

Abstract
Using molecular phylogenetic data and methods we inferred divergence times and diversification patterns for the weevil subfamily Ceutorhynchinae in the context of host‐plant associations and global climate over evolutionary time. We detected four major diversification shifts that correlate with both host shifts and major climate events. Ceutorhynchinae experienced an increase in diversification rate at ∼53 Ma, during the Early Eocene Climate Optimum, coincident with a host shift to Lamiaceae. A second major diversification phase occurred at the end of the Eocene (∼34 Ma). This contrasts with the overall deterioration in climate equability at the Eocene‐Oligocene boundary, but tracks the diversification of important host plant clades in temperate (higher) latitudes, leading to increased diversification rates in the weevil clades infesting temperate hosts. A third major phase of diversification is correlated with the rising temperatures of the Late Oligocene Warming Event (∼26.5 Ma); diversification rates then declined shortly after the Middle Miocene Climate Transition (∼14.9 Ma). Our results indicate that biotic and abiotic factors together explain the evolution of Ceutorhynchinae better than each of these drivers viewed in isolation.
Keywords: Climate variation, Cenozoic, diversification, evolution, insect‐plant associations, weevils
The conspicuously unbalanced species richness in clades across the tree of life raises the question of why some groups of animals or plants underwent an enormous taxonomic radiation, proliferating into hundreds or thousands of species, whereas sometimes very closely related groups remain species‐poor. Although both ultimate and proximate causes for this imbalance might be very specific in each single case, macroevolutionary hypotheses have been proposed based on mechanisms that generate differential diversification rates. For herbivore insects, coevolutionary interactions with their host‐plants have been recognized as a fundamental mechanism of insect radiation by Ehrlich and Raven (1964). The association of Pierinae butterfly caterpillars with their hosts (crucifers, caper family, and other families containing mustard oil glucosides as defense against herbivores) is a key example of this “escape and radiate” model of reciprocal coevolution. Recent studies inferring the timing and pattern of the crucifers’ chemical defense mechanisms (the mustard oil glucosides) and the counter adaptations of Pierinae (the nitrile‐specifier protein gene) provide strong evidence for an ancient arms race between these butterflies and crucifers that escalated in complexity over time (Wheat et al. 2007; Winde and Wittstock 2011; Edger et al. 2015). Differential host‐plant associations can consequently lead to a pattern of non‐random differential diversification among herbivorous insect lineages, with accelerated diversification rates associated with particular host shifts into novel “adaptive zones” (cf. Winkler and Mitter 2008).
Besides these biotic or intrinsic mechanisms, changes in the physical or abiotic environment have also been postulated as potential drivers of extraordinary taxonomic radiations in animals and plants, meaning that (extrinsic) factors, such as tectonic movements or changes in climate, exert a strong effect on macroevolution. During the Cenozoic, for example, vascular plant species diversity was closely linked to global temperatures, with high species richness during the warm and humid Early Eocene, followed by a decline in diversity in the cooler Late Eocene and Oligocene (Jaramillo et al. 2006, 2010; Xing et al. 2014; Antonelli et al. 2015). This trend is not limited to plants. The dramatic turnover in the European Mammalian fauna at the Eocene‐Oligocene boundary has been explained by a significant temperature drop at the end of the Eocene (e.g., Ivany et al. 2000; Zhang et al. 2012). However, evidence is scarce to date for similar long‐term responses to these climate changes in insects (McKenna and Farrell 2006; Peña and Wahlberg 2008; Winkler et al. 2009; Condamine et al. 2012, 2018).
Across evolutionary timescales, insect diversity dynamics may depend upon interactions between biotic and abiotic factors, meaning that climatic variation, such as changes in temperature, atmospheric carbon dioxide concentrations or precipitation, drive the diversity of plant clades, but might also affect ecological opportunities for phytophagous insects (Condamine et al. 2012; Nyman et al. 2012).
Here, we assess the phylogeny and evolution–including the potential impact of abiotic and biotic factors on diversification–of the (primarily) temperate‐zone weevil subfamily Ceutorhynchinae (Curculionidae, Coleoptera; ∼1362 described species), using molecular phylogenetic data. The Ceutorhynchinae (Fig. 1) comprise a morphologically homogeneous subfamily of Curculionidae (true weevils), easily recognized by their robust appearance and ability to fold their rostrum (anterior prolongation of the head, containing the mouthparts) underneath the body, between the coxae (Korotyaev 2006). Although their monophyly is not questioned, their taxonomic status is currently disputed and they are sometimes considered a tribe within the subfamily Conoderinae (Prena et al. 2014) rather than a subfamily. Ceutorhynchinae display an almost worldwide distribution, with a notable “hot spot” of species richness in the western Palearctic, followed by the Nearctic and the Afrotropical regions. Ceutorhynchinae collectively feed on at least 55 plant families (Colonnelli 2004). Among known hosts are plants protected by potent allelochemicals (e.g., glucosinolates and alkaloids) such as garlic (Allium), wolfsbane (Aconitum), poppy (Papaver), joint‐pine (Ephedra), and many species of crucifers (Brassicaceae). The numerically most important host plant families are the composite family (Asteraceae, host for 80 species), the borage family (Boraginaceae, host for 75 species), the deadnettle family (Lamiaceae, host for 57 species), the buckwheat family (Polygonaceae, host for 55 species), the joint‐pine family (Ephedraceae, host for 52 species), and crucifers (Brassicaceae, host for ∼400 species). Most Ceutorhynchinae that feed on Brassicaceae belong to the type genus Ceutorhynchus, which is by far the largest genus of Ceutorhynchinae and alone represents about one quarter of all known species in the subfamily. Several species in the genus Ceutorhynchus are notorious pests of cabbage, rape seed, or garden crucifers, and have a considerable impact on agriculture (Alford et al. 2003). In contrast, species of the genera Hadroplontus and Euhrychiopsis are used as bio‐control agents against pest plants (Muller et al. 2011; Cline et al. 2013), such as Eurasian watermilfoil (Myriophyllum spicatum) and Canada thistle (Cirsium arvense).
Figure 1.

Members of Ceutorhynchinae on their host plants. (A) Ceutorhynchus resedae, (Ceutorhynchini) on Reseda luteola; (B) Mogulones cruciger (Ceutorhynchini) on Cynoglossum officinale; (C) Micrelus ericae (Ceutorhynchini) on Erica tetralix; (D) Tapeinotus sellatus (Scleropterini) on Lysimachia vulgaris; (E) Ceutorhynchus barbareae (Ceutorhynchini) on Barbarea vulgaris; (F) Mogulones geographicus (Ceutorhynchini) on Echium vulgare. Photo copyright: Frank Köhler, Bornheim, Germany, used with permission.
In Coleoptera, the leaf beetle genera Psylliodes and Phyllotreta (Chrysomelidae) also radiated on crucifers (Beran et al. 2014), but ceutorhynch weevils are probably the only other radiation of phytophagous insects feeding on crucifers that is comparable in species richness to the Pierinae butterflies. However, an important difference is that the entire life cycle of ceutorhynchine weevils (including the adult stage) is bound to their host plants, whereas pierine butterflies are only affiliated with crucifers as larvae. It is thus tempting to assess whether timing and patterns of radiation among these weevils with intimate life‐long host associations parallel those observed among the pierine butterflies–the only crucifer‐feeding butterfly clade.
To assess the relative impact of intrinsic ecological opportunities and extrinsic abiotic forces on potentially nonrandom differential diversification events in Ceutorhynchinae, we evaluated whether or not particular host‐plant affiliations, or switches to certain host plant taxa, are associated with enhanced species richness. Additionally, we investigated temporal coincidence between established Cenozoic climate changes and potentially associated shifts in diversification rate during the weevils’ evolutionary history. For these purposes, we evaluated a series of macroevolutionary models.
Materials and Methods
TAXON AND GENE SAMPLING
Two hundred four ingroup species were sampled, representing nine of the eleven currently recognized tribes in the subfamily Ceutorhynchinae. The species‐poor African tribes Lioxyonychini and Hypohypurini (Colonnelli 2004) were not sampled. In total, the dataset represents ∼15% of all recognized species of Ceutorhynchinae. Summary statistics for taxon sampling (organized by genus) are provided in Table S1. Of the 204 species sampled, 148 species were obtained from the Molecular Weevil Identification Project (MWI), which also provided COI barcode sequences (Schütte et al. 2013; Stüben et al. 2015). Twenty‐two species were collected between 2010 and 2015 in Israel, Austria, and Japan. Sequences for an additional 34 species were obtained from National Center for Biotechnology Information (NCBI). Thirty‐one species of weevils, including exemplars from all other weevil families, were used as outgroups. Fragments of the mitochondrial (mt) 16S rRNA and Cytochrome‐c‐oxidase I (COI) genes, as well as fragments of the nuclear (nc) 28S rRNA (segments D1–D3) and Elongation factor 1‐alpha (Ef1α) genes were amplified and sequenced. Laboratory protocols follow Winter et al. (2017). GenBank accession numbers are provided in Table S2.
