

Summary
Here we present the identification and characterization of the H3K4‐specific histone methyltransferase Set1 and its counterpart, the Jumonji C demethylase Kdm5, in the rice pathogen Fusarium fujikuroi. While Set1 is responsible for all detectable H3K4me2/me3 in this fungus, Kdm5 antagonizes the H3K4me3 mark. Notably, deletion of both SET1 and KDM5 mainly resulted in the upregulation of genome‐wide transcription, also affecting a large set of secondary metabolite (SM) key genes. Although H3K4 methylation is a hallmark of actively transcribed euchromatin, several SM gene clusters located in subtelomeric regions were affected by Set1 and Kdm5. While the regulation of many of them is likely indirect, H3K4me2 levels at gibberellic acid (GA) genes correlated with GA biosynthesis in the wild type, Δkdm5 and OE::KDM5 under inducing conditions. Whereas Δset1 showed an abolished GA3 production in axenic culture, phytohormone biosynthesis was induced in planta, so that residual amounts of GA3 were detected during rice infection. Accordingly, Δset1 exhibited a strongly attenuated, though not abolished, virulence on rice. Apart from regulating secondary metabolism, Set1 and Kdm5 function as activator and repressor of conidiation respectively. They antagonistically regulate H3K4me3 levels and expression of the major conidiation‐specific transcription factor gene ABA1 in F. fujikuroi.
Introduction
Fusarium fujikuroi is a phytopathogenic ascomycete and the founding member of the Fusarium (Gibberella) fujikuroi species complex (Nirenberg and O'Donnell, 1998; Leslie and Summerell, 2006). F. fujikuroi is the causative agent of the so‐called bakanae (foolish‐seedling) disease of rice plants which was first described over 100 years ago in Japan (Hori, 1890). Since then, the fungus has gained considerable attention due to the fact that it is a major threat to rice‐growing countries worldwide. Its secretion of the bioactive phytohormones gibberellic acids (GAs) results in thin, chlorotic and hyper‐elongated rice internodes, oftentimes sterile grains, and therefore full harvest losses (Sun and Snyder, 1981; Bömke and Tudzynski, 2009). On the contrary, F. fujikuroi is exploited as efficient producer of GAs for the use as plant growth regulators in agriculture and horticulture (Rademacher, 1997; Sponsel and Hedden, 2004).
GAs belong to the group of secondary metabolites (SMs) that are per definition not required for fungal growth in general, but might pose an advantage to the fungus under certain environmental conditions (Fox and Howlett, 2008). Recent sequencing of the F. fujikuroi genome revealed 47 predicted SM key genes (Wiemann et al., 2013). Next to the GA gene cluster, our group has identified several additional SMs and the respective biosynthetic genes in F. fujikuroi over the last years (Janevska and Tudzynski, 2018). Among those are the red pigments bikaverin (BIK) and fusarubins (FSR), two polyketide synthase (PKS)‐derived SMs (Wiemann et al., 2009; Studt et al., 2012). The mycotoxins apicidin F (APF) and beauvericin are produced by non‐ribosomal peptide synthetases (NRPSs) (Niehaus et al., 2014a, 2016), while two other mycotoxins, fusarins (FUS) and fusaric acid (FSA), are generated by a hybrid PKS‐NRPS enzyme and by two separate key enzymes respectively (Niehaus et al., 2013, 2014b; Studt et al., 2016a).
Fungal secondary metabolism is regulated by a complex network functioning on different hierarchical levels, including so‐called pathway‐/cluster‐specific transcription factors (TFs) that are often encoded within the respective gene cluster (Brakhage, 2013), as well as global regulators. The latter are TFs which affect a larger set of genes of both primary and secondary metabolism in response to different environmental cues, such as the availability of carbon and nitrogen sources or the presence of certain pH and light conditions (Macheleidt et al., 2016). On a higher level of regulation, SM biosynthesis underlies the controlled change of the chromatin state which can be either tightly condensed (i.e., heterochromatin) or more loosely packed (i.e., euchromatin), depending on the specific set of post‐translational histone modifications deposited in response to environmental cues (Gacek and Strauss, 2012).
Nearly all of the identified histone (H) modifications are reversible and dynamic. The acetylation of lysine (K) residues is associated with an active transcription, while the methylation of lysine or arginine residues gives a more complex output, depending on associated reader proteins (Brosch et al., 2008; Gacek and Strauss, 2012). In general, the methylation of H3K9 and H3K27 is involved in the formation of constitutive and facultative heterochromatin, respectively (Rando and Chang, 2009; Wiles and Selker, 2017), regions predominantly found at centromeres and subtelomeres in F. fujikuroi (Wiemann et al., 2013; Studt et al., 2016b). In contrast, methylation of H3K4 and H3K36 is considered as hallmark of euchromatin in yeast and higher eukaryotes (Rando and Chang, 2009; Wagner and Carpenter, 2012). However, the picture seems to be more diverse in filamentous fungi, as recent data revealed the ubiquitous presence of the H3K36 trimethylation (me3) mark and accordingly, no correlation with active transcription in both F. fujikuroi and Fusarium graminearum (Connolly et al., 2013; Janevska et al., 2018).
H3K4 methylation has been indeed found in transcribed euchromatic regions in F. fujikuroi (Wiemann et al., 2013), and has been clearly linked to actively expressed genes in budding yeast and filamentous fungi, that is, Saccharomyces cerevisiae, Aspergillus nidulans and F. graminearum (Pokholok et al., 2005; Connolly et al., 2013; Gacek‐Matthews et al., 2016). In 5′ promoter regions, H3K4 methylation is associated with the initiating form of RNA polymerase II: all euchromatic genes were found to be decorated with H3K4me2, while H3K4me3 marked actively or recently transcribed genes in S. cerevisiae (Santos‐Rosa et al., 2002; Ng et al., 2003). Noteworthy, S. cerevisiae chromatin does not harbour the repressive marks H3K9me3 and H3K27me3, and H3K4 methylation has been implicated in the silencing of subtelomeric genes (Briggs et al., 2001; Krogan et al., 2002; Bryk et al., 2002; Fingerman et al., 2005). However, the exact mechanism remains to be elucidated.
The methylation of H3K4 has been studied in a number of different organisms from yeast to humans, and has been shown to depend on the conserved Su(var)3‐9, Enhancer‐of‐zeste, Trithorax (SET) domain‐containing methyltransferase Set1 (Briggs et al., 2001; Lee and Skalnik, 2005; Freitag, 2017). The genomes of yeast and filamentous fungi, such as A. nidulans, F. graminearum and Fusarium verticillioides, encode one Set1 homologue that is responsible for all three methylation states H3K4me1/me2/me3 (Briggs et al., 2001; Fingerman et al., 2005; Harting et al., 2013; Govindaraghavan et al., 2014; Liu et al., 2015; Gu et al., 2017). As a counterpart, the Jumonji C (JmjC) domain‐containing demethylase Jhd2 has been identified in S. cerevisiae (Liang et al., 2007; Seward et al., 2007), and later also in A. nidulans (Gacek‐Matthews et al., 2016). S. cerevisiae Jhd2 and the A. nidulans homolog KdmB have been shown to counteract H3K4me3 in vivo (Liang et al., 2007; Gacek‐Matthews et al., 2016).
Set1 is part of COMPASS (Complex of Proteins Associated with Set1). Recently, we identified and functionally characterized the COMPASS component Ccl1 in F. fujikuroi and F. graminearum. In both fungi, Ccl1 is a critical factor for efficient genome‐wide H3K4 trimethylation by COMPASS and has great impact on SM biosynthesis (Studt et al., 2017). However, the exact mechanism of how COMPASS affects the expression of subtelomeric SM genes in both fungi remains unclear so far.
In the present work, we focused on the catalytic subunit Set1 of COMPASS, and on the H3K4me3‐specific demethylase Kdm5 as counterpart of Set1. We found a great impact on genome‐wide transcription, especially upon SET1 deletion, as well as a considerable effect on SM production for Δset1, Δkdm5 and OE::KDM5 mutants. Notably, albeit completely abolished GA production levels of Δset1 in axenic culture, the mutant induced – though strongly attenuated – bakanae symptoms on rice. This phenotype goes in line with residual GA3 levels found in planta. Next to secondary metabolism, Set1 and Kdm5 function as activator and repressor of conidiation respectively. Gene knock‐out, chromatin immunoprecipitation (ChIP) and/or gene expression analyses revealed the conidiation‐specific TF‐encoding gene ABA1 as major target of Set1, Kdm5 and also other regulators, that is, Csm1, Flb3 and Flb4.
Results
Identification of the H3K4 methyltransferase Set1 and demethylase Kdm5 in F. fujikuroi
After analyzing the COMPASS component Ccl1 in F. fujikuroi (Studt et al., 2017), we have now identified the putative methyltransferase of this complex by determining the ortholog using QuartetS (Yu et al., 2011). The predicted protein FFUJ_02475 is the ortholog of the well‐characterized H3K4‐specific methyltransferases Set1 in S. cerevisiae and SetA in A. nidulans (Briggs et al., 2001; Govindaraghavan et al., 2014). FFUJ_02475 contains the catalytically active SET (IPR001214), N‐SET (IPR024657) and Post‐SET (IPR003616) domains as well as an RNA‐binding (IPR035979) domain also present in Set1 and SetA from S. cerevisiae and A. nidulans, respectively (Fig. 1A), and was therefore designated F. fujikuroi Set1.
Figure 1.
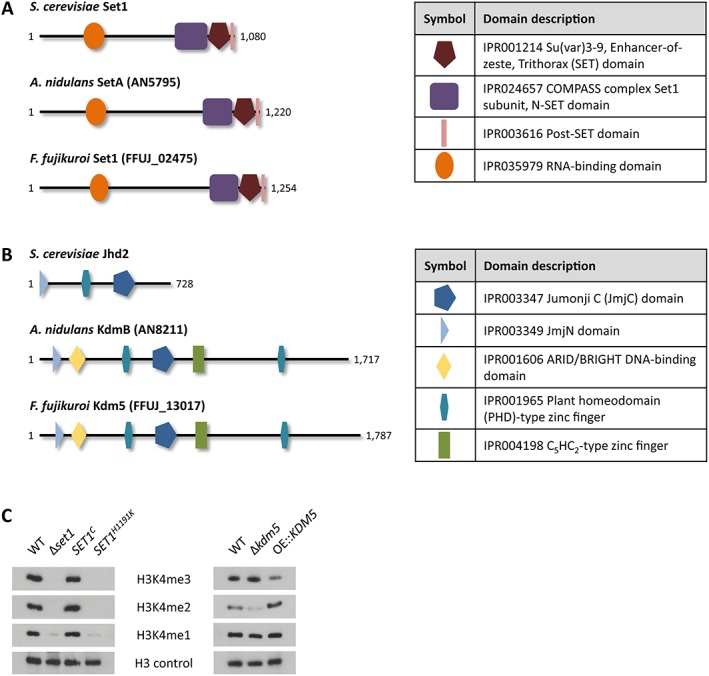
Fusarium fujikuroi Set1 and Kdm5 are a H3K4‐specific histone methyltransferase and demethylase respectively. A,B. Schematic representation of the domain structure of Set1 and Kdm5 homologues conserved in S. cerevisiae, A. nidulans and F. fujikuroi. The domain description includes the respective InterPro accession numbers. C. Western blot analysis using the H3K4me3/me2/me1 and H3 antibodies. Indicated strains were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to protein extraction. For the detection of H3K4me3 in Δkdm5 and OE::KDM5 mutants, 10 μg of the protein extract was loaded on to the gel, while 15 μg was loaded for the rest.
In addition to Set1, FFUJ_13017 was determined by QuartetS as the A. nidulans KdmB ortholog (Gacek‐Matthews et al., 2016; Yu et al., 2011), a putative H3K4‐specific demethylase and possible antagonist to F. fujikuroi Set1. Hence, FFUJ_13017 was designated F. fujikuroi Kdm5 according to the general nomenclature (Allis et al., 2007). Both F. fujikuroi Kdm5 and A. nidulans KdmB show an identical domain structure which is more complex than that of the S. cerevisiae ortholog Jhd2 (Liang et al., 2007). All three share the catalytically active JmjC (IPR003347) and JmjN (IPR003349) domains, while KdmB and Kdm5 harbor not only one but two plant homeodomain‐type zinc fingers (IPR001965). In addition, the latter have ARID/BRIGHT DNA‐binding (IPR001606) and C5HC2‐type zinc finger (IPR004198) domains (Fig. 1B).
Single deletion mutants have been successfully generated for F. fujikuroi SET1 and KDM5, and KDM5 was additionally overexpressed using the constitutive oliC promoter from A. nidulans. While abundant in the F. fujikuroi wild type (WT) IMI58289, all detectable global H3K4me3 and H3K4me2 signals were lost in Δset1 mutants as identified by western blot analyses (Fig. 1C). Contrary to S. cerevisae and A. nidulans (Fingerman et al., 2005; Govindaraghavan et al., 2014), but in agreement with F. graminearum (Liu et al., 2015), a faint band was still detectable for H3K4me1. However, it remains unclear at this moment whether there are indeed residual monomethylation levels in Δset1 or whether this band is due to insufficient antibody specificity. The methylation defect was completely restored upon in loco complementation of Δset1 with the full‐length gene, gaining SET1 C strains (Fig. 1C). In contrast, the in loco integration of the point‐mutated variant SET1 H1191K, in which the catalytically active histidine of the SET domain has been mutated to a lysine residue (Tanaka et al., 2007; Janevska et al., 2018), did not complement as expected (Fig. 1C).
Next, F. fujikuroi Kdm5 was identified as a true counterpart of Set1, functioning as an H3K4me3‐specific demethylase. The total level of H3K4me3 was increased in Δkdm5 and decreased in OE::KDM5, while the opposite was true for H3K4me2 (Fig. 1C). More precisely, H3K4me3 cannot be removed in the Δkdm5 mutant, which results in a shift of H3K4me2 to H3K4me3 genome‐wide (Fig. 1C; Supporting Information Fig. S1), as H3K4me2 is still continuously converted into the trimethylation state by Set1. Something very similar has also been observed for S. cerevisiae JHD2 and A. nidulans kdmB deletion mutants (Liang et al., 2007; Gacek‐Matthews et al., 2016).
In summary, Set1 is responsible for H3K4 tri‐, di‐ and possibly also monomethylation in F. fujikuroi, while Kdm5 was found to counteract Set1‐mediated H3K4me3.
