 medium (CORESTEM Inc.) containing 10% fetal bovine serum (Life Technologies, Grand Island, NY), 2.5mM L‐alanyl‐L‐glutamine (Biochrom, Berlin, Germany), and 1% penicillin–streptomycin (Biochrom) in a humidified incubator at 37°C with 7% CO2. The nonadherent cells were removed after the initial plating, and fresh medium was replaced twice per week. Cells were harvested at 80% confluency using 0.125% trypsin–ethylenediaminetetraacetic acid (Life Technologies). To confirm sterility, the samples were cultured for bacteria, fungi, viruses, and mycoplasma, and real‐time polymerase chain reaction was also performed to detect contaminating mycoplasma. No evidence of bacterial, fungal, viral, or mycoplasmal contamination was found. BM‐MSC cultures were characterized by phenotypic analyses of cell surface antigens using flow cytometry. Cultures displayed >98% of CD29 (BD Pharmingen, Franklin Lakes, NJ; catalog 555443), CD44 (BD Biosciences, Franklin Lakes, NJ; catalog 550989), CD73 (BD Pharmingen, catalog 550257), and CD105 (BD Pharmingen, catalog 560839), and <0.1% of CD34 (BD Biosciences, catalog 348057) and CD45 (BD Pharmingen, catalog 555483) expression (BD FACS Canto II).10 In addition to evidence of sterility and MSC characteristics, a minimum requirement of therapeutic efficacy of BM‐MSCs was confirmed by VEGF (R&D Systems, Minneapolis, MN; catalog DVE00, Human VEGF Immunoassay) level > 170.0pg per 1 × 104 cells, as determined using an enzyme‐linked immunosorbent assay assay and quantitate with a Bio‐Rad Laboratories (Hercules, CA) xMark Microplate Absorbance Spectrophotometer.
One day before each BM‐MSC injection, each MSC group participant's CSF (approximately 20–30ml) was collected via lumbar puncture for the purpose of suspension of BM‐MSCs and post hoc analysis of cytokines. At that time, BM‐MSCs were isolated after repeated washing out procedures of cultured media with phosphate‐buffered saline, and these cells were suspended with CSF in the GMP facility. BM‐MSCs were supplied with different volumes as a suspended state in a 5ml syringe with a concentration of 1 × 107 cells per ml of CSF and total number of MSCs adjusted by body weight (1 × 106 cells per kg).
BM‐MSCs were delivered to the hospital at 2 to 8°C and were administered to the participant within 12 hours from completion of suspension (BM‐MSC characteristics are shown in Supplementary Table 2; injected volume and number of BM‐MSCs are shown in Supplementary Table 3). Using a standard lumbar puncture at the level of L2–L4, MSCs were slowly injected over a period of approximately 2 minutes. Subsequently, participants remained in the Trendelenburg position with the application of a mechanical vibrator on their hip bone for 2 hours.
Safety Assessments
The safety of the treatment was evaluated based on the occurrence of adverse events (AEs), serious AEs (SAEs), and laboratory abnormalities, as defined by the CONSORT (Consolidated Standards of Reporting Trials) group.15 We monitored AEs and SAEs monthly for 4 months from Visit 5 to Visit 9, and for an additional 2 months at Visit 10. The Common Terminology Criteria for Adverse Events (v3.0) was used to evaluate the AEs as grade I–V.16 Vital signs and physical examinations were performed at 6‐hour intervals for 48 hours after BM‐MSC injection. The patient was discharged if there were no SAEs or if AEs persisted only transiently. Physical and neurological examinations were performed at every visit. Assessment of laboratory abnormalities was performed at Visit 1, Visit 5, and Visit 9 and included complete blood counts, blood chemistry, renal function, liver function, and urine content.
Efficacy Outcomes Assessments
The mean change of ALSFRS‐R score from Visit 5 (baseline, 0 months) to Visit 9 (+4 months) was considered the primary efficacy outcome for functional assessment. After the trial was approved, the KMFDS recommended adding the mean change of the ALSFRS‐R score from Visit 5 to Visit 10 (+6 months) as an additional primary efficacy outcome. The ALSFRS‐R score (48 [normal] to 0 [maximally impaired]) was assessed throughout a 3‐month lead‐in period and 6‐month follow‐up period as shown in Figure 1A.
Secondary efficacy outcomes consisted of the changes in slope (monthly rate of decline) of ALSFRS‐R between the lead‐in and follow‐up periods, and responder analyses at 4 and 6 months after treatment. Additional secondary efficacy outcomes were the mean change of AALS (30 [normal] to 164 [maximally impaired]) scores at 4 months, mean changes of FVC at 4 months, mean changes of 36‐Item Short‐Form Health Survey (SF‐36) at 4 months, and changes in slope of AALS score and FVC between the lead‐in and follow‐up periods. AALS score, FVC, and SF‐36 were assessed at Visit 1, Visit 5, and Visit 9. The percentage changes in ALSFRS‐R slope between the lead‐in and follow‐up periods (4 and 6 months) were calculated to identify responders to the MSC treatment. Good responders were defined as those whose decline in ALSFRS‐R slope showed ≥50% improvement compared to the slope of the lead‐in period according to the modified concept of the survey of the Northeast ALS Consortium,17 whereas poor responders were defined as participants with <50% improvement. In our analysis, we included deceased participants in the classification of poor responders.
The dates of percutaneous endoscopic gastrostomy, noninvasive ventilation, and tracheostomy were also recorded. In some participants, the clinical status was acquired over the telephone.
