

Summary
Recognition of the AVRPM3A2/F2 avirulence protein from powdery mildew by the wheat PM3A/F immune receptor induces a hypersensitive response after co‐expression in Nicotiana benthamiana. The molecular determinants of this interaction and how they shape natural AvrPm3 a2/f2 allelic diversity are unknown.
We sequenced the AvrPm3 a2/f2 gene in a worldwide collection of 272 mildew isolates. Using the natural polymorphisms of AvrPm3 a2/f2 as well as sequence information from related gene family members, we tested 85 single‐residue‐altered AVRPM3A2/F2 variants with PM3A, PM3F and PM3FL 456P/Y458H (modified for improved signaling) in Nicotiana benthamiana for effects on recognition.
An intact AvrPm3 a2/f2 gene was found in all analyzed isolates and the protein variant recognized by PM3A/F occurred globally at high frequencies. Single‐residue alterations in AVRPM3A2/F2 mostly disrupted, but occasionally enhanced, the recognition response by PM3A, PM3F and PM3FL 456P/Y458H. Residues enhancing hypersensitive responses constituted a protein domain separate from both naturally occurring polymorphisms and positively selected residues of the gene family.
These results demonstrate the utility of using gene family sequence diversity to screen residues for their role in recognition. This approach identified a putative interaction surface in AVRPM3A2/F2 not polymorphic in natural alleles. We conclude that molecular mechanisms besides recognition drive AvrPm3 a2/f2 diversification.
Keywords: avirulence gene, Blumeria graminis, gene synthesis, natural diversity, Nicotiana benthamiana, Pm3, site‐directed mutagenesis, wheat
Introduction
Innate immune responses are crucial for early and efficient detection of infectious pathogens. Plant defense responses against pathogen‐caused diseases rely heavily on immune receptors that perceive infection and elicit a localized cell death response to prevent pathogen growth and proliferation. The immunity conferred by such receptors represents an important source of resistance in agriculture. In particular, intracellular nucleotide‐binding, leucine‐rich repeat receptors (NLRs) mediate resistance by detecting secreted pathogen effectors and activating effector‐triggered immune responses (effector‐triggered immunity, ETI) (Qi & Innes, 2013; Zipfel, 2014). ETI is an induced localized cell death, or hypersensitive response (HR), that is especially effective against biotrophic fungal and oomycete pathogens, which need living host tissue to survive.
To complete their life cycle, biotrophic pathogens must establish close associations with the host to acquire nutrients and avoid triggering the immune system (Koeck et al., 2011). To this end, filamentous fungal and oomycete pathogens secrete small effector proteins to inhibit host defense responses or hijack the cellular metabolism (Lo Presti et al., 2015). Fungal and oomycete effectors typically have no predicted homology to known proteins and can be structurally related, but often are not sequence related (Win et al., 2012; Maqbool et al., 2015; Lu et al., 2016; Praz et al., 2017). Pathogen fitness can depend heavily on effector function, where the loss or inactivation of important effectors reduces virulence (Huang et al., 2006; Bos et al., 2010). The recognition of specific effectors by host immune receptors also compromises fitness. Accordingly, effectors that are recognized by resistance (R) genes become avirulence factors (Avrs) and experience strong selection pressure to evade host recognition.
Interactions between host R genes and pathogen Avrs are highly specific. This specificity of recognition is considered the main driver of Avr allelic diversification. Examination of Avr natural diversity and the effects of individual polymorphisms on recognition has been highly informative for the study of R gene specificity. For example, resistance against Leptosphaeria maculans is mediated by the dual recognition of the AvrLm4‐7 effector by both Rlm4 and Rlm7 resistance genes (Parlange et al., 2009). Isolates virulent on Rlm4 contain AvrLm4‐7 alleles with a single common residue alteration which has been functionally validated as specifically disrupting Rlm4 recognition (Parlange et al., 2009; Blondeau et al., 2015). Following gain‐of‐virulence mutations in pathogens, reciprocal evolution of host R genes may re‐establish recognition and resistance. This has also been found in the step‐wise evolution of specificity between Avr‐Pik alleles from Magnaporthe oryzae and Pik alleles from rice (Oryza sativa) (Kanzaki et al., 2012). In contrast with gain‐of‐virulence on Rlm4, L. maculans isolates virulent on Rlm7 primarily contain either deletions or several polymorphisms in AvrLm4‐7 that lead to inactivation of the protein (Daverdin et al., 2012; Blondeau et al., 2015). This demonstrates that, in addition to diversification of the AVR protein sequence, successful evasion of recognition can also be mediated by Avr deletion or gene inactivation, despite potential fitness consequences (Huang et al., 2006, 2010).
Studies of recognition specificity have focused extensively on information from natural polymorphisms in both Avr and R genes. Variants of the avirulence factor ATR1 from Hyaloperonospora arabidopsis are recognized by different alleles of the RPP1 immune receptor from Arabidopsis (Krasileva et al., 2010). An informed selection of mutations from among 69 polymorphisms in ATR1 variants from eight isolates revealed that distributed recognition surfaces on ATR1 determine the specificity of different RPP1 alleles (Krasileva et al., 2010; Chou et al., 2011). Similarly, in the study of the recognition specificity of flax (Linum usitatissimum) L5 and L6 alleles for AvrL567 variants of the flax rust pathogen (Melampsora lini), tests combining several mutations from among 35 naturally polymorphic sites at the AvrL567 locus in six rust strains revealed their additive effect on recognition, suggesting that multiple amino acid contact points also determine specificity in this interaction (Ravensdale et al., 2012). However, only four AVR‐Pik variants were identified in a worldwide screen of 39 M. oryzae isolates (Kanzaki et al., 2012), demonstrating that, even in larger worldwide diversity screens, limited sequence diversity is often observed in fungal Avr genes. Therefore, in vitro‐created sequence diversity would be useful for determining the basis of recognition specificity for fungal Avr–R interactions.
Active R genes are known to coevolve with their cognate pathogen Avrs under strong diversifying selection or, more rarely, by balancing selection. They often form larger gene families and complex clusters of gene paralogs, but rarely form true allelic series conferring race specificity (Ellis et al., 1999; Rose et al., 2004; Seeholzer et al., 2010; Kanzaki et al., 2012). Early descriptions of the cognate Avrs of multiallelic race‐specific R genes indicated that allelic variants recognize naturally occurring AVR variants in the pathogen (Dodds et al., 2006; Krasileva et al., 2010; Kanzaki et al., 2012). However, recent advances in the study of multiallelic R genes conferring resistance to the cereal powdery mildews (Blumeria graminis ff. spp.), specifically the Pm3 gene in wheat (Triticum aestivum) and the Mla gene in barley (Hordeum vulgare), suggest that distinct resistance alleles recognize highly sequence diverse effectors (Bourras et al., 2015; Lu et al., 2016).
Powdery mildew of wheat is caused by the obligate biotrophic fungal pathogen, Blumeria graminis f. sp. tritici (B.g. tritici). B.g. tritici followed the spread of wheat cultivation to all major wheat‐growing regions worldwide. The multiallelic Pm3 gene from wheat encodes a coiled‐coiled (CC)‐NLR protein that confers race‐specific resistance against B.g. tritici. The 17 functionally distinct alleles of Pm3 in the hexaploid bread wheat gene pool share particularly high sequence identity (>97%) (Yahiaoui et al., 2004; Srichumpa et al., 2005; Bhullar et al., 2010). The Pm3a and Pm3f alleles share overlapping recognition spectra towards powdery mildew races, where the spectrum of races recognized by Pm3a includes those recognized by Pm3f (Brunner et al., 2010). This overlap is the result of their shared specificity for the AvrPm3 a2/f2 gene encoded by B.g. tritici. The AvrPm3 a2/f2 gene belongs to a family of 24 sequence‐divergent, but structurally related, secreted effectors (Bourras et al., 2015). In transient expression assays in Nicotiana benthamiana, it was demonstrated that AVRPM3A2/F2 is recognized specifically by the Pm3a and Pm3f alleles. The HR elicited by PM3A recognition is much stronger than that elicited by the PM3F allele; however, it was demonstrated that two substitutions (L456P, Y458H) in the ARC2 subdomain of the nucleotide‐binding site domain (NBS) of PM3F are sufficient to enhance this response to levels comparable with those of PM3A (Stirnweis et al., 2014). In contrast with gene‐for‐gene interactions (Flor, 1971), segregating phenotypes among the progeny of a genetic cross of isolates hinted at the involvement of a second pathogen‐encoded factor, the suppressor‐of‐recognition SvrPm3 a1/f1, which was cloned and functionally validated. Expression analyses of both fungal effector genes suggested that the SvrPm3 a1/f1 suppressor acts quantitatively to suppress AvrPm3 a2/f2 recognition mediated by PM3A/F (Bourras et al., 2015). Altogether, these findings led to the development of the Avr‐R‐Svr model of interaction in the wheat–powdery mildew pathosystem, where R specificity for Avr recognition is modified by the action of an Svr (Bourras et al., 2016).
In this study, we examined the basis of Pm3a/f recognition specificity using the natural sequence diversity of AvrPm3 a2/f2 from a worldwide collection of B.g. tritici and B.g. triticale. We found that AvrPm3 a2/f2 is present in all isolates and shows limited natural diversity worldwide. We provide further evidence of the role of the suppressor SvrPm3 a2/f2 in increasing the virulence of isolates from Europe that express the active AvrPm3 a2/f2. Using the sequence diversity from the structurally related AvrPm3 a2/f2 effector family, we identified a region of AVRPM3A2/F2 in which mutations strongly influence recognition and specificity. This putative interaction domain does not overlap with residues under positive selection in the effector family or residues polymorphic in the natural isolates. In light of our results, we conclude that using sequence diversity from a related gene family is informative for studies of recognition specificity, and propose that, for the AvrPm3 a2/f2–Pm3a/f interaction, selection pressure from recognition is not the primary source of AvrPm3 a2/f2 allelic diversity.
Materials and Methods
Fungal collection, propagation and virulence tests
Blumeria graminis isolates were maintained on detached leaves of the appropriate susceptible cereal cultivar (‘Kanzler’ (Triticum aestivum), ‘Inbar’ (Triticum durum) or ‘Matador’ (Triticosecale)) on benzimidazole agar, as described by Parlange et al. (2011). The worldwide collection contained 272 isolates: 55 isolates collected in 13 states in the USA (Cowger et al., 2018), 101 collected in 12 provinces in China (Zeng et al., 2014), 61 collected in four eco‐geographic regions of Israel (Ben‐David et al., 2016), 51 collected primarily in Switzerland as well as in five other European countries (our collection), two in Japan and two in Australia. All isolates from Europe, Japan and Australia, and a subset of isolates from the USA, China and Israel, were maintained in‐house. Spore samples for DNA extraction and sequencing by PCR were obtained primarily for US and Israeli isolates, whereas complete genomes for a subset of Chinese isolates were used for in silico extraction (Praz et al., 2017). European, Japanese and Australian isolates were collected in the field and single spore isolated twice, as described by Brown & Wolfe (1990). Virulence tests on Pm3a (cv. Asosan/8*Chancellor) and Pm3f (cv. Michigan Amber/8*Chancellor) of isolates in‐house were performed as described by Brunner et al. (2010), and scored as described in Bourras et al. (2015). External Israeli, US and Chinese isolates were collected, maintained and phenotyped as described in Ben‐David et al. (2016) (Israeli and US) and Zeng et al. (2014) (Chinese).