Deletion of SET1 and overexpression of KDM5 affect vegetative growth
Next, we performed a plate assay using complex (complete medium, CM and vegetable juice agar, V8) as well as minimal (Czapek Dox, CD and synthetic ICI with 6 mM NH4NO3) media to assess the impact of H3K4 methylation on vegetative growth. Deletion of SET1 resulted in a growth defect on all tested media, which was restored in SET1 C but not in SET1 H1191K strains (Fig. 2A). Therefore, it is likely that the loss of H3K4 methylation is the sole reason for the poor vegetative growth of Δset1.
Figure 2.
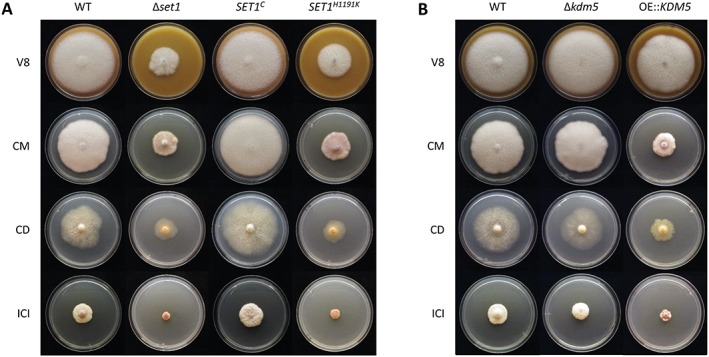
Influence of SET1 deletion as well as KDM5 deletion and overexpression on vegetative growth. A. The WT, Δset1, SET1 C and SET1 H1191K mutants were grown on solid complex (V8, CM) and minimal (CD, ICI + 6 mM NH4NO3) media for 7 days in the dark in triplicates. B. The WT, Δkdm5 and OE::KDM5 mutants were grown under above described conditions.
While Δkdm5 mutants showed a WT‐like colony diameter, OE::KDM5 mutants exhibited an attenuated growth (Fig. 2B), thereby resembling Δset1 and SET1 H1191K. In conclusion, strains with reduced (OE::KDM5) or abolished (Δset1, SET1 H1191K) H3K4me3 levels (Fig. 1C) showed retarded hyphal growth, suggesting a correlation between H3K4me3 and vegetative growth in F. fujikuroi.
Only a small set of genes is antagonistically affected in Δset1 and Δkdm5
In order to compare the genome‐wide impact of SET1 and KDM5 deletion on transcription, we performed a microarray expression analysis. For this, the WT, Δset1 and Δkdm5 mutants were cultivated in synthetic ICI liquid medium with limiting (6 mM, N−) or saturating (60 mM, N+) amounts of glutamine (Gln), conditions that were previously shown to either induce or repress the expression of several SM genes in F. fujikuroi (Wiemann et al., 2013; Niehaus et al., 2017a). Based on the selection criteria of a fourfold change in expression (log2 fold change ≥ 2 or ≤ −2) at the 95% confidence interval (false discovery rate < 0.05), 2282 of the 14,816 annotated genes (15.4%) were affected in a Set1‐ and/or Kdm5‐dependent manner in at least one condition. Among these, 1,656 and 1,191 genes were differentially expressed (compared with the WT) in the presence of 6 and 60 mM Gln, respectively, giving an overlap of 565 genes affected under both nitrogen conditions (Fig. 3).
Figure 3.
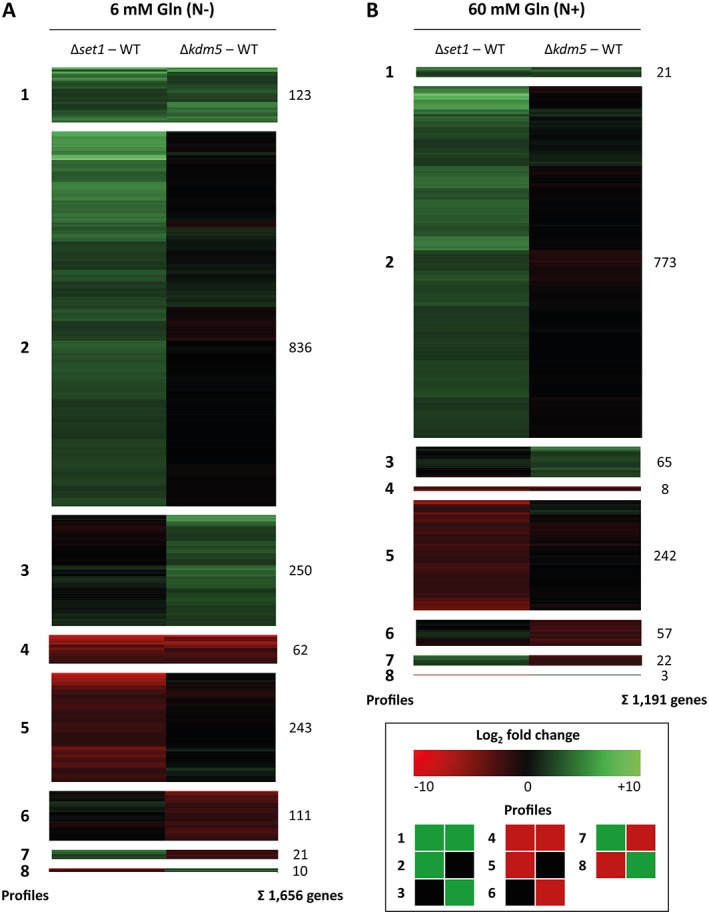
Microarray expression analysis of differentially regulated genes in Δset1 and Δkdm5. The WT and the two deletion mutants were grown in ICI liquid culture in the presence of (A) limiting (6 mM, N−) and (B) saturating (60 mM, N+) amounts of Gln for 3 days prior to RNA extraction. Data are mean values (n = 2). Genes upregulated in the deletion mutants compared with the WT are green (log2 fold change ≥ 2), downregulated genes are red (log2 fold change ≤ −2), and not differentially expressed genes are black (between −2 and 2). The eight profiles were extracted first, and then the genes were clustered for each profile.
The majority of genes was regulated (directly or indirectly) by Set1, while only a smaller set of genes was affected by Kdm5, and an even smaller set of genes was regulated by both Set1 and Kdm5. More precisely, under nitrogen limitation, 1,079 genes (65.2%) were only affected in Δset1, 361 genes (21.8%) were only affected in Δkdm5 and 216 genes (13.0%) were up‐ and/or downregulated in both Δset1 and Δkdm5 (Fig. 3A). Notably, the majority of genes was upregulated upon SET1 and/or KDM5 deletion (73.0% in Fig. 3A), whereas fewer genes were downregulated (25.1% in Fig. 3A). In contrast to our expectation, only a small set of genes was regulated in an antagonistic manner by Set1 and Kdm5 (1.9% in Fig. 3A and 2.1% in Fig. 3B). At this point, no direct regulation mechanisms can be concluded from these data, as the observed changes in gene expression can still be mediated by downstream regulators, and not via direct binding to the loci by Set1 and Kdm5.
Under both nitrogen conditions, the largest group comprised genes upregulated in Δset1 but not affected in Δkdm5: this profile 2 contained 50.5% and 64.9% of all differentially regulated genes in the presence of 6 and 60 mM Gln respectively (Fig. 3). A Functional Catalogue (FunCat) analysis (Ruepp et al., 2004) to identify significantly enriched protein functions in this profile identified the FunCat groups ‘01.20 secondary metabolism’ and ‘01 metabolism’ under both nitrogen conditions (Supporting Information Table S1). Furthermore, in the presence of 6 mM Gln, four additional FunCat groups were enriched with a high significance in profile 2: ‘01.05 C‐compound and carbohydrate metabolism’, ‘01.06.05 fatty acid metabolism’, ‘02.07 pentose‐phosphate pathway’ and ‘32.07 detoxification’ (Supporting Information Table S1).
In summary, the genome‐wide transcriptome analysis yielded interesting insights: the majority of genes is up‐ but not downregulated upon deletion of SET1, including several SM‐related genes. Set1 and Kdm5 act only in a minority of cases as true antagonists, either directly or indirectly on a transcriptional level.
WT‐like SM biosynthesis depends on both Set1 and Kdm5
Based on the microarray analysis, several SM key enzyme‐encoding genes are differentially expressed in Δset1 and/or Δkdm5 in comparison to the WT. Strikingly, 15 out of the 21 affected key genes are responsive to nitrogen in the WT, and are preferentially expressed under either nitrogen limitation or surplus conditions (Supporting Information Table S2). In agreement with the genome‐wide transcriptome analysis (Fig. 3), several SM key genes were found to be upregulated in Δset1. In fact, 8 out of 12 and 5 out of 8 genes were upregulated in the Δset1 mutant compared with the WT under N− and N+ conditions respectively (Supporting Information Table S2). Among those are several so far cryptic SM key genes without yet assigned products, that is, PKS‐NRPS9, PKS type III, NRPS4, NRPS23 and DMATS3, rendering this strain an interesting target for future product analyses. Fewer SM genes were affected by the loss of KDM5. Here, only 8 SM key genes were deregulated in Δkdm5 with the majority of them (5 out of 8) being downregulated (Supporting Information Table S2). Noteworthy, only two of them, that is, PKS4 and NRPS31, involved in the biosynthesis of BIK and APF, respectively, were found to be regulated in an antagonistic manner by Set1 and Kdm5. Indeed, not only the key enzyme‐encoding genes, but all 6 BIK cluster genes and 11 APF cluster genes were similarly affected by SET1 and KDM5 deletion (Supporting Information Fig. S2).
Among the known SMs, which can be readily quantified in liquid cultures via high‐performance liquid chromatography coupled to diode array detection (HPLC‐DAD), production of the two red pigments BIK and FSR was shown to be increased in Δset1 and OE::KDM5, and decreased in Δkdm5 compared with the WT under their respective producing conditions (Fig. 4A,B). A similar pattern was observed for the mycotoxin FUS: FUS biosynthesis was downregulated in Δkdm5, and upregulated in the other two strains (Fig. 4C). In contrast, production of the mycotoxin FSA was only slightly enhanced in Δset1, slightly decreased in OE::KDM5 and not affected in Δkdm5 (Fig. 4D). Notably, the biosynthesis of the bioactive phytohormone gibberellic acid GA3 showed a different pattern: GA3 was not detectable in Δset1, its levels were reduced to about 20% in Δkdm5, and not affected in the KDM5 overexpression strain (Fig. 4E). Therefore, Set1 and Kdm5 were found to antagonize BIK, FSR, FUS (via HPLC) and possibly APF biosynthesis (according to the microarray). Not all of the described effects on SM production were reflected as significant changes in the microarray expression analysis: the GA key gene was only downregulated in Δset1 but not in Δkdm5 (6 mM Gln), and the changes for the FUS key gene were not significant (60 mM Gln; Supporting Information Table S2). Furthermore, the producing condition for FSR (6 mM NaNO3) was not included in the microarray. The discrepancy between SM gene expression and SM production levels can likely be explained by the fact that the two analyses were performed at different time points, that is, after 3 and 7 days of cultivation respectively. While production levels show the accumulation of a metabolite within an extended time period, gene expression levels stand for one (representative) time point.
Figure 4.
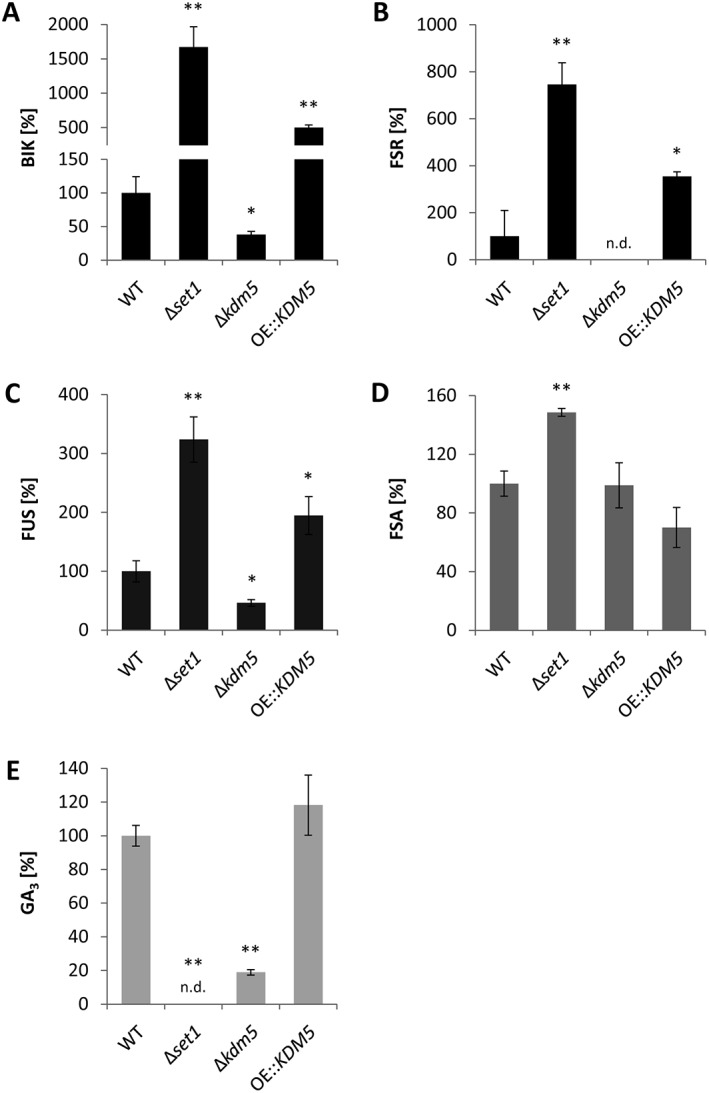
SM biosynthesis is deregulated in Δset1, Δkdm5 and OE::KDM5 mutants. Indicated strains were grown in ICI liquid culture for 7 days and analysed via HPLC‐DAD. The cultivation was done in the presence of 6 mM Gln for bikaverin (BIK) and gibberellic acid GA3 (A, E), 6 mM NaNO3 for fusarubins (FSR) (B) or 60 mM Gln for fusarins (FUS) and fusaric acid (FSA) (C, D). The production was related to the dry weight of the strains and the production level of the WT was arbitrarily set to 100%. Data are mean values ± SD (n = 3). For statistical analysis, the mutants were compared with the WT using the student's t‐test: *, p < 0.05; **, p < 0.01.; n.d., not detected.