PHYLOGENETIC ANALYSES
All genes were separately aligned using the software MAFFT v7.2.7 (Katoh and Standley 2013). Codon positions of each protein‐coding gene, as well as the paired and unpaired regions of the mt 16S and nc 28s rRNA genes, were defined as separate partitions. Consensus secondary structure information for rRNA sequences was inferred with RNAsalsa v0.81 (Stocsits et al. 2009) and ambiguously aligned positions in rRNA genes were excluded using the software aliscore v2.0 (Misof and Misof 2009). The resulting dataset comprised a total of 3502 aligned nucleotides and had ten partitions. Phylogenetic analyses were performed using maximum likelihood (ML) inference implemented in the software IQ‐TREE v.1.4.3 (Nguyen et al. 2015). For each partition, the GTR substitution model with a FreeRate heterogeneity model and four rate categories was used (Soubrier et al. 2012). To increase the chance of finding the global maximum likelihood tree, we increased the number of the starting trees for the likelihood tree search to 1000 (default: 100). Nodal support was inferred using parametric ultrafast bootstrap analysis (UFBoot) (Minh et al. 2013).
DIVERGENCE TIME ESTIMATION
Divergence time estimates were inferred with a Bayesian Markov Chain Monte Carlo (MCMC) analysis, implemented in the software BEAST v1.8.2 (Drummond et al. 2012). An uncorrelated lognormal relaxed‐clock model was assumed, and MCMC analyses were conducted on the fixed topology obtained by the IQ‐TREE analyses. Two runs were conducted, and each was run for 150 million generations (sampling every 10,000 generations). The first 30 million generations were discarded as burn‐in, following the indication provided by the Tracer software v1.6 (Rambaut et al. 2014) that was used to monitor parameter development. Post burn‐in samples were combined across runs to summarize parameter estimates and used to construct a maximum clade credibility tree with median node heights using TreeAnnotator v1.7.5 (Drummond et al. 2012).
To test whether different diversification process priors have an impact on age estimates, we performed two different BEAST analyses, one using a Yule (pure‐birth) prior (BEAST‐I), and another using a birth‐death prior (BEAST‐II). The Yule prior is traditionally used in most studies evaluating diversification times, but an impact of these priors has been shown in previous analyses (Condamine et al. 2015). We used Bayes factors (BF) to compare the support of both the Yule and the birth‐death priors. To calculate the marginal likelihood estimations (MLE), we used the stepping‐stone sampling (SS) approach (Baele et al. 2012), implemented in BEAST. SS was applied with default parameters. To test the potential impact of alternative topologies (and divergence times) on the subsequent diversification analyses, we conducted a tree search in an additional BEAST analysis (BEAST‐III), where only nodes with BS support ≥95 in the ML analyses were constrained prior to the analysis (cf. Fig. S2C).
To calibrate the divergence time estimates, we used the fossil species Axelrodiellus† (Zherikhin and Gratshev 2004) from the Aptian (126.3–112.9 Ma; Santana formation in Brazil) to calibrate the clade Brentidae, and the ceutorhynch fossil Ceutorhynchus succinus† (Legalov 2013) from the Priabonian (37.5–33.9 Ma; Prussian formation near Koenigsberg, Russia) to calibrate the genus Ceutorhynchus. For both nodes, a uniform prior estimate of the divergence date was applied, with the lower bound indicated by the minimum age of the fossil layer interval and the upper bound representing either the Triassic‐Jurassic boundary (201.3 Ma: Brentidae), or the minimum age of the Brentidae (112.9 Ma: Ceutorhynchus). We deliberately refrained from using more weevil fossils for calibration since the assignment of many fossils to extant families remains equivocal (Oberprieler et al. 2014; Gunter et al. 2016; Shin et al. 2017). BEAST analyses were conducted using the CIPRES portal (Miller et al. 2015).
ANCESTRAL HOST‐PLANT RECONSTRUCTION
Ancestral host associations were reconstructed using stochastic character mapping (Huelsenbeck and Bollback 2001), via the make.simmap command in the R‐package phytools v0.5‐14 (Revell 2012). The posterior distribution of the transition rate matrix was determined using a Markov chain Monte Carlo (MCMC) simulation, which ran for 10,000 generations and was sampled every 100 generations. Stochastic mapping was simulated 100 times and applied to 100 random trees from the results of the maximum likelihood bootstrap analyses in IQ‐TREE. Host‐plant associations were reconstructed using plant families as entities for the host‐plant character states. Plant families (rather than some other taxonomic level) were used because they are principally characterized by morphological and/or ecological similarities, including consistent chemical defense systems against herbivorous insects. A list of host plant taxa and families for each ceutorhynch species and the according references (Korotyaev and Anderson 2002; Colonnelli 2004; Yoshitake et al. 2008; Rheinheimer and Hassler 2010; San Vicente and Salgueira 2010; Yoshitake and Ito 2011; Stüben and Schütte 2014; Krátky 2015, 2016; Stüben 2017) can be found in Table S3.
TREE‐WIDE CHANGES IN DIVERSIFICATION RATES
To determine whether potential tree‐wide changes in diversification rates coincide with changes in climate, we applied a birth‐death model, where diversification rates were allowed to vary episodically through time (Stadler 2011; Höhna et al. 2015). This model assumes that speciation and extinction rates are piecewise constant through time, but may change at some events. Between these events or rate shifts, the rates are assumed to remain constant. The episodic birth‐death model (EBD) was implemented in a Bayesian framework using the software RevBayes v1.0.2 (Höhna et al. 2016).
Our dataset represents about 15% of all described ceutorhynch species. As shown in Table S1, the sampling of the included genera is non‐random, with ratios spanning from 8 to 100%. Sampling of the larger genera (>30 species, including Thamiocolus s.l., Mogulones s.l., Ceutorhynchus s.l., as well as the tribe Oxyonychini) is more consistent, spanning from 17 to 27% of described species. Missing species and taxon sampling are potential sources of error when inferring diversification rates (Cusimano and Renner 2010; Brock et al. 2011; Maddison and FitzJohn 2015). The methods we applied all provide one or more strategies to account for the impact of incomplete and non‐random taxon sampling. For the EBD analyses, we applied a uniform and two empirical taxon sampling approaches. For the uniform sampling strategy, we specified the ratio of sampled species in the tree over the total species in the group. For the empirical sampling strategies, we provide the numbers of missing species for specific taxonomic groups. The first empirical sampling scheme distinguishes between the subfamily Ceutorhynchinae, the tribe Ceutorhynchini, and the genus Ceutorhynchus. The second empirical sampling scheme distinguishes between all genera included. EBD analyses were applied on the BEAST chronogram and run for one million generations, sampling every 1000 generations. Convergence, mixing of parameters and the effective sample size ESS were inspected using Tracer v1.6 (Rambaut et al. 2014). Results were visualized using the R‐package RevGadgets v1.0 (Höhna et al. 2016). Additionally, we performed analyses to fit an episodic birth‐death model with mass extinction, to test if the Cretaceous‐Paleogene (K‐Pg) extinction event (66 Ma) and the Eocene‐Oligocene extinction event (33.9 Ma) impacted ceutorhynch diversification. These analyses were conducted with the R‐Package TESS v2.1.0 (Höhna et al. 2015) by using the CPP on Mass Extinction Times model (CoMET: May et al. 2016). This models tree‐wide mass extinction events as independent compound Poisson processes (CPP). The sampling fraction was evaluated with a uniform sampling scheme, similar to the EBD analyses.
CLADE‐SPECIFIC CHANGES IN DIVERSIFICATION
To determine whether diversification rates vary significantly across ceutorhynchine clades, we used a birth‐death model where diversification rates are allowed to depart from a model with constant rates across a chronogram. We first conducted analyses where potential diversification rate shifts were estimated automatically without the definition of specific clades. This was implemented using the software BAMM v2.5.0 (Rabosky et al. 2013), which considers rate shift configurations across clades of a given chronogram in proportion to their joint posterior probability. We accounted for incomplete taxon sampling as in the EBD analyses. Subsequently, BAMM was run with eight chains for ten million generations, sampling every 10,000 generations. The evaluation of ESS of both the log‐likelihood and the number of shift events, and the visualizations of rates and shifts were conducted with the R‐package BAMMtools v2.1.0 (Rabosky et al. 2014). Rate shifts were visualized with a phylorate plot, which indicates distinct speciation rates by mapping colors to rates on all branches and the maximum shift credibility (MSC) configuration, which maximizes the marginal probability of rate shifts along individual branches (Fig. 3A and Figs. S4). Prior distributions on speciation (lambda) and extinction (mu) rates were calculated with BAMMtools (lambdaInitPrior = 2.606, lambdaShiftPrior = 0.013, muInitPrior = 2.606).
Figure 3.