DNA/RNA isolation and construction of plasmid vectors
High‐molecular‐weight DNA for PCR amplification was extracted as described previously by Bourras et al. (2015). RNA samples were extracted from infected detached ‘Chancellor’ leaves using the Qiagen miRNeasy Mini Kit (Qiagen) according to the manufacturer. Full‐length cDNA was prepared using the Superscript III RT kit (Invitrogen) according to the manufacturer. Molecular cloning into the Gateway‐compatible entry vector was performed using the pENTR/D‐TOPO Cloning Kit (Invitrogen) according to the manufacturer. Gene synthesis including Gateway‐compatible cloning sites was performed by gen9 (https://www.gen9bio.com/), resulting in a Gateway‐compatible cloning vector (pG9m‐2) containing the synthesized gene. The synthesized gene was then cloned directly into the pIPKb004 expression vector (Himmelbach et al., 2007) used for transient expression in N. benthamiana. Site‐directed mutagenesis was performed by PCR amplification using overlapping primers containing desired mutations on the Gateway‐compatible pENTR/D‐TOPO vector containing AvrPm3 a2/f2. Recombination of the mutant PCR product into the binary vector pIPKb004 was performed as described previously by Stirnweis et al. (2014). Primers used for gene amplification and site‐directed mutagenesis are listed in Supporting Information Table S1. Constructs are listed in Table S2. All AvrPm3 a2/f2 and SvrPm3 a1/f1 genomic sequences used for this study are available at the GenBank database under the accession numbers MG739404–MG739429.
Genetic analyses and tests for selection
The Templeton, Crandall and Sing (TCS) network was visualized using the popart software (Leigh & Bryant, 2015). Maximum likelihood computations and estimation of the average nonsynonymous/synonymous (d N /d S) rate ratio for AvrPm3 a2/f2 haplotypes were conducted using the hyphy software package (Pond et al., 2005) in the mega7 program (Kumar et al., 2016). All multiple alignments were performed with muscle 3.8.31 (Edgar, 2004). The protein alignments were back‐translated to nucleotide alignments using translatorx v1.1 (Abascal et al., 2010). To test for positive selection, we estimated the likelihood of the maximum likelihood tree under the M8a and M8 models (Yang & Nielsen, 2000) with paml 4.8 (Yang, 2007) and using the Bayes empirical Bayes method (Yang et al., 2005).
Quantitative real‐time PCR experiments
Quantitative real‐time PCR experiments were performed as described in Praz et al. (2017) with the modification of a 20‐s extension time. Three independent biological replicates were sampled at 2 d after inoculation. Glyceraldehyde 3‐phosphate dehydrogenase (Gapdh) was used as an internal control, as described in Bourras et al. (2015). Gene expression was normalized to that of Gapdh. Quantitative real‐time PCR primers for reference and target genes have been described previously (Bourras et al., 2015).
Transient protein expression assays in N. benthamiana
Transient expression by agroinfiltration in N. benthamiana was conducted according to the protocols of Ma et al. (2012) and Dugdale et al. (2014), and with the modifications described by Bourras et al. (2015). To test for AvrPm3–Pm3 interaction, Agrobacterium expressing the AvrPm3 constructs or the Pm resistance allele were mixed in a 4 : 1 ratio of Avr : R. At least three replications were performed and the HR was visualized as described by Praz et al. (2017). HR intensity was calculated using the mean gray value estimated by ImageJ (Schneider et al., 2012) and normalizing against the noninfiltrated background. Normalized values were compared between HR elicited by mutant AvrPm3 a2/f2 constructs and wild‐type AvrPm3 a2/f2 constructs, and statistical significance was estimated using the Student's t‐test.
Results
A worldwide survey reveals the ubiquitous presence and limited sequence diversity of AvrPm3 a2/f2
To study the natural sequence diversity of AVRPM3A2/F2 and to characterize the variants eliciting recognition by Pm3a/f, we sequenced the complete AvrPm3 a2/f2 gene in an unprecedented worldwide collection of 272 isolates with diverse genetic backgrounds and a balanced representation of global geographic origins (Table S3). The 251 B.g. tritici and 22 B.g. triticale (powdery mildew of triticale) isolates form six geographically distinct populations: USA, 56 isolates from 13 states; China, 101 isolates from 12 provinces (Zeng et al., 2014); Europe, 51 isolates from six countries; Israel, 61 isolates (Ben‐David et al., 2016); Australia, two isolates; Japan, two isolates. All sampled isolates within this collection encoded a complete AvrPm3 a2/f2 gene, with no case of nonsense mutation, truncation or complete gene deletion. Although it is impossible to identify identical gene duplications by sequencing of PCR products, our whole gene sequencing data yielded no evidence of mixed gene sequences that would indicate multiple divergent AvrPm3 a2/f2 sequences encoded by a single isolate. In addition, de novo analysis of 41 available whole genome assemblies using iterative blast searches of the AvrPm3 a2/f2 gene sequence revealed no evidence of duplications or multiple haplotypes encoded in a single isolate. We identified 11 novel AvrPm3 a2/f2 haplotypes in addition to the two reported previously (Bourras et al., 2015). The 13 haplotypes share 95–99% identity, and 12 encode unique protein sequences (labelled A–H, J–M, Table 1). All novel protein encoding haplotypes were produced by site‐directed mutagenesis or gene synthesis and tested in transient assays in N. benthamiana for recognition by PM3A, PM3F and PM3FL456P/Y458H, as described in Bourras et al. (2015). We used the previously validated AVRPM3A2/F2‐A as the positive control for HR induction upon recognition by PM3A and PM3FL456P/Y458H. By visual inspection and fluorescence imaging (Praz et al., 2017) of the assayed N. benthamiana leaves at 5 d post‐infiltration, we observed that none of the new variants induced HR in the presence of PM3A or PM3FL456P/Y458H, suggesting that they encode inactive AVR proteins (Fig. S1). These results indicate that AVRPM3A2/F2‐A (Bourras et al., 2015) is the only active AVR variant in natural isolates.
Table 1.
Disrupting recognition by polymorphisms in the amino acid sequences of the AVRPM3A2/F2 variants from wheat powdery mildew
| No. | 21 | 24 | 25 | 26 | 27 | 31 | 38 | 52 | 66 | 69 | 80 | 86 | 89 | 91 | 93 | 95 | 109 | 119 | 122 | 123 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AVRPM3A2/F2‐Aa | 122 | A | S | G | ‐ | ‐ | N | H | E | N | R | N | G | N | K | E | F | A | Y | T | E |
| AVRPM3A2/F2‐C | 28 | . | . | . | ‐ | ‐ | . | . | . | . | . | . | . | . | . | . | L | . | . | . | . |
| AVRPM3A2/F2‐E | 16 | . | . | . | ‐ | ‐ | . | . | . | . | . | . | . | . | . | D | . | . | . | . | . |
| AVRPM3A2/F2‐Fb | 15 | . | . | . | ‐ | ‐ | . | . | . | K | S | . | E | . | . | . | . | . | . | . | . |
| AVRPM3A2/F2‐D | 24 | . | . | . | ‐ | ‐ | . | . | . | K | . | S | . | . | T | K | . | . | . | . | . |
| AVRPM3A2/F2‐B | 52 | . | . | . | ‐ | ‐ | . | Q | . | . | . | . | E | . | . | . | . | . | . | . | D |
| AVRPM3A2/F2‐G | 4 | V | N | S | P | V | E> | Q | . | K | . | . | E | Y | . | . | . | V | H | . | . |
| AVRPM3A2/F2‐M | 1 | . | . | . | ‐ | ‐ | E | Q | N | . | . | . | E | . | . | . | . | . | . | . | D |
| AVRPM3A2/F2‐K | 2 | . | . | . | ‐ | ‐ | . | Q | . | . | . | . | E | . | . | . | . | . | . | . | . |
| AVRPM3A2/F2‐H | 4 | . | . | . | ‐ | ‐ | . | . | . | . | . | . | E | . | . | . | . | . | . | . | D |
| AVRPM3A2/F2‐J | 3 | . | . | . | ‐ | ‐ | . | . | . | . | . | . | E | . | . | . | . | . | . | R | D |
| AVRPM3A2/F2‐La | 1 | . | . | . | ‐ | ‐ | . | . | . | . | . | . | E | . | . | . | . | V | . | . | . |
| Total | 272 |
Polymorphisms compared with the avirulent variant are depicted using the one‐letter code for amino acids. Residue numbering includes the signal peptide. The number of isolates encoding each variant in the global collection of 272 isolates is given (No.). Residues that individually disrupt recognition by PM3A and PM3FL456P/Y458H are indicated with black boxes. Untested residues are in gray boxes. Dashes indicate gaps and dots indicate identical residues.
Bourras et al. (2015).
Variant found in isolates collected on Triticum dicoccoides (wild emmer).
Genotyping the mildew collection revealed that the AvrPm3 a2/f2 ‐A haplotype is the most common worldwide (122 isolates, 45%), and is present in all six geographic populations at varying frequencies (15–88%, excluding Australia and Japan; Fig. 1a). We obtained phenotypic data on both Pm3a and Pm3f wheat for 166 isolates in the collection (Table 2). Comparing AvrPm3 a2/f2 genotypes and isolate phenotypes on Pm3a and Pm3f, we observed that, of the isolates encoding any of the inactive AVRPM3A2/F2 variants B–H, J–M, 72% (65/90) were virulent on Pm3a, whereas 78% (70/90) were virulent on Pm3f (Table 2). However, all isolates encoding variant ‘F’ (also not recognized in our transient assay) were consistently avirulent on Pm3a and Pm3f, with one exception. We also tested the hypothesis that variant ‘F’ is recognized by other Pm3 alleles using the same assays in N. benthamiana. We found no evidence of interaction with Pm3b, Pm3c, Pm3d and Pm3e alleles, or with the mildew resistance gene Pm8 (Fig. S2). Evidence of a third factor (AvrPm3 a3) recognized by Pm3a was found previously in a genetic cross of two mildew isolates segregating on Pm3a and Pm3f (Bourras et al., 2015). There is also genetic evidence from one UV‐mutagenized mildew strain (EXP3) that a fourth factor regulates AvrPm3 a2/f2 gene expression (Bourras et al., 2015; Parlange et al., 2015). Therefore, it is likely that additional Avr factors or Avr modifiers polymorphic in global populations could explain the avirulence of isolates harboring AVRPM3A2/F2‐F on Pm3a/f. Another possibility is that AvrPm3 a2/f2 is recognized by another NLR in wheat. Although evidence from our genetic mapping populations indicates that AvrPm3 a2/f2 is not recognized by Pm1a, Pm2, Pm3b‐e, Pm3g, Pm4a, Pm4b, Pm5a, Pm8 and Pm17, we cannot exclude possible recognition by NLRs other than those we have tested (Parlange et al., 2015; Bourras et al., 2015; Praz et al., 2017; Notes S1; Tables [Link], [Link], [Link]). In isolates encoding the recognized AvrPm3 a2/f2 ‐A haplotype, we observed that 71% (54/76) were avirulent on Pm3a wheat, and 17 of these (22%) were also avirulent on Pm3f wheat (Table 2). Together, these results suggest that additional pathogen‐encoded factors besides AvrPm3 a2/f2 are contributing to recognition by Pm3a/f in natural mildew populations.