Post hoc Analysis on CSF Cytokines, Genetics, and Long‐Term Effectiveness
After completion of the clinical trial, a series of post hoc analyses were conducted for evaluation of the following: (1) the relationship between changes of CSF cytokine profiles and the responsiveness to stem cell therapy; (2) genetics of the common causative genes of ALS to exclude the possibility of genetic heterogeneity in enrolled participants with sporadic ALS; and (3) long‐term safety and effectiveness of single cycle of BM‐MSC treatment, focusing on the survival analysis, with a view to facilitate optimal design of future large‐scale clinical trials. CSF biomarkers were analyzed to understand the immunomodulatory effects of BM‐MSCs as described previously.10 Remnant CSF was collected just before the first (Visit 5) and second (Visit 6) treatment and immediately stored at −80°C until measurement of cytokines was performed by multiplex assay (Bio‐Rad Laboratories). The levels of TGF‐β1, TGF‐β2, TGF‐β3, IL‐6, IL‐10, IL‐1β, monocyte chemoattractant protein‐1 (MCP‐1), and tumor necrosis factor (TNF)‐α were measured before and after treatment. To evaluate genetic heterogeneity, the C9orf72 repeat expansions were tested by using a 2‐step polymerase chain reaction protocol.18 Furthermore, the pathogenic variants in SOD1, FUS, TARDBP, ANG, and OPTN genes were screened by conventional Sanger sequencing as described previously.19 Lastly, we evaluated the survival analysis using the Kaplan–Meier method. Survival times (in months) were considered from the initial screening (Visit 1) to death or censoring date of February 28, 2018, for up to a maximum of 75 months. Tracheostomy‐free survival was also analyzed. An exploratory post hoc subgroup analysis was performed to determine which subgroups have a possible survival benefit. The subgroups selected were based on important clinical variables (sex, age at screening, site of onset). Age at screening was dichotomized according to its mean value.
Statistical Analysis
A sample size of 32 participants per group was needed to detect a 2.28 difference (≥43%) in the mean change of ALSFRS‐R total score at 4 months from baseline between the 2 groups with 80% power, 1‐sided type I error of 2.5, and a 10% dropout rate. The statistical parameters were based on a previous investigator‐initiated trial.20
All participants were included in the safety analysis. All AEs were categorized according to their grade, the affected organ system, and the specific event. The efficacy analyses were based on the intention‐to‐treat principle and included the full analysis set, defined as all randomized participants with a baseline and at least 1 follow‐up efficacy value. Baseline characteristics, efficacy, and safety outcomes were analyzed using the chi‐square (or Fisher exact) test for categorical variables and Student t tests for continuous variables. Continuous variables were summarized as means and standard deviation (SD), whereas categorical variables were described with absolute value and relative frequency.
Post hoc analysis was conducted to support primary efficacy outcomes using the piecewise linear mixed model. These linear mixed models included 4 mixed effects to consider within‐patient deviations, in addition to the fixed effect, the intercept, and the slope, from the lead‐in period and after treatment for each group.21
In the CSF biomarker analysis, data were represented as the mean and standard error (SE). Differences in cytokine levels between the 2 time points were analyzed using paired t tests. The receiver operating characteristic (ROC) curves and area under the ROC curve (AUC) were used to assess the sensitivity and specificity of the CSF biomarkers. Linear regression analysis was applied for correlation between CSF cytokines. As a post hoc analysis, survival analysis used the Kaplan–Meier method with the log‐rank test to compare the distributions of the time to an event between the MSC group and the control group. Participants not reaching the survival endpoint or participants who enrolled in another MSC therapy after completion of the follow‐up period of this trial were censored.
SAS 9.4 (SAS Institute, Cary, NC) was used for efficacy and safety analyses, whereas SPSS 17.0 (SPSS, Chicago, IL) and Prism 6.0 (GraphPad Software, San Diego, CA) were used for survival and CSF biomarker analyses.
Results
Between December 5, 2011 and November 22, 2012, a total of 71 participants with ALS were screened, of whom 7 were excluded (see Fig 1B). Of the remaining 64 participants (mean age = 53.3 years; 33 men and 31 women), 33 were allocated to receive BM‐MSCs with riluzole treatment (MSC group) and 31 received riluzole alone (control group). A total of 59 participants at 4 months and 56 participants at 6 months were included in the primary efficacy analysis (full analysis set). Four participants of the control group (withdrawal, 2; death, 1; SAE, 1) and 1 patient of the MSC group (withdrawal) were excluded from the full analysis set because these events occurred before Visit 5 (baseline; see Fig 1B).
Both the control and the MSC group had similar baseline characteristics (Table 1) and laboratory findings (Supplementary Table 4). All participants were diagnosed with sporadic ALS without family history.
Table 1.
Demographics and Baseline Characteristics of the Full Analysis Set Population
| Characteristic | MSC Group, n = 32 | Control Group, n = 27 | p |
|---|---|---|---|
| Male/Female, n (%) | 18 (56%)/14 (44%) | 11 (41%)/16 (59%) | 0.235 |
| Age at screening, yr | 53.7 (7.7) | 52.5 (9.4) | 0.598 |
| Duration of symptoms, mo | 22.4 (10.6) | 24.0 (11.5) | 0.581 |
| Time from diagnosis to baseline, mo | 11.7 (8.8) | 10.8 (6.9) | 0.667 |
| Limb/bulbar onset, n (%) | 23 (72%)/9 (28%) | 21 (78%)/6 (22%) | 0.604 |
| Family history of amyotrophic lateral sclerosis, n (%) | 0 (0%) | 0 (0%) | — |
| ALSFRS‐R at baseline | 35.5 (4.2) | 34.7 (5.5) | 0.538 |
| AALS at baseline | 80.8 (17.4) | 89.2 (23.8) | 0.124 |
| FVC at baseline, % | 73.1 (21.1) | 71.3 (16.1) | 0.727 |
| SF‐36 at baseline | 42.4 (14.8) | 41.8 (17.2) | 0.893 |
| Slope in ALSFRS‐R during lead‐in period, per month | −1.55 (1.21) | −1.20 (1.35) | 0.294 |
| Slope in AALS during lead‐in period, per month | 4.21 (2.98) | 4.47 (3.60) | 0.759 |
| Slope in FVC during lead‐in period, per month | −1.39 (2.33) | −1.89 (3.04) | 0.493 |
| Concomitant riluzole, n (%) | 32 (100%) | 27 (100%) | — |
Data are mean (standard deviation) for continuous variables, or n (%) for categorical variables. Baseline was obtained at Visit 5 (just prior to the first MSC injection).