BIK, FSR, FSA and GA clusters are located within, and the FUS cluster is located in close proximity to subtelomeric regions of facultative heterochromatin, indicated by the heterochromatic mark H3K27me3 (Supporting Information Fig. S3; Studt et al., 2016b). Previous genome‐wide ChIP‐sequencing analysis revealed the absence of H3K4me2 at most of the here analyzed SM genes, except for the GA gene cluster. Here, two out of the seven GA cluster genes were shown to be decorated with H3K4me2 in the WT background only under inducing conditions (Supporting Information Fig. S3C; Wiemann et al., 2013). To gain a deeper insight into the H3K4 methylation pattern, we performed ChIP with subsequent quantitative real‐time polymerase chain reaction (qPCR) to determine H3K4me3 and H3K4me2 levels at GA cluster genes in Δset1, Δkdm5 and OE::KDM5 mutants under GA‐inducing conditions. The key gene CPS/KS as well as P450‐1 were chosen as highly expressed GA cluster genes, while P450‐2 and P450‐4 were the ones decorated with H3K4me2 under inducing conditions (Supporting Information Fig. S3C).
As expected, lack of SET1 resulted in an overall decrease of H3K4me3 and H3K4me2 at all analysed GA genes (Fig. 5A,B), a phenotype that goes in line with the global loss of H3K4me3/me2 as shown by western blot (Fig. 1C). While H3K4me3 levels followed a similar pattern in Δset1 and OE::KDM5, and were reduced in the ChIP and western blot analyses (Figs. 1C and 5A), a significant increase in H3K4me3 was only observed for CPS/KS and P450‐1, but not P450‐2 and P450‐4, in Δkdm5 (Fig. 5A). This might be due to relatively high amounts of H3K4me3 at these two genes in the WT background, which cannot be elevated any further in Δkdm5.
Figure 5.
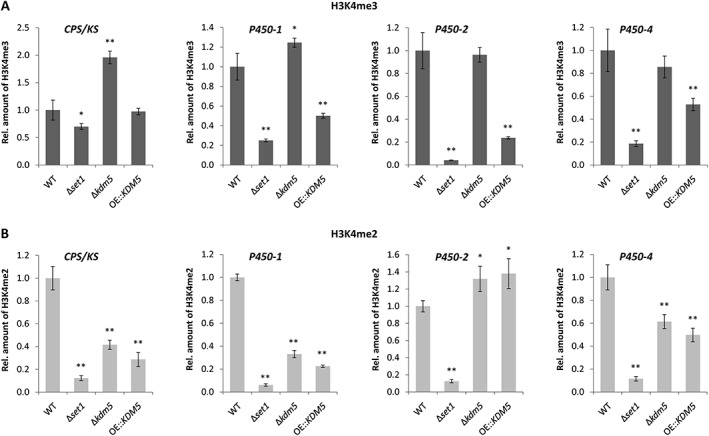
H3K4me3 and H3K4me2 levels at gibberellic acid (GA) cluster genes in Δset1, Δkdm5 and OE::KDM5 mutants. Indicated strains were grown in liquid culture (ICI + 6 mM Gln) for 3 days prior to ChIP‐qPCR using the (A) H3K4me3 or (B) H3K4me2 antibody. Therefore, the enriched samples (precipitated by antibody) were normalized to the respective input samples (initially applied chromatin). The WT was arbitrarily set to 1, and the data are mean values ± SD (n = 4). For statistical analysis, the mutants were compared with the WT using the student's t‐test: *, p < 0.05; **, p < 0.01.
Contrary to H3K4me3, H3K4me2 levels were comparable in Δkdm5 and OE::KDM5 at GA cluster genes (Fig. 5B), while both H3K4 methylation marks behaved antagonistically genome‐wide as shown by western blot (Fig. 1C). The effects were more obvious for the GA cluster genes P450‐2 and P450‐4, which exhibit a strong accumulation of H3K4me2 in the WT background (Supporting Information Fig. S3C), and followed a similar trend for CPS/KS and P450‐1 (Fig. 5B). Bearing in mind that Δkdm5 and OE::KDM5 showed a similar phenotype and both produced significant amounts of GA3 in axenic culture (Fig. 4C), this might rely on comparable levels of H3K4me2, but not H3K4me3, in these two strains (Fig. 5A,B).
To conclude, secondary metabolism in F. fujikuroi was found to depend on functional Set1 and Kdm5: the biosynthesis of several SMs was deregulated in the analyzed mutants Δset1, Δkdm5 and OE::KDM5. However, the exact regulatory circuits remain to be elucidated.
Deletion of SET1 results in an attenuated virulence on rice
Most F. fujikuroi strains cause bakanae disease when infecting rice plants as a result of their secretion of GAs, a group of highly bioactive phytohormones (Niehaus et al., 2017a). Since the GA3 production in axenic culture was fully abolished in Δset1 and significantly decreased in Δkdm5 (Fig. 4E), we next performed infection studies of the analysed mutant strains. Therefore, surface‐sterilized and pre‐germinated rice seedlings were infected with the WT, Δset1, Δkdm5 and OE::KDM5 strains (Fig. 6; Supporting Information Fig. S4). Both Δkdm5 and OE::KDM5 resulted in a WT‐like phenotype, characterized by chlorotic and hyper‐elongated rice internodes (Fig. 6A,B; Supporting Information Fig. S4), albeit the significantly reduced GA3 levels found in Δkdm5 strains during axenic growth (Fig. 4E). Notably, even the rice plants infected with Δset1, a strain that did not produce detectable GA3 levels during axenic growth (Fig. 4E), showed slightly chlorotic and extended internodes (Fig. 6A,B). Subsequent chemical quantification of GA3 levels in planta revealed that GA3 levels were in fact not abolished in Δset1 during the infection. Rice samples infected with Δset1 contained 24% of the GA3 level of WT‐infected rice samples (Fig. 6C). Notably, GA3 levels in Δkdm5‐infected plant material were also higher compared with axenic growth (78% of WT‐infected samples instead of 20% in axenic culture) (Figs. 4E and 6C), thereby likely explaining the WT‐like infection pattern of this strain (Fig. 6A,B).
Figure 6.
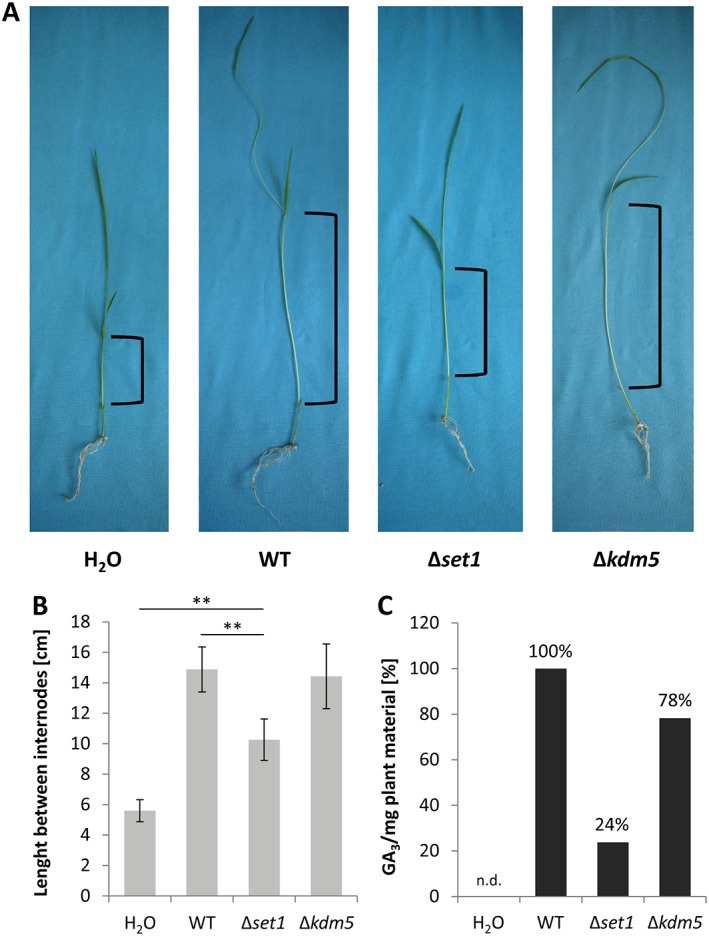
Determination of virulence and GA3 levels of Δset1‐ and Δkdm5‐infected rice plants. A. Germinated rice seedlings were infected with the WT, the Δset1 and Δkdm5 strains. Mock infection was achieved through the addition of H2O and served as negative control. Progression of infection was determined after 7 days. B. The length between rice internodes was measured (indicated in A). Data are mean values ± SD (n = 5). For statistical analysis, Δset1 was compared with the WT and mock control using the student's t‐test: **, p < 0.01. C. Gibberellic acid GA3 levels in planta were quantified via HPLC‐MS/MS and normalized to the input plant material. The 5 (infected) rice plants were combined, and then divided again to yield a technical replicate for the SM extraction and quantification. The WT was arbitrarily set to 100%. n.d., not detected.
Set1 and Kdm5 are positive and negative regulators of conidiation, respectively
The formation of asexual spores is an important way for filamentous, pathogenic fungi to proliferate and spread. The F. fujikuroi WT IMI58289 used in this work produces only microconidia (here simply referred to as ‘conidia’), while other strains produce micro‐ and macroconidia (with several septa), or predominantly macroconidia (Niehaus et al., 2017a). As conidiation‐related genes are part of the conserved fungal genome, we assumed an effect on conidiogenesis upon perturbation of the euchromatic H3K4 methylation mark in SET1 and KDM5 mutants.
To assess the ability of Δset1, Δkdm5 and OE::KDM5 mutants to reproduce asexually, we next quantified conidia formation. While both Δset1 and OE::KDM5 produced only very few conidia, Δkdm5 showed elevated conidia formation in comparison to the WT, suggesting that Set1 and Kdm5 indeed antagonize conidiogenesis (Fig. 7A).
Figure 7.
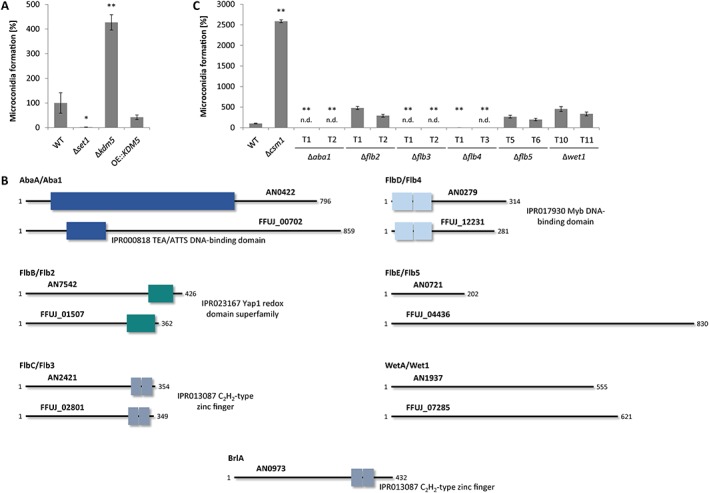
Conidiation is affected by SET1 and KDM5 deletion, and is regulated by the conserved TFs Aba1, Flb3 and Flb4. A. Indicated strains were grown on V8 agar in the presence of a 12 h light/12 h dark cycle for 14 days prior to analysis of conidia formation. Data are mean values ± SD (n = 3). For statistical analysis, the mutants were compared with the WT using the student's t‐test: *, p < 0.05; **, p < 0.01. B. The A. nidulans regulators AbaA, FlbB‐FlbE and WetA, but not BrlA, are conserved in F. fujikuroi. The domain description includes the respective InterPro accession numbers. C. Indicated strains were grown and analysed as described above. n.d., not detected.
Intrigued by these findings, we searched the F. fujikuroi genome for conidiation‐related genes, which have already been well‐characterized in A. nidulans (Krijgsheld et al., 2013). The upstream cascade is mainly transmitted by the so‐called ‘fluffy’ proteins, many of which are TFs, such as FlbB (basic leucine zipper TF), FlbC (C2H2 TF) and FlbD (cMyb TF), while FlbE does not harbor a conserved domain (Fig. 7B). Both FlbC and a heterodimer of FlbB/FlbD (FlbD is released from a heterodimer with FlbE) are required for efficient brlA (bristle) expression, which encodes the central regulator of asexual development in A. nidulans. BrlA further activates the TF gene abaA (abacus), which in turn activates wetA (wet white), the two being required for conidia formation and maturation respectively (Krijgsheld et al., 2013).
Orthologs for A. nidulans flbB‐flbD were determined by QuartetS (Yu et al., 2011), while no entry was found for abaA, flbE and wetA. Thus, here putative orthologs were identified as ‘Best‐Hits’ by BlastP analyses. No putative ortholog for brlA could be identified in the genome of F. fujikuroi (Fig. 7B). BrlA seems to be absent from the genus Fusarium (Son et al., 2014a; Niehaus et al., 2017a). Single deletion mutants were generated for the F. fujikuroi orthologs, designated ABA1, FLB2‐FLB5 and WET1, and the mutants were tested for their ability to produce conidia. In Fig. 7C, the novel mutants are shown in comparison to Δcsm1 (regulator of conidiation and secondary metabolism). The TF‐encoding gene CSM1 encodes a major repressor of conidiation in F. fujikuroi, and the respective deletion strain is characterized by a strongly elevated conidia formation relative to the WT (Niehaus et al., 2017b). Indeed, conidiogenesis was abolished in Δaba1, Δflb3 and Δflb4 mutants, while WT‐like amounts of conidia were produced in Δflb2, Δflb5 and Δwet1 (Fig. 7C). A plate assay performed under conidiation‐inducing conditions did not show an altered phenotype, concerning the colony diameter or morphology, for any of the generated deletion mutants (Supporting Information Fig. S5A). Furthermore, a microscopic analysis revealed that Wet1 is probably also involved in conidia separation and maturation in F. fujikuroi, as Δwet1 mutants did not form longer strings of conidia as the WT, but formed smaller nests of conidia that remained close to the vegetative hyphae (Supporting Information Fig. S5B).
As indicated above, F. fujikuroi IMI58289 analyzed so far produces only microconidia, while the sequenced strain F. fujikuroi E282 produces macroconidia (Niehaus et al., 2017a). To elucidate whether Aba1 might be also required for macroconidia formation in E282, the respective ABA1 ortholog FFE2_00769 was deleted also in the E282 background. Indeed, macroconidia formation was fully abolished in E282/Δaba1 mutants (data not shown), suggesting that the synthesis of micro‐ and macroconidia underlies similar transcriptional regulation mechanisms in Fusarium spp.