Figure 1.
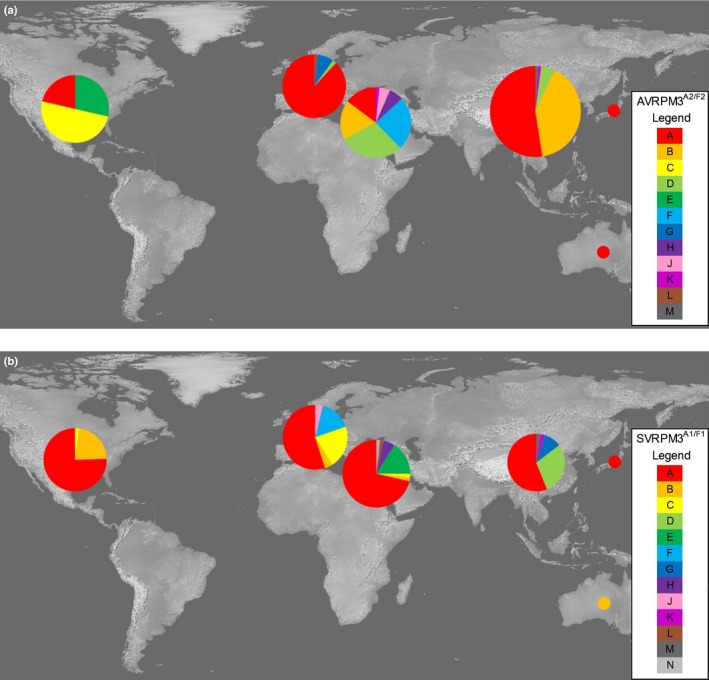
Geographic distribution of the AVRPM3A2/F2 and SVRPM3A1/F1 variants. (a) Geographic distribution of the 12 AVRPM3A2/F2 variants (A–H, J–M) encoded by Blumeria graminis f. sp. tritici and triticale isolates from the USA, Europe, Israel, China, Japan and Australia. (b) Geographic distribution of the SVRPM3A1/F1 variants (A–H, J–N) encoded by Blumeria graminis f. sp. tritici and triticale isolates from the USA, Europe, Israel, China, Japan and Australia. The areas of the circles are proportional to the number of isolates from that region in the tested collection.
Table 2.
Summary of AVRPM3A2/F2 variants and phenotypes on Pm3a and Pm3f wheat observed in natural isolates

AVRPM3A2/F2 variants (A–H, J–M) found in natural Blumeria graminis f. sp. tritici and triticale isolates are listed, together with the observed phenotypes on Pm3a and Pm3f wheat. Variant A (red) is recognized by PM3A and PM3FL456P/Y458H in functional assays. Phenotypes (avirulent, A (green); intermediate, I (yellow); virulent, V (pink)) are colored for ease of reading.
To summarize, our results reveal limited natural diversity of AvrPm3 a2/f2, and the prevalence of the AVRPM3A2/F2‐A protein variant, suggesting an important role of avr/AvrPm3 a2/f2 and, in particular, the active AVRPM3A2/F2‐A allele in pathogen fitness. In addition, our phenotypic data suggest that other factors are involved in the interaction. For example, an as yet uncharacterized AvrPm3 a3 avirulence gene might be rendering isolates without the active AVRPM3A2/F2‐A variant avirulent on Pm3a, whereas the suppressor of recognition, SvrPm3 a1/f1, is probably increasing the virulence in isolates encoding the active AVRPM3A2/F2‐A variant.
Virulence in isolates encoding the recognized AVRPM3A2/F2‐A is associated with the SVRPM3A1/F1 suppressor
Our phenotypic data indicate that, of the 54 of 76 isolates encoding the AVRPM3A2/F2‐A variant that are virulent on Pm3f, 16 are also virulent on Pm3a despite the presence of the recognized AvrPm3 a2/f2 allele (Table 2). Previously, a suppressor of recognition, SvrPm3 a1lf1, was functionally validated as quantitatively suppressing the recognition of AvrPm3 a2/f2 by Pm3a/f in a genetic cross of two European mildew isolates (Bourras et al., 2015). Polymorphic presence of the suppressor might cause the high variability of virulence in isolates encoding the active AVRPM3A2/F2‐A variant on Pm3a and Pm3f wheat. To determine whether the presence of the active suppressor is associated with virulence, we obtained sequences of the SvrPm3 a1/f1 gene from 201 isolates from the worldwide collection (for which genomic DNA or genome sequences were available), and compared the Svr genotypes and phenotypes of the subset of 76 isolates encoding the active AvrPm3 a2/f2. As with AvrPm3 a2/f2, we found a complete SvrPm3 a1/f1 gene in all isolates, suggesting that it also contributes to pathogen fitness. We identified seven new haplotypes in addition to the six haplotypes described previously (Parlange et al., 2015), for a total of 13 unique, but highly similar, protein variants (98–99% sequence identity; labeled A–H, J–N, Table 3). The previously reported active suppressor variant, SVRPM3A1/F1‐A, is also the most predominant (130 isolates) globally (Fig. 1b). For 155 isolates, we obtained both AvrPm3 a2/f2 and SvrPm3 a1/f1 sequences, as well as phenotypic data on Pm3a and Pm3f wheat (Table 4). Of these isolates, 96 encode the active suppressor and, among these, 91 are virulent on Pm3f and 68 are virulent on Pm3a. Strikingly, with only one exception, all isolates encoding the recognized AVRPM3A2/F2‐A variant and the active suppressor variant SVRPM3A1/F1‐A (42 isolates) show virulence or intermediate virulence on Pm3f. By contrast, all isolates encoding AVRPM3A2/F2‐A and suppressor variants other than SVRPM3A1/F1‐A (32 isolates) are avirulent on Pm3a, with two exceptions (Table 4), suggesting that most SVR variants other than ‘A’ do not function as active suppressors. The SVRPM3A1/F1‐B variant might have low suppressor activity, as we found eight isolates encoding AVRPM3A2/F2‐A and SVRPM3A1/F1‐B that were virulent on Pm3f wheat. These results indicate that Pm3f resistance can be suppressed by SVRPM3A1/F1‐A independently of the mildew isolate genetic background.
Table 3.
Amino acid sequences of the SVRPM3A1/F1 variants in wheat powdery mildew
| No. | 22 | 31 | 32 | 70 | 71 | 102 | 118 | 125 | 127 | |
|---|---|---|---|---|---|---|---|---|---|---|
| SVRPM3A1/F1‐Aa | 130 | I | K | P | A | R | L | M | Y | T |
| SVRPM3A1/F1‐Ba | 16 | . | . | . | . | H | . | . | . | . |
| SVRPM3A1/F1‐Ca | 13 | . | . | . | . | H | P | . | . | . |
| SVRPM3A1/F1‐Na | 1 | . | . | H | . | H | P | . | . | . |
| SVRPM3A1/F1‐Fa | 8 | . | . | . | . | H | P | . | . | K |
| SVRPM3A1/F1‐J | 2 | . | N | . | . | H | P | . | . | . |
| SVRPM3A1/F1‐H | 3 | T | . | . | . | H | P | . | . | . |
| SVRPM3A1/F1‐Ea, b | 9 | . | . | . | S | H | S | . | . | . |
| SVRPM3A1/F1‐D | 12 | . | . | . | . | H | F | . | . | . |
| SVRPM3A1/F1‐M | 1 | . | . | . | . | H | F | V | . | . |
| SVRPM3A1/F1‐K | 1 | . | . | . | . | . | F | . | . | . |
| SVRPM3A1/F1‐G | 4 | . | . | . | . | . | . | V | . | . |
| SVRPM3A1/F1‐L | 1 | . | . | . | . | . | . | . | C | . |
| Total | 201 |
Polymorphisms compared with the known active suppressor variant are depicted using the one‐letter code for amino acids. Residue numbering includes the signal peptide. Dots indicate identical residues.
Parlange et al. (2015).
Variant found in isolates collected on wild emmer (Triticum dicoccoides).
Table 4.
Summary of unique AVRPM3A2/F2 and SVRPM3A1/F1 variant combinations and isolate phenotypes on Pm3a and Pm3f observed in natural isolates
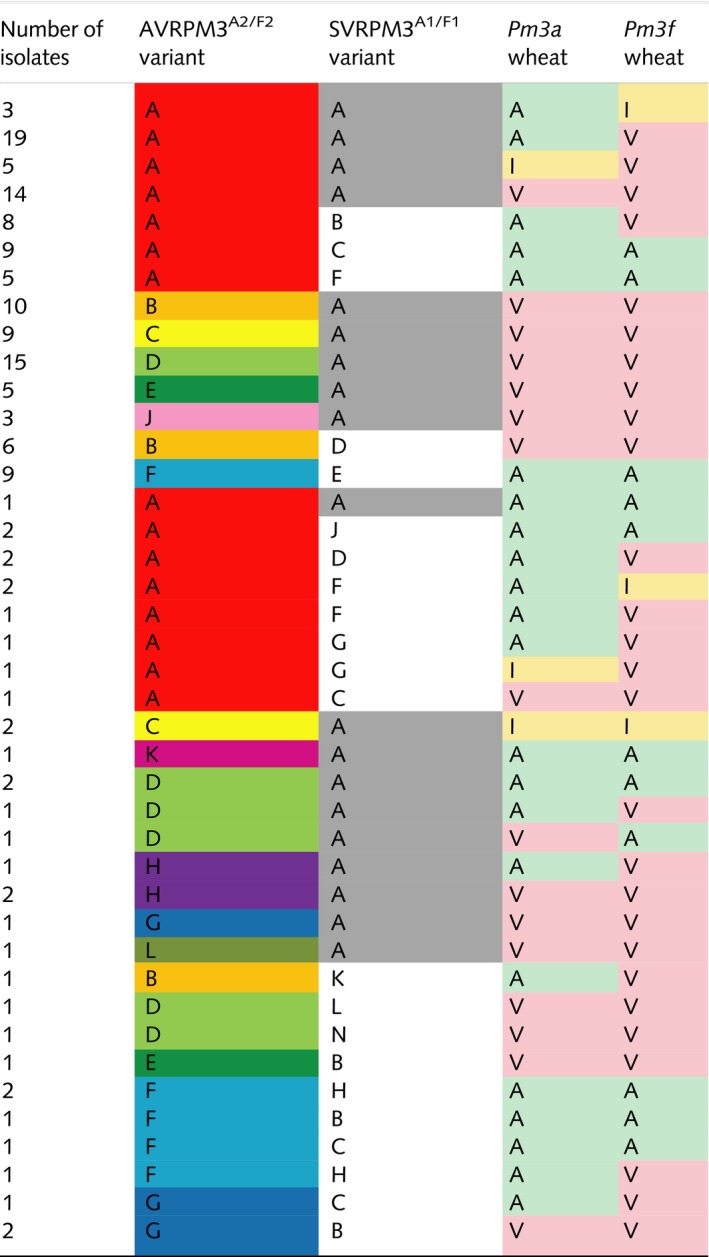
Unique AVR–SVR variant combinations from Blumeria graminis f. sp. tritici and triticale isolates and their observed phenotypes on Pm3a and Pm3f are shown. AVRPM3A2/F2 variants (A–H, J) and SVRPM3A1/F1 variants (A–F, H and J) are indicated. AVRPM3A2/F2‐A is the only recognized AVR variant (red), and SVRPM3A1/F1‐A is the known active suppressor variant (gray, Bourras et al., 2015). Phenotypes (avirulent, A (green); intermediate, I (yellow); virulent, V (pink)) are colored for ease of reading. Variant–phenotype combinations observed in less than three isolates are listed separately for clarity.