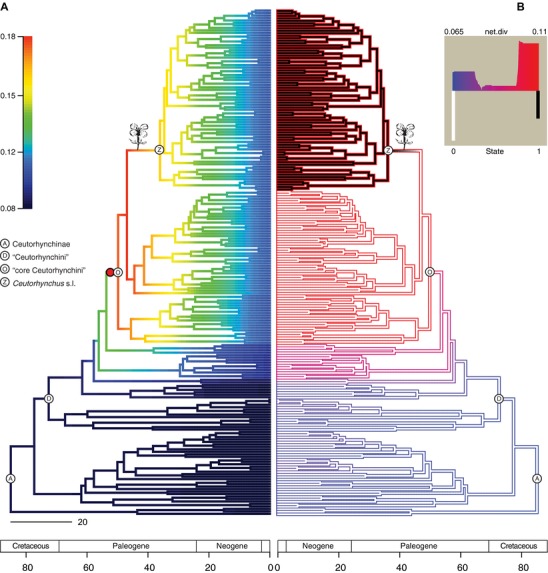
Branch‐specific and character‐dependent diversification rates in Ceutorhynchinae. The conventionalized crucifer flower at the branch leading to Ceutorhynchus s.l. indicates the host shift to Brassicaceae. (A) Phylorate plot indicates the results of the evolutionary rate analyses in BAMM, based on a uniform sampling strategy. Colors indicate relative speciation rates along each branch on the chronogram (increasing from blue to red). The red circle on the branch leading to the “core Ceutorhynchini” indicates the position of a regime shift in the maximum shift credibility (MSC) configuration. (B) Net diversification rates (increasing from blue to red) and host associations (black branches indicate crucifer feeding) using hidden state speciation and extinction (HiSSE) under the best model (HiSSE Model 22, see Materials and Methods for details) based on the model‐averaged parameter estimates from the set of all 44 models. The bottom‐left panel shows the distribution of net diversification rates along the phylogeny (state 0: non‐crucifer feeding; state 1: crucifer feeding).
The reliability of BAMM has recently been the subject of considerable discussion (Moore et al. 2016; Rabosky 2017; Rabosky et al. 2017; Meyer and Wiens 2018). Therefore, we conducted clade specific diversification rate analyses with alternative approaches (to cross‐validate the BAMM results). The software BayesRate v1.6.5 (Silvestro et al. 2011) can estimate the fit of different models in which the rates can vary between predefined clades. BayesRate employs a thermodynamic integration approach to calculate marginal likelihoods of different diversification models and uses Bayes Factors (BF) to evaluate the best model. We compared a model A with one common diversification rate for the whole tree with three models in which different rates are applied to specific clades: model B applies rates that differ between the “core Ceutorhynchini” and the remaining species, model C applies different rates between Ceutorhynchus and the remaing species, and finally model D applies five different rates for each of the following groups: Ceutorhynchus, Mogulones sensu lato, Thamiocolus sensu lato, Oxyonychini, and all remaining species (Table 2). Prior to these analyses, we estimated parameters for different model setups (pure‐birth vs birth‐death, linked vs unlinked rates). For all models a setup with a birth‐death model for the background rate and pure‐birth models for the radiating taxa performed best. After parameter selection for each of the four models, we calculated BF to indicate the best fitting model. For all analyses, we used flat priors and clade‐specific taxon sampling proportions to account for missing taxa. We ran all MCMC analyses for one million generations, sampling every 100 generations and discarding the first 100,000 generations as burn‐in. ESS and parameter convergence were checked with Tracer. Diagrams showing the 95% credibility intervals for individual (post burn‐in) net diversification rates were produced with the R‐package diversitree v0.9.9 (FitzJohn 2012).
Table 2.
Results of Bayes factor tests in the BayesRate analyses
| Model | dF | Clades | LM | BF |
|---|---|---|---|---|
| A | 1 | Ceutorhynchinae | –830.5 | 44.8 |
| B | 3 | (Rest|cC) | –812.3 | 8.2 |
| C | 3 | (Rest|Ce) | –818.5 | 20.7 |
| D | 6 | (Rest|Ce|Th|Mo|Ox) | –808.2 | 0 |
For each different model (A–D), the marginal likelihoods (LM) and the relative Bayes Factors (BF) are presented. Core Ceutorhynchini (cC), Ceutorhynchus (Ce), Mogulones s.l. (Mo), Thamiocolus s.l. (Th) Oxyonychini (Ox), remaining taxa (Rest).
Finally, we used the Hidden State Speciation and Extinction (HiSSE) model (Beaulieu and O'Meara 2016) to infer whether diversification rates are faster in the crucifer‐feeding genus Ceutorhynchus. HiSSE is an extension of the state‐dependent speciation and extinction (SSE) model family, which calculates whether the evolution of a certain character is associated with increased speciation rates and/or decreased extinction rates. The HiSSE model employs a “hidden” character that represents an unknown associated character and thus allows estimating speciation and extinction rates for observed and hidden states, while also allowing transition rates to vary among these states. We accounted for incomplete taxon sampling by providing a uniform sampling fraction for both crucifer‐feeding and non‐crucifer‐feeding ceutorhynchine clades. HiSSE analyses were conducted with the R‐package hisse v1.8.2 (Beaulieu and O'Meara 2016). We fitted a total of 44 models (Table S4) and selected the best‐fit model with the Akaike Information Criterion (AICc), corrected for small sample sizes.
Results
PHYLOGENY AND DIVERGENCE TIMES
The Bayes factor values, calculated with the marginal likelihood estimates of the stepping‐stone analyses, significantly favored the Yule prior (BEAST‐I) over the birth‐death prior (BEAST‐II) for the diversification process (BF = 49.24). We therefore relied on the results of the analyses with the Yule prior in subsequent analyses (reported below). Detailed results of tree reconstruction and both divergence time estimations, including highest probability density (95% HPDs), computed from the post burn‐in posterior topologies, as well as nodal support values (Bootstrap support, BS), are provided in Table 1 and in Figures S1, S2A, and S2B.
Table 1.
Summary of the tree reconstructions and divergence time estimates
| Support | Age estimations (Ma) | |||
|---|---|---|---|---|
| Clade | BS* | BEAST‐I | BEAST‐II | BEAST‐III |
| Clade A (Ceutorhynchinae) | 99 | 85.0 (74.4–96.3) | 85.1 (75.7–95.7) | 88.5 (77.6–101.2) |
| Clade B | 97 | 77.3 (68.0–87.4) | 77.0 (68.8–86.3) | 79.4 (70.9–90.2) |
| Clade C | 100 | 61.9 (53.4–71.3) | 61.8 (53.0–70.4) | 62.7 (54.7–67.2) |
| Clade D (“Ceutorhynchini”) | 70 | 72.6 (63.7–82.9) | 72.2 (64.3–81.0) | 74.3 (65.6–84.0) |
| Clade E | 71 | 67.2 (57.4–77.8) | 66.7 (57.7–75.9) | 68.8 (58.9–79.1) |
| Clade F | 80 | 58.0 (44.5–71.3) | 57.5 (44.6–69.7) | 59.0 (45.4–72.7) |
| Clade G | 98 | 59.9 (48.3–71.3) | 59.4 (48.5–69.6) | 61.7 (50.7–73.1) |
| Clade H | 100 | 61.6 (53.9–70.5) | 61.0 (54.3–68.4) | 62.9 (56.0–71.1) |
| Clade I | 100 | 45.7 (35.7–56.7) | 44.9 (35.6–54.5) | 46.1 (36.6–56.5) |
| Clade J | 99 | 57.8 (50.2–66.6) | 57.4 (51.4–64.9) | 58.1 (52.0–66.0) |
| Clade K | 90 | 56.2 (49.0–64.2) | 55.8 (49.8–62.5) | – |
| Clade L (Oxyonychini) | 100 | 29.5 (22.4–37.1) | 28.7 (23.8–35.5) | 30.1 (25.0–35.6) |
| Clade M | 99 | 53.7 (46.8–61.3) | 53.3 (47.4–59.6) | 54.7 (48.7–62.0) |
| Clade N (Phrydiuchus) | 100 | 38.1 (28.7–46.3) | 38.0 (30.1–45.7) | 38.7 (31.5–46.8) |
| Clade O (“core Ceutorhynchini”) | 71 | 49.8 (43.5–56.7) | 49.4 (44.4–55.5) | – |
| Clade P | 62 | 45.6 (39.0–52.9) | 45.2 (39.1–51.4) | 45.9 (40.0–52.9) |
| Clade Q | 43 | 40.9 (32.3–49.1) | 40.4 (32.5–47.8) | – |
| Clade R | 100 | 36.0 (30.0–42.3) | 35.7 (30.2–41.9) | 35.8 (30.5–42.5) |
| Clade S | 93 | 46.9 (41.1–53.6) | 46.6 (41.6–52.4) | – |
| Clade T | 92 | 44.5 (38.5–51.1) | 44.2 (39.0–50.0) | – |
| Clade U | 100 | 41.3 (35.7–48.1) | 41.0 (36.1–46.4) | 42.2 (37.0–48.3) |
| Clade V (Thamiocolus s.l.) | 100 | 31.5 (25.7–37.6) | 31.3 (26.3–36.8) | 32.5 (27.1–39.4) |
| Clade W | 88 | 38.7 (32.4–45.2) | 38.5 (33.3–44.0) | 38.8 (33.4‐45.2) |
| Clade X (Hadroplontus + Microplontus) | 100 | 31.9 (26.1–39.2) | 31.6 (25.6–37.2) | 32.6 (26.6–39.2) |
| Clade Y (Mogulones s.l.) | 98 | 31.1 (25.5–37.7) | 30.7 (26.2–36.2) | 32.5 (25.8–38.6) |
| Clade Z (Ceutorhynchus s.l.) | 100 | 36.0 (33.9–40.2) | 35.8 (33.9–40.0) | 35.9 (33.9–39.8) |
*Bootstrap support values refer only to BEAST‐I and BEAST‐II analyses.