To elucidate whether Set1 and Kdm5 regulate any of the identified conidiation‐specific TF‐encoding genes, ABA1, FLB3 or FLB4, the relative expression of these three genes was assessed in Δset1 and Δkdm5 mutants in comparison to the respective WT IMI58289 under conidiation‐inducing light conditions. Moreover, ChIP‐qPCR was performed to identify changes in H3K4me3 at these genes, which are located in conserved euchromatic regions of chromosome I (ABA1), chromosome III (FLB3) and chromosome VIII (FLB4). Only the expression of ABA1, but not of FLB3 or FLB4, fitted to the conidia production levels of the mutants at the time point chosen: ABA1 was downregulated in Δset1 and upregulated in Δkdm5 after 3 days of incubation (Fig. 8A), correlating with decreased and increased conidia formation in these mutants respectively (Fig. 7A). Indeed, the deposition of H3K4me3 correlated with the detected ABA1 expression and conidia production levels of the mutants (Fig. 8B). The distribution of H3K4me3 at the genes FLB3 and FLB4 showed the same trend (Fig. 8B), and it cannot be excluded that their expression exhibited the same profile at an earlier time point, with H3K4me3 indicating recently performed gene expression.
Figure 8.
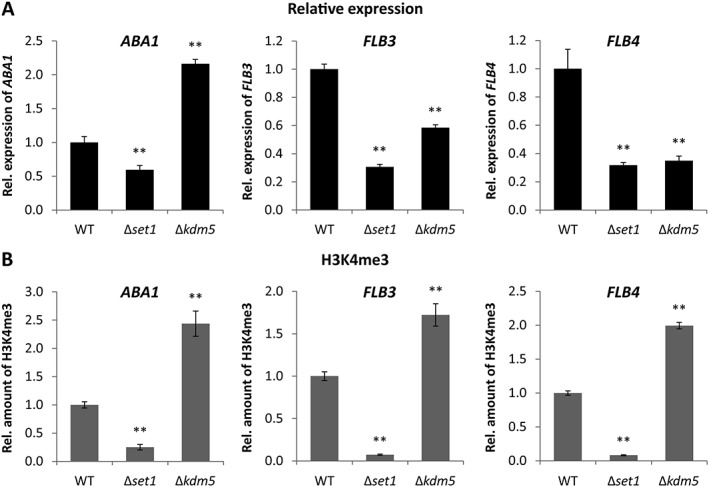
Relative expression as well as H3K4me3 levels of conidiation‐related TF genes in Δset1 and Δkdm5 mutants. Indicated strains were grown on V8 agar in the presence of a 12 h light/12 h dark cycle for 3 days to induce conidiation. A. RNA was extracted and the relative expression of ABA1, FLB3 and FLB4 was determined by RT‐qPCR. B. ChIP‐qPCR was performed for the same genes using the H3K4me3 antibody. Therefore, the enriched samples (precipitated by antibody) were normalized to the respective input samples (initially applied chromatin). The WT was arbitrarily set to 1, and the data are mean values ± SD (n = 4). For statistical analysis, the mutants were compared with the WT using the student's t‐test: *, p < 0.05; **, p < 0.01.
To summarize, Set1 and Kdm5 were identified as activator and repressor of conidiation, respectively, which likely regulate ABA1 expression via deposition of H3K4me3. In addition to Aba1, Flb3 and Flb4 act as positive TFs in conidia formation, while Wet1 is probably involved in conidia maturation.
Fusarium fujikuroi ABA1 is the major conidiation‐specific TF gene targeted by global regulators
In order to gain a deeper insight into the hierarchical network of regulation, we performed a qPCR expression analysis of Δaba1, Δflb3, Δflb4 and Δwet1, in comparison to the WT and Δcsm1, grown under conidiation‐inducing conditions. The analysis revealed that ABA1 is likely the major target of these regulators: its expression is induced by Flb3 and Flb4 (downregulated in the deletion mutants), whereas it is strongly repressed by Csm1 in the WT background (upregulated in Δcsm1) (Fig. 9A). Furthermore, WET1 was neither significantly deregulated in Δaba1 nor in any other mutant strain at the time point chosen (Fig. 9B).
Figure 9.
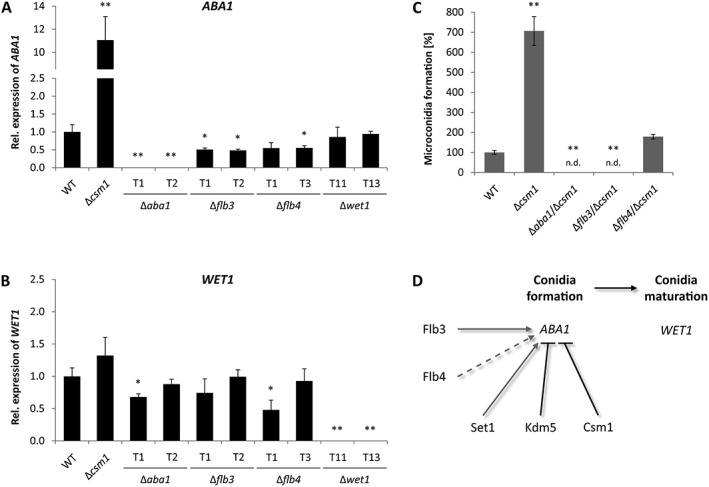
Regulatory network of conidiation. A,B. The strains were grown on V8 agar in the presence of a 12 h light/12 h dark cycle for 3 days. Then, RNA was extracted and the relative expression of ABA1 and WET1 was determined by RT‐qPCR. Data are mean values (n = 3). C. Indicated strains were grown for 14 days under above described conditions prior to analysis of conidia formation. Data are mean values ± SD (n = 4). For statistical analysis, the mutants were compared with the WT using the student's t‐test: *, p < 0.05; **, p < 0.01; n.d., not detected. D. Current model of conidia formation and maturation in F. fujikuroi, with ABA1 as major target of positive and negative regulators of conidiation. Dashed lines indicate that Flb4 can be overruled by Csm1.
Earlier investigations have revealed several global regulators with an influence on asexual development in F. fujikuroi. Thus, the fungus‐specific velvet protein Vel1 and the GATA‐type TF Csm1 were shown to be an activator and repressor of conidiation respectively (Wiemann et al., 2010; Niehaus et al., 2017b) (Fig. 9A). CSM1 deletion in a Δvel1 background (no conidia) restored conidiogenesis (Niehaus et al., 2017b). To analyze if lack of the repressor gene CSM1 also restores conidiogenesis in Δaba1, Δflb3 and Δflb4, CSM1 was deleted in the respective mutant strains in the present work. Both Aba1 and Flb3, but not Flb4, were strictly required for asexual development, and the loss of conidia formation in these mutants was not overcome by deleting CSM1 (Fig. 9C).
Based on these data, we have established the following working model that is depicted in Fig. 9D: Flb3 is strictly required, but also Flb4 is a positive regulator of ABA1 expression. In contrast, Csm1 represses the expression of ABA1, which is responsible for conidia formation. Subsequently, Wet1 induces conidia maturation. Furthermore, we have identified two novel regulators of conidiation: Set1 as activator and Kdm5 as repressor of ABA1 expression.
Discussion
In the present work, we analyzed F. fujikuroi Set1, the methyltransferase of the H3K4‐specific COMPASS complex, which was shown to be responsible for all detectable H3K4me2/me3 genome‐wide (and for the majority, if not all, of H3K4me1). As a counterpart, we generated single deletion and overexpression mutants of F. fujikuroi KDM5, encoding the ortholog to S. cerevisiae Jhd2 and A. nidulans KdmB (Liang et al., 2007; Gacek‐Matthews et al., 2016). Similarly to Jhd2 and KdmB, Kdm5 was shown to remove H3K4me3 in vivo, resulting in a genome‐wide increase and decrease of H3K4me3 in Δkdm5 and OE::KDM5 mutants respectively. The remaining H3K4me2/me1 marks are likely to be demethylated by an Lsd1‐type amine oxidase in F. fujikuroi, which was first described for Schizosaccharomyces pombe (Shi et al., 2004).
Set1 and Kdm5 affect genome‐wide transcription and vegetative growth
A transcriptome analysis of Δset1 and Δkdm5 mutants in comparison to the F. fujikuroi WT revealed that the two enzymes act as true antagonists only in the minority of cases, using the strong selection criterion of a fourfold change in expression. However, we did find more subtle antagonistic roles of Set1 and Kdm5 in SM biosynthesis and conidiation, as further discussed below. In the genome‐wide expression analysis, the majority of affected genes was upregulated upon both SET1 and KDM5 deletion. This was surprising regarding the fact that the euchromatic, activating mark H3K4me3 was antagonistically affected in western blot analyses, being decreased in Δset1 and increased in Δkdm5 mutants respectively. Of course, many of the observed effects on gene expression are likely to be secondary, and to be mediated by unknown downstream regulators, such as Set1‐dependent repressors in this case. Similarly, genome‐wide transcription was both up‐ and downregulated upon deletion of F. fujikuroi GCN5 that is responsible for the majority of the activating acetylation (ac) marks, for example, H3K9ac and H3K27ac (Rösler et al., 2016). This was also true for the deletion of the two histone deacetylase (HDAC) genes HDA1 and HDA2 in this fungus (Studt et al., 2013).
In the performed microarray, the largest group was profile 2 under both nitrogen quantities tested: genes upregulated in Δset1 and not differentially expressed in Δkdm5 (compared with the WT in each case). Several FunCat groups were identified to be significantly enriched in this profile, including genes of primary and secondary metabolism as well as of detoxification pathways. Also, a significant number of cryptic SM key genes was shown to be upregulated in Δset1. Thus, either the perturbation of carbohydrate and fatty acid metabolism, or the upregulation of possibly toxic SMs could be the reason for the slow growth of Δset1 mutants in plate assays. Something similar was suggested for the crippled F. graminearum Δkmt6 mutant that exhibited a downregulation of the repressive mark H3K27me3 and as a consequence, an upregulation of cryptic SM gene clusters (Connolly et al., 2013). In the performed microarray, PKS‐NRPS1, encoding the key enzyme in trichosetin biosynthesis, was upregulated in Δset1 under nitrogen limitation. The tetramic acid trichosetin is a potent mycotoxin, which was shown to inhibit vegetative growth when overproduced by F. fujikuroi (Janevska et al., 2017). Noteworthy, PKS‐NRPS1 expression was similarly induced in F. fujikuroi KMT6 knock‐down mutants, also exhibiting poor vegetative growth (Studt et al., 2016b).
The in loco complementation of Δset1 with the point‐mutated variant SET1 H1191K did not restore H3K4 methylation levels and notably, the observed growth defect, suggesting that the two are correlated. OE::KDM5 mutants were also characterized by a slow growth, indicating that the depletion of H3K4me3 in Δset1, SET1 H1191K and OE::KDM5 mutants could be associated with this phenotype. A slight reduction in colony diameter was also observed for Δccl1 in both F. fujikuroi and F. graminearum (Studt et al., 2017), further strengthening the assumption that reduced H3K4me3 levels are responsible for growth retardation. Whether the strong phenotype of OE::KDM5 might also be correlated with an overexpression of PKS‐NRPS1, and therefore partially be attributed to trichosetin overproduction, remains elusive at this point. Noteworthy, colony diameters of F. graminearum and F. verticillioides Δset1 mutants were also reduced in comparison to the respective WT strains (Liu et al., 2015; Gu et al., 2017).
Set1 and Kdm5 regulate SM biosynthesis mostly in an indirect manner
As indicated above, secondary metabolism was strongly affected upon SET1 deletion, but also upon KDM5 deletion and overexpression. The strength of the present study is that we were able to compare the different and possibly antagonizing phenotypes with each other. Notably, production of the two red pigments BIK and FSR as well as of the mycotoxin FUS was regulated in an antagonistic manner by Set1 and Kdm5: all three SMs were upregulated in Δset1 and OE::KDM5, but downregulated in Δkdm5 in comparison to the WT. This suggests a repressive influence of H3K4me3 on the respective SM gene clusters in the WT background, because this mark was decreased in both Δset1 and OE::KDM5, and accordingly increased in Δkdm5 genome‐wide. These results are in accordance with a previous study, where we have observed an increase in BIK and FUS biosynthesis (FSR was not tested) upon deletion of CCL1, also showing reduced levels of H3K4me3 genome‐wide (Studt et al., 2017). However, in Δccl1, H3K4me2/me3 levels were not affected at FUS cluster genes, and H3K4me2 was elevated at BIK cluster genes with unchanged H3K4me3 levels (Studt et al., 2017). Contrary to this, the Δset1 mutant showed a complete loss of all detectable H3K4me2 and H3K4me3. Therefore, the alterations in H3K4me2 at the BIK genes observed in Δccl1 cannot be the reason for the comparable BIK phenotype in Δset1 and Δccl1 mutants. Overall this suggests that the observed phenotypes concerning BIK, FSR and FUS biosynthesis are indeed mediated by the globally changing H3K4me3 levels, however, by an as yet unknown regulatory circuit.
Generally, H3K4 methylation is found within euchromatic regions and is absent from the majority of subtelomeric SM gene clusters in the F. fujikuroi WT, also under inducing conditions (Wiemann et al., 2013). Something very similar was described also for F. graminearum and A. nidulans (Connolly et al., 2013; Gacek‐Matthews et al., 2016), suggesting that most SM gene clusters are rather regulated in an indirect manner, and are not targeted by Set1 or Kdm5, in F. fujikuroi and likely also in other fungi. Early work on Set1 and H3K4 methylation in S. cerevisiae has revealed a HDAC‐independent silencing mechanism of rDNA, mating type and subtelomeric loci (Briggs et al., 2001; Krogan et al., 2002; Bryk et al., 2002). However, the exact mechanism is still not well understood, and recruitment of the HDAC Sir2 to subtelomeric loci and subsequent hypoacetylation is generally accepted as main silencing mechanism in budding yeast (Erlendson et al., 2017). As Fusarium species employ H3K9 and especially H3K27 methylation to silence vast stretches of constitutive and facultative heterochromatin, respectively (Connolly et al., 2013; Wiemann et al., 2013; Studt et al., 2016b), it is questionable whether H3K4 methylation further contributes as a silencing mechanism in F. fujikuroi. The fact that H3K4 methylation is predominantly absent from facultative heterochromatin at subtelomeres in general, and from silenced SM genes in particular (Wiemann et al., 2013), would argue against this hypothesis.
The only subtelomeric SM gene cluster analysed here that harbors H3K4me2 under inducing conditions is the GA gene cluster. Here, two out of the seven cluster genes, P450‐2 and P450‐4, are decorated with H3K4me2 under inducing but not under repressing conditions (Wiemann et al., 2013). Indeed, ChIP‐qPCR analyses revealed that these genes are decorated with H3K4me2 in the WT, Δkdm5 and OE::KDM5 mutants, possibly correlating with GA biosynthesis in these strains, while the distribution of H3K4me3 did not fit the production levels.