AALS = Appel ALS Rating Scale; ALSFRS‐R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (48 [normal] to 0 [maximally impaired]); FVC = forced vital capacity; MSC = mesenchymal stem cell; SF‐36 = 36‐Item Short‐Form Health Survey.
Safety Assessments
The overall incidence of AEs during the 4‐ and 6‐month follow‐up periods was not significantly different between groups (4 months: 20 [61%] of 33 in MSC vs 22 [71%] of 31 in controls, p = 0.383; 6 months: 21 [64%] of 33 in MSC vs 23 [74%] of 31 in controls, p = 0.363; Supplementary Table 5). The common AEs in the MSC group at 6 months were influenzalike illness (n = 7), back pain (n = 5), and musculoskeletal pain (n = 5).
The incidence of adverse drug reactions (ADRs) was 9% (3/33, 4 events) in the MSC group (Table 2), and included headache (2 events), pyrexia (1 event), and pain (1 event); these ADRs were mild and transient, occurred within 48 hours postinjection, and were self‐limited or subsided within 48 hours of treatment with simple analgesics. Additionally, there were no treatment‐related AEs during the extended 2‐month follow‐up. The incidence of SAEs during the entire follow‐up period was 9% (3/33, 3 events) in the MSC group versus 19% (6/31, 6 events) in the control group; SAEs in the MSC group were not considered to be treatment‐related. Four deaths occurred during the 6‐month follow‐up: 1 in the MSC group (respiratory failure at 5 months postinjection, related to disease progression) and 3 in the control group (2 of respiratory failure and 1 of sudden cardiac arrest before Visit 3). There were no clinically significant changes in the laboratory tests after BM‐MSC treatment (see Supplementary Table 4).
Table 2.
Summary of Adverse Drug Reaction and Serious Adverse Events for All Enrolled Participants
| 4 Months | 6 Months | |||||||
|---|---|---|---|---|---|---|---|---|
| MSC Group, n = 33 | Control Group, n = 31 | MSC Group, n = 33 | Control Group, n = 31 | |||||
| Adverse Reaction/Event | Event | Participants, n (%) | Event | Participants, n (%) | Event | Participants, n (%) | Event | Participants, n (%) |
| Adverse drug reactions, total | 4 | 3 (9) | 0 | 0 (0) | 4 | 3 (9) | 0 | 0 (0) |
| General disorders and administration site conditions | 2 | 2 (6) | 0 | 0 (0) | 2 | 2 (6) | 0 | 0 (0) |
| Pyrexia | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Pain | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Nervous system disorders | 2 | 2 (6) | 0 | 0 (0) | 2 | 2 (6) | 0 | 0 (0) |
| Headache | 2 | 2 (6) | 0 | 0 (0) | 2 | 2 (6) | 0 | 0 (0) |
| Serious adverse events, total | 2 | 2 (6) | 3 | 3 (9) | 3 | 3 (9) | 6 | 6 (19) |
| Musculoskeletal and connective tissue disorders | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 1 | 1 (3) |
| Rhabdomyolysis | 0 | 0 (0) | 0 | 0 (0) | 0 | 0 (0) | 1 | 1 (3) |
| Infections and infestations | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Pyelonephritis, acute | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Injury, poisoning, and procedural complications | 0 | 0 (0) | 2 | 2 (7) | 0 | 0 (0) | 2 | 2 (6) |
| Contusion | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) |
| Ankle fracture | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) |
| Respiratory, thoracic, and mediastinal disorders | 0 | 0 (0) | 0 | 0 (0) | 1 | 1 (3) | 2 | 2 (6) |
| Respiratory failure | 0 | 0 (0) | 0 | 0 (0) | 1 | 1 (3) | 2 | 2 (6) |
| Metabolism and nutrition disorders | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Hyponatremia | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) |
| Cardiac disorders | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) |
| Cardiac arrest | 0 | 0 (0) | 1 | 1 (3) | 0 | 0 (0) | 1 | 1 (3) |
Four deaths occurred during the entire follow‐up period: 1 in the MSC group (5 months after the first bone marrow–derived MSC injection) and 3 in the control group. Of these 4 participants, 3 (1 MSC‐treated participants and 2 control participants) died of respiratory failure related to disease progression. The other one in the control group died of sudden cardiac arrest (before Visit 3, excluded in full analysis set). Adverse events were coded by MedRA by SOC (system organ classes) and PT (preferred term).
MSC = mesenchymal stem cell.
Efficacy Outcome Assessments
The mean changes in the ALSFRS‐R total scores at Visit 9 (+4 months) were −1.69 (SD = 2.51) in the MSC group versus −4.67 (SD = 3.25) in the control group. The primary efficacy outcome was 2.98 (SE = 0.75, 95% confidence interval [CI] = 1.48–4.47, p < 0.001). The group difference of the mean changes of ALSFRS‐R total score at Visit 10 (+6 months) was 3.38 (SE = 1.07, 95% CI = 1.23–5.54), which was significant (p = 0.003, Table 3 and Fig 2A).
Table 3.