Ultrafast bootstrap support (BS) and median ages of the divergence time estimates with two different priors for the diversification process are presented. Yule, pure‐birth branching prior; BD, birth‐death branching prior (upper and lower bounds of the 95% highest posterior density (HPD) in parentheses).
We recovered the origin of crown Ceutorhynchinae at 85.0 Ma (median crown age; 95% Height posterior density, HPD: 74.4–96.3 Ma), which is in the late Cretaceous (Fig. 2A, and Figs. S1 and S2). The traditionally recognized tribes Scleropterini, Phytobiini, and the most speciose Ceutorhynchini appear polyphyletic. Micrelus + Cyphosenus sp. appear as sister group to all other Ceutorhynchinae (BS = 97). A highly supported clade (BS = 100) comprises the tribes Scleropterini, Phytobiini, Mecysmoderini, Cnemogonini, and Hypurini (61.9 Ma; HPD: 95% 53.4–71.3 Ma). The remaining Ceutorhynchini form a third group (BS = 71) including two species of the tribe Scleropterini and also the monogeneric tribe Mononychini. Within this group, the “core Ceutorhynchini” (BS = 99) appear at 49.8 Ma (95% HPD: 43.5–56.7 Ma). The constrained tree search in the BEAST‐III analysis (Table 1 and Fig. S2C) differs mainly in the position of Cyphosenus sp., which is not sister to Micrelus, but forms a weakly supported clade with Curculio camelliae, as sister to the remaining Ceutorhynchinae (posterior Probability, pP = 0.73). Nedyus quadrimaculatus, which is sister to a clade consisting of Thamiocolus sl.l., Hadroplontus + Microplontus, and Mogulones s.l. in the original ML tree reconstruction (BS = 92), appears as sister to the remaining “core Ceutorhynchini” (pP = 0.94).
Figure 2.
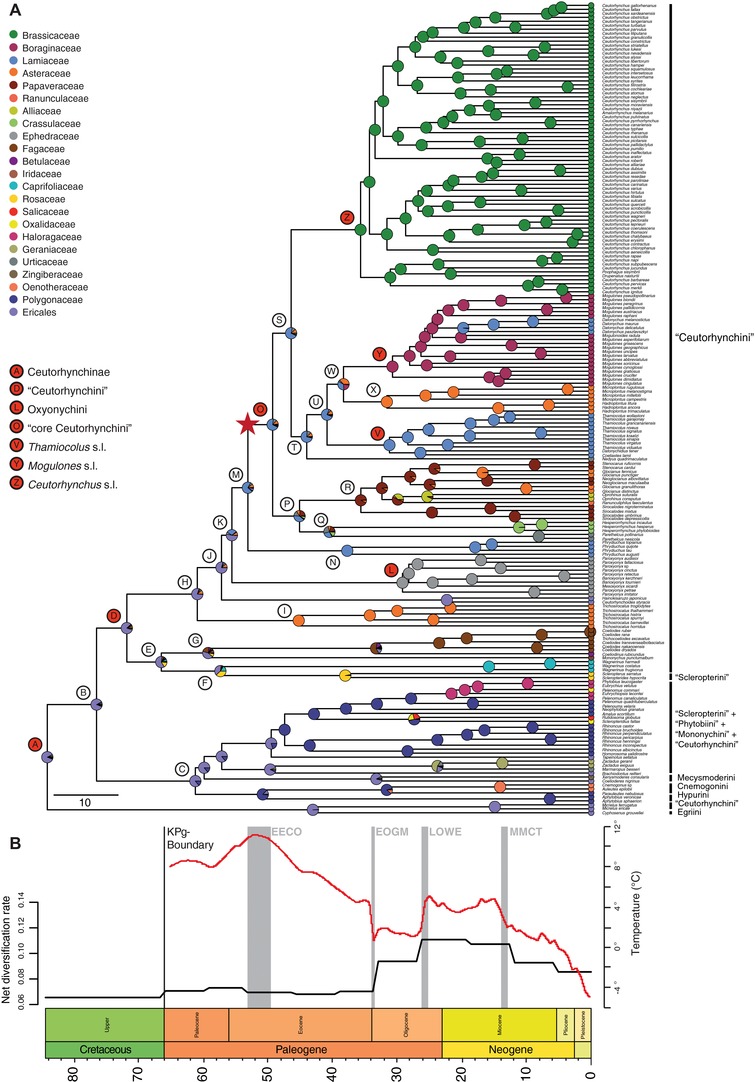
Timing, diversification, and host‐plant associations of Ceutorhynchinae. (A) Chronogram showing divergence times of ceutorhynch weevils and their host‐plant associations. Predicted ancestral host‐plant associations (viz. host families) are indicated as colored pie charts at the inner nodes, with the proportion of each color representing the relative likelihood of each ancestral host‐plant association. The red star indicates the increased diversification rate calculated in BAMM (uniform sampling strategy). Thamiocolus sensu lato includes the species Datonychidius tener and Coeliastes lamii and Mogulones sensu lato includes the genus Datonychus. (B) Results of the diversification rate through time analysis (black) in RevBayes (uniform sampling strategy), superimposed on a time‐averaged record of high‐latitude sea‐surface temperatures (red; adapted from Zachos et al. 2001) as a proxy for global climate. Gray bars refer to climatic events in the Cenozoic: EECO, Early Eocene climate optimum; EOGM, Early Oligocene glacial maximum; LOWE, Late Oligocene warming event; MMCT, Middle Miocene climatic transition. KPg‐Boundary: Cretaceous–Paleogene boundary.
ANCESTRAL HOST‐PLANT ASSOCIATIONS
The reconstruction of ancestral host‐plant associations suggests that the plant families Asteraceae, Lamiaceae, and Polygonaceae have each been colonized at least twice by Ceutorhynchinae, whereas Boraginaceae and Papaveraceae have each been colonized only once. However, paraphyletic grades of taxa are associated with the latter two plant families, and some ceutorhynch species initially associated with them later colonized other host‐plant families as well. The only ceutorhynch clades strictly limited to single host‐plant families are those associated with Ephedraceae and Brassicaceae, respectively.
TREE‐WIDE CHANGES IN DIVERSIFICATION
Results of the diversification rate through time analyses were congruent among all three taxon sampling schemes. EBD analyses show a conspicuous increase in tree‐wide net diversification rate at the beginning of the Oligocene (∼34 Ma), reaching a peak at ∼26 Ma in the late Oligocene and decreasing again at ∼12 Ma in the middle Miocene (Fig. 2B and Figs. S3). The analyses based on the tree inferred with the additional BEAST‐III approach also indicated an increase in net‐diversification at ∼63 Ma, shortly after the K‐Pg boundary (Fig. S3D). However, the additional CoMET analyses in TESS could not detect significant evidence for mass extinction events in the history of Ceutorhynchinae (Figs. S4).
CLADE‐SPECIFIC CHANGES IN DIVERSIFICATION
BAMM analyses provided similar estimates of clade‐specific diversification rates (Fig. 3A and Figs. S5). In the “core Ceutorhynchini,” BAMM analyses indicated one significant rate shift ∼53 Ma in the Paleogene (Figs. 2A and 3A). This was supported by the results of both HiSSE analyses. In the analysis based on the BEAST‐I tree, model M22 had the lowest AICc and the two models with a ΔAICc of <2 (M10, M22) had a combined Akaike weight (AICw) of 0.77. In the additional analysis based on the BEAST‐III tree, model M5 had the lowest AICc and also two models had a ΔAICc of <2 (M5, M16) and their combined Akaike weight (AICw) was 0.64. All models suggest that a rate shift occurred independently from the colonization of crucifers, at the node of the “core Ceutorhynchini” (Fig. 3B). In BayesRate (Table 2, Fig. 4), the model with equal rates for all ceutorhynch clades (Fig. 4A) was strongly rejected in favor of variable rate models with different rates assigned to certain genera and tribes. These were defined according to specific host‐plant associations and to the diversification rate shift found in the BAMM analysis. The highest marginal likelihood was obtained for a model assigning different rates to the five groups Ceutorhynchus, Mogulones s.l., Thamiocolus s.l., Oxyonychini, and the remaining species, respectively (Fig. 4D). Alternative models, which assign different rates to only “core Ceutorhynchini” and the remaining species (Fig. 4C), as well as only Ceutorhynchus and the remaining species (Fig. 4B), are also strongly preferred over the equal rate model, but not over the best model. In all models with distinct rates for Ceutorhynchus, the latter shows the highest diversification rate among all clades that were compared.