Accordingly, loss of H3K4me2 in Δset1 was accompanied with an abolished GA3 production in vitro, which correlated with a reduced virulence on rice. Similarly, Set1 was required in both F. graminearum and F. verticillioides for full virulence on wheat and maize respectively (Liu et al., 2015; Gu et al., 2017). In F. graminearum, the SM deoxynivalenol (DON) is the virulence factor essential for successful progression of wheat infection. While H3K4me3 was mostly absent from DON cluster genes, H3K4me2 levels correlated well with DON gene expression and product formation in the F. graminearum WT (producer) and SET1 mutant (non‐producer) under axenic conditions (Liu et al., 2015). Therefore, in both F. fujikuroi and F. graminearum, H3K4me2 might be important for GA and DON biosynthesis respectively.
While Set1 was required for full virulence of F. fujikuroi on its preferred host plant rice, Δkdm5‐infected rice seedling resembled WT‐infected samples. It is noteworthy that Δset1 strains still showed bakanae symptoms on infected rice samples, though strongly attenuated. This contradictory phenotype can be explained by the finding that although abolished during axenic growth, residual GA3 levels of about 20% were detectable in Δset1‐infected rice samples. For Δkdm5, GA3 levels were reduced to about 20% in axenic culture, but elevated to about 80% during in planta growth, likely explaining the WT‐like virulence of this strain. Similar phenotypes have also been observed for Δccl1 (Studt et al., 2017) and Δsge1 (Michielse et al., 2015) mutants before, with SGE1 encoding a global SM regulator in F. fujikuroi. This induction of GA3 biosynthesis in planta may be mediated by a specific, possibly plant‐derived signal, thereby suggesting a more complex regulation during the infection process as opposed to axenic growth.
In summary, a significant influence of H3K4 methylation on SM biosynthesis was observed, however, further research is required to determine the mode of action of COMPASS and Kdm5, and how exactly subtelomeric SM gene clusters are affected by these histone modifiers.
Set1 and Kdm5 regulate the conidiation‐specific TF gene ABA1
One of the most intriguing phenotypes of the Δset1 and Δkdm5 mutants is the abolished and enhanced conidiation respectively. In the present work, we identified the orthologues to the A. nidulans conidiation‐related genes abaA, flbB‐flbE and wetA, which were designated F. fujikuroi ABA1, FLB2‐FLB5 and WET1. We showed that Aba1, Flb3 and Flb4 are essential for conidia formation, while Flb2 and Flb5 are dispensable in F. fujikuroi. In A. nidulans, the ΔflbB‐ΔflbE mutants were characterized by a fluffy phenotype and a low brlA expression, and therefore an abolished or delayed conidiogenesis (Wieser et al., 1994; Krijgsheld et al., 2013). Additionally, asexual development has been studied in F. graminearum. In this fungus, only AbaA and FlbD, but not the FlbC homolog, were required for macroconidia formation (Son et al., 2013, 2014a). Indeed in F. fujikuroi, Aba1 is essential for both micro‐ and macroconidia formation in the strains IMI58289 and E282 respectively.
Furthermore, a microscopic analysis revealed that F. fujikuroi Wet1 is likely involved in conidia maturation, as already established for the A. nidulans and F. graminearum WetA homologs. Thus, Aspergillus WetA affects the conidial cell wall composition as well as trehalose accumulation (Sewall et al., 1990; Tao and Yu, 2011), while F. graminearum ΔwetA mutants had malformed and longer conidia with fewer septa that were sensitive to stressors (Son et al., 2014b). Although the exact role of F. fujikuroi Wet1 remains to be elucidated, one can deduce a conserved role of this protein in conidia separation and maturation for all three filamentous fungi.
We propose a working model in which different global regulators affect ABA1 expression, and therefore we suggest that Aba1 is the central conidiation‐specific TF in F. fujikuroi. Flb3 and Flb4 are positive regulators, while Csm1 is a repressor of ABA1 expression. Only Flb3 cannot be overruled by Csm1, so that this TF is strictly required for ABA1 expression (Fig. 9D). Of course, the time‐dependent expression of these regulator genes was not taken into consideration, so that a larger hierarchical network cannot be excluded at this point.
Finally, we identified Set1 as activator and Kdm5 as repressor of ABA1 expression. Noteworthy, conidiation was not affected upon deletion of F. graminearum and F. verticillioides SET1 (Liu et al., 2015; Gu et al., 2017). However, in F. fujikuroi, conidia formation was nearly abolished in Δset1 and increased in Δkdm5, respectively, which correlated with down‐ and upregulated H3K4me3 levels at the 5′ end of ABA1.
In conclusion, we present a comprehensive analysis of the H3K4‐specific histone modifiers Set1 and Kdm5 in the rice pathogen F. fujikuroi. We show that they antagonize H3K4me3 genome‐wide, affecting genes of primary and secondary metabolism as well as asexual conidia formation. While their influence on subtelomeric SM gene clusters remains to be resolved in the future, a direct regulation of the conidiation‐specific TF gene ABA1 could be suggested, giving a direct link between H3K4me3 levels, ABA1 expression and conidia formation.
Experimental procedures
Fungal strains, media and growth conditions
F. fujikuroi IMI58289 (Commonwealth Mycological Institute, Kew, UK) (Wiemann et al., 2013) was used as WT strain for the generation of deletion, complementation, point‐mutation and overexpression mutants. Furthermore, ABA1 was deleted in F. fujikuroi E282 (Niehaus et al., 2017a).
Vegetative growth was assessed in triplicates on solid V8 (30 mM CaCO3, 20%, v/v, vegetable juice; Campbell Food, Puurs, Belgium), CM (Pontecorvo et al., 1953), CD (Czapek Dox; Sigma‐Aldrich, Steinheim, Germany) and ICI + 6 mM NH4NO3 (Imperial Chemical Industries Ltd., London, UK) (Geissman et al., 1966) media after incubation for 7 days in the dark (28°C). Furthermore, conidiation was assessed on V8 agar in the presence of a 12 h light/12 h dark cycle (18°C) after incubation for 3 or 14 days for ChIP, gene expression or conidia formation respectively. Light microscopy with an Axio Scope.A1 (Carl Zeiss, Jena, Germany) was performed using colonies grown on conidiation‐inducing KCl plates (8 g l−1 KCl, 15 g l−1 agar) for 10 days in the dark (28°C). For the isolation of DNA and RNA from solid cultures, the WT and mutant strains were incubated on solidified medium covered with a layer of cellophane.
For submersed cultures, a pre‐culture was done, consisting of 100 ml Darken medium (Darken et al., 1959) in 300 ml‐Erlenmeyer flasks, which was shaken at 180 rpm and 28°C for 3 days in the dark. Then, 0.5% (v/v) of this pre‐culture was transferred to the main culture, consisting of 100 ml ICI medium with the appropriate nitrogen source (6 or 60 mM Gln to gain acidic, 6 mM NaNO3 to gain alkaline pH conditions) in 300 ml‐Erlenmeyer flasks, being shaken under above described conditions for 3 or 7 days for ChIP, gene expression or SM analyses respectively. Protoplast transformation of F. fujikuroi was achieved using a main culture of 100 ml ICI medium with 10 g l−1 fructose instead of glucose as well as 0.5 g l−1 (NH4)2SO4 as nitrogen source, which was shaken for a maximum of 16 h under above described conditions.
Plasmid constructions
Yeast recombinational cloning (Colot et al., 2006; Schumacher, 2012) was applied for the generation of deletion, complementation, point‐mutation and overexpression vectors. For deletion vectors, about 1 kb large fragments upstream and downstream of the respective gene of interest were amplified, 5′ flanks with 5F/5R and 3′ flanks with 3F/3R primer pairs (Supporting Information Table S3). The hygromycin B resistance cassette hphR (hygromycin B phosphotransferase gene under the control of the trpC promoter from A. nidulans) was amplified with hph_F/hph_R (Supporting Information Table S3) from the template pCSN44 (Staben et al., 1989). Additionally for CSM1 deletion, the nourseothricin resistance cassette natR (nourseothricin resistance gene under control of A. nidulans PtrpC) was gained from the template pZPnat1 (GenBank AY631958). S. cerevisiae FY834 (Winston et al., 1995) was transformed with the obtained PCR products (5′ flank, 3′ flank, resistance cassette) as well as with the EcoRI/XhoI digested shuttle vector pRS426 (Christianson et al., 1992), gaining the respective deletion vectors.
SET1 complementation and point‐mutation vectors were generated as follows. The full‐length gene SET1 including its 5′ sequence was amplified in two overlapping fragments with primer pairs set1_5F/set1_c_R1 and set1_c_F2/set1_c_R2 (Supporting Information Table S4). For the point‐mutated variant, three overlapping fragments were generated to insert the base pair substitutions using primer pairs set1_5F/set1_c_R1, set1_c_F2/set1_mut_R and set1_mut_F/set1_c_R2 (Supporting Information Table S4). The Tgluc terminator sequence was amplified from Botrytis cinerea B05.10 genomic DNA with BcGlu_Term_F2/Tgluc_Nat1_R (Supporting Information Table S4), while the resistance cassette natR and the SET1 3′ flank were generated as described above. Then, S. cerevisiae FY834 was transformed with the obtained fragments and with the EcoRI/XhoI digested plasmid pRS426, yielding pSET1 C and pSET1 H1191K (Supporting Information Fig. S6A).
For overexpression of KDM5, the first 1.7 kb of this gene was amplified with OE_kdm5_F/OE_kdm5_R (Supporting Information Table S4) and was cloned into the NcoI/NotI restricted plasmid pNDH‐OGG (Schumacher, 2012), gaining pOE::KDM5 (Supporting Information Fig. S6B). All complementation, point‐mutation and overexpression vectors were verified by sequencing using primers listed in Supporting Information Table S4.
Fungal transformations and analysis of transformants
Fusarium fujikuroi protoplast transformation was carried out as described earlier (Tudzynski et al., 1999). Deletion cassettes were amplified from the deletion vectors with primer pairs 5F/3R (Supporting Information Table S3), while 10–40 μg of the ApaI/XbaI digested vectors pSET1 C and pSET1 H1191K (Supporting Information Fig. S6A) as well as of the circular vector pOE::KDM5 (Supporting Information Fig. S6B) were applied for transformation. The transformants were selected with 100 μg ml−1 hygromycin B (Calbiochem, Darmstadt, Germany) and/or 100 μg ml−1 nourseothricin (Werner‐Bioagents, Jena, Germany) resistance markers.
Single and double deletion mutants were verified by Southern blot and/or diagnostic PCR, showing the homologous integration of deletion cassettes as well as the absence of WT genes. Diagnostic PCRs for three independent Δset1 and three independent Δkdm5 mutants can be found in Supporting Information Figs. S7A and S8A respectively. Furthermore, Supporting Information Fig. S9A–G shows diagnostic PCRs for two Δaba1 (IMI58289 WT background), two E282/Δaba1, two Δflb2, three Δflb3, two Δflb4, three Δflb5 and three Δwet1 mutants. And Supporting Information Fig. S10A–C depicts diagnostic PCRs for two Δaba1/Δcsm1, one Δflb3/Δcsm1 and two Δflb4/Δcsm1 double mutants.
Moreover, the in loco integration of SET1 C and SET1 H1191K constructs was verified with the amplification of 5′ and 3′ flanks and the absence of untransformed nuclei for three independent transformants each (Supporting Information Fig. S6C), and the in loco integration of pOE::KDM5 was shown for three independent transformants (Supporting Information Fig. S6D). A northern blot expression analysis verified the successful overexpression of KDM5 in the mutants (Supporting Information Fig. S6E). For the complementation and point‐mutation mutants, it was checked that they were unable to grow on hygromycin B (deletion phenotype), but were only able to grow on nourseothricin (complementation phenotype), while the point‐mutation in SET1 H1191K mutants was verified by sequencing.
DNA analysis via Southern blot and PCR
Plasmid DNA from S. cerevisiae FY834 and E. coli Top10F’ (Invitrogen, Darmstadt, Germany) was isolated using the NucleoSpin Plasmid Kit (Macherey‐Nagel, Düren, Germany). Moreover, F. fujikuroi genomic DNA from lyophilised and ground mycelium was extracted as described elsewhere (Cenis, 1992). The analysis of ectopically integrated deletion cassettes via Southern blot (Southern, 1975) started with the digestion of genomic DNA of WT and deletion mutants with an appropriate restriction enzyme (Thermo Fisher Scientific, Schwerte, Germany). The DNA was separated in a 1% (w/v) agarose gel, transferred to a nylon membrane (Nytran SPC; Whatman, Sanford, FL) via downward alkali blotting (Ausubel et al., 1987) and hybridized with 32P‐labelled probes. Probes were generated with the random oligomer‐primer method (Sambrook et al., 1989) using gene flanks as templates (Supporting Information Table S3). Southern blots for Δset1 and Δkdm5 mutants can be found in Supporting Information Figs. S7B/C and S8B/C respectively. For the amplification of DNA by PCR, BioTherm DNA Polymerase (GeneCraft, Lüdinghausen, Germany), TaKaRa LA Taq DNA Polymerase (Takara Bio, Saint‐Germain‐en‐Laye, France) or Phusion High‐Fidelity DNA Polymerase (Finnzymes, Vantaa, Finland) were used.
Expression analysis via northern blot and qPCR
RNA from lyophilised and ground mycelium was extracted with the TRI Reagent (Sigma‐Aldrich, Steinheim, Germany). For northern blot expression analysis (Church and Gilbert, 1984) of OE::KDM5 mutants, 20 μg of total RNA was separated in a 1% (w/v) denaturating agarose gel (Sambrook et al., 1989). Blotting and hybridisation were done as described above. For probe generation, the KDM5 WT fragment (kdm5_WT_F/kdm5_WT_R; Supporting Information Table S3) was used as template.
For expression analysis via qPCR, 1 μg of DNase I‐treated (Thermo Fisher Scientific, Schwerte, Germany) total RNA was transcribed into cDNA with oligo dT primers and SuperScript II Reverse Transcriptase (Invitrogen, Darmstadt, Germany). Furthermore, iQ SYBR Green Supermix (Bio‐Rad, München, Germany) was used for qPCRs in a C1000 Touch Thermal Cycler with a CFX96 Real‐Time System (Bio‐Rad, München, Germany). Expression levels of ABA1, FLB3, FLB4 and WET1 as well as of the constitutively expressed reference genes (FFUJ_07710, GDP mannose transporter gene GMT; FFUJ_05652, related actin gene RAC; FFUJ_08398, ubiquitin gene UBI) were determined in triplicates or quadruplicates with primer pairs listed in Supporting Information Table S5. Annealing temperatures of 60°C yielded primer efficiencies between 90% and 110% and the expression levels were calculated with the ΔΔCt‐method (Pfaffl, 2001).