Primary and Secondary Efficacy Outcome
| MSC Group | Control Group | |||||
|---|---|---|---|---|---|---|
| Efficacy Outcomes | n | Mean (SD) | n | Mean (SD) | Mean Difference between Group (SE, 95% CI) | p |
| Primary efficacy outcome | ||||||
| ALSFRS‐R score change from baseline to 4 months | 32 | −1.69 (2.51) | 27 | −4.67 (3.25) | 2.98 (0.75, 1.48 to 4.47) | <0.001 |
| ALSFRS‐R score change from baseline to 6 months | 31 | −3.10 (3.51) | 25 | −6.48 (4.53) | 3.38 (1.07, 1.23 to 5.54) | 0.003 |
| Secondary efficacy outcome | ||||||
| Changes in ALSFRS‐R slope between lead‐in and follow‐up perioda | 32 | 1.13 (1.33) | 27 | 0.03 (1.33) | 1.10 (0.35, 0.40 to 1.79) | 0.003 |
| Changes in ALSFRS‐R slope between lead‐in and follow‐up period,b up to 6 months | 32 | 1.08 (1.34) | 25 | 0.16 (1.46) | 0.92 (0.38, 0.16 to 1.67) | 0.018 |
| AALS score change from baseline to 4 months | 32 | 10.44 (9.24) | 25 | 17.96 (11.78) | −7.18 (2.76, −12.71 to −1.64) | 0.009 |
| Changes in AALS slope between lead‐in and follow‐up period | 31 | −1.51 (3.52) | 25 | 0.29 (5.06) | −1.80 (1.14, −4.08 to 0.48) | 0.119 |
| FVC % change from baseline to 4 months | 31 | −11.28 (10.06) | 25 | −10.75 (8.40) | −0.53 (2.52, −5.58 to 4.51) | 0.833 |
| Changes in FVC % slope between lead‐in and follow‐up period | 31 | −1.54 (3.38) | 25 | −0.80 (3.18) | −0.74 (0.89, −2.52 to 1.03) | 0.406 |
| SF‐36 change from baseline to 4 months | 32 | −9.06 (12.83) | 25 | −11.83 (11.28) | 2.78 (12.18, −3.74 to 9.29) | 0.397 |
| 4 Months | 6 Months | |||||
|---|---|---|---|---|---|---|
| Responder Analysis | MSC, n = 32 | Control, n = 27 | p | MSC, n = 32 | Control, n = 27 | p |
| <50% improvement in ALSFRS‐R slope, poor responders, n (%) | 10 (31%) | 22 (82%) | <0.001 | 12 (38%) | 21 (78%) | 0.002 |
| ≥50% improvement in ALSFRS‐R slope, good responders, n (%) | 22 (69%) | 5 (19%) | <0.001 | 20 (63%) | 6 (22%) | 0.002 |
| ≥75% improvement in ALSFRS‐R slope, n (%) | 18 (56%) | 2 (7%) | <0.001 | 17 (53%) | 3 (11%) | 0.001 |
| ≥100% improvement in ALSFRS‐R slope, n (%) | 13 (41%) | 1 (4%) | 0.001 | 8 (25%) | 1 (4%) | 0.031 |
Baseline was obtained at Visit 5 (just prior to the first MSC injection).
Changes in ALSFRS‐R slope = ([V5 − V1]/3 − [V9 − V5]/4).
Changes in ALSFRS‐R slope = ([V5 − V1]/3 − [V10 − V5]/6).
AALS = Appel ALS Rating Scale; ALSFRS‐R = Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised; CI = confidence interval; FVC = forced vital capacity; MSC = mesenchymal stem cell; SD = standard deviation; SE = standard error; SF‐36 = 36‐Item Short‐Form Health Survey; V = visit.
Figure 2.
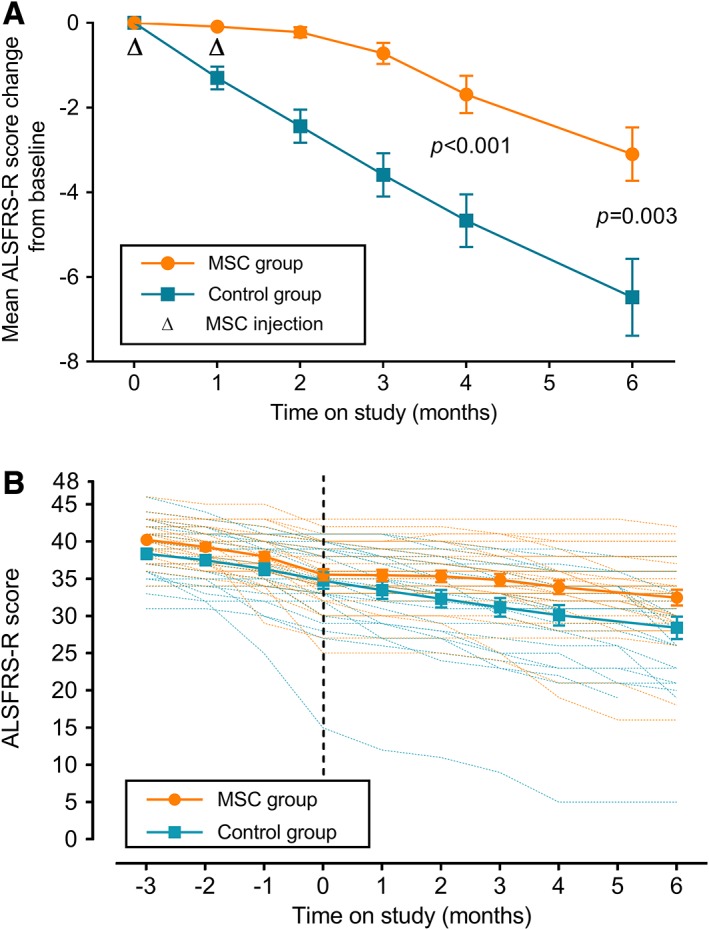
Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised (ALSFRS‐R) score in the full analysis set. (A) Changes from baseline in the mean ALSFRS‐R score change during the follow‐up period. (B) Adjusted mean ALSFRS‐R score during the 3‐month lead‐in and the 6‐month follow‐up period (piecewise linear mixed model over time). Data are given as least squares mean with standard error, and p value is for control group versus mesenchymal stem cell (MSC) group.
As one of the secondary efficacy outcomes, the difference in the change of the ALSFRS‐R slope (monthly decline rate) between the lead‐in and follow‐up period was significantly reduced in the MSC group in comparison to the control group (4 months: 1.10, SE = 0.35, 95% CI = 0.40–1.79, p = 0.003; 6 months: 0.92, SE = 0.38, 95% CI = 0.16–1.67, p = 0.018).
Table 3 summarizes the other secondary efficacy outcomes. The group difference of the mean change in the AALS score between Visit 5 and Visit 9 was significant (−7.18, 95% CI = −12.71 to −1.64, p = 0.009). FVC and SF‐36 were not significantly different between groups during the 4‐month follow‐up period.