Most of the phenotypic variability of isolates encoding both the active AVRPM3A2/F2 and SVRPM3A1/F1 is observed in the European population in which the suppressor was originally described. It is also the population in which the combination of both active AVRPM3A2/F2 and SVRPM3A1/F1 is most frequent (24/50 isolates, 48%, Fig. S3). Within the European population, we identified 12 isolates that combine both active variants, but are avirulent on Pm3a wheat (Table S3). To test whether this phenotype can be explained by low expression of the suppressor gene, we compared the gene expression levels of AvrPm3 a2/f2 ‐A and SvrPm3 a1/f1 ‐A in two Pm3a virulent European B.g. tritici isolates (7004 and 07296) and two Pm3a avirulent European B.g. tritici isolates (07237 and JIW2) at 2 d post‐infection (Fig. 2). The isolates virulent on Pm3a wheat (7004 and 07296) showed similar (07296) or higher (7004) expression levels of SvrPm3 a1/f1 ‐A relative to AvrPm3 a2/f2 ‐A, consistent with the hypothesis that virulence in these isolates is mediated by relatively higher Svr expression. In the Pm3a avirulent isolates, we found that SvrPm3 a1/f1 ‐A was expressed at significantly lower levels than AvrPm3 a2/f2 ‐A, and also relative to the expression of the suppressor in the Pm3a virulent isolates, 7004 and 07296 (Fig. 2; Table S7). These results substantiate our hypothesis that avirulence on Pm3a, in the subset of 12 European isolates combining active AVRPM3A2/F2 and SVRPM3A1/F1 variants, can be explained by lower expression levels of SvrPm3 a1/f1 ‐A. In addition, we compared gene expression levels of AvrPm3 a2/f2 ‐A and SvrPm3 a1/f1 ‐A in six European B.g. triticale isolates, three Israeli B.g. tritici isolates and one Japanese B.g. tritici isolate with various phenotypes on Pm3a and Pm3f (Fig. 2). We observed some association between SvrPm3 a1/f1 ‐A expression relative to AvrPm3 a2/f2 ‐A expression and virulence in the B.g. triticale isolates, but no association in isolates from Israel and Japan. These data suggest that the quantitative action of a suppressor might be most observable in the European population, and that other factors known to be involved in the interaction (e.g. AvrPm3 a3) might prevent observable associations between phenotype and SvrPm3 a1/f1 ‐A expression in other populations. In an analysis of the expression of AvrPm3 a2/f2 ‐A with other SvrPm3 a1/f1 haplotypes (B, C, D, F, G and J), we did not observe any similar association between expression and virulence, although expression of these variants was overall lower than SvrPm3 a1/f1 ‐A, and appeared to be more consistent within isolates and less polymorphic between isolates encoding the same haplotype (Fig. S4). Taken together, our results demonstrate that recognition of AvrPm3 a2/f2 ‐A encoding isolates by Pm3a/f wheat is strongly associated with the presence and expression level of SvrPm3 a1/f1 ‐A in European isolates, thus providing additional evidence for the Avr‐R‐Svr genetic model at the population level.
Figure 2.
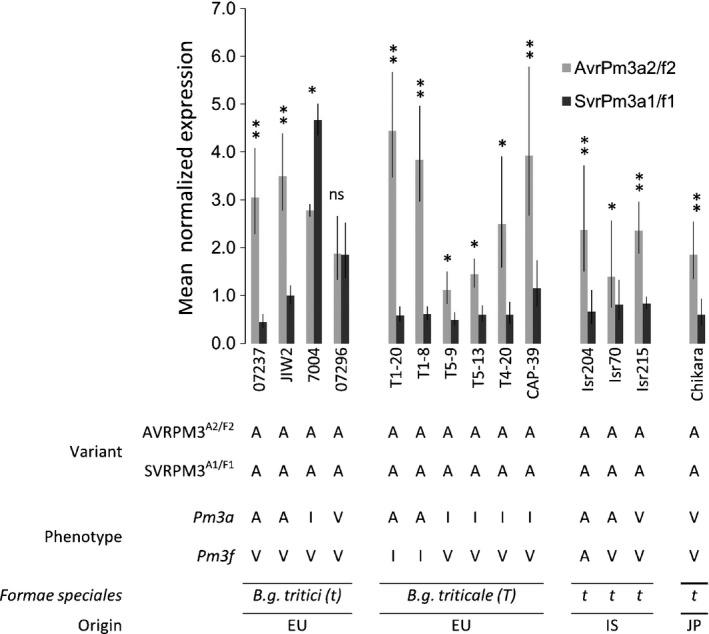
Expression analysis of isolates encoding the AVRPM3A2/F2‐A and SVRPM3A1/F1‐A variant combination which give different phenotypes on Pm3a and Pm3f wheat. The mean normalized expression of AvrPm3 a2/f2 (light gray histograms) and SvrPm3 a1/f1 (dark gray histograms) at 2 d post‐infection. Leaf segments from the susceptible recurrent parent line ‘Chancellor’ were infected with Blumeria graminis f. sp. tritici (t) and B.g. triticale (T) isolates from diverse geographic origins (EU, Europe; IS, Israel; JP, Japan) that contain the same Avr‐Svr genotype, but have different virulences (avirulent, A; intermediate, I; virulent, V) on Pm3a and Pm3f wheat. The results of a t‐test comparing AvrPm3 a2/f2 and SvrPm3 a1/f1 expression are given by * and **, indicating P < 0.05 and P < 0.01, respectively (ns, not significant). The error bars indicate ± SEM.
Single‐residue changes in AVRPM3A2/F2 are sufficient to abolish, enhance or alter recognition by PM3A, PM3F and PM3F L456P/Y458H
In the study of the recognition specificity of L5 and L6 for AvrL567, Ravensdale et al. (2012) chose a subset of four residues among 35 polymorphic sites associated with differences in specificity among AvrLm567 alleles, and tested their individual contribution towards specificity. More recently, Maqbool et al. (2015) used protein structural information to design four mutations in AVR‐PikD which they predicted would disturb binding to the PikP cognate NLR. As neither excessive natural polymorphisms nor protein structural information was available for AVRPM3A2/F2, our strategy was to combine information from the 17 natural polymorphisms with an analysis of sequence diversity from the previously described AvrPm3 a2/f2 effector family (Bourras et al., 2015). AVRPM3A2/F2 belongs to a family of 24 small effectors (90–111 residues in the mature peptide) sharing very little sequence similarity, but an overall structural homology (Bourras et al., 2016). We wanted to select single‐residue alterations across the entire AVRPM3A2/F2 protein to identify amino acid residues affecting recognition specificity by Pm3a and Pm3f. We assumed that highly conserved residues among the 24 family members were important for structure and that substitutions in AVRPM3A2/F2 with residues conserved in many other family members would be structurally conservative. Following this strategy for large‐scale mutagenesis that conserved protein structure, we individually altered residues in AVRPM3A2/F2 towards residues shared by at least six (25%) family members (Fig. 3). In addition, the three most highly conserved residues in the family, Y35, C37 and C118, were individually changed to glycine to test their role in recognition, and mutations at residues 102, 128 and 132 were included for even coverage using either the next most frequent residue or the residue encoded by the close relative, Pu_23.
Figure 3.
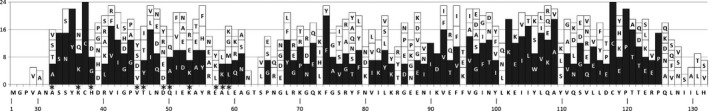
Conserved residues and motifs within the AVRPM3A2/F2 family from wheat powdery mildew. The protein sequences of 24 family members without the signal peptide were aligned and compared with the AVRPM3A2/F2 sequence. Residues present in at least three family members (white histograms) and the most frequent residues conserved in more than six of 24 family members (black histograms) are shown. Asterisks indicate residues under diversifying selection (probability (ω > 1) of > 0.95).
Including the two single mutations described previously (Bourras et al., 2015), we generated 85 constructs individually mutating 66/106 residues (62.3%) with an average of 1.3 mutations per amino acid (Fig. 4a). We co‐infiltrated each of these constructs with constructs expressing PM3A, PM3F and the modified PM3FL456P/Y458H in N. benthamiana and assessed HR at 5 d post‐infiltration. Of the 85 single‐residue‐altered constructs, 25 elicited HR to levels comparable with the wild‐type (‘neutral’ alterations), whereas a majority of alterations (50/85, 59%) elicited no HR with any construct (‘disruptive’ alterations, Fig. 4a). For the alterations that were disruptive or neutral on PM3A and PM3FL456P/Y458H, we did not observe HR when co‐expressed with the natural PM3F allele. At four positions, amino acid changes were either neutral or disruptive, depending on the residue chosen for replacement (positions 52, 91, 106 and 119; Fig. 4a). Interestingly, we identified a single mutation (H132D) that abolished recognition by PM3A, whilst maintaining the wild‐type‐level HR when co‐expressed with PM3FL456P/Y458H (Fig. 4a,b). This is surprising as this is the final C‐terminal residue of the AVRPM3A2/F2 protein, and residues at the ends of proteins are not often implicated in protein–protein interactions or recognition specificity (Bos et al., 2010). Overall, we observed disruptive alterations across the AVRPM3A2/F2 protein, whereas neutral alterations were concentrated in residues 60–75 (Fig. 4a).
Figure 4.