Expression analysis via microarray
The WT, Δset1 and Δkdm5 mutants were grown in liquid ICI medium with 6 or 60 mM Gln for 3 days in duplicates. After total RNA isolation as described above, an additional clean‐up step was done with the NucleoSpin RNA Clean‐up Kit (Macherey‐Nagel, Düren, Germany). Agilent Technologies (Santa Clara, CA) designed the microarrays, while Arrows Biomedical (Münster, Germany) prepared the hybridisation according to the manufacturer's protocol. For the heatmaps, the eight expression profiles were extracted first, and were then clustered in a second step using the programme Perseus 1.5.8.5 (Max Planck Institute of Biochemistry, Martinsried, Germany) (Tyanova et al., 2016) with standard settings. Genes upregulated in the mutants compared with the WT showed a log2 fold change of ≥ 2 (green), downregulated genes of ≤ −2 (red).
The Functional Catalogue (FunCat) for protein sequences (Ruepp et al., 2004) was used to identify significantly enriched protein functions. To correct for multiple comparisons in multiple hypotheses testing for the 845 taxonomically allowed FunCat groups, we calculated the Bonferroni correction as well as the false discovery rate with an experiment‐wide significance level of 0.05. The microarray data as well as additional information on sample preparation and data processing are available at the NCBI Gene Expression Omnibus under the accession number GSE90948.
Western blot analysis
The WT, Δset1, Δkdm5 and OE::KDM5 mutants were grown in liquid ICI medium with 60 mM Gln for 3 days. Western blot detection of K4‐methylated H3 proteins in a whole‐protein extract was done as described elsewhere (Janevska et al., 2018). The following primary antibodies were used (Active Motif, La Hulpe, Belgium): anti‐H3K4me1 (#39298; 1:10 000), anti‐H3K4me2 (#39141; 1:10 000), anti‐H3K4me3 (#39159; 1:10 000) and anti‐H3 C‐terminal (#39163; 1:10 000). Donkey anti‐rabbit IgG‐HRP served as secondary antibody (sc‐2317; 1:10 000; Santa Cruz Biotechnology, Heidelberg, Germany).
ChIP analysis
For SM‐related genes, the WT, Δset1, Δkdm5 and OE::KDM5 were grown in liquid ICI medium with 6 mM for 3 days. For conidiation‐related genes, the WT, Δset1 and Δkdm5 were incubated on solid V8 agar with cellophane in the presence of a 12 h light/12 h dark cycle for 3 days. Crosslinking was performed with 1% (v/v) formaldehyde for 15 min at 28°C and 90 rpm, followed by quenching with 125 mM glycine for 5 min at 37°C. Next, the mycelium was separated from the liquid, was shock‐frozen with liquid nitrogen and ground to a fine powder. Further sample preparation was essentially performed as described elsewhere (Gacek‐Matthews et al., 2015) using the Bioruptor Plus (Diagenode, Seraing, Belgium) for sonication. The ChIPs were done with the anti‐H3K4me2 (#39141; Active Motif, La Hulpe, Belgium) and anti‐H3K4me3 (#39159; Active Motif, La Hulpe, Belgium) antibodies, followed by treatment with Dynabeads Protein A (Thermo Fisher Scientific, Schwerte, Germany) to precipitate the chromatin‐antibody‐conjugate. ChIP‐qPCR of CPS/KS, P450‐1, P450‐2, P450‐4, ABA1, FLB3 and FLB4 genes was performed in quadruplicates with primers that bind at the 5′ gene ends (Supporting Information Table S5).
Chemical analysis of SM production levels
The WT, Δset1, Δkdm5 and OE::KDM5 mutants were grown in liquid ICI medium with 6 mM Gln (BIK, GA3), 6 mM NaNO3 (FSR) or 60 mM Gln (FUS, FSA) for 7 days in triplicates. BIK, FSR, FUS and FSA were directly analysed after filter‐sterilization of the supernatant to remove the mycelium (0.45 μm membrane filters; BGB Analytik, Schloßböckelheim, Germany). The four SMs were measured via HPLC‐DAD which was essentially performed as described elsewhere (Studt et al., 2012), using a VWR Hitachi Chromaster HPLC system (VWR International, Darmstadt Germany) with an EZChrome Elite 3.3.2 SP2 software (Agilent Technologies, Santa Clara, CA). GA3 was extracted and concentrated from 20 ml supernatant applying Sep‐Pak C18 cartridges (Waters, Eschborn, Germany) and 2 ml 55% (v/v) acetonitrile for elution. The HPLC‐DAD analysis of GA3 was essentially performed as described earlier (Wiemann et al., 2012) using the same HPLC system from above. In each case, the production of the SMs was related to the dry weight of the strains which was determined after harvesting and freeze‐drying.
SM extraction and subsequent quantification of GA3 levels in planta via HPLC coupled to tandem mass spectrometry (HPLC‐MS/MS) were performed as described previously (Studt et al., 2017). Mock infection was achieved through the addition of H2O and served as negative control. Quantified metabolite levels were normalized to the input sample weights.
Rice virulence assay
The infection of surface‐sterilized and pre‐germinated seedlings of Oryza sativa spp. japonica cv. Nipponbare (kindly provided by the USDA ARS Dale Bumpers National Rice Research Center, Arkansas, USA) with mycelial plugs of the WT, Δset1, Δkdm5 and OE::KDM5 mutants was performed for 7 days as described elsewhere (Janevska et al., 2017). Addition of 100 ppm GA3 and H2O served as positive and negative control respectively.
Supporting information
Fig. S1. Western blot analysis of the Δkdm5 mutant. The western blot was done using the H3K4me3 and H3K4me2 antibodies. Indicated strains were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to protein extraction. 15 μg of the protein extract was loaded on to the gel, and an unspecific band served as loading control.
Fig. S2. Microarray expression analysis of differentially regulated A) bikaverin (BIK) and B) apicidin F (APF) cluster genes in Δset1 and Δkdm5. The WT and the two deletion mutants were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to RNA extraction. Data are mean values (n = 2). Genes upregulated in the deletion mutants compared with the WT are green (significant when log2 fold change ≥2), downregulated genes are red (significant when log2 fold change ≤ −2), and not differentially expressed genes are white (between −2 and 2). The tables show the gene accession numbers and the respective cluster genes, while non‐cluster genes are highlighted in grey. The cluster organization is depicted schematically below, with arrows indicating the direction of transcription and white bars indicating introns.
Fig. S3. Chromosomal location of the analysed SM gene clusters. The distribution of H3K27me3 is shown for A) chromosome II, B) chromosome III, C) chromosome V, and D) chromosome IX (taken from Studt et al., 2016b). The location of the respective SM key genes is indicated on the chromosomes. Furthermore, the distribution of H3K4me2 is shown for the gibberellic acid (GA) gene cluster (taken from Wiemann et al., 2013). For both ChIP‐Seq analyses, the WT was grown in liquid culture (ICI + 6 mM Gln) for 3 days.
Fig. S4. Virulence on rice of Δset1, Δkdm5 and OE::KDM5 mutants. Germinated rice seedlings were infected with 100 ppm gibberellic acid GA3 (positive control), H2O (negative control), and indicated strains for 7 days. Data are mean values ±SD (n = 3). For statistical analysis, the mutants were compared with the WT using the student's t‐test: **, P < 0.01.
Fig. S5. Plate assay and microscopic analysis of Δaba1, Δflb2‐Δflb5 and Δwet1 mutants. A) Indicated strains were grown on V8 agar in the presence of a 12 h light/12 h dark cycle for 14 days to induce conidiation. B) Microscopic analysis of microconidia formation in the F. fujikuroi WT IMI58289 and the respective Δwet1 deletion mutant grown on KCl agar for 10 days. For both strains, the same microscopic magnification was applied.
Fig. S6. Generation of SET1 C, SET1 H1191K and OE::KDM5 mutants. A) The deletion mutant Δset1 T34 was transformed with ApaI/XbaI digested pSET1 C or pSET1 H1191K vectors harbouring the WT or point‐mutated SET1 gene, respectively, driven by its native promoter as well as the nourseothricin resistance cassette natR. The nucleotide substitutions resulting in the point mutation are indicated. B) KDM5 was overexpressed with the constitutive PoliC promoter from A. nidulans. The first 1.7 kb of KDM5 was cloned into NcoI/NotI restricted pNDH‐OGG conferring hygromycin B resistance (hphR). C) The in loco integration of the constructs in three independent SET1 C and SET1 H1191K mutants, respectively, was verified using primer pairs set1_5diag/set1_c_diag (5′ flank; 1.40 kb), set1_3diag/nat1_R1 (3′ flank; 1.57 kb) and set1_5diag/trpC_T (untransformed nuclei; 1.25 kb for Δset1). The respective deletion mutant (Δ) and the WT were used as controls. D) The in loco integration of pOE::KDM5 in the three transformants was checked using primer pair PoliC_Seq_F2/OE_kdm5_diag (1.95 kb). M, GeneRuler DNA Ladder Mix. E) The WT and OE::KDM5 mutants were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to RNA extraction from the harvested mycelium. The northern blot was probed with DNA corresponding to KDM5, and the ribosomal RNA was visualized for the respective gel as loading control.
Fig. S7. Verification of Δset1 deletion mutants by diagnostic PCR and Southern blot. A) Deletion via homologous recombination with the hygromycin B resistance cassette (hphR) was verified with the amplification of 5′ (set1_5diag/trpC_T) and 3′ (set1_3diag/trpC_P2) flanks but no amplification of WT (set1_WT_F/set1_WT_R) signal for three independent transformants. B) For analysing ectopic integration of deletion constructs, genomic DNA of transformants and WT was digested with ClaI, while the 5′ flank was applied for probing. C) Detected signals match the expected 10.66 kb for the WT and 3.11 kb for Δset1. λ, λ/HindIII; M, GeneRuler 1 kb Plus DNA Ladder.
Fig. S8. Verification of Δkdm5 deletion mutants by diagnostic PCR and Southern blot. A) Deletion via homologous recombination with the hygromycin B resistance cassette (hphR) was verified with the amplification of 5′ (kdm5_5diag/trpC_T) and 3′ (kdm5_3diag/trpC_P2) flanks but no amplification of WT (kdm5_WT_F/kdm5_WT_R) signal for three independent transformants. B) For analysing ectopic integration of deletion constructs, genomic DNA of transformants and WT was digested with SalI, while the 3′ flank was applied for probing. C) Detected signals match the expected 7.07 kb for the WT and 2.70 kb for Δkdm5. λ, λ/HindIII; M, GeneRuler DNA Ladder Mix.
Fig. S9. Verification of Δaba1, Δflb2‐Δflb5 and Δwet1 single deletion mutants. Deletion via homologous recombination with the hygromycin B resistance cassette was verified with the amplification of 5′ (5diag/trpC_T) and 3′ (3diag/trpC_P2) flanks but no amplification of WT (WT_F/WT_R) signal for A) two independent Δaba1 mutants (IMI58289 WT background), B) two independent E282/Δaba1 mutants, C) two independent Δflb2 mutants, D) three independent Δflb3 mutants, E) two independent Δflb4 mutants, F) three independent Δflb5 mutants and G) three independent Δwet1 mutants. λ, λ/HindIII.
Fig. S10. Deletion of CSM1 in Δaba1, Δflb3 and Δflb4 backgrounds. Deletion of CSM1 via homologous recombination with the nourseothricin resistance cassette was verified with the amplification of 5′ (csm1_5diag/nat1_hiF) and 3′ (csm1_3diag/trpC_P2) flanks but no amplification of WT (csm1_WT_F/csm1_WT_R) signal for A) two independent Δaba1/Δcsm1 double mutants, B) one Δflb3/Δcsm1 double mutant and C) two independent Δflb4/Δcsm1 double mutants. λ, λ/HindIII.
Table S1. Significantly enriched protein functions of genes deregulated in Δset1 and/or Δkdm5 in the microarray expression analysis. Genes upregulated in the deletion mutants compared with the WT have a log2 fold change ≥2, downregulated genes have a log2 fold change ≤ −2. The table is sorted by the P‐value and the filters rely on the following criteria: B, Bonferroni correction <0.05; F, P‐value < FDR; *, P‐value <0.05. abs., absolute; rel., relative; FDR, false discovery rate.
Table S2. Microarray expression analysis of differentially regulated SM key genes in Δset1 and Δkdm5. The WT and the two deletion mutants were grown in ICI liquid culture in the presence of limiting (6 mM, N‐) and saturating (60 mM, N+) amounts of Gln for 3 days prior to RNA extraction. Data are mean values (n = 2). Genes upregulated in the deletion mutants compared with the WT are green (log2 fold change ≥2), and downregulated genes are red (log2 fold change ≤ −2). Shown are the gene accession numbers, the encoded SM key genes as well as the produced SMs. PKS, polyketide synthase; NRPS, non‐ribosomal peptide synthetase; STC, sesquiterpene cyclase; DTC, diterpene cyclase; TeTC, tetraterpene cyclase; DMATS, dimethylallyltryptophan synthase.
Table S3. Primer sequences used for the generation of deletion constructs and for the verification of their homologous integration. Introduced overhangs required for yeast recombinational cloning are underlined.
Table S4. Primer sequences used for the generation and analysis of complementation, point‐mutation and overexpression vectors. Introduced overhangs required for yeast recombinational cloning are underlined.
Table S5. Primer sequences for analysing relative expression (qPCR) and ChIP‐qPCR (5′ChIP). Reference genes for relative expression: GMT, GDP mannose transporter gene; RAC, related actin gene; UBI, ubiquitin gene.
Acknowledgements
We thank Eva‐Maria Niehaus, Kathleen Huß and Sabine Huber for generating the conidiation‐related mutant strains. Furthermore, we thank Leonie Baumann for generating the Δkdm5 transformants, Lena Rindermann for the Δkdm5 Southern blot and Simone Bachleitner for her help with the infection assay. Additionally, Martin Münsterkötter helped us with the microarray raw data. This work was supported by the German Research Foundation (project numbers TU101/16‐2 and ME 1682/6‐2 as a project part (LAP3714) of the special research area ‘SFB Fusarium’ of the Austrian Science Fund, FWF) as well as by the FWF (Lise Meitner grant M 2149‐B22 to LS). The authors declare that no conflict of interest exists.