Based on the responder analysis, the MSC group exhibited significantly greater functional stability at 4 and 6 months after treatment (4 months: 69% vs 19%, p = 0.002; 6 months: 63% vs 22%, p = 0.002; see Table 3). The numbers of responders in the MSC group at 4 months with ≥75% and ≥100% improvement were 18 (56%) and 13 (41%), respectively, and the responders at 6 months were 17 (53%) and 8 (25%), respectively.
Post Hoc Analysis on CSF Cytokines, Genetics, and Long‐Term Effectiveness
In the post hoc analysis using the piecewise linear mixed model, mean estimates of decline in ALSFRS‐R slope (monthly rate of decline), from baseline through the 6‐month follow‐up, were −0.55 (SE = 0.11) and −1.13 (SE = 0.15) in the MSC group and control group, respectively (p = 0.003; see Fig 2B). These results provided robust evidence of the clinical effectiveness of BM‐MSCs.
In the post hoc analysis, CSF cytokines were measured in 28 of 32 participants treated with BM‐MSCs. As shown in Figure 3A, the mean levels of TGF‐β1–3, IL‐6, and IL‐10 were significantly increased between just before the first (Visit 5) and second (Visit 6) BM‐MSC injections, whereas the mean levels of TNF‐α and MCP‐1 were significantly decreased. Each CSF cytokine levels of the MSC group quantitated at the point of before and after MSC treatment are detailed in Supplementary Table 7. ROC curve revealed a high AUC value for TGF‐β1, TGF‐β3, IL‐6, MCP‐1, and TNF‐α. In good responders, the increased level of TGF‐β1 significantly correlated with decreased level of MCP‐1 (4 months: R 2 = 0.259, p = 0.011; 6 months: R 2 = 0.259, p = 0.016; see Fig 3B). In contrast, an inversely correlated pattern of that found in good responders was not noted in poor responders (4 months: R 2 = 0.062, p = 0.276; 6 months: R 2 = 0.102, p = 0.185). Thus, inverse correlation of these 2 cytokines could be used as a potential biomarker to predict treatment responsiveness.
Figure 3.
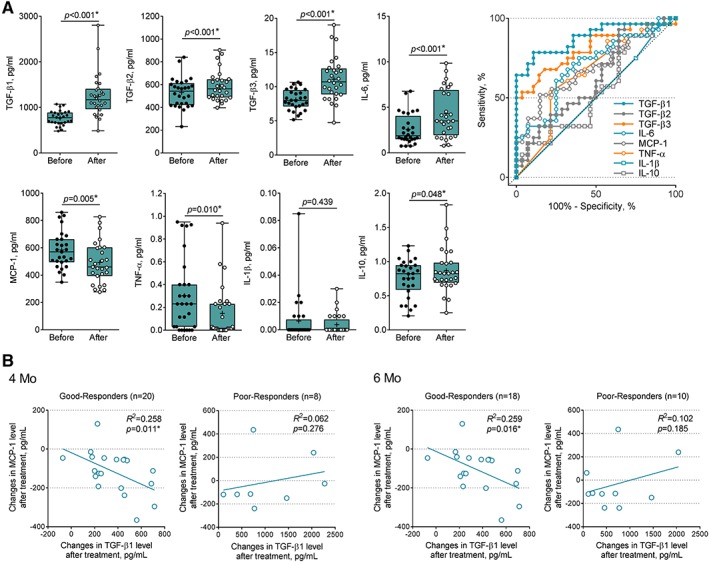
Changing patterns of cerebrospinal fluid (CSF) cytokine levels in the mesenchymal stem cell (MSC) treatment group. (A) Comparison of cytokine levels in CSF from patients before and after MSC treatment. Median and first and third quartiles (black bars) are shown, and each dot (black dot, before treatment; white dot, after treatment) represents individual data. Probability values were calculated using paired t test. Receiver operating characteristic analyses of individual cytokines compare before versus after MSC treatment in the test cohort. (B) Subgroup analysis in good responders and poor responders after MSC treatment. Correlation is shown between changes of transforming growth factor (TGF)‐β1 and monocyte chemoattractant protein (MCP)‐1 by MSC treatment in good responders at 4 and at 6 months; black lines indicate R 2. After MSC treatment, participants with ≥50% improvement in Amyotrophic Lateral Sclerosis Functional Rating Scale–Revised slope were defined as good responders and those with <50% improvement were defined as poor responders. *Statistically significant. TNF = tumor necrosis factor.
In the post hoc genetic analysis, all participants were negative for genetic screening of major ALS genes including C9orf72, SOD1, FUS, TARDBP, ANG, and OPTN (Supplementary Table 6).
Supplementary Table 8 summarizes survival analysis data of participants. Estimated mean survival time was 48 (SE = 6) months in the control group and 55 (SE = 4) months in the MSC group with no significance (p = 0.487). A Kaplan–Meier plot of the survival data shows a separation of the survival curves, although the curves crossed slightly at around 45 months. The cumulative probabilities of tracheostomy or death were not significantly different in the full analysis set (p = 0.318).
An exploratory post hoc subgroup analysis was performed to find out which subgroups have a possible survival benefit (see Supplementary Table 8). None of the subgroup analyses yielded statistically significant results. However, estimated survival and tracheostomy‐free survival time favored MSC treatment performed in the younger age group (<54 years; survival: 65 vs 48 months, p = 0.101; tracheostomy‐free survival: 48 vs 25 months, p = 0.103). Events of long‐term observation of each participant are shown in Supplementary Table 3.