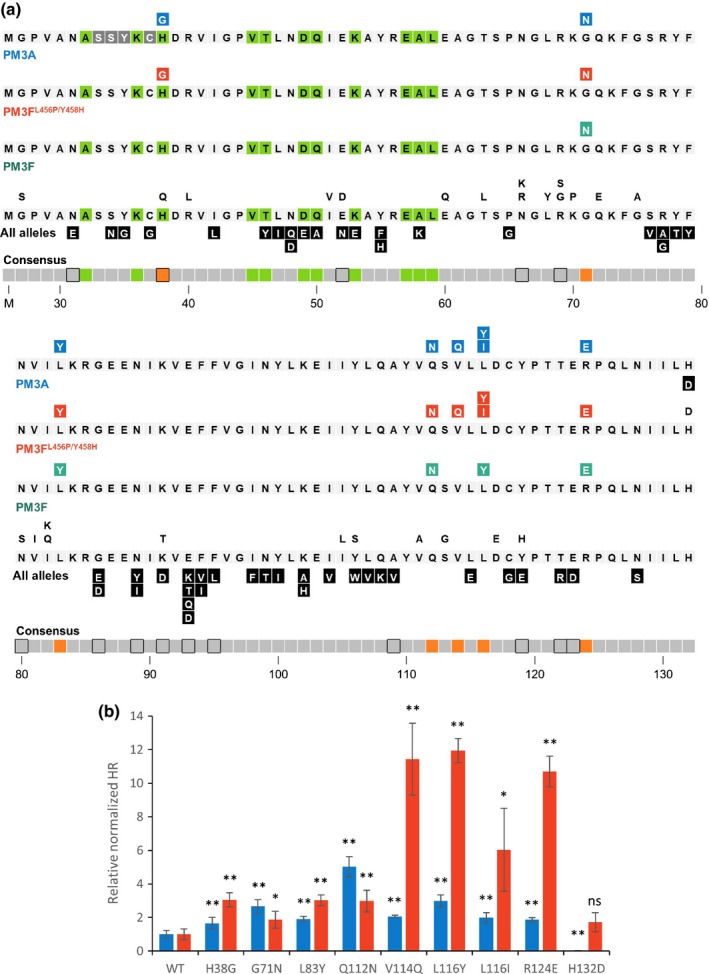
Single‐residue alterations in the wheat powdery mildew AVRPM3A2/F2 avirulence protein tested by co‐expression in Nicotiana benthamiana with the nucleotide‐binding, leucine‐rich repeat receptors (NLRs) PM3A, PM3F and PM3FL 456P/Y458H. Single‐residue changes derived from natural polymorphisms in the variants and sequence variation in the effector family span the complete AVRPM3A2/F2 protein sequence without the signal peptide. These were tested by transient co‐expression in N. benthamiana with constructs expressing PM3A, PM3F and PM3FL 456P/Y458H. (a) The sequence of the AVRPM3A2/F2 protein without signal peptide (replaced by methionine) is depicted, with the residues under diversifying selection in the family (green boxes) indicated. Results are shown on three lines separately for PM3A, PM3FL 456P/Y458H and PM3F. Single‐residue alterations that do not affect recognition are not colored, whereas mutations that disrupt recognition (black boxes), enhance the hypersensitive response (HR) with PM3A (blue boxes), PM3FL 456P/Y458H (red boxes) or gain a visible HR with PM3F (teal boxes) are indicated. A consensus diagram is given of the residues under positive selection in the family (green boxes), polymorphic residues in the variants (boxes outlined in black) and residues that result in enhanced HR when co‐expressed with PM3A, PM3F or PM3FL 456P/Y458H compared with the wild‐type (orange boxes). (b) The subset of constructs selected for quantification based on their enhancement or altered patterns of HR when co‐expressed with PM3A or PM3FL 456P/Y458H. Normalized quantification of the HR of mutants co‐expressed with PM3A (blue histograms) or PM3FL 456P/Y458H (red histograms) is shown relative to the wild‐type (WT). Asterisks indicate significant deviation (*P < 0.05 and **P < 0.01) from the HR elicited by the wild‐type construct individually for each leaf assay using a two‐tailed Student's t‐test. The error bars indicate ± SEM.
Surprisingly, we identified eight mutations that enhanced the HR (‘enhancing’ alterations) with either PM3A, PM3FL456P/Y458H, or both (H38G, G71N, L83Y, Q112N, V114Q, L116Y, L116I and R124E; Fig. 4a). As a result of the rapid HR responses, these reactions were quantified at 3 d post‐infiltration using fluorescence scanning (Praz et al., 2017; Fig. S5), and we observed differences in the effects of each of the mutations on the cell death response by PM3A and PM3FL456P/Y458H (Fig. 4b). The alterations H38G, G71N and L83Y enhanced the cell death response by both PM3A and PM3FL456P/Y458H by two‐ to three‐fold (Fig. 4b). For the Q112N construct, the increase in HR was stronger with PM3A (five‐fold) than with PM3FL456P/Y458H (three‐fold). The opposite was observed for the V114Q, L116Y, L116I and R124E mutations, where the increase in HR was stronger with PM3FL456P/Y458H than PM3A, and up to 12‐fold higher than the wild‐type for the V114Q, L116Y and R124E alterations (Fig. 4b). It is interesting to note that two positions identified as enhancing also gave different results depending on the residue alteration, where the change from histidine to glycine at position 38 enhanced recognition, but the change to glutamine gave no discernible effect (Fig. 4a). Altering leucine at position 116 to tyrosine enhanced recognition by both PM3A and PM3FL456P/Y458H by 12‐fold, whereas changing it to isoleucine specifically enhanced PM3FL456P/Y458H recognition by six‐fold, and showed no effect on recognition by PM3A.
Previously, it has been shown that the wild‐type AVRPM3A2/F2‐A variant induces only weak and occasional HR when co‐expressed with the natural PM3F in N. benthamiana (Bourras et al., 2015). Therefore, we tested all single‐residue‐altered constructs with PM3F to observe similarly enhanced recognition with the natural allele. We found five mutations that enhanced recognition with PM3A and PM3FL456P/Y458H and also elicited HR when co‐expressed with PM3F to levels similar to the positive control (G71N, L83Y, Q112N, L116Y and R124E; Fig. 5; Fig. 4a, teal boxes). A complete deletion of the region from Q112N to the L116Y mutation resulted in a disruption of recognition by PM3A, PM3F and PM3FL456P/Y458H (Fig. S6). Surprisingly, two mutations which gave a 12‐fold increase in the response by PM3FL456P/Y458H (L116Y and R124E) also elicited HR with PM3F; however, the Q112N alteration that was also recognized by natural PM3F was not so strongly recognized by the modified PM3FL456P/Y458H, suggesting that the effect of these residues was specifically dependent on the L456P and Y458H modifications in the NBS domain of the PM3FL456P/Y458H protein.
Figure 5.
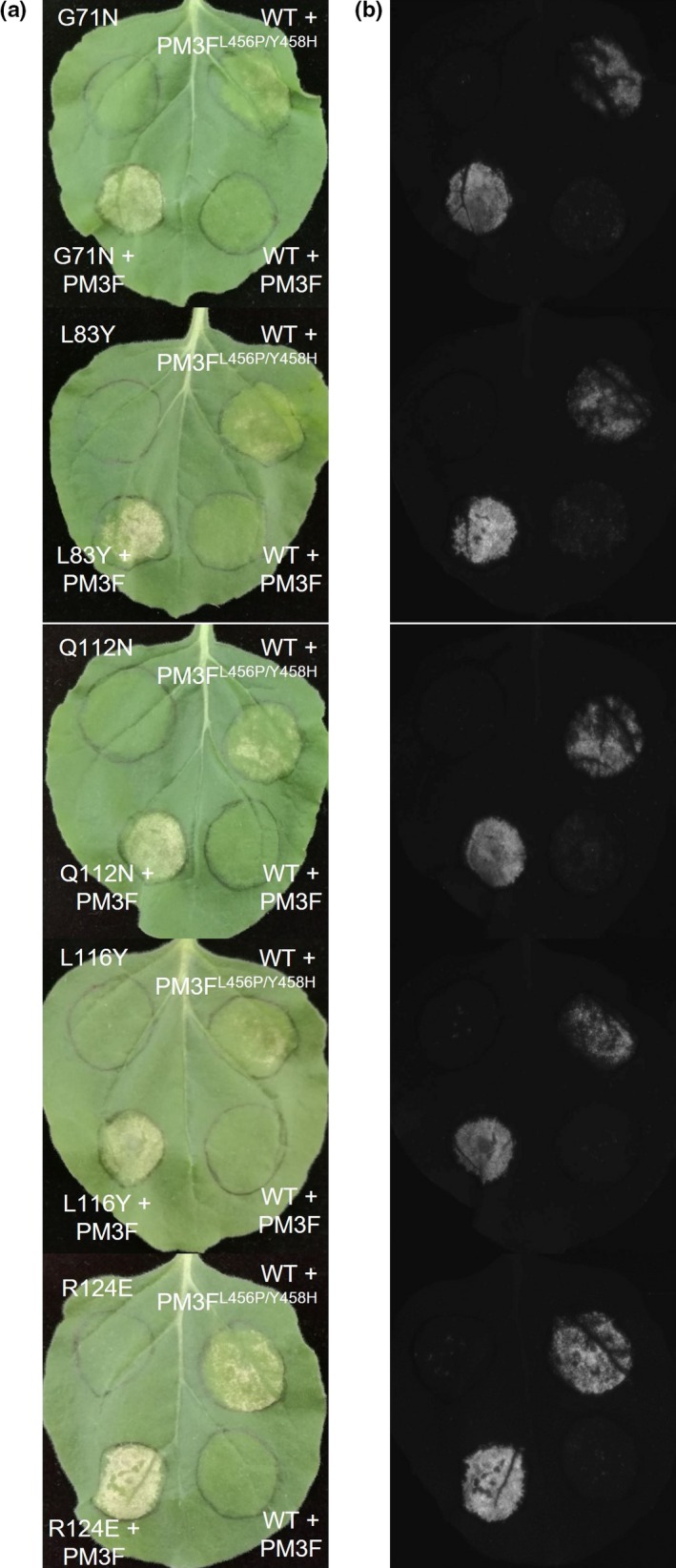
Single‐site mutations in the wheat powdery mildew AVRPM3A2/F2 avirulence protein confer a gain of recognition when co‐infiltrated with the natural PM3F allele. (a) Leaf images taken at 3 d post‐infiltration (dpi) demonstrate the strong hypersensitive response (HR) of the five single‐residue alterations that give enhanced recognition with the natural PM3F allele compared with the wild‐type (WT) AVRPM3A2/F2. (b) Fluorescence imaging of the leaves visualizes the weak HR of the WT + PM3F control.
We conclude that the cell death response following recognition of the AVRPM3A2/F2 effector by PM3A, PM3F and the modified PM3FL456P/Y458H receptor is highly sensitive to single‐residue alterations in AVRPM3A2/F2. We observed differences across the protein, including a neutral region from residues 60 to 75, and a concentration of residue alterations resulting in an enhanced cell death response in the region from residue 112 to 116.