References
- Allis, C. D. , Berger, S. L. , Cote, J. , Dent, S. , Jenuwien, T. , Kouzarides, T. , et al (2007) New nomenclature for chromatin‐modifying enzymes. Cell 131: 633–636. [DOI] [PubMed] [Google Scholar]
- Ausubel, F. M. , Brent, R. , Kingston, R. E. , Moore, D. D. , Seidman, J. G. , Smith, J. A. , and Struhl, K. (1987) Current Protocols in Molecular Biology. New York, NY: Wiley. [Google Scholar]
- Bömke, C. , and Tudzynski, B. (2009) Diversity, regulation, and evolution of the gibberellin biosynthetic pathway in fungi compared to plants and bacteria. Phytochemistry 70: 1876–1893. [DOI] [PubMed] [Google Scholar]
- Brakhage, A. A. (2013) Regulation of fungal secondary metabolism. Nat Rev Microbiol 11: 21–32. [DOI] [PubMed] [Google Scholar]
- Briggs, S. D. , Bryk, M. , Strahl, B. D. , Cheung, W. L. , Davie, J. K. , Dent, S. Y. R. , et al (2001) Histone H3 lysine 4 methylation is mediated by Set1 and required for cell growth and rDNA silencing in Saccharomyces cerevisiae . Genes Dev 15: 3286–3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosch, G. , Loidl, P. , and Graessle, S. (2008) Histone modifications and chromatin dynamics: a focus on filamentous fungi. FEMS Microbiol Rev 32: 409–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryk, M. , Briggs, S. D. , Strahl, B. D. , Curcio, M. J. , Allis, C. D. , and Winston, F. (2002) Evidence that Set1, a factor required for methylation of histone H3, regulates rDNA silencing in S. cerevisiae by a Sir2‐independent mechanism. Curr Biol 12: 165–170. [DOI] [PubMed] [Google Scholar]
- Cenis, J. L. (1992) Rapid extraction of fungal DNA for PCR amplification. Nucleic Acids Res 20: 2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson, T. W. , Sikorski, R. S. , Dante, M. , Shero, J. H. , and Hieter, P. (1992) Multifunctional yeast high‐copy‐number shuttle vectors. Gene 110: 119–122. [DOI] [PubMed] [Google Scholar]
- Church, G. M. , and Gilbert, W. (1984) Genomic sequencing. Proc Natl Acad Sci U S A 81: 1991–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colot, H. V. , Park, G. , Turner, G. E. , Ringelberg, C. , Crew, C. M. , Litvinkova, L. , et al (2006) A high‐throughput gene knockout procedure for Neurospora reveals functions for multiple transcription factors. Proc Natl Acad Sci U S A 103: 10352–10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly, L. R. , Smith, K. M. , and Freitag, M. (2013) The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genet 9: e1003919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darken, M. A. , Jensen, A. L. , and Ahu, P. (1959) Production of gibberellic acid by fermentation. Appl Microbiol 7: 301–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlendson, A. A. , Friedman, S. , and Freitag, M. (2017) A matter of scale and dimensions: chromatin of chromosome landmarks in the fungi. Microbiol Spectr 5: 1–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingerman, I. M. , Wu, C. , Wilson, B. D. , and Briggs, S. (2005) Global loss of Set1‐mediated H3 Lys4 trimethylation is associated with silencing defects in Saccharomyces cerevisiae . J Biol Chem 280: 28761–28765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox, E. M. , and Howlett, B. J. (2008) Secondary metabolism: regulation and role in fungal biology. Curr Opin Microbiol 11: 481–487. [DOI] [PubMed] [Google Scholar]
- Freitag, M. (2017) Histone methylation by SET domain proteins in fungi. Annu Rev Microbiol 71: 413–439. [DOI] [PubMed] [Google Scholar]
- Gacek, A. , and Strauss, J. (2012) The chromatin code of fungal secondary metabolite gene clusters. Appl Microbiol Biotechnol 95: 1389–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek‐Matthews, A. , Noble, L. M. , Gruber, C. , Berger, H. , Sulyok, M. , Marcos, A. T. , et al (2015) KdmA, a histone H3 demethylase with bipartite function, differentially regulates primary and secondary metabolism in Aspergillus nidulans . Mol Microbiol 96: 839–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacek‐Matthews, A. , Berger, H. , Sasaki, T. , Wittstein, K. , Gruber, C. , Lewis, Z. A. , and Strauss, J. (2016) KdmB, a Jumonji histone H3 demethylase, regulates genome‐wide H3K4 trimethylation and is required for normal induction of secondary metabolism in Aspergillus nidulans . PLoS Genet 12: e1006222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissman, T. A. , Verbiscar, A. J. , Phinney, B. O. , and Cragg, G. (1966) Studies on the biosynthesis of gibberellins from (−)‐kaurenoic acid in cultures of Gibberella fujikuroi . Phytochemistry 5: 933–947. [Google Scholar]
- Govindaraghavan, M. , Anglin, S. L. , Osmani, A. H. , and Osmani, S. A. (2014) The Set1/COMPASS histone H3 methyltransferase helps regulate mitosis with the CDK1 and NIMA mitotic kinases in Aspergillus nidulans . Genetics 197: 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, Q. , Tahir, H. A. S. , Zhang, H. , Huang, H. , Ji, T. , Sun, X. , et al (2017) Involvement of FvSet1 in fumonisin B1 biosynthesis, vegetative growth, fungal virulence, and environmental stress responses in Fusarium verticillioides . Toxins 9: 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harting, R. , Bayram, O. , Laubinger, K. , Valerius, O. , and Braus, G. H. (2013) Interplay of the fungal sumoylation network for control of multicellular development. Mol Microbiol 90: 1125–1145. [DOI] [PubMed] [Google Scholar]
- Hori, S. (1890) Injurious fungi attacking rice plants. Bot Mag 4: 425–427. [Google Scholar]
- Janevska, S. , and Tudzynski, B. (2018) Secondary metabolism in Fusarium fujikuroi: strategies to unravel the function of biosynthetic pathways. Appl Microbiol Biotechnol 102: 615–630. [DOI] [PubMed] [Google Scholar]
- Janevska, S. , Arndt, B. , Baumann, L. , Apken, L. H. , Marques, L. M. M. , Humpf, H. , and Tudzynski, B. (2017) Establishment of the inducible Tet‐on system for the activation of the silent trichosetin gene cluster in Fusarium fujikuroi . Toxins 9: 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janevska, S. , Baumann, L. , Sieber, C. M. K. , Münsterkötter, M. , Ulrich, J. , Kämper, J. , et al (2018) Elucidation of the two H3K36me3 histone methyltransferases Set2 and Ash1 in Fusarium fujikuroi unravels their different chromosomal targets and a major impact of Ash1 on genome stability. Genetics 208: 153–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijgsheld, P. , Bleichrodt, R. , van Veluw, G. J. , Wang, F. , Müller, W. H. , Dijksterhuis, J. , and Wösten, H. A. B. (2013) Development in Aspergillus . Stud Mycol 74: 1–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan, N. J. , Dover, J. , Khorrami, S. , Greenblatt, J. F. , Schneider, J. , Johnston, M. , and Shilatifard, A. (2002) COMPASS, a histone H3 (lysine 4) methyltransferase required for telomeric silencing of gene expression. J Biol Chem 277: 10753–10755. [DOI] [PubMed] [Google Scholar]
- Lee, J. , and Skalnik, D. G. (2005) CpG‐binding protein (CXXC finger protein 1) is a component of the mammalian Set1 histone H3‐Lys4 methyltransferase complex, the analogue of the yeast Set1/COMPASS complex. J Biol Chem 280: 41725–41731. [DOI] [PubMed] [Google Scholar]
- Leslie, J. F. , and Summerell, B. A. (2006) Fusarium laboratory workshops ‐ a recent history. Mycotoxin Res 22: 73–74. [DOI] [PubMed] [Google Scholar]
- Liang, G. , Klose, R. J. , Gardner, K. E. , and Zhang, Y. (2007) Yeast Jhd2p is a histone H3 Lys4 trimethyl demethylase. Nat Struct Mol Biol 14: 243–245. [DOI] [PubMed] [Google Scholar]
- Liu, Y. , Liu, N. , Yin, Y. , Chen, Y. , Jiang, J. , and Ma, Z. (2015) Histone H3K4 methylation regulates hyphal growth, secondary metabolism and multiple stress responses in Fusarium graminearum . Environ Microbiol 17: 4615–4630. [DOI] [PubMed] [Google Scholar]
- Macheleidt, J. , Mattern, D. J. , Fischer, J. , Netzker, T. , Weber, J. , Schroeckh, V. , et al (2016) Regulation and role of fungal secondary metabolites. Annu Rev Genet 50: 371–392. [DOI] [PubMed] [Google Scholar]
- Michielse, C. B. , Studt, L. , Janevska, S. , Sieber, C. M. K. , Arndt, B. , Espino, J. J. , et al (2015) The global regulator FfSge1 is required for expression of secondary metabolite gene clusters but not for pathogenicity in Fusarium fujikuroi . Environ Microbiol 17: 2690–2708. [DOI] [PubMed] [Google Scholar]
- Ng, H. H. , Robert, F. , Young, R. A. , and Struhl, K. (2003) Targeted recruitment of Set1 histone methylase by elongating pol II provides a localized mark and memory of recent transcriptional activity. Mol Cell 11: 709–719. [DOI] [PubMed] [Google Scholar]
- Niehaus, E. , Kleigrewe, K. , Wiemann, P. , Studt, L. , Sieber, C. M. K. , Connolly, L. R. , et al (2013) Genetic manipulation of the Fusarium fujikuroi fusarin gene cluster yields insight into the complex regulation and fusarin biosynthetic pathway. Chem Biol 20: 1055–1066. [DOI] [PubMed] [Google Scholar]
- Niehaus, E. , Janevska, S. , von Bargen, K. W. , Sieber, C. M. K. , Harrer, H. , Humpf, H. , and Tudzynski, B. (2014a) Apicidin F: characterization and genetic manipulation of a new secondary metabolite gene cluster in the rice pathogen Fusarium fujikuroi . PLoS ONE 9: e103336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus, E. , von Bargen, K. W. , Espino, J. J. , Pfannmüller, A. , Humpf, H. , and Tudzynski, B. (2014b) Characterization of the fusaric acid gene cluster in Fusarium fujikuroi . Appl Microbiol Biotechnol 98: 1749–1762. [DOI] [PubMed] [Google Scholar]
- Niehaus, E. , Studt, L. , von Bargen, K. W. , Kummer, W. , Humpf, H. , Reuter, G. , and Tudzynski, B. (2016) Sound of silence: the beauvericin cluster in Fusarium fujikuroi is controlled by cluster‐specific and global regulators mediated by H3K27 modification. Environ Microbiol 18: 4282–4302. [DOI] [PubMed] [Google Scholar]
- Niehaus, E. , Kim, H. , Münsterkötter, M. , Janevska, S. , Arndt, B. , Kalinina, S. A. , et al (2017a) Comparative genomics of geographically distant Fusarium fujikuroi isolates revealed two distinct pathotypes correlating with secondary metabolite profiles. PLoS Pathog 13: e1006670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehaus, E. , Schumacher, J. , Burkhardt, I. , Rabe, P. , Münsterkötter, M. , Güldener, U. , et al (2017b) The GATA‐type transcription factor Csm1 regulates conidiation and secondary metabolism in Fusarium fujikuroi . Front Microbiol 8: 1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg, H. I. , and O'Donnell, K. (1998) New Fusarium species and combinations within the Gibberella fujikuroi species complex. Mycologia 90: 434–458. [Google Scholar]
- Pfaffl, M. W. (2001) A new mathematical model for relative quantification in real‐time RT‐PCR. Nucleic Acids Res 29: e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pokholok, D. K. , Harbison, C. T. , Levine, S. , Cole, M. , Hannett, N. M. , Tong, I. L. , et al (2005) Genome‐wide map of nucleosome acetylation and methylation in yeast. Cell 122: 517–527. [DOI] [PubMed] [Google Scholar]
- Pontecorvo, G. , Roper, J. A. , Chemmons, L. M. , Macdonald, K. D. , and Bufton, A. W. J. (1953) The genetics of Aspergillus nidulans . Adv Genet 5: 141–238. [DOI] [PubMed] [Google Scholar]
- Rademacher, W. (1997)Gibberellins . In Fungal Biotechnology, Anke T. (ed). London: Chapman and Hall, pp. 193–205. [Google Scholar]
- Rando, O. J. , and Chang, H. Y. (2009) Genome‐wide views of chromatin structure. Annu Rev Biochem 78: 245–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rösler, S. M. , Kramer, K. , Finkemeier, I. , Humpf, H. , and Tudzynski, B. (2016) The SAGA complex in the rice pathogen Fusarium fujikuroi: structure and functional characterization. Mol Microbiol 102: 951–974. [DOI] [PubMed] [Google Scholar]
- Ruepp, A. , Zollner, A. , Maier, D. , Albermann, K. , Hani, J. , Mokrejs, M. , et al (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32: 5539–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E. F. , and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual, 2nd ed. New York, NY: Cold Spring Harbor. [Google Scholar]
- Santos‐Rosa, H. , Schneider, R. , Bannister, A. J. , Sherriff, J. , Bernstein, B. E. , Emre, N. C. T. , et al (2002) Active genes are tri‐methylated at K4 of histone H3. Nature 419: 407–411. [DOI] [PubMed] [Google Scholar]
- Schumacher, J. (2012) Tools for Botrytis cinerea: new expression vectors make the gray mold fungus more accessible to cell biology approaches. Fungal Genet Biol 49: 483–497. [DOI] [PubMed] [Google Scholar]
- Sewall, T. C. , Mims, C. W. , and Timberlake, W. E. (1990) Conidium differentiation in Aspergillus nidulans wild‐type and wet‐white (wetA) mutant strains. Dev Biol 138: 499–508. [DOI] [PubMed] [Google Scholar]
- Seward, D. J. , Cubberley, G. , Kim, S. , Schonewald, M. , Zhang, L. , Tripet, B. , and Bentley, D. L. (2007) Demethylation of trimethylated histone H3 Lys4 in vivo by JARID1 JmjC proteins. Nat Struct Mol Biol 14: 240–242. [DOI] [PubMed] [Google Scholar]
- Shi, Y. , Lan, F. , Matson, C. , Mulligan, P. , Whetstine, J. R. , Cole, P. A. , et al (2004) Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 119: 941–953. [DOI] [PubMed] [Google Scholar]
- Son, H. , Kim, M. , Min, K. , Seo, Y. , Lim, J. Y. , Choi, G. J. , et al (2013) AbaA regulates conidiogenesis in the ascomycete fungus Fusarium graminearum . PLoS ONE 8: e72915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son, H. , Kim, M. , Chae, S. , and Lee, Y. (2014a) FgFlbD regulates hyphal differentiation required for sexual and asexual reproduction in the ascomycete fungus Fusarium graminearum . J Microbiol 52: 930–939. [DOI] [PubMed] [Google Scholar]