Discussion
This is the first randomized, controlled trial designed to evaluate the safety and efficacy of repeated intrathecal BM‐MSC therapy in ALS patients, combined with post hoc analysis of candidate biological markers related with the response to MSC therapy. In addition, we analyzed post hoc genetic screening and long‐term safety and effectiveness using survival curves after 1 cycle of treatment with MSCs. With the advancement of stem cell research, diverse types of stem cell therapy have been conducted; however, randomized controlled trials on cell‐based therapies for ALS were not published until early 2016. To date, most clinical trials of MSCs for ALS were single‐arm phase 122, 23 or phase 224 trials focusing on the safety, feasibility, and possible efficacy, whereas only a few compared natural historical data.25, 26
In this study, 2 repeated treatments with intrathecal autologous BM‐MSCs (1 × 106 cells per kg with a 26‐day interval) showed significant therapeutic benefit with safety in patients with ALS. The decline in ALSFRS‐R was significantly reduced in the MSC group in comparison to the control group after 4‐ and 6‐month follow‐up. Also, secondary efficacy outcomes including the change in ALSFRS‐R slope (monthly rate of decline) and the mean changes of AALS score show significant positive treatment effect. Moreover, the proportion of patients who exhibited a ≥50% improvement in the ALSFRS‐R slope at 6 months in the MSC group was almost 3 times higher than in the control group (63% in the MSC group vs 22% in the control group, p = 0.002). ALS progression as measured by ALSFRS‐R has individual variability. According to a study using PRO‐ACT (Pooled Resource Open‐Access ALS Clinical Trials) database, the proportion of ALS patients who did not decline in ALSFRS‐R for 6 months was 25%.27 A similar trend was observed in the control group of the current study.
The incidence of AEs and SAEs was not different between the groups, indicating that 2 repeated intrathecal BM‐MSC treatments were safe. None of the 32 participants in the MSC group experienced any procedure‐ or treatment‐related SAEs for up to 6 months after the BM‐MSC injection.
Post hoc survival analysis did not show a significant difference between the 2 groups. Despite the positive effect on ALSFRS‐R lasting at least 6 months, the lack of long‐term survival benefit may be associated with the number of MSC treatments, with 2 limited injections in this trial protocol. The potential therapeutic effect of BM‐MSCs would not be long‐lasting because BM‐MSCs gradually disappear over time in CSF. Considering the immunomodulatory effect of BM‐MSC treatment using less invasive procedures, serial additional BM‐MSC treatments after 6 months might improve long‐term efficacy. Interestingly, estimated survival and tracheostomy‐free survival time favored MSC in those at younger age at screening (<54 years). This result suggests that greater clinical benefit of MSC treatment might be possible in young patients.
Our focus on BM‐MSC stem cell therapy for ALS, starting from the initial pilot trial and preclinical study in 2006,12 has extended to an investigator‐initiated clinical trial (KMFDS ID, Bio‐medicine Dept.‐16002),11 a phase I clinical trial,10 and finally, this study, which describes a completed phase 2 trial along with post hoc analysis on CSF biomarkers, genetic screening, and long‐term effectiveness in follow‐up until February 2018, for up to a maximum of 75 months. This study was enriched in that it considered clinical findings but also genetic aspects by excluding known common mutations that might be seen even in sporadic ALS. Furthermore, relevance of CSF biomarkers was evaluated for depth of understanding underlying immunomodulatory mechanism of BM‐MSCs. The initial pilot study revealed that single intrathecal BM‐MSC therapy was only slightly effective at the early or moderate stages of ALS (ALSFRS‐R score > 30), and not at all effective against advanced stage ALS. Based on these results and the available preclinical data, we conducted the open‐label, single‐arm, investigator‐initiated trial (IIT)11 to evaluate reliable biological markers to predict the effectiveness of BM‐MSC treatment to be used in a future phase 1–2 study. With this series of preclinical, pilot, and IIT clinical data, optimal dosages of BM‐MSCs, selection of the intrathecal method as a delivery route, numbers of injections, enrichment design for selection of early to moderate stage of ALS patients, minimal requirement of BM‐MSCs for the secretion of neurotrophic factors, and selection of biological markers to be used in post hoc analysis were defined for phase 1 and this phase 2 trial.
The anatomical substrates affected in ALS are related to long motor neural axes and their networks. Therefore, direct injections of MSCs into spinal cord or brain parenchyma could not overcome this limitation by providing differentiated new motor neurons, as the extensive connections among cortical neurons or between specific cortical neurons and their spinal counterparts that have already been lost would not be reproduced.28 In contrast, intrathecal injections of MSCs have the advantage of being less invasive in nature, which permits repeated treatments, in addition to the capacity of MSCs to secrete neurotrophic factors and modulate immunoinflammation, potentially leading to neuroprotective effects.
In previous studies, we proposed that intrathecal BM‐MSC injection in ALS possibly results in increased peripheral and central Tregs with IL‐4, IL‐10, and TGF‐β elevation in peripheral blood mononuclear cells of ALS patients, and switches microglia functional phenotypes toward anti‐inflammatory type.29 This leads to an anti‐inflammatory environment in the central nervous system in agreement with results of CSF biomarker analysis in a previous phase 1 study10 and the present phase 2 trial. Decreased numbers/activity of Tregs and decreased FoxP3 expression level in peripheral lymphocytes are biomarkers that predict rapid disease progression and attenuated survival in ALS patients.30 Another mechanism of BM‐MSCs could be an anti‐inflammatory M2 phenotypic “switch” away from the toxic or proinflammatory microglial form (M1), known to play an important role in accelerating neuronal death, thus increasing the rate of disease progression in patients with ALS.31 In addition, we previously reported that TGF‐β, secreted by BM‐MSCs, increases the phagocytic activity and anti‐inflammatory functions of microglial cells.29 Collectively, the plausible mechanisms of BM‐MSC therapy for slowing the progression of ALS shown in this trial may include increase of CSF anti‐inflammatory cytokines, the known paracrine effect of secreting neurotrophic factors, and other undetermined positive actions of BM‐MSCs.
Previous reports have shown that MCP‐1 is increased in the CSF of ALS patients, and its high expression in glial cells accelerates the disease progression rate.32, 33 In pericytes, MCP‐1 negatively correlated with TGF‐β,34 and anti‐inflammatory milieu such as increased state of CSF TGF‐β by MSC treatment may contribute to the reduction of MCP‐1 in ALS patients' CSF, as shown in Figure 3A. One interesting finding in this study was that IL‐6, known as a proinflammatory cytokine, was elevated in posttreated CSF. However, subgroup analysis between good responders and poor responders showed that significantly increased IL‐6 levels in posttreated state was remarkable in the poor responders group. The inverse correlation of TGF‐β1 and MCP‐1 levels shown in Figure 3B, that is, the higher TGF‐β1 with lowered level of MCP‐1 after the BM‐MSC therapy, was noted in good responders, which could be potential biomarker to predict effectiveness.