Figure 4.
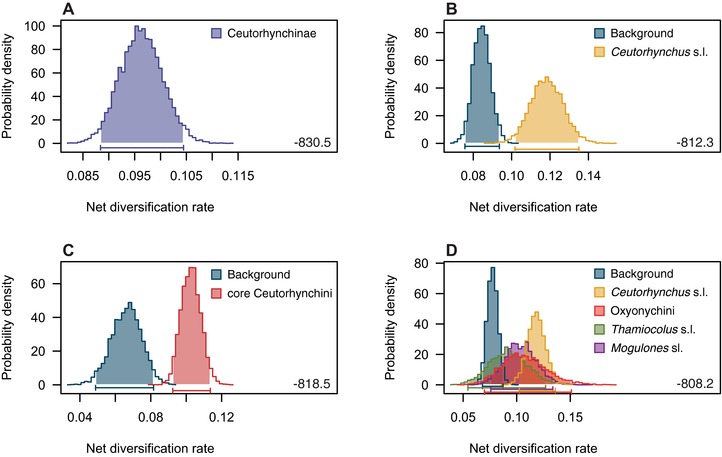
Net diversification rates (r = speciation rate λ – extinction rate μ) between distinct ceutorhynch clades as indicated by different BayesRate models. (A) Model assuming one diversification rate common to all subordinated weevil groups (r = 0.096). (B) Model with different rates for the taxa Ceutorhynchus (r = 0.118), Thamiocolus (r = 0.093), Mogulones (r = 0.104), Oxyonychini (r = 0.107), and all others (r = 0.069). (C) Model with different rates for “core Ceutorhynchini” (r = 0.103) and all others (r = 0.066). (D) Model with different rates for the genus Ceutorhynchus (r = 0.118) and all others (r = 0.085). 95% credibility intervals are shaded and indicated by bars along the x‐axis. Numbers show marginal likelihoods.
Discussion
HOST‐PLANT AFFILIATIONS
The reconstruction of ancestral host associations reveals a generally high conservatism of ceutorhynch host‐plant affiliations. Thus weevil species feeding on plants of the same family are generally closely related. However, the strength of these associations depends on the colonized plant hosts. In the larger clades, we found the strongest host conservatism in the crucifer‐feeding weevils, where the morphologically distinct genera Amalorrhynchus, Drupenatus, and Poophagus are nested within Ceutorhynchus, rendering this species‐rich genus paraphyletic if maintaining the aforementioned genera as distinct entities. Furthermore, Ceutorhynchinae feeding on Ephedraceae are all members of the xerophilic tribe Oxyonychini and are exclusively associated with plants of the genus Ephedra. This indicates that the colonization of both Brassicaceae and Ephedraceae constituted some kind of evolutionary constraint, which on one hand allowed a considerable radiation on these plants, but also prohibited later switches to other hosts. The lack of plasticity in crucifer associations is similar to the monophyly of the crucifer‐feeding leaf beetle genus Phyllotreta, but contrasts with the situation in the chrysomelid genus Psylliodes, where colonization of Brassicaceae has evolved twice (F. Beran, pers. comm.), and also in Pierinae butterflies, where the colonization of Brassicales was later succeeded by colonization of different host plant families (Edger et al. 2015). It is also contrasted by the genus Mogulones s.l., where a subsequent switch from Boraginaceae in Mogulones s.str. to Lamiaceae in Datonychus occurred, a switch back to the potential primary host of “core Ceutorhynchinae.” The colonization of Papaveraceae has also occurred just once, but was later succeeded by switches to completely unrelated hosts in the families Asteraceae, Ranunculaceae, and Alliaceae. Of the plant families housing larger groups of ceutorhynchs, species feeding on Asteraceae and Polygonaceae are less conservative in their use of different host plants over evolutionary time; these plant families have been colonized four and three times, respectively. This pattern is remarkably similar to the evolution of host‐plant use in the brentid tribe Apionini, where only these plant families have been colonized more than one time (Winter et al. 2017).
DIFFERENTIAL DIVERSIFICATION IN CEUTORHYNCHINAE
According to our analyses, ceutorhynchine weevils experienced four major phases of non‐constant diversification rates during the Paleogene and Neogene. The clade‐specific diversification shift associated with the emergence of the “core Ceutorhynchini” in the early Eocene ∼53 Ma coincides with the colonization of plants in the family Lamiaceae (mints and relatives). The “Early Eocene Climatic Optimum” (EECO; 50–53 Ma) was the overall warmest period in the Cenozoic Era (Zachos et al. 2001, 2008). This warm and humid climate was probably responsible for overall high floral species richness (Jaramillo et al. 2006, 2010; Antonelli et al. 2015) and might have also promoted the radiation of Lamiaceae. Consequently, the early radiation in “core Ceutorhynchini” might be an adaptation to feeding on Lamiaceae. The emergence of Lamiaceae has recently been localized in the Paleocene (Magallón et al. 2015; D.C. Tank and R.G. Olmstead, pers. comm.), indicating an early colonization of Lamiaceae by Ceutorhynchinae and thus a potential ancient codiversification pattern. Overall, these patterns are consistent with the results of McKenna et al. (2009), who proposed that Curculionidae diversified alongside angiospermous plants, particularly core eudicots.
A second major phase of ceutorhynchine diversification occurred at the end of the Eocene (∼34 Ma). A significant temperature drop at the end of Eocene escalated the general cooling trend after the EECO. Floral species richness globally decreased and higher latitudes became dominated by deciduous temperate forests (Wolfe 1992; Morley 2003). Besides a general decrease in floral diversity, the retraction of tropical forests toward the equator induced the speciation of plant taxa adapted to cool and dry environments (Jacobs et al. 1999; Bouchenak‐Khelladi et al. 2014). Important weevil host plant clades congruently emerged or diversified in the temperate zones of Asia or the Mediterranean. The dry‐adapted genus Ephedra radiated in the Mediterranean and Asia in the early Oligocene (Ickert‐Bond et al. 2009). Several clades of Boraginaceae (Chacón et al. 2017) and Papaveraceae (Kadereit et al. 2011; Pérez‐Gutiérrez et al. 2015) adapted to more temperate and dry climates in the Early Oligocene, and radiated in the Mediterranean and/or Asia, and the cool‐adapted core Brassicaceae began their radiation in the Northern hemisphere at the Eocene‐Oligocene boundary (Couvreur et al. 2009; Edger et al. 2015; Huang et al. 2015). According to our phylogenetic analyses, these radiations were quickly tracked by colonizing ceutorhynchine weevils, which in several cases also led to an increased rate of diversification in the relevant weevil clades (Fig. 4D). The diversification of Ceutorhynchinae declined shortly after the Middle Miocene Climate Transition (MMCT, ∼14.9 Ma). In the middle Miocene and the early Pliocene temperature and atmospheric carbon dioxide concentrations declined (Edwards et al. 2010), leading to a global expansion of arid and semiarid environments, which further promoted the radiation of succulent plants and the spread of C4 grasslands (Edwards et al. 2010; Arakaki et al. 2011). Ceutorhynchinae weevils use none of these plant groups as hosts and their diversification rate decreased.
The inferred specific timings of weevil and host‐plant radiations also indicate that the observed Cenozoic climate changes most probably indirectly influenced weevil radiations by shaping their biotic environment, that is the specific diversification of host‐plant taxa. This was especially pronounced in the late Eocene and Oligocene, when the climatic favor of a flora adapted to temperate climate was associated with the diversification of important weevil host‐plant groups such as Brassicaceae, Boraginaceae, and Ephedra. The rise of these new “adaptive zones” then potentially facilitated contemporaneous climate‐linked host shifts in several taxa of “core Ceutorhynchini” (Fig. 4D). The complex pattern of interactions observed among the radiations of Ceutorhynchinae, variation in climate and the emergence of plant taxa as potential hosts strengthens the view that a hierarchical interplay of abiotic (climate) and biotic (host associations) factors best explains the evolution of ceutorhynch weevils: the long‐term tracking of mean annual temperatures in the Cenozoic by ceutorhynch diversification rates indicates climate changes in the temperate Northern higher latitudes as the ultimate driver for the radiation of Ceutorhynchinae. However, single radiations in genera such as Ceutorhynchus and Mogulones s.l. are more likely the result of individual temporal and spatial adaptations to their plant hosts, where major radiations in Brassicaceae and Boraginaceae in the Holarctic are indicative of temperate‐zone adaptations of these plants in the late Eocene and early Oligocene (Couvreur et al. 2009; Chacón et al. 2017). Consequently, ceutorhynch weevils appear to have responded to the abiotic environmental changes in the Northern hemisphere in the Cenozoic via biotic interactions with their specific hosts, which were in turn shaped by their own adaptation to these environmental changes.