Comparing residues under selection pressure with the effects of single‐residue alterations on recognition by PM3A/F distinguishes putative AVRPM3A2/F2 functional domains
To determine whether selection to evade recognition is a dominant driver of evolution among the AvrPm3 a2/f2 haplotypes, we analyzed the selection acting on the AvrPm3 a2/f2 gene and the AvrPm3 a2/f2 effector family. Among the 13 AvrPm3 a2/f2 haplotype sequences are a 6‐bp insertion and 23 single nucleotide polymorphisms (SNPs), all but one of which are nonsynonymous (Table S8). Excluding the AvrPm3 a2/f2 ‐G haplotype, which is probably the result of a gene conversion event, the 278.4 nonsynonymous and 89.3 synonymous sites represent a significant excess of nonsynonymous substitutions relative to neutral expectation (P = 0.016). Tests for positive selection within the gene family using the Bayes empirical Bayes procedure (Yang et al., 2005) to compare M8a and M8 models (see the Materials and Methods section) identified 11 positions under significant positive selection (Fig. 4a, green boxes). This region of the protein (residues 32–59) shares no overlap with the domains identified by single‐residue alterations as being distinctly neutral (residues 60–75) or enhancing recognition (residues 112–116). The positively selected residues also do not share substantial overlap with natural polymorphisms in AvrPm3 a2/f2 (Fig. 4a, black framed boxes). The single exception in both cases is position 38, where some haplotypes encode a neutral alteration to glutamic acid (Q). The alteration to glycine (G) at this position, which is common within the effector family but was not found among natural AVRPM3A2/F2 variants, caused two‐fold HR enhancement with PM3FL456P/Y458H and three‐fold HR enhancement with PM3A (Fig. 4b).
Thus, we have identified a region under positive selection in the effector family (residues 32–59) and a protein domain where natural polymorphisms are concentrated (residues 86–95). Both regions are distinct from the two functionally identified domains, including the ‘enhancing’ region (residues 112–116), which might constitute an interaction domain. These data indicate that positive selection acting on the AvrPm3 a2/f2 effector family is distinct from the selection pressure driving diversity of AvrPm3 a2/f2 in natural isolates. This suggests that mutations to evade recognition by Pm3a and Pm3f would not necessarily interfere with effector function.
Discussion
Maintenance of a recognized effector at high frequencies in natural populations is possibly caused by the presence of a suppressor
The characterization of the natural diversity of fungal or oomycete avirulence genes has generally been limited to relatively small sample sizes (Allen et al., 2008; Barrett et al., 2009; Kanzaki et al., 2012), or has been primarily marker based (Parlange et al., 2009; Oliva et al., 2015), a strategy which focuses exclusively on known polymorphisms and therefore cannot detect novel diversity. Our genotyping approach of sequencing complete genes enabled us to comprehensively identify the natural diversity of AvrPm3 a2/f2, globally. From this large collection, we could observe that the recognized AVRPM3A2/F2‐A variant is maintained at high frequencies in all regions despite the presence of Pm3a/f wheat in cultivation. This contrasts with studies of AvrLm4‐7 diversity following prolonged exposure of a population of L. maculans to selection pressure from Rlm7 resistance (Daverdin et al., 2012). During the course of 4 yr, the frequency of avirulent AvrLm7 alleles sharply decreased and, accordingly, Rlm7‐mediated resistance was all but defeated (Daverdin et al., 2012). In our study, we observed the recognized AVRPM3A2/F2‐A variant at high frequencies, and many of these isolates also encoded the previously functionally validated variant of the suppressor of recognition, SvrPm3 a1/f1 ‐A (Bourras et al., 2015; Table 4; Fig. S3). The association observed between active expressed suppressor and virulence on Pm3a and Pm3f in these isolates fits with a model in which the quantitative action of SvrPm3 a1/f1 shelters AvrPm3 a2/f2 from recognition by Pm3a and Pm3f.
However, our data also show that there are a considerable number of isolates that defy the three‐component Avr‐R‐Svr model (Table 4). First, we observed 24 isolates that do not encode the recognized AVRPM3A2/F2‐A variant, but that nevertheless exhibit avirulence on Pm3a, Pm3f or both (Table 2). More than one‐half of these isolates (14) encode the AVRPM3A2/F2‐F variant and were originally collected on wild emmer wheat, hinting at the possible association between host diversity at the crop center of origin and pathogen diversity (Ben‐David et al., 2016). They have a genetic background closely related, but distinct from, isolates collected on domestic durum or bread wheat. There are probably additional factors in this genetic background that restrict virulence on the domesticated Pm3a/f wheat cultivars. The other ten exceptions might be explained by the presence of a second avirulence factor in the pathogen, recognized by Pm3a. This AvrPm3 a3 was previously genetically identified in the cross of two European isolates (Bourras et al., 2015), where segregating phenotypes in the progeny implied an additional recognized factor. It is likely that this factor is polymorphic in natural isolates, and leads to avirulent phenotypes in the absence of a recognized AVRPM3A2/F2 variant.
In addition, we observed 16 isolates encoding AVRPM3A2/F2‐A and an SVRPM3A1/F1 variant other than the functionally validated SVRPM3A1/F1‐A suppressor that exhibited some level of virulence on Pm3a, Pm3f or both (Table 4). It is as yet unclear whether alternative variants of SVRPM3A1/F1, such as the ‘B’ variant, function as active suppressors. Follow‐up studies testing the suppressor activity of these variants and expanding expression studies to natural isolates from populations outside of Europe should determine the variability in suppressor activity of sequence‐diverse variants and in natural populations globally.
Information from effector family sequence diversity can inform studies of recognition specificity
Previous mutagenesis approaches to investigate the basis of recognition specificity have focused primarily on limited polymorphisms in the natural diversity (Allen et al., 2008; Parlange et al., 2009; Kanzaki et al., 2012) or random mutagenesis (Leonelli et al., 2011), or have relied on information from already solved protein structures (Chou et al., 2011; Blondeau et al., 2015; Maqbool et al., 2015). To reduce bias in selecting additional residue alterations outside the natural allelic diversity of the AvrPm3 a2/f2 gene, we identified the most common residue at each position within the structurally related, sequence‐divergent family. Comparatively, alanine screens have been widely used to test the role of specific residues in function and recognition (Joosten & de Wit, 1999; Whisson et al., 2007). In theory, alanine screening answers the question of the specific role of an amino acid side‐chain, but the drastic alteration of the biochemical properties of residues is likely to also affect protein structure and stability. By contrast, by selecting common residues within the gene family towards which we altered single residues in the AVRPM3A2/F2 protein, we reduced the chances of disturbing the overall structure of the protein. This is an important aspect to consider in a large‐scale mutant screen in which the measurement of protein stability for each tested construct is not feasible. This is particularly crucial for studies of the AVRPM3A2/F2 protein, as attempts to tag the protein whilst maintaining functionality have thus far been unsuccessful. Ultimately, testing residues common within the effector family, which represents the pool of functional diversity available to the effector, not only reduces the probability of selecting residue alterations that may disrupt overall protein structure, but is also informative with regard to the specificity of the receptor for the AvrPm3 a2/f2 gene.
Utilizing an effector family‐informed mutagenesis approach to study recognition specificity allowed us to identify single‐residue alterations that enhance the HR on recognition by the cognate R protein. To our knowledge, an enhanced HR resulting from alterations in the AVR protein has not been described previously. None of the enhancing residue alterations we identified exist as polymorphisms in the natural diversity of the AvrPm3 a2/f2 gene. Thus, a study limited to the natural sequence diversity, similar to AvrLm4‐7 (Blondeau et al., 2015), AvrL567 (Ravensdale et al., 2012) and ATR1 (Krasileva et al., 2010; Chou et al., 2011), would have found no enhanced recognition effects.
In addition, compared with random mutagenesis, our approach provided clues to which of the shared structural characteristics within the family are important for recognition. It enabled us to identify even conservative residue changes in the AVRPM3A2/F2 protein that affect recognition specificity, and also revealed unforeseen variation in the strength of the immune response by Pm3a and Pm3f. This was exemplified by the single biochemically conservative L116I alteration that resulted in a 10‐fold increase in the HR elicited by the PM3FL456P/Y458H construct. It is possible that disruption and enhancement of the HR may be related to protein stability; however, attempts to tag a functional AVRPM3A2/F2‐A have thus far been unsuccessful. Furthering our understanding of the role of specific residues in recognition will be a useful tool for the potential development of synthetic R genes with enhanced recognition abilities. Future studies of residues in the NLR that interact with the functional residues we have identified in this study might allow us to design a PM3 variant that recognizes the most common features or residues shared by the AvrPm3 a2/f2 family. The development of NLRs with such expanded structural recognition features would be a robust source of resistance in breeding.
Analysis of additional sequence diversity can distinguish effector function domains from sites determining recognition specificity
By characterizing the natural diversity, testing single‐residue alterations for recognition and estimating selection acting on the effector family, we have identified four distinct domains in the AVRPM3A2/F2 protein (Fig. 6b): (1) residues under positive diversifying selection in the effector family (residues 32–59; Fig. 6b, green), which overlap with the highly variable region and the YxC motif conserved in the effector family (Fig. 6a); (2) the neutral domain (residues 60–75; Fig. 6b, dark gray), which also overlaps with the highly variable region; (3) the domain that is most polymorphic in the AvrPm3 a2/f2 natural alleles (Fig. 6b, black frame); and (4) the domain in which residue alterations enhanced and/or altered the specificity of the HR elicited by PM3A/F (residues 112–116; Fig. 6b, orange). Assuming direct recognition of AVRPM3A2/F2 by PM3A/F, the enhancing domain might constitute a putative interaction site. Traditionally, selection pressure from direct recognition by the NLR drives Avr diversification, often at the site of their protein–protein interactions (Ravensdale et al., 2012; Maqbool et al., 2015); however, we observed no naturally occurring polymorphisms at the putative interaction site. We did observe that most of the alterations in the naturally polymorphic domain disrupted recognition, but it is unclear why this region shows a high level of diversification or whether this diversity is related to recognition, as most residue alterations across the protein are sufficient to disrupt recognition (Fig. 4a, black boxes). As both of these domains are distinct from the domain under positive selection in the effector family, we conclude that the latter is probably involved in effector function, perhaps co‐evolving with a target protein in the host.
Figure 6.
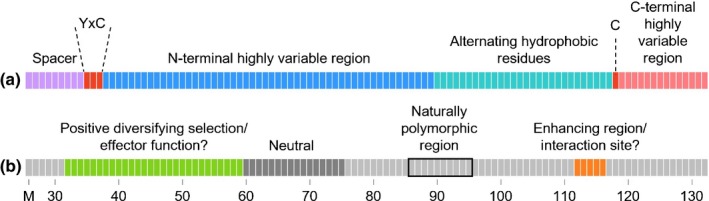
Overview of structural and functional domains of the wheat powdery mildew AVRPM3A2/F2 avirulence protein. Schematic depiction of (a) structural domains conserved among the AVRPM3A2/F2 effector family adapted from Bourras et al. (2016), and (b) the functional domains identified in this study.