This study has some limitations. First, this study was not a double‐blinded trial, and did not include a sham procedure in the control group. Although this trial was a randomized clinical trial, we did not choose a double‐blinded format to avoid performing sham procedures in a control group for ethical reasons. We considered the risk of procedures35 and the possibility of compromised recruitment.36 Second, the follow‐up period was only 6 months postinjection. Thus, it did not establish long‐term effectiveness of BM‐MSC treatment. ALSFRS‐R might not be the best outcome measure in a small sample size and in a short‐duration clinical trial. A late‐stage clinical trial with long‐term follow‐up and study of other biomarkers is needed to confirm the efficacy. Third, the mean age of participants was relatively low when compared to other studies. One of the possible explanations is that younger patients have a tendency to actively seek new clinical trials or specialized treatment. Fourth, this study was unable to determine a significant difference of FVC. This trial might lack power to detect a difference in FVC.
During the preparation of manuscript, the draft of guidelines for Clinical Trials in ALS/MND37 was released by the ALS Clinical Trials Workshop, which took place at the Airlie Conference in Warrenton, Virginia (March 2016). It emphasizes the importance of excluding genetic and clinical heterogeneity when enrolling subjects and post hoc analysis on biological markers to identify the subgroup of patients who appear to respond better to the specific treatment. Although our trial did not completely cover all the ideal requirements of this updated guideline, our 10‐year experience with studying BM‐MSC therapy for ALS with a series of stepwise modified enriched model may serve as a good example for approaching the complicated evaluation of effectiveness of stem cell therapy in ALS.
In conclusion, this study provides preliminary evidence that 2 repeated intrathecal, autologous BM‐MSC injections are safe and effective in reducing the decline of ALSFRS‐R for at least 6 months. Although this approach is feasible and well tolerated, a future randomized, double‐blind, sham‐procedure–controlled, large‐scale phase 3 clinical trial is required to confirm the long‐term safety and efficacy of the treatment, and to delineate other plausible mechanisms of action of BM‐MSCs.
Author Contributions
Conception and design of the study: K.‐W.O., H.Y.K., and S.H.K. Acquisition and analysis of data: all authors. Drafting the text or preparing the figures: K.‐W.O., M.‐Y.N., and S.H.K. All authors critically revised the manuscript and approved the final article.
Potential Conflicts of Interest
S.H.K. received funding for the postmarketing survey of Autologous Bone Marrow‐Derived Mesenchymal Stem Cells (HYNR‐CS) from CORESTEM Inc. according to the safety guideline of KMFDS after conditional approval of HYNR‐CS as an orphan drug from KMFDS.
Supporting information
eTable 1. Study schedule.
eTable 2. Characteristics of mesenchymal stem cell.
eTable 3. Clinical events of long‐term observation and mesenchymal stem cell dose of each injection in participant.
eTable 4. Laboratory parameters of the MSC group and control group.
eTable 5. Adverse events and serious adverse events for all participants from the time of the first MSC injection to 4 months and at 6 months.
eTable 6. Post‐hoc genetic analysis of participants.
eTable 7. CSF cytokines levels between before and after MSCs treatment.
eTable 8. Post‐hoc survival analysis and subgroup comparison.
Acknowledgment
This study was supported by the Ministry for Health and Welfare Affairs, Republic of Korea (HI10C1673 and HI15C0876), and CORESTEM Inc.. The Korean Ministry for Health and Welfare Affairs and CORESTEM Inc. had no role in the study design, data collection, or writing the report. The role of CORESTEM Inc. was providing manufactured BM‐MSCs.
Sun‐hye Kim (Biostatistician, Dream CIS) performed the main statistical analysis and Dr Byong Ho Nam (Herings Global, Seoul, Republic of Korea) evaluated and confirmed the overall statistical analysis. This study would not have been possible without the participation of the patients and their families.
References
- 1. Kiernan MC, Vucic S, Cheah BC, et al. Amyotrophic lateral sclerosis. Lancet 2011;377:942–955. [DOI] [PubMed] [Google Scholar]
- 2. Appel SH, Beers DR, Henkel JS. T cell‐microglial dialogue in Parkinson's disease and amyotrophic lateral sclerosis: are we listening? Trends Immunol 2010;31:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature 2016;539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sreedharan J, Brown RH. Amyotrophic lateral sclerosis: problems and prospects. Ann Neurol 2013;74:309–316. [DOI] [PubMed] [Google Scholar]
- 5. Uccelli A, Laroni A, Freedman MS. Mesenchymal stem cells for the treatment of multiple sclerosis and other neurological diseases. Lancet Neurol 2011;10:649–656. [DOI] [PubMed] [Google Scholar]
- 6. Keating A. Mesenchymal stromal cells: new directions. Cell Stem Cell 2012;10:709–716. [DOI] [PubMed] [Google Scholar]
- 7. Lunn JS, Sakowski SA, Hur J, Feldman EL. Stem cell technology for neurodegenerative diseases. Ann Neurol 2011;70:353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kwon M‐S, Noh M‐Y, Oh K‐W, et al. The immunomodulatory effects of human mesenchymal stem cells on peripheral blood mononuclear cells in ALS patients. J Neurochem 2014;131:206–218. [DOI] [PubMed] [Google Scholar]
- 9. Bernardo ME, Fibbe WE. Mesenchymal stromal cells: sensors and switchers of inflammation. Cell Stem Cell 2013;13:392–402. [DOI] [PubMed] [Google Scholar]