PLANT SECONDARY COMPOUNDS
Notably, all major host plant groups of “core Ceutorhynchini” are protected against herbivores by strong allelochemical defense mechanisms. Thus, the exploitation of these plants must have been facilitated by specific counter‐adaptations in weevils, contributing to their increased diversification. Multiple genera of leaf beetles, grasshoppers, butterflies, and moths are able to detoxify and partly sequester the pyrrolizidine alkaloids of Boraginaceae and other plant families, whereas others can cope with the deterrent terpene derivatives of Lamiaceae, for example iridoid glycosides (Dobler et al. 2000, 2011; Sehlmeyer et al. 2010; Opitz et al. 2012; Wang et al. 2012). However, with our current dataset, we cannot yet ascertain if the proposed coevolutionary patterns observed in ceutorhynch weevils either simply support a “sequential evolution” model, where a single colonization of a new host plant group promoted diversification of the colonizing insect taxa, or if the inferred insect‐host associations may constitute a reciprocal system of coevolutionary radiations, as predicted by the “escape‐and‐radiate” model. The pattern of ceutorhynch diversification in the Oligocene shows two successive escalation periods, thus indicating some degree of an escalating evolutionary arms race with hosts evolving novel chemical defenses, and weevils adapting by developing new strategies to overcome these defenses. To infer potentially escalating weevil‐plant (counter‐) adaptation patterns in Ceutorhynchus or in any other taxa, more detailed studies on the evolutionary history of hosts and weevils are needed. Furthermore, information on the specific mechanisms involved, that is the genetics and physiology of the plants’ defenses and the weevils’ detoxification system, are also needed to conclusively disentangle the evolutionary history of these intricate interactions.
Associate Editor: D. Rabosky
Handling Editor: P. Tiffin
Supporting information
Table S1: Sampling ratio of all included genera of Ceutorhynchinae.
Table S2: Taxon sampling including NCBI genbank accession numbers.
Table S2.1: Voucher IDs of all specimens provided by the Molecular Weevil Identification project.
Table S3: Host plant associations of all included ceutorhynchine weevils.
Table S4A: Results of the character‐dependent diversification analysis in HiSSe, based on the BEAST‐I analysis.
Table S4B: Results of the character‐dependent diversification analysis in HiSSe, based on the BEAST‐III analysis.
Figure S1. Results of the maximum likelihood phylogenetic tree reconstruction in IQ‐Tree.
Figures S2. Results from the divergence time analyses in BEAST.
Figures S3. Results of the diversification rate through time analyses in RevBayes.
Figure S4: Results of the diversification rate through time analyses with TESS.
Figures S5: Results of the clade‐specific divergence time analyses in BAMM.
AUTHOR CONTRIBUTIONS
H.L. designed the experiments and performed the analyses; B.G. and C.M. conducted the laboratory work; J.A. and A.F. contributed samples. H.L. wrote the manuscript. A.F., B.G., D.M., J.A., and K.F. critically revised and contributed content to the manuscript.
ACKNOWLEDGMENTS
We are grateful for the comments by Dan Rabosky, Fabien Condamine, and one anonymous reviewer on an earlier versions of this study. We thank Hiraku Yoshitake, National Institute for Agro‐Environmental Sciences, Japan for providing Asian specimens and discussions on an earlier version of this article. We thank the Molecular Weevil Identification project (jointly implemented by Curculio Institute and Museum Koenig, Germany) for providing the bulk of the material used in this project, namely Peter Stüben, André Schütte, Hannah Janssen, and the Curci collecting team. We thank Jan Schnitzler, University of Leipzig, Germany for help with BayesRate. We are grateful that the CIPRES science gateway (http://www.phylo.org) provided computational time. We specially acknowledge Frank Köhler, Bornheim, Germany for contributing pictures of Ceutorhynchinae. The work was funded by the Faculty of Life Sciences, University Vienna, Austria. D.M. acknowledges support from the United States National Science Foundation DEB1355169.
DATA ARCHIVING
Datasets, including alignments, tree search, and dating results as well as information on host plants are provided at Dryad: https://doi.org/10.5061/dryad.9mj8mt5.
Literature cited
- Alford, D. V. , Nilsson C., and Ulber B.. 2003. Insect pests of oilseed rape crops Pp. 9–42 in Alford D. V., ed. Biocontrol of oilseed rape pests. Blackwell Science, Hoboken, New Jersey. [Google Scholar]
- Antonelli, A. , Zizka A., Silvestro D., Scharn R., Cascales‐Miñana B., and Bacon C. D.. 2015. An engine for global plant diversity: highest evolutionary turnover and emigration in the American tropics. Front. Genet. 6:130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arakaki, M. , Christin P.‐A., Nyffeler R., Lendel A., Eggli U., Ogburn R. M., Spriggs E., Moore M. J., and Edwards E. J.. 2011. Contemporaneous and recent radiations of the world's major succulent plant lineages. Proc. Natl. Acad. Sci. 108:8379–8384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baele, G. , Lemey P., Bedford T., Rambaut A., Suchard M. A., and Alekseyenko A. V.. 2012. Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29:2157–2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu, J. M. , and O'Meara B. C.. 2016. Detecting hidden diversification shifts in models of trait‐dependent speciation and extinction. Syst. Biol. 65:583–601. [DOI] [PubMed] [Google Scholar]
- Beran, F. , Pauchet Y., Kunert G., Reichelt M., Wielsch N., Vogel H., Reinecke A., Svatos A., Mewis I., Schmid D., et al. 2014. Phyllotreta striolata flea beetles use host plant defense compounds to create their own glucosinolate‐myrosinase system. Proc. Natl. Acad. Sci. 111:7349–7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchenak‐Khelladi, Y. , Slingsby J. A., Verboom G. A., and Bond W. J.. 2014. Diversification of C4 grasses (Poaceae) does not coincide with their ecological dominance. Am. J. Bot. 101:300–307. [DOI] [PubMed] [Google Scholar]
- Brock, C. D. , Harmon L. J., and Alfaro M. E.. 2011. Testing for temporal variation in diversification rates when sampling is incomplete and nonrandom. Syst. Biol. 60:410–419. [DOI] [PubMed] [Google Scholar]
- Chacón, J. , Luebert F., and Weigend M.. 2017. Biogeographic events are not correlated with diaspore dispersal modes in Boraginaceae. Front. Ecol. Evol. 5:26. [Google Scholar]
- Cline, A. R. , Villegas B., Pitcairn M. J., and O'Brien C. W.. 2013. The status of Euhrychiopsis lecontei (Dietz) (Coleoptera: Curculionidae) in California, with notes on other weevils associated with milfoil. Coleopt. Bull. 67:75–80. [Google Scholar]
- Colonnelli, E. 2004. Catalogue of ceutorhynchinae of the world, with a key to genera. (Insecta: Coleoptera: Curculionidae). Argania editio, Barcelona, 124 p.