- Son, H. , Kim, M. , Min, K. , Lim, J. Y. , Choi, G. J. , Kim, J. , et al (2014b) WetA is required for conidiogenesis and conidium maturation in the ascomycete fungus Fusarium graminearum . Eukaryot Cell 13: 87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southern, E. M. (1975) Detection of specific sequences among DNA fragments separated by gel electrophoresis. J Mol Biol 98: 503–517. [DOI] [PubMed] [Google Scholar]
- Sponsel, V. M. , and Hedden, P. (2004) Gibberellin biosynthesis and inactivation . In Plant Hormones. Biosynthesis, Signal Transduction, Action! P, Davies J. (ed). Dordrecht, The Netherlands: Kluwer Academic Publishers, pp. 63–94. [Google Scholar]
- Staben, C. , Jensen, B. , Singer, M. , Pollock, J. , Schechtmann, M. , Kinsey, J. , and Selker, E. (1989) Use of a bacterial hygromycin B resistance gene as a dominant selectable marker in Neurospora crassa transformation. Fungal Genet Newslett 36: 79–81. [Google Scholar]
- Studt, L. , Wiemann, P. , Kleigrewe, K. , Humpf, H. , and Tudzynski, B. (2012) Biosynthesis of fusarubins accounts for pigmentation of Fusarium fujikuroi perithecia. Appl Environ Microbiol 78: 4468–4480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt, L. , Schmidt, F. J. , Jahn, L. , Sieber, C. M. K. , Connolly, L. R. , Niehaus, E. , et al (2013) Two histone deacetylases, FfHda1 and FfHda2, are important for Fusarium fujikuroi secondary metabolism and virulence. Appl Environ Microbiol 79: 7719–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt, L. , Janevska, S. , Niehaus, E. , Burkhardt, I. , Arndt, B. , Sieber, C. M. K. , et al (2016a) Two separate key enzymes and two pathway‐specific transcription factors are involved in fusaric acid biosynthesis in Fusarium fujikuroi . Environ Microbiol 18: 936–956. [DOI] [PubMed] [Google Scholar]
- Studt, L. , Rösler, S. M. , Burkhardt, I. , Arndt, B. , Freitag, M. , Humpf, H. , et al (2016b) Knock‐down of the methyltransferase Kmt6 relieves H3K27me3 and results in induction of cryptic and otherwise silent secondary metabolite gene clusters in Fusarium fujikuroi . Environ Microbiol 18: 4037–4054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt, L. , Janevska, S. , Arndt, B. , Boedi, S. , Sulyok, M. , Humpf, H. , et al (2017) Lack of the COMPASS component Ccl1 reduces H3K4 trimethylation levels and affects transcription of secondary metabolite genes in two plant‐pathogenic Fusarium species. Front Microbiol 7: 2144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S. , and Snyder, W. C. (1981)The bakanae disease of the rice plant . In Fusarium: Diseases, Biology and Taxonomy, Nelson P. E., Toussoun T. A., and Cook R. J. (eds). University Park: The Pennsylvania State University Press, pp. 104–113. [Google Scholar]
- Tanaka, Y. , Katagiri, Z. , Kawahashi, K. , Kioussis, D. , and Kitajima, S. (2007) Trithorax‐group protein ASH1 methylates histone H3 lysine 36. Gene 397: 161–168. [DOI] [PubMed] [Google Scholar]
- Tao, L. , and Yu, J. (2011) AbaA and WetA govern distinct stages of Aspergillus fumigatus development. Microbiology 157: 313–326. [DOI] [PubMed] [Google Scholar]
- Tudzynski, B. , Homann, V. , Feng, B. , and Marzluf, G. A. (1999) Isolation, characterization and disruption of the areA nitrogen regulatory gene of Gibberella fujikuroi . Mol Gen Genet 261: 106–114. [DOI] [PubMed] [Google Scholar]
- Tyanova, S. , Temu, T. , Sinitcyn, P. , Carlson, A. , Hein, M. Y. , Geiger, T. , et al (2016) The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods 13: 731–740. [DOI] [PubMed] [Google Scholar]
- Wagner, E. J. , and Carpenter, P. B. (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13: 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann, P. , Willmann, A. , Straeten, M. , Kleigrewe, K. , Beyer, M. , Humpf, H. , and Tudzynski, B. (2009) Biosynthesis of the red pigment bikaverin in Fusarium fujikuroi: genes, their function and regulation. Mol Microbiol 72: 931–946. [DOI] [PubMed] [Google Scholar]
- Wiemann, P. , Brown, D. W. , Kleigrewe, K. , Bok, J. W. , Keller, N. P. , Humpf, H. , and Tudzynski, B. (2010) FfVel1 and FfLae1, components of a velvet‐like complex in Fusarium fujikuroi, affect differentiation, secondary metabolism and virulence. Mol Microbiol 77: 972–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann, P. , Albermann, S. , Niehaus, E. , Studt, L. , von Bargen, K. W. , Brock, N. L. , et al (2012) The Sfp‐type 4′‐phosphopantetheinyl transferase Ppt1 of Fusarium fujikuroi controls development, secondary metabolism and pathogenicity. PLoS ONE 7: e37519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann, P. , Sieber, C. M. K. , von Bargen, K. W. , Studt, L. , Niehaus, E. , Espino, J. J. , et al (2013) Deciphering the cryptic genome: genome‐wide analyses of the rice pathogen Fusarium fujikuroi reveal complex regulation of secondary metabolism and novel metabolites. PLoS Pathog 9: e1003475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser, J. , Lee, B. N. , Fondon, J. W., III , and Adams, T. H. (1994) Genetic requirements for initiating asexual development in Aspergillus nidulans . Curr Genet 27: 62–69. [DOI] [PubMed] [Google Scholar]
- Wiles, E. T. , and Selker, E. U. (2017) H3K27 methylation: a promiscuous repressive chromatin mark. Curr Opin Genet Dev 43: 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston, F. , Dollard, C. , and Ricupero‐Hovasse, S. L. (1995) Construction of a set of convenient Saccharomyces cerevisiae strains that are isogenic to S288C. Yeast 11: 53–55. [DOI] [PubMed] [Google Scholar]
- Yu, C. , Zavaljevski, N. , Desai, V. , and Reifman, J. (2011) QuartetS: a fast and accurate algorithm for large‐scale orthology detection. Nucleic Acids Res 39: e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Western blot analysis of the Δkdm5 mutant. The western blot was done using the H3K4me3 and H3K4me2 antibodies. Indicated strains were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to protein extraction. 15 μg of the protein extract was loaded on to the gel, and an unspecific band served as loading control.
Fig. S2. Microarray expression analysis of differentially regulated A) bikaverin (BIK) and B) apicidin F (APF) cluster genes in Δset1 and Δkdm5. The WT and the two deletion mutants were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to RNA extraction. Data are mean values (n = 2). Genes upregulated in the deletion mutants compared with the WT are green (significant when log2 fold change ≥2), downregulated genes are red (significant when log2 fold change ≤ −2), and not differentially expressed genes are white (between −2 and 2). The tables show the gene accession numbers and the respective cluster genes, while non‐cluster genes are highlighted in grey. The cluster organization is depicted schematically below, with arrows indicating the direction of transcription and white bars indicating introns.
Fig. S3. Chromosomal location of the analysed SM gene clusters. The distribution of H3K27me3 is shown for A) chromosome II, B) chromosome III, C) chromosome V, and D) chromosome IX (taken from Studt et al., 2016b). The location of the respective SM key genes is indicated on the chromosomes. Furthermore, the distribution of H3K4me2 is shown for the gibberellic acid (GA) gene cluster (taken from Wiemann et al., 2013). For both ChIP‐Seq analyses, the WT was grown in liquid culture (ICI + 6 mM Gln) for 3 days.
Fig. S4. Virulence on rice of Δset1, Δkdm5 and OE::KDM5 mutants. Germinated rice seedlings were infected with 100 ppm gibberellic acid GA3 (positive control), H2O (negative control), and indicated strains for 7 days. Data are mean values ±SD (n = 3). For statistical analysis, the mutants were compared with the WT using the student's t‐test: **, P < 0.01.
Fig. S5. Plate assay and microscopic analysis of Δaba1, Δflb2‐Δflb5 and Δwet1 mutants. A) Indicated strains were grown on V8 agar in the presence of a 12 h light/12 h dark cycle for 14 days to induce conidiation. B) Microscopic analysis of microconidia formation in the F. fujikuroi WT IMI58289 and the respective Δwet1 deletion mutant grown on KCl agar for 10 days. For both strains, the same microscopic magnification was applied.
Fig. S6. Generation of SET1 C, SET1 H1191K and OE::KDM5 mutants. A) The deletion mutant Δset1 T34 was transformed with ApaI/XbaI digested pSET1 C or pSET1 H1191K vectors harbouring the WT or point‐mutated SET1 gene, respectively, driven by its native promoter as well as the nourseothricin resistance cassette natR. The nucleotide substitutions resulting in the point mutation are indicated. B) KDM5 was overexpressed with the constitutive PoliC promoter from A. nidulans. The first 1.7 kb of KDM5 was cloned into NcoI/NotI restricted pNDH‐OGG conferring hygromycin B resistance (hphR). C) The in loco integration of the constructs in three independent SET1 C and SET1 H1191K mutants, respectively, was verified using primer pairs set1_5diag/set1_c_diag (5′ flank; 1.40 kb), set1_3diag/nat1_R1 (3′ flank; 1.57 kb) and set1_5diag/trpC_T (untransformed nuclei; 1.25 kb for Δset1). The respective deletion mutant (Δ) and the WT were used as controls. D) The in loco integration of pOE::KDM5 in the three transformants was checked using primer pair PoliC_Seq_F2/OE_kdm5_diag (1.95 kb). M, GeneRuler DNA Ladder Mix. E) The WT and OE::KDM5 mutants were grown in liquid culture (ICI + 60 mM Gln) for 3 days prior to RNA extraction from the harvested mycelium. The northern blot was probed with DNA corresponding to KDM5, and the ribosomal RNA was visualized for the respective gel as loading control.
Fig. S7. Verification of Δset1 deletion mutants by diagnostic PCR and Southern blot. A) Deletion via homologous recombination with the hygromycin B resistance cassette (hphR) was verified with the amplification of 5′ (set1_5diag/trpC_T) and 3′ (set1_3diag/trpC_P2) flanks but no amplification of WT (set1_WT_F/set1_WT_R) signal for three independent transformants. B) For analysing ectopic integration of deletion constructs, genomic DNA of transformants and WT was digested with ClaI, while the 5′ flank was applied for probing. C) Detected signals match the expected 10.66 kb for the WT and 3.11 kb for Δset1. λ, λ/HindIII; M, GeneRuler 1 kb Plus DNA Ladder.
Fig. S8. Verification of Δkdm5 deletion mutants by diagnostic PCR and Southern blot. A) Deletion via homologous recombination with the hygromycin B resistance cassette (hphR) was verified with the amplification of 5′ (kdm5_5diag/trpC_T) and 3′ (kdm5_3diag/trpC_P2) flanks but no amplification of WT (kdm5_WT_F/kdm5_WT_R) signal for three independent transformants. B) For analysing ectopic integration of deletion constructs, genomic DNA of transformants and WT was digested with SalI, while the 3′ flank was applied for probing. C) Detected signals match the expected 7.07 kb for the WT and 2.70 kb for Δkdm5. λ, λ/HindIII; M, GeneRuler DNA Ladder Mix.
Fig. S9. Verification of Δaba1, Δflb2‐Δflb5 and Δwet1 single deletion mutants. Deletion via homologous recombination with the hygromycin B resistance cassette was verified with the amplification of 5′ (5diag/trpC_T) and 3′ (3diag/trpC_P2) flanks but no amplification of WT (WT_F/WT_R) signal for A) two independent Δaba1 mutants (IMI58289 WT background), B) two independent E282/Δaba1 mutants, C) two independent Δflb2 mutants, D) three independent Δflb3 mutants, E) two independent Δflb4 mutants, F) three independent Δflb5 mutants and G) three independent Δwet1 mutants. λ, λ/HindIII.
Fig. S10. Deletion of CSM1 in Δaba1, Δflb3 and Δflb4 backgrounds. Deletion of CSM1 via homologous recombination with the nourseothricin resistance cassette was verified with the amplification of 5′ (csm1_5diag/nat1_hiF) and 3′ (csm1_3diag/trpC_P2) flanks but no amplification of WT (csm1_WT_F/csm1_WT_R) signal for A) two independent Δaba1/Δcsm1 double mutants, B) one Δflb3/Δcsm1 double mutant and C) two independent Δflb4/Δcsm1 double mutants. λ, λ/HindIII.
Table S1. Significantly enriched protein functions of genes deregulated in Δset1 and/or Δkdm5 in the microarray expression analysis. Genes upregulated in the deletion mutants compared with the WT have a log2 fold change ≥2, downregulated genes have a log2 fold change ≤ −2. The table is sorted by the P‐value and the filters rely on the following criteria: B, Bonferroni correction <0.05; F, P‐value < FDR; *, P‐value <0.05. abs., absolute; rel., relative; FDR, false discovery rate.
Table S2. Microarray expression analysis of differentially regulated SM key genes in Δset1 and Δkdm5. The WT and the two deletion mutants were grown in ICI liquid culture in the presence of limiting (6 mM, N‐) and saturating (60 mM, N+) amounts of Gln for 3 days prior to RNA extraction. Data are mean values (n = 2). Genes upregulated in the deletion mutants compared with the WT are green (log2 fold change ≥2), and downregulated genes are red (log2 fold change ≤ −2). Shown are the gene accession numbers, the encoded SM key genes as well as the produced SMs. PKS, polyketide synthase; NRPS, non‐ribosomal peptide synthetase; STC, sesquiterpene cyclase; DTC, diterpene cyclase; TeTC, tetraterpene cyclase; DMATS, dimethylallyltryptophan synthase.
Table S3. Primer sequences used for the generation of deletion constructs and for the verification of their homologous integration. Introduced overhangs required for yeast recombinational cloning are underlined.
Table S4. Primer sequences used for the generation and analysis of complementation, point‐mutation and overexpression vectors. Introduced overhangs required for yeast recombinational cloning are underlined.
Table S5. Primer sequences for analysing relative expression (qPCR) and ChIP‐qPCR (5′ChIP). Reference genes for relative expression: GMT, GDP mannose transporter gene; RAC, related actin gene; UBI, ubiquitin gene.