- 10. Oh K‐W, Moon C, Kim HY, et al. Phase I trial of repeated intrathecal autologous bone marrow‐derived mesenchymal stromal cells in amyotrophic lateral sclerosis. Stem Cells Transl Med 2015;4:590–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim HY, Kim H, Oh K‐W, et al. Biological markers of mesenchymal stromal cells as predictors of response to autologous stem cell transplantation in patients with amyotrophic lateral sclerosis: an investigator‐initiated trial and in vivo study. Stem Cells 2014;32:2724–2731. [DOI] [PubMed] [Google Scholar]
- 12. Kim H, Kim HY, Choi MR, et al. Dose‐dependent efficacy of ALS‐human mesenchymal stem cells transplantation into cisterna magna in SOD1‐G93A ALS mice. Neurosci Lett 2010;468:190–194. [DOI] [PubMed] [Google Scholar]
- 13. Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 14. Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006;8:315–317. [DOI] [PubMed] [Google Scholar]
- 15. Schulz KF, Altman DG, Moher D, CONSORT Group . CONSORT 2010 statement: updated guidelines for reporting parallel group randomized trials. Ann Intern Med 2010;152:726–732. [DOI] [PubMed] [Google Scholar]
- 16. Trotti A, Colevas AD, Setser A, et al. CTCAE v3.0: development of a comprehensive grading system for the adverse effects of cancer treatment. Semin Radiat Oncol 2003;13:176–181. [DOI] [PubMed] [Google Scholar]
- 17. Castrillo‐Viguera C, Grasso DL, Simpson E, et al. Clinical significance in the change of decline in ALSFRS‐R. Amyotroph Lateral Scler 2010;11:178–180. [DOI] [PubMed] [Google Scholar]
- 18. Jang J‐H, Kwon M‐J, Choi WJ, et al. Analysis of the C9orf72 hexanucleotide repeat expansion in Korean patients with familial and sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2013;34:1311.e7–1311.e9. [DOI] [PubMed] [Google Scholar]
- 19. Kwon MJ, Baek W, Ki CS, et al. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol Aging 2012;33:1017.e17–1017.e23. [DOI] [PubMed] [Google Scholar]
- 20. Kim HY, Paek J, Park HK, et al. Efficacy and safety of autologous bone marrow‐derived mesenchymal stem cell treatment in patients with amyotrophic lateral sclerosis. J Korean Neurol Assoc 2009;27:163–169. [Google Scholar]
- 21. Naumova EN. Tutorial in biostatistics: evaluating the impact of “critical periods” in longitudinal studies of growth using piecewise mixed effects models. Int J Epidemiol 2001;30:1332–1341. [DOI] [PubMed] [Google Scholar]
- 22. Staff NP, Madigan NN, Morris J, et al. Safety of intrathecal autologous adipose‐derived mesenchymal stromal cells in patients with ALS. Neurology 2016;87:2230–2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mazzini L, Gelati M, Profico DC, et al. Human neural stem cell transplantation in ALS: initial results from a phase I trial. J Transl Med 2015;13:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Petrou P, Gothelf Y, Argov Z, et al. Safety and clinical effects of mesenchymal stem cells secreting neurotrophic factor transplantation in patients with amyotrophic lateral sclerosis. JAMA Neurol 2016;73:337. [DOI] [PubMed] [Google Scholar]
- 25. Glass JD, Hertzberg VS, Boulis NM, et al. Transplantation of spinal cord–derived neural stem cells for ALS. Neurology 2016;87:392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Abdul Wahid SF, Law ZK, Ismail NA, et al. Cell‐based therapies for amyotrophic lateral sclerosis/motor neuron disease. Cochrane Database Syst Rev 2016;11:CD011742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bedlack RS, Vaughan T, Wicks P, et al. How common are ALS plateaus and reversals? Neurology 2016;86:808–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Appel SH, Armon C. Stem cells in amyotrophic lateral sclerosis. Neurology 2016;87:348–349. [DOI] [PubMed] [Google Scholar]
- 29. Noh MY, Lim SM, Oh K‐W, et al. Mesenchymal stem cells modulate the functional properties of microglia via TGF‐β secretion. Stem Cells Transl Med 2016;5:1538–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Henkel JS, Beers DR, Wen S, et al. Regulatory T‐lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO Mol Med 2013;5:64–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Frakes AE, Ferraiuolo L, Haidet‐Phillips AM, et al. Microglia induce motor neuron death via the classical NF‐κB pathway in amyotrophic lateral sclerosis. Neuron 2014;81:1009–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Henkel JS, Engelhardt JI, Siklós L, et al. Presence of dendritic cells, MCP‐1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 2004;55:221–235. [DOI] [PubMed] [Google Scholar]
- 33. Kawaguchi‐Niida M, Yamamoto T, Kato Y, et al. MCP‐1/CCR2 signaling‐mediated astrocytosis is accelerated in a transgenic mouse model of SOD1‐mutated familial ALS. Acta Neuropathol Commun 2013;1:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rustenhoven J, Aalderink M, Scotter EL, et al. TGF‐beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J Neuroinflammation 2016;13:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Savitz SI, Cramer SC, Wechsler L, et al. Stem cells as an emerging paradigm in stroke 3: enhancing the development of clinical trials. Stroke 2014;45:634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Cohen PD, Isaacs T, Willocks P, et al. Sham neurosurgical procedures: the patients' perspective. Lancet Neurol 2012;11:1022. [DOI] [PubMed] [Google Scholar]
- 37. The final draft of the ALS clinical trials guidelines. 2016. Available at: http://www.alsa.org/advocacy/fda/assets/als-drug-development-guidance-for-public-comment-5-2-16.pdf. Accessed August 16, 2016.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
eTable 1. Study schedule.
eTable 2. Characteristics of mesenchymal stem cell.
eTable 3. Clinical events of long‐term observation and mesenchymal stem cell dose of each injection in participant.
eTable 4. Laboratory parameters of the MSC group and control group.
eTable 5. Adverse events and serious adverse events for all participants from the time of the first MSC injection to 4 months and at 6 months.
eTable 6. Post‐hoc genetic analysis of participants.
eTable 7. CSF cytokines levels between before and after MSCs treatment.
eTable 8. Post‐hoc survival analysis and subgroup comparison.