- Condamine, F. L. , Nagalingum N. S., Marshall C. R., and Morlon H.. 2015. Origin and diversification of living cycads: a cautionary tale on the impact of the branching process prior in Bayesian molecular dating. BMC Evol. Biol. 15:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condamine, F. L. , Rolland J., Höhna S., Sperling F. A. H., and Sanmartín I.. 2018. Testing the role of the red queen and court jester as drivers of the macroevolution of apollo butterflies. Syst. Biol. 10.1093/sysbio/syy009. [DOI] [PubMed] [Google Scholar]
- Condamine, F. L. , Sperling F. A., Wahlberg N., Rasplus J.‐Y., and Kergoat G. J.. 2012. What causes latitudinal gradients in species diversity? Evolutionary processes and ecological constraints on swallowtail biodiversity. Ecol. Lett. 15:267–277. [DOI] [PubMed] [Google Scholar]
- Couvreur, T. L. , Franzke A., Al‐Shehbaz I. A., Bakker F. T., Koch M. A., and Mummenhoff K.. 2009. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 27:55–71. [DOI] [PubMed] [Google Scholar]
- Cusimano, N. , and Renner S. S.. 2010. Slowdowns in diversification rates from real phylogenies may not be real. Syst. Biol. 59:458–464. [DOI] [PubMed] [Google Scholar]
- Dobler, S. , Haberer W., Witte L., and Hartmann T.. 2000. Selective sequestration of pyrrolizidine alkaloids from diverse host plants by Longitarsus flea beetles. J. Chem. Ecol. 26:1281–1298. [Google Scholar]
- Dobler, S. , Petschenka G., and Pankoke H.. 2011. Coping with toxic plant compounds–the insect's perspective on iridoid glycosides and cardenolides. Phytochemistry 72:1593–1604. [DOI] [PubMed] [Google Scholar]
- Drummond, A. J. , Suchard M. A., Xie D., and Rambaut A.. 2012. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29:1969–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edger, P. P. , Heidel‐Fischer H. M., Bekaert M., Rota J., Glöckner G., Platts A. E., Heckel D. G., Der J. P., Wafula E. K., Tang M., et al. 2015. The butterfly plant arms‐race escalated by gene and genome duplications. Proc. Natl. Acad. Sci. 112:8362–8366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards, E. J. , Osborne C. P., Strömberg C. A., Smith S. A., and Consortium C4 Grasses. 2010. The origins of C4 grasslands: integrating evolutionary and ecosystem science. Science 328:587–591. [DOI] [PubMed] [Google Scholar]
- Ehrlich, P. R. , and Raven P. H.. 1964. Butterflies and plants: a study in coevolution. Evolution 18:586–608. [Google Scholar]
- FitzJohn, R. G. 2012. Diversitree: comparative phylogenetic analyses of diversification in R. Methods Ecol. Evol. 3:1084–1092. [Google Scholar]
- Gunter, N. L. , Oberprieler R. G., and Cameron S. L.. 2016. Molecular phylogenetics of Australian weevils (Coleoptera: Curculionoidea): exploring relationships in a hyperdiverse lineage through comparison of independent analyses. Austral. Entomol. 55:217–233. [Google Scholar]
- Höhna, S. , Landis M. J., Heath T. A., Boussau B., Lartillot N., Moore B. R., Huelsenbeck J. P., and Ronquist F.. 2016. RevBayes: Bayesian phylogenetic inference using graphical models and an interactive model‐specification language. Syst. Biol. 65:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höhna, S. , May M. R., and Moore B. R.. 2015. TESS: an R package for efficiently simulating phylogenetic trees and performing Bayesian inference of lineage diversification rates. Bioinformatics 32:789–791. [DOI] [PubMed] [Google Scholar]
- Huang, C.‐H. , Sun R., Hu Y., Zeng L., Zhang N., Cai L., Zhang Q., Koch M. A., Al‐Shehbaz I., Edger P. P., et al. 2015. Resolution of Brassicaceae phylogeny using nuclear genes uncovers nested radiations and supports convergent morphological evolution. Mol. Biol. Evol. 33:394–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck, J. P. , and Bollback J. P.. 2001. Empirical and hierarchical Bayesian estimation of ancestral states. Syst.Biol. 50:351–366. [PubMed] [Google Scholar]
- Ickert‐Bond, S. M. , Rydin C., and Renner S. S.. 2009. A fossil‐calibrated relaxed clock for Ephedra indicates an Oligocene age for the divergence of Asian and New World clades and Miocene dispersal into South America. J. Syst. Evol. 47:444–456. [Google Scholar]
- Ivany, L. C. , Patterson W. P., and Lohmann K. C.. 2000. Cooler winters as a possible cause of mass extinctions at the Eocene/Oligocene boundary. Nature 407:887–890. [DOI] [PubMed] [Google Scholar]
- Jacobs, B. F. , Kingston J. D., and Jacobs L. L.. 1999. The origin of grass‐dominated ecosystems. Ann. Mo. Bot. Gard. 86:590–643. [Google Scholar]
- Jaramillo, C. , Ochoa D., Contreras L., Pagani M., Carvajal‐Ortiz H., Pratt L. M., Krishnan S., Cardona A., Romero M., Quiroz L., et al. 2010. Effects of rapid global warming at the Paleocene‐Eocene boundary on neotropical vegetation. Science 330:957–961. [DOI] [PubMed] [Google Scholar]
- Jaramillo, C. , Rueda M. J., and Mora G.. 2006. Cenozoic plant diversity in the Neotropics. Science 311:1893–1896. [DOI] [PubMed] [Google Scholar]
- Kadereit, J. W. , Preston C. D., and Valtueña F. J.. 2011. Is Welsh poppy, Meconopsis cambrica (L.) Vig. (Papaveraceae), truly a Meconopsis? New J. Bot. 1:80–88. [Google Scholar]
- Katoh, K. , and Standley D. M.. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30:772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotyaev, B. A. 2006. A review of the weevil genus Rhinoncomimus Wagner (Coleoptera: Curculionidae: Ceutorhynchinae). Entomol. Abh. 63:99–122. [Google Scholar]
- Korotyaev, B. A. , and Anderson R. S.. 2002. Ceutorhynchinae American Beetles, Volume II: Polyphaga: Scarabaeoidea through Curculionoidea. CRC Press, Boca Raton, FL, p. 861. [Google Scholar]
- Krátky, J. 2015. Aphytobius veronicae (Frivaldszky, 1884) (Coleoptera, Curculionidae, Ceutorhynchinae, Hypurini)–species status revised. Snudebiller 15:8. [Google Scholar]
- Krátky, J. 2016. Ceutorhynchus paroliniae sp.n. from Gran Canaria (Coleoptera, Curculionidae, Ceutorhynchinae). Snudebiller 17:6. [Google Scholar]
- Legalov, A. A. 2013. New and little known weevils (Coleoptera: Curculionoidea) from the Paleogene and Neogene. Hist. Biol. 25:59–80. [Google Scholar]
- Maddison, W. P. , and FitzJohn R. G.. 2015. The unsolved challenge to phylogenetic correlation tests for categorical characters. Syst. Biol. 64:127–136. [DOI] [PubMed] [Google Scholar]
- Magallón, S. , Gómez‐Acevedo S., Sánchez‐Reyes L. L., and Hernández‐Hernández T.. 2015. A metacalibrated time‐tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207:437–453. [DOI] [PubMed] [Google Scholar]
- May, M. R. , Höhna S., and Moore B. R.. 2016. A Bayesian approach for detecting the impact of mass‐extinction events on molecular phylogenies when rates of lineage diversification may vary. Methods Ecol. Evol. 7:947–959. [Google Scholar]
- McKenna, D. D. , and Farrell B. D.. 2006. Tropical forests are both evolutionary cradles and museums of leaf beetle diversity. Proc. Natl. Acad. Sci. 103:10947–10951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, D. D. , Sequeira A. S., Marvaldi A. E., and Farrell B. D.. 2009. Temporal lags and overlap in the diversification of weevils and flowering plants. Proc. Natl. Acad. Sci. 106:7083–7088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, A. L. , and Wiens J. J.. 2018. Estimating diversification rates for higher taxa: BAMM can give problematic estimates of rates and rate shifts. Evolution 72:39–53. [DOI] [PubMed] [Google Scholar]
- Miller, M. A. , Schwartz T., Pickett B. E., He S., Klem E. B., Scheuermann R. H., Passarotti M., Kaufman S., and O'Leary M. A.. 2015. A RESTful API for access to phylogenetic tools via the CIPRES science gateway. Evol. Bioinforma. 11:EBO–S21501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minh, B. Q. , Nguyen M. A. T., and von Haeseler A.. 2013. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30:1188–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misof, B. , and Misof K.. 2009. A monte carlo approach successfully identifies randomness in multiple sequence alignments: a more objective means of data exclusion. Syst. Biol. 58:21–34. [DOI] [PubMed] [Google Scholar]
- Moore, B. R. , Höhna S., May M. R., Rannala B., and Huelsenbeck J. P.. 2016. Critically evaluating the theory and performance of Bayesian analysis of macroevolutionary mixtures. Proc. Natl. Acad. Sci. 113:9569–9574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morley, R. J. 2003. Interplate dispersal paths for megathermal angiosperms. Perspect. Plant Ecol. Evol. Syst. 6:5–20. [Google Scholar]
- Muller, F. J. , Mason P. G., Dosdall L. M., and Kuhlmann U.. 2011. European ectoparasitoids of two classical weed biological control agents released in North America. Can. Entomol. 143:197–210. [Google Scholar]
- Nguyen, L.‐T. , Schmidt H. A., von Haeseler A., and Minh B. Q.. 2015. IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum‐likelihood phylogenies. Mol. Biol. Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyman, T. , Linder H. P., Peña C., Malm T., and Wahlberg N.. 2012. Climate‐driven diversity dynamics in plants and plant‐feeding insects. Ecol. Lett. 15:889–898. [DOI] [PubMed] [Google Scholar]
- Oberprieler, R. G. , Anderson R. S., and Marvaldi A. E.. 2014. Curculionoidea Latreille, 1802: Introduction, phylogeny Pp. 285–300 in Leschen R. A. B. and Beutel R. G. eds. Handbook of Zoology Arthropoda Insecta. Berlin: Walter de Gruyter. [Google Scholar]
- Opitz, S. E. W. , Boevé J.‐L., Nagy Z. T., Sonet G., Koch F., and Müller C.. 2012. Host shifts from Lamiales to Brassicaceae in the Sawfly Genus Athalia. PLOS ONE 7:e33649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peña, C. , and Wahlberg N.. 2008. Prehistorical climate change increased diversification of a group of butterflies. Biol. Lett. 4:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Gutiérrez, M. A. , Romero‐García A. T., Fernández M. C., Blanca G., Salinas‐Bonillo M. J., and Suárez‐Santiago V. N.. 2015. Evolutionary history of fumitories (subfamily Fumarioideae, Papaveraceae): an old story shaped by the main geological and climatic events in the Northern Hemisphere. Mol. Phylogenet. Evol. 88:75–92. [DOI] [PubMed] [Google Scholar]
- Prena, J. , Colonnelli E., Hespenheide H. A., Leschen R. A. B., and Beutel R. G.. 2014. Conoderinae Schoenherr, 1833. Handb. Zool. Arthropoda Insecta Coleopt. Beetles 3:577–589. [Google Scholar]
- Rabosky, D. L. 2017. How to make any method “fail”: BAMM at the kangaroo court of false equivalency. ArXiv171103253 Q‐Bio.