In a model of indirect recognition, mutations that enhance effector function would increase the signal of a modified decoy or guardee that is detected by PM3A/F, leading to a stronger HR. Alternatively, if the effector function of AVRPM3A2/F2 involves suppression of host defense responses, and this activity is maintained in the N. benthamiana system, mutations in residues important for this virulence function might reduce the ability of the effector to suppress the HR, but still allow for PM3A/F recognition. This would effectively enhance the visible HR in transient assays. Increased HR could also be the result of a change in the rate of effector protein turnover. Fine tuning of the molecular interaction of an R protein that directly recognizes a cognate AVR or enhancing the indirect recognition of modifications to effector targets are both potential strategies for the engineering of enhanced resistance in this interaction.
Selection pressures other than recognition can also drive Avr allele diversification
Comparing the number of disruptive alterations based on natural diversity (10/16, 63%) with those designed using information from the effector family (50/74, 67.8%), we observed that polymorphisms identified in naturally occurring AVRPM3A2/F2 variants are equally as likely to disturb recognition as a selection of alterations from our screen. This contrasts strongly with the observation that the additive effects of multiple mutations are required to disrupt the direct recognition of AvrL567 by L5 and L6 alleles (Ravensdale et al., 2012). In the evolutionary context, one mutation that abolishes recognition is sufficient to eliminate host resistance as a source of selection pressure. Eight of 12 natural AVRPM3A2/F2 variants contain the G86E mutation, which is sufficient to disrupt recognition. Six of these variants have additional mutations that alone are sufficient to abolish recognition by PM3A/F (Table 1). The G86E polymorphism is therefore more likely to be ancestral than a gain‐of‐virulence mutation. In the remaining three variants (AVRPM3A2/F2‐C, AVRPM3A2/F2‐E and AVRPM3A2/F2‐D), we find three single polymorphisms (F95L, E93D and E93K, respectively) representing putative gain‐of‐virulence mutations. The remaining six natural polymorphisms most likely either existed before Pm3a/f recognition, are the product of random genetic drift or are the result of selection pressure from other sources, such as effector function. This hypothesis is supported by the absence of significant positive selection acting at positions within the putative interaction domain (residues 112–116). We detected, however, significant positive selection at residues in the effector family, which should result from selection for effector function and not recognition. Novel effector functions have been described in Cladosporium fulvum, where both Avr4 and CfEcp6 effectors bind chitin through their lysine (LysM) domains, but only Avr4 has been demonstrated to have the additional function of protecting fungal hyphae from hydrolysis by plant enzymes (van den Burg et al., 2006; de Jonge et al., 2010). All of the mildew isolates tested here encode the AvrPm3 a2/f2 gene, suggesting that loss of the gene might result in fitness costs. In addition, the region experiencing positive diversifying selection in the effector family, characteristic of shared effector function, is not the domain experiencing diversification in the AvrPm3 a2/f2 alleles. We conclude that neither recognition nor shared effector family function, but rather an important novel function that increases pathogen fitness, is potentially the main driver of diversity within the AvrPm3 a2/f2 gene.
Author contributions
K.E.M., S.B. and B.K. designed the study. K.E.M., L.L. and L.K. performed the experiments. K.E.M., F.M. and C.R.P. performed the analyses. K.E.M., C.R.P, M.C.M, R.B‐D., K.C., A.D. and E.M. collected the data. K.E.M., S.B. and B.K. wrote and edited the manuscript. R.B‐D., C.C., M.X., F.Z., S.G. and D.Y. contributed the data.
Supporting information
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Recognition tests for AVRPM3A2/F2 variants.
Fig. S2 Recognition tests for AVRPM3A2/F2‐F.
Fig. S3 Geographic distribution of AVRPM3A2/F2–SVRPM3A1/F1 variant combinations.
Fig. S4 Expression analysis of isolates encoding the active AVRPM3A2/F2‐A variant and a SVRPM3A1/F1 variant with unknown activity.
Fig. S5 Single‐site mutations in AVRPM3A2/F2 that enhance recognition when co‐infiltrated with PM3A and PM3FL456P/Y458H.
Fig. S6 Recognition test of a five‐amino‐acid deletion of the putative enhancing region (residues 112–116) of AVRPM3A2/F2‐A.
Table S1 Primers used for specific amplification of AvrPm3 a2/f2 (pu7) and SvrPm3 a1/f1 (bcg1)
Table S7 Student's t‐test of significant differences in expression
Table S8 Nucleotide polymorphisms in the AvrPm3 a2/f2 haplotypes
Notes S1 Genetic evidence indicating that AvrPm3 a2/f2 is not recognized by Pm1a, Pm2, Pm3b‐e, Pm3g, Pm4a, Pm4b, Pm5a, Pm8 and Pm17.
Table S2 Single‐residue‐altered AVRPM3A2/F2 constructs
Table S3 The worldwide collection of mildew isolates, their AVRPM3A2/F2 and SVRPM3A1/F1 genotypes, and phenotypes on Pm3a/f wheat
Table S4 Phenotypes of progeny from the 96224 × 94202 population on Pm1a, Pm4a and Pm4b wheat
Table S5 Phenotypes of progeny from the 96224 × 94202 population on Pm5a wheat
Table S6 Phenotypes of progeny from the 96224 × 94202 population on Pm17 wheat
Acknowledgements
This work was supported by the Swiss National Science Foundation (grant 310030‐163260). We would like to acknowledge Prof. Dr Kentaro Yoshida (Kobe University, Kobe, Japan), Prof. Dr Tomohiro Ban (Yokohama City University, Yokohama, Japan) and Dr Simon Ellwood (Curtin University, Perth, Australia) for collecting and shipping mildew isolates. We would also like to acknowledge Simon Flückiger and Michael Schläfli for their technical support throughout the revisions.
Contributor Information
Dazhao Yu, Email: dazhaoyu@china.com.
Salim Bourras, Email: s.bourras@botinst.uzh.ch.
Beat Keller, Email: bkeller@botinst.uzh.ch.
References
- Abascal F, Zardoya R, Telford MJ. 2010. TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Research 38: W7–W13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen RL, Meitz JC, Baumber RE, Hall SA, Lee SC, Rose LE, Beynon JL. 2008. Natural variation reveals key amino acids in a downy mildew effector that alters recognition specificity by an Arabidopsis resistance gene. Molecular Plant Pathology 9: 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett LG, Thrall PH, Dodds PN, Van der Merwe M, Linde CC, Lawrence GJ, Burdon JJ. 2009. Diversity and evolution of effector loci in natural populations of the plant pathogen Melampsora lini . Molecular Biology and Evolution 26: 2499–2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben‐David R, Parks R, Dinoor A, Kosman E, Wicker T, Keller B, Cowger C. 2016. Differentiation among Blumeria graminis f. sp. tritici isolates originating from wild versus domesticated Triticum species in Israel. Phytopathology 106: 861–870. [DOI] [PubMed] [Google Scholar]
- Bhullar NK, Zhang Z, Wicker T, Keller B. 2010. Wheat gene bank accessions as a source of new alleles of the powdery mildew resistance gene Pm3: a large scale allele mining project. BMC Plant Biology 10: 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondeau K, Blaise F, Graille M, Kale SD, Linglin J, Ollivier B, Labarde A, Lazar N, Daverdin G, Balesdent MH et al 2015. Crystal structure of the effector AvrLm4–7 of Leptosphaeria maculans reveals insights into its translocation into plant cells and recognition by resistance proteins. Plant Journal 83: 610–624. [DOI] [PubMed] [Google Scholar]
- Bos JI, Armstrong MR, Gilroy EM, Boevink PC, Hein I, Taylor RM, Zhendong T, Engelhardt S, Vetukuri RR, Harrower B et al 2010. Phytophthora infestans effector AVR3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proceedings of the National Academy of Sciences, USA 107: 9909–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourras S, McNally KE, Ben‐David R, Parlange F, Roffler S, Praz CR, Oberhaensli S, Menardo F, Stirnweis D, Frenkel Z et al 2015. Multiple avirulence loci and allele‐specific effector recognition control the Pm3 race‐specific resistance of wheat to powdery mildew. Plant Cell 27: 2991–3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourras S, McNally KE, Müller MC, Wicker T, Keller B. 2016. Avirulence genes in cereal powdery mildews: the gene‐for‐gene hypothesis 2.0. Frontiers Plant Science 7: 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JKM, Wolfe MS. 1990. Structure and evolution of a population of Erysiphe graminis f. sp. hordei . Plant Pathology 39: 376–390. [Google Scholar]
- Brunner S, Hurni S, Streckeisen P, Mayr G, Albrecht M, Yahiaoui N, Keller B. 2010. Intragenic allele pyramiding combines different specificities of wheat Pm3 resistance alleles. Plant Journal 64: 433–445. [DOI] [PubMed] [Google Scholar]
- van den Burg HA, Harrison SJ, Joosten MH, Vervoort J, de Wit PJ. 2006. Cladosporium fulvum Avr4 protects fungal cell walls against hydrolysis by plant chitinases accumulating during infection. Molecular Plant–Microbe Interactions 19: 1420–1430. [DOI] [PubMed] [Google Scholar]
- Chou S, Krasileva KV, Holton JM, Steinbrenner AD, Alber T, Staskawicz BJ. 2011. Hyaloperonospora arabidopsidis ATR1 effector is a repeat protein with distributed recognition surfaces. Proceedings of the National Academy of Sciences, USA 108: 13323–13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowger C, Mehra L, Arellano C, Meyers E, Murphy JP. 2018. Virulence differences in Blumeria graminis f. sp. tritici from the central and eastern United States. Phytopathology. doi: 10.1094/PHYTO-06-17-0211-R. [DOI] [PubMed] [Google Scholar]
- Daverdin G, Rouxel T, Gout L, Aubertot JN, Fudal I, Meyer M, Parlange F, Carpezat J, Balesdent MH. 2012. Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathogens 8: e1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds PN, Lawrence GJ, Catanzariti AM, Teh T, Wang CI, Ayliffe MA, Kobe B, Ellis JG. 2006. Direct protein interaction underlies gene‐for‐gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proceedings of the National Academy of Sciences, USA 103: 8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugdale B, Mortimer CL, Kato M, James TA, Harding RM, Dale JL. 2014. Design and construction of an in‐plant activation cassette for transgene expression and recombinant protein production in plants. Nature Protocols 9: 1010–1027. [DOI] [PubMed] [Google Scholar]
- Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis JG, Lawrence GJ, Luck JE, Dodds PN. 1999. Identification of regions in alleles of the flax rust resistance gene L that determine differences in gene‐for‐gene specificity. Plant Cell 11: 495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor HH. 1971. Current status of the gene‐for‐gene concept. Annual Review of Phytopathology 9: 275–296. [Google Scholar]
- Himmelbach A, Zierold U, Hensel G, Riechen J, Douchkov D, Schweizer P, Kumlehn J. 2007. A set of modular binary vectors for transformation of cereals. Plant Physiology 145: 1192–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YJ, Balesdent MH, Li ZQ, Evans N, Rouxel T, Fitt BD. 2010. Fitness cost of virulence differs between the AvrLm1 and AvrLm4 loci in Leptosphaeria maculans (phoma stem canker of oilseed rape). European Journal of Plant Pathology 126: 279. [Google Scholar]
- Huang YJ, Li ZQ, Evans N, Rouxel T, Fitt BD, Balesdent MH. 2006. Fitness cost associated with loss of the AvrLm4 avirulence function in Leptosphaeria maculans (phoma stem canker of oilseed rape). European Journal of Plant Pathology 114: 77–89. [Google Scholar]
- de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MH, Thomma BP. 2010. Conserved fungal LysM effector Ecp6 prevents chitin‐triggered immunity in plants. Science 329: 953–955. [DOI] [PubMed] [Google Scholar]
- Joosten MH, de Wit PJ. 1999. The tomato–Cladosporium fulvum interaction: a versatile experimental system to study plant–pathogen interactions. Annual Review of Phytopathology 37: 335–367. [DOI] [PubMed] [Google Scholar]
- Kanzaki H, Yoshida K, Saitoh H, Fujisaki K, Hirabuchi A, Alaux L, Fournier E, Tharreau D, Terauchi R. 2012. Arms race co‐evolution of Magnaporthe oryzae AVR‐Pik and rice Pik genes driven by their physical interactions. Plant Journal 72: 894–907. [DOI] [PubMed] [Google Scholar]
- Koeck M, Hardham AR, Dodds PN. 2011. The role of effectors of biotrophic and hemibiotrophic fungi in infection. Cellular Microbiology 13: 1849–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasileva KV, Dahlbeck D, Staskawicz BJ. 2010. Activation of an Arabidopsis resistance protein is specified by the in planta association of its leucine‐rich repeat domain with the cognate oomycete effector. Plant Cell 22: 2444–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Stecher G, Tamura K. 2016. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Molecular Biology and Evolution 33: 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leigh JW, Bryant D. 2015. Popart: full‐feature software for haplotype network construction. Methods in Ecology and Evolution 6: 1110–1116. [Google Scholar]
- Leonelli L, Pelton J, Schoeffler A, Dahlbeck D, Berger J, Wemmer DE, Staskawicz B. 2011. Structural elucidation and functional characterization of the Hyaloperonospora arabidopsidis effector protein ATR13 . PLoS Pathogens 7: e1002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti L, Lanver D, Schweizer G, Tanaka S, Liang L, Tollot M, Zuccaro A, Reissmann S, Kahmann R. 2015. Fungal effectors and plant susceptibility. Annual Review of Plant Biology 66: 513–545. [DOI] [PubMed] [Google Scholar]
- Lu X, Kracher B, Saur IM, Bauer S, Ellwood SR, Wise R, Yaeno T, Maekawa T, Schulze‐Lefert P. 2016. Allelic barley MLA immune receptors recognize sequence‐unrelated avirulence effectors of the powdery mildew pathogen. Proceedings of the National Academy of Sciences, USA 4: E6486–E6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Lukasik E, Gawehns F, Takken FL. 2012. The use of agroinfiltration for transient expression of plant resistance and fungal effector proteins in Nicotiana benthamiana leaves In: Bolton MD, Thomma BPHJ, eds. Plant fungal pathogens: methods and protocols. New York, NY, USA: Humana Press, 61–74. [DOI] [PubMed] [Google Scholar]
- Maqbool A, Saitoh H, Franceschetti M, Stevenson CEM, Uemura A, Kanzaki H, Kamoun S, Terauchi R, Banfield MJ. 2015. Structural basis of pathogen recognition by an integrated HMA domain in a plant NLR immune receptor. eLife 4: e08709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva RF, Cano LM, Raffaele S, Win J, Bozkurt TO, Belhaj K, Oh SK, Thines M, Kamoun S. 2015. A recent expansion of the RXLR effector gene Avrblb2 is maintained in global populations of Phytophthora infestans indicating different contributions to virulence. Molecular Plant–Microbe Interactions 28: 901–912. [DOI] [PubMed] [Google Scholar]
- Parlange F, Daverdin G, Fudal I, Kuhn ML, Balesdent MH, Blaise F, Grezes‐Besset B, Rouxel T. 2009. Leptosphaeria maculans avirulence gene AvrLm4‐7 confers a dual recognition specificity by the Rlm4 and Rlm7 resistance genes of oilseed rape, and circumvents Rlm4‐mediated recognition through a single amino acid change. Molecular Microbiology 71: 851–863. [DOI] [PubMed] [Google Scholar]
- Parlange F, Oberhaensli S, Breen J, Platzer M, Taudien S, Šimková H, Wicker T, Doležel J, Keller B. 2011. A major invasion of transposable elements accounts for the large size of the Blumeria graminis f. sp. tritici genome. Functional & Integrative Genomics 11: 671–677. [DOI] [PubMed] [Google Scholar]
- Parlange F, Roffler S, Menardo F, Ben‐David R, Bourras S, McNally KE, Oberhaensli S, Stirnweis D, Buchmann G, Wicker T et al 2015. Genetic and molecular characterization of a locus involved in avirulence of Blumeria graminis f. sp. tritici on wheat Pm3 resistance alleles. Fungal Genetics and Biology 82: 181–192. [DOI] [PubMed] [Google Scholar]
- Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21: 676–679. [DOI] [PubMed] [Google Scholar]
- Praz CR, Bourras S, Zeng F, Sánchez‐Martín J, Menardo F, Xue M, Yang L, Roffler S, Böni R, Herren G et al 2017. AvrPm2 encodes an RNase‐like avirulence effector which is conserved in the two different specialized forms of wheat and rye powdery mildew fungus. New Phytologist 213: 1301–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi D, Innes RW. 2013. Recent advances in plant NLR structure, function, localization, and signaling. Frontiers in Immunology 4: 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravensdale M, Bernoux M, Ve T, Kobe B, Thrall PH, Ellis JG, Dodds PN. 2012. Intramolecular interaction influences binding of the Flax L5 and L6 resistance proteins to their AvrL567 ligands. PLoS Pathogens 8: e1003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose LE, Bittner‐Eddy PD, Langley CH, Holub EB, Michelmore RW, Beynon JL. 2004. The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana . Genetics 166: 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9: 671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeholzer S, Tsuchimatsu T, Jordan T, Bieri S, Pajonk S, Yang W, Jahoor A, Shimizu KK, Keller B, Schulze‐Lefert P. 2010. Diversity at the Mla powdery mildew resistance locus from cultivated barley reveals sites of positive selection. Molecular Plant–Microbe Interactions 23: 497–509. [DOI] [PubMed] [Google Scholar]
- Srichumpa P, Brunner S, Keller B, Yahiaoui N. 2005. Allelic series of four powdery mildew resistance genes at the Pm3 locus in hexaploid bread wheat. Plant Physiology 139: 885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnweis D, Milani SD, Jordan T, Keller B, Brunner S. 2014. Substitutions of two amino acids in the nucleotide‐binding site domain of a resistance protein enhance the hypersensitive response and enlarge the PM3F resistance spectrum in wheat. Molecular Plant–Microbe Interactions 27: 265–276. [DOI] [PubMed] [Google Scholar]
- Whisson SC, Boevink PC, Moleleki L, Avrova AO, Morales JG, Gilroy EM, Armstrong MR, Grouffaud S, Van West P, Chapman S et al 2007. A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature 450: 115–118. [DOI] [PubMed] [Google Scholar]
- Win J, Krasileva KV, Kamoun S, Shirasu K, Staskawicz BJ, Banfield MJ. 2012. Sequence divergent RXLR effectors share a structural fold conserved across plant pathogenic oomycete species. PLoS Pathogens 8(1): e1002400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahiaoui N, Srichumpa P, Dudler R, Keller B. 2004. Genome analysis at different ploidy levels allows cloning of the powdery mildew resistance gene Pm3b from hexaploid wheat. Plant Journal 37: 528–538. [DOI] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Molecular Biology and Evolution 24: 1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R. 2000. Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Molecular Biology and Evolution 17: 32–43. [DOI] [PubMed] [Google Scholar]
- Yang Z, Wong WS, Nielsen R. 2005. Bayes empirical Bayes inference of amino acid sites under positive selection. Molecular Biology and Evolution 22: 1107–1118. [DOI] [PubMed] [Google Scholar]
- Zeng FS, Yang LJ, Gong SJ, Shi WQ, Zhang XJ, Wang H, Xiang LB, Xue MF, Yu DZ. 2014. Virulence and diversity of Blumeria graminis f. sp. tritici populations in China. Journal of Integrative Agriculture 13: 2424–2437. [Google Scholar]
- Zipfel C. 2014. Plant pattern‐recognition receptors. Trends in Immunology 35: 345–351. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Please note: Wiley Blackwell are not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Fig. S1 Recognition tests for AVRPM3A2/F2 variants.
Fig. S2 Recognition tests for AVRPM3A2/F2‐F.
Fig. S3 Geographic distribution of AVRPM3A2/F2–SVRPM3A1/F1 variant combinations.
Fig. S4 Expression analysis of isolates encoding the active AVRPM3A2/F2‐A variant and a SVRPM3A1/F1 variant with unknown activity.
Fig. S5 Single‐site mutations in AVRPM3A2/F2 that enhance recognition when co‐infiltrated with PM3A and PM3FL456P/Y458H.
Fig. S6 Recognition test of a five‐amino‐acid deletion of the putative enhancing region (residues 112–116) of AVRPM3A2/F2‐A.
Table S1 Primers used for specific amplification of AvrPm3 a2/f2 (pu7) and SvrPm3 a1/f1 (bcg1)
Table S7 Student's t‐test of significant differences in expression
Table S8 Nucleotide polymorphisms in the AvrPm3 a2/f2 haplotypes
Notes S1 Genetic evidence indicating that AvrPm3 a2/f2 is not recognized by Pm1a, Pm2, Pm3b‐e, Pm3g, Pm4a, Pm4b, Pm5a, Pm8 and Pm17.
Table S2 Single‐residue‐altered AVRPM3A2/F2 constructs
Table S3 The worldwide collection of mildew isolates, their AVRPM3A2/F2 and SVRPM3A1/F1 genotypes, and phenotypes on Pm3a/f wheat
Table S4 Phenotypes of progeny from the 96224 × 94202 population on Pm1a, Pm4a and Pm4b wheat
Table S5 Phenotypes of progeny from the 96224 × 94202 population on Pm5a wheat
Table S6 Phenotypes of progeny from the 96224 × 94202 population on Pm17 wheat