- Rabosky, D. L. , Grundler M., Anderson C., Shi J. J., Brown J. W., Huang H., Larson J. G., et al. 2014. BAMMtools: an R package for the analysis of evolutionary dynamics on phylogenetic trees. Methods Ecol. Evol. 5:701–707. [Google Scholar]
- Rabosky, D. L. , Mitchell J. S., and Chang J.. 2017. Is BAMM flawed? Theoretical and practical concerns in the analysis of multi‐rate diversification models. Syst. Biol. 66:477–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabosky, D. L. , Santini F., Eastman J., Smith S. A., Sidlauskas B., Chang J., and Alfaro M. E.. 2013. Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nat. Commun. 4:1958. [DOI] [PubMed] [Google Scholar]
- Rambaut, A. , Suchard M. A., Xie D., and Drummond A. J.. 2014. Tracer v1.6.
- Revell, L. J. 2012. phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3:217–223. [Google Scholar]
- Rheinheimer, J. , and Hassler M.. 2010. Die Rüsselkäfer Baden‐Württembergs. Verlag Regionalkultur.
- San Vicente, I. U. , and Salgueira C.. 2010. New records of Phrydiuchus quijote Sánchez Ruiz & Alonso‐Zarazaga, 1995 and P. topiarius (Germar, 1824) from the Iberian Peninsula, with some taxonomic comments (Coleoptera: Curculionidae, Ceutorhynchinae). Boletin SEA 311–314.
- Schütte, A. , Stüben P., and Sprick P.. 2013. The molecular weevil identification project (Coleoptera: Curculionoidea), part I. A contribution to integrative taxonomy and phylogenetic systematics. Snudebiller 14:77. [Google Scholar]
- Sehlmeyer, S. , Wang L., Langel D., Heckel D. G., Mohagheghi H., Petschenka G., and Ober D.. 2010. Flavin‐dependent monooxygenases as a detoxification mechanism in insects: new insights from the Arctiids (Lepidoptera). PLOS ONE 5:e10435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin, S. , Clarke D. J., Lemmon A. R., Lemmon E. M., Aitken A. L., Haddad S., Farrell B. D., Marvaldi A. E., Oberprieler R. G., and McKenna D. D.. 2017. Phylogenomic data yield new and robust insights into the phylogeny and evolution of weevils. Mol. Biol. Evol. 35:823–836. [DOI] [PubMed] [Google Scholar]
- Silvestro, D. , Schnitzler J., and Zizka G.. 2011. A Bayesian framework to estimate diversification rates and their variation through time and space. BMC Evol. Biol. 11:311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubrier, J. , Steel M., Lee M. S. Y., Sarkissian C. D., Guindon S., Ho S. Y. W., and Cooper A.. 2012. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Mol. Biol. Evol. 29:3345–3358. [DOI] [PubMed] [Google Scholar]
- Stadler, T. 2011. Mammalian phylogeny reveals recent diversification rate shifts. Proc. Natl. Acad. Sci. 108:6187–6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocsits, R. R. , Letsch H., Hertel J., Misof B., and Stadler P. F.. 2009. Accurate and efficient reconstruction of deep phylogenies from structured RNAs. Nucleic Acids Res. 37:6184–6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stüben, P. E. 2017. The Macaronesian islands—An encyclopedia of curculionoidea (Coleoptera). Available online under https://www.curci.de/institute/lecharancon/catalogue_4/catalogue_4 Accessed on [10th September 2014].
- Stüben, P. E. , Schütte A., Bayer C., and Astrin J. J.. 2015. The molecular weevil indentification project (Coleoptera: Curculionoidea), part II‐towards and integrative taxonomy. Snudebiller 16:1–294. [Google Scholar]
- Stüben, P. , and Schütte A.. 2014. Zwei neue Arten aus dem Thamiocolus wollastoni – Komplex von den Kanarischen Inseln (Coleoptera: Curculionidae: Ceutorhynchinae). Snudebiller 15:10. [Google Scholar]
- Wang, L. , Beuerle T., Timbilla J., and Ober D.. 2012. Independent recruitment of a flavin‐dependent monooxygenase for safe accumulation of sequestered pyrrolizidine alkaloids in grasshoppers and moths. PLOS ONE 7:e31796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wheat, C. W. , Vogel H., Wittstock U., Braby M. F., Underwood D., and Mitchell‐Olds T.. 2007. The genetic basis of a plant–insect coevolutionary key innovation. Proc. Natl. Acad. Sci. 104:20427–20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winde, I. , and Wittstock U.. 2011. Insect herbivore counteradaptations to the plant glucosinolate–myrosinase system. Phytochemistry 72:1566–1575. [DOI] [PubMed] [Google Scholar]
- Winkler, I. S. , and Mitter C.. 2008. The Phylogenetic Dimension of Insect‐Plant Interactions: A Review of Recent Evidence. In Tilmon P. K., ed. Specialization, Speciation, and Radiation: The Evolutionary Biology of Herbivorous Insects. University of California Press, Berkeley. [Google Scholar]
- Winkler, I. S. , Mitter C., and Scheffer S. J.. 2009. Repeated climate‐linked host shifts have promoted diversification in a temperate clade of leaf‐mining flies. Proc. Natl. Acad. Sci. 106:18103–18108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter, S. , Friedman A. L. L., Astrin J. J., Gottsberger B., and Letsch H.. 2017. Timing and host plant associations in the evolution of the weevil tribe Apionini (Apioninae, Brentidae, Curculionoidea, Coleoptera) indicate an ancient co‐diversification pattern of beetles and flowering plants. Mol. Phylogenet. Evol. 107:179–190. [DOI] [PubMed] [Google Scholar]
- Wolfe, J. A. 1992. Climatic, floristic, and vegetational changes near the Eocene/Oligocene boundary in North America. Pp. 421–436 in Prothero D. R. and Berggren W. A., eds. Eocene‐Oligocene Biotic and Climatic Evolution. Princeton University Press, NJ. [Google Scholar]
- Xing, Y. , Onstein R. E., Carter R. J., Stadler T., and Peter Linder H.. 2014. Fossils and a large molecular phylogeny show that the evolution of species richness, generic diversity, and turnover rates are disconnected. Evolution 68:2821–2832. [DOI] [PubMed] [Google Scholar]
- Yoshitake, H. , and Ito M.. 2011. First host record for Wagnerinus harmandi (Hustache) (Coleoptera, Curculionidae, Ceutorhynchinae). Elytra 1:343–344. [Google Scholar]
- Yoshitake, H. , Kato T., Jinbo U., and Ito M.. 2008. A new Wagnerinus (Coleoptera: Curculionidae) from northern Japan: description including a DNA barcode. Zootaxa 1740:15–27. [Google Scholar]
- Zachos, J. C. , Dickens G. R., and Zeebe R. E.. 2008. An early Cenozoic perspective on greenhouse warming and carbon‐cycle dynamics. Nature 451:279–283. [DOI] [PubMed] [Google Scholar]
- Zachos, J. , Pagani M., Sloan L., Thomas E., and Billups K.. 2001. Trends, rhythms, and aberrations in global climate 65 Ma to present. Science 292:686–693. [DOI] [PubMed] [Google Scholar]
- Zhang, R. , Kravchinsky V. A., and Yue L.. 2012. Link between global cooling and mammalian transformation across the Eocene–Oligocene boundary in the continental interior of Asia. Int. J. Earth Sci. 101:2193–2200. [Google Scholar]
- Zherikhin, V. V. , and Gratshev V. G.. 2004. Fossil curculionoid beetles (Coleoptera, Curculionoidea) from the Lower Cretaceous of northeastern Brazil. Paleontol. J. CC Paleontol. ZHURNAL 38:528–537. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Sampling ratio of all included genera of Ceutorhynchinae.
Table S2: Taxon sampling including NCBI genbank accession numbers.
Table S2.1: Voucher IDs of all specimens provided by the Molecular Weevil Identification project.
Table S3: Host plant associations of all included ceutorhynchine weevils.
Table S4A: Results of the character‐dependent diversification analysis in HiSSe, based on the BEAST‐I analysis.
Table S4B: Results of the character‐dependent diversification analysis in HiSSe, based on the BEAST‐III analysis.
Figure S1. Results of the maximum likelihood phylogenetic tree reconstruction in IQ‐Tree.
Figures S2. Results from the divergence time analyses in BEAST.
Figures S3. Results of the diversification rate through time analyses in RevBayes.
Figure S4: Results of the diversification rate through time analyses with TESS.
Figures S5: Results of the clade‐specific divergence time analyses in BAMM.