

Abstract
Simultaneous analysis of multiple genes using next‐generation sequencing (NGS) technology has become widely available. Copy‐number variations (CNVs) in disease‐associated genes have emerged as a cause for several hereditary disorders. CNVs are, however, not routinely detected using NGS analysis. The aim of this study was to assess the diagnostic yield and the prevalence of CNVs using our panel of Hereditary Thoracic Aortic Disease (H‐TAD)‐associated genes. Eight hundred ten patients suspected of H‐TAD were analyzed by targeted NGS analysis of 21 H‐TAD associated genes. In addition, the eXome hidden Markov model (XHMM; an algorithm to identify CNVs in targeted NGS data) was used to detect CNVs in these genes. A pathogenic or likely pathogenic variant was found in 66 of 810 patients (8.1%). Of these 66 pathogenic or likely pathogenic variants, six (9.1%) were CNVs not detectable by routine NGS analysis. These CNVs were four intragenic (multi‐)exon deletions in MYLK, TGFB2, SMAD3, and PRKG1, respectively. In addition, a large duplication including NOTCH1 and a large deletion encompassing SCARF2 were detected. As confirmed by additional analyses, both CNVs indicated larger chromosomal abnormalities, which could explain the phenotype in both patients. Given the clinical relevance of the identification of a genetic cause, CNV analysis using a method such as XHMM should be incorporated into the clinical diagnostic care for H‐TAD patients.
Keywords: copy‐number variations, eXome hidden Markov model, genetics, thoracic aortic aneurysm, thoracic aortic dissection
1. BACKGROUND
Over the last decade, advances in clinical genetics have led to the identification of disease‐associated genes at a rapid pace. Especially when surveillance, early detection, and/or treatment provide health benefits for the index patient and at‐risk relatives, identification of an underlying genetic cause is highly relevant. Therefore, recommendations for genetic counseling and DNA testing are increasingly being incorporated into clinical guidelines (Ackerman et al., 2011; Eccles et al., 2016). Thoracic aortic aneurysms and aortic dissections (TAAD) are a significant cause of sudden death at young age and is an example of a disease where screening of at‐risk relatives can be lifesaving (Hoyert, Arias, Smith, Murphy, & Kochanek, 2001; Olsson, Thelin, Stahle, Ekbom, & Granath, 2006). Because aortic aneurysms are often asymptomatic and aortic dissections are often fatal and preventable by timely surgical intervention, the identification and clinical screening of at‐risk relatives are clinically highly relevant and recommended (Hiratzka et al., 2010). In the majority of cases, TAAD is a sporadic occurrence, associated with, among others, hypertension, bicuspid aortic valve, and older age. However, in approximately 20% of cases TAAD is reported to be familial (FTAAD), often with an autosomal dominant pattern of inheritance with incomplete penetrance (Biddinger, Rocklin, Coselli, & Milewicz, 1997; Coady et al., 1999; Robertson et al., 2016). TAAD that is caused by a pathogenic variant in one of the disease‐associated genes (Hereditary Thoracic Aortic Disease (H‐TAD)) can be subdivided in nonsyndromic and syndromic aortic disease. The phenotypic manifestations of both syndromic and nonsyndromic H‐TAD are highly variable, both within and between families. Syndromic H‐TAD is only diagnosed in a minority of cases and includes, among others, Marfan syndrome (MIM# 154700), Loeys–Dietz syndrome (MIM# 609192, MIM# 610168, MIM# 613795, MIM# 614816, and MIM# 615582), and vascular Ehlers–Danlos syndrome (MIM# 130050). The genes most frequently associated with nonsyndromic H‐TAD are involved in smooth‐muscle cell function (ACTA2, MIM# 611788, MYH11, MIM# 132900, and MYLK, MIM# 613780). Of note, variants in genes originally associated with syndromic H‐TAD have also been reported in patients presenting with apparently nonsyndromic H‐TAD (Gago‐Diaz et al., 2014; Regalado et al., 2011, 2016). Given the incomplete penetrance and the highly variable age of onset within both heritable and sporadic TAAD (Campens et al., 2015; Coady et al., 1999; Khalique et al., 2009; Robertson et al., 2016), follow‐up of at‐risk relatives with normal aortic diameters at initial cardiologic screening is important. The identification of a pathogenic variant in a TAAD patient allows for targeted screening of relatives and enables prenatal and preimplantation genetic diagnosis. In addition, specific recommendations on imaging, surgical, and pharmacological treatment based on the underlying genetic cause are emerging (den Hartog et al., 2016; Franken et al., 2015; D. Milewicz et al., 2016). A causative variant can be identified in approximately 20% of FTAAD families (D. M. Milewicz, Regalado, Shendure, Nickerson, & Guo, 2014). Next‐generation sequencing (NGS) allows for the rapid analysis of multiple genes in a diagnostic setting at relatively low costs. Therefore, DNA testing is increasingly offered to TAAD patients. The majority of the detected variants are single‐nucleotide changes. CNVs have emerged as a relevant cause for several genetic disorders including cancer, intellectual disability, and neuropsychiatric disorders (Pollack et al., 2002; Shlien & Malkin, 2010; Thapar & Cooper, 2013). Routine diagnostic variant‐calling analysis by (short reads‐)NGS technology is not suitable for detecting CNVs. Therefore, CNVs may be missed unless additional testing is performed, for example, by multiplex ligation‐dependent probe amplification (MLPA) or targeted array analysis. However, these tests are often not routinely performed and/or do not include all the relevant genes. The detection of CNVs in NGS sequencing data using statistical and computational tools is an alternative approach. The eXome hidden Markov model (XHMM) is one of several algorithms developed for the detection of CNVs through NGS data (Fromer & Purcell, 2014; Fromer et al., 2012). XHMM has identified (potential) causative CNVs in, for example, patients with Parkinson's disease, autism spectrum disorders, and rare diseases like Joubert syndrome and very early onset inflammatory bowel disease (Kelsen et al., 2015; Koyama et al., 2017; Poultney et al., 2013; Spataro et al., 2017). The aim of this study was to assess both the diagnostic yield of our panel of H‐TAD‐associated genes and the prevalence of CNVs in these genes. Here, we present the results of routine NGS analysis (variant‐calling analysis) and XHMM analysis on the NGS sequencing data of the largest series of TAAD patients described so far (n = 810) referred for analyses of the H‐TAD panel. In addition, we provide an overview of the clinical data of patients with a pathogenic or likely pathogenic variant, with a special focus on patients with CNVs. The results of this study underline the importance of CNV analysis in routine diagnostic testing in patients with H‐TAD.
2. METHODS
2.1. Genetic data
DNA diagnostics was performed at the Department of Clinical Genetics at the VU University Medical Center (VUmc, Amsterdam, the Netherlands) from March 2015 to June 2017. The routine NGS panel included ACTA2, COL3A1, EFEMP2, ELN, FBN1, FBN2, MYH11, MYLK, NOTCH1, PLOD1, PRKG1, SCARF2, SKI, SLC2A10, SMAD2, SMAD3, SMAD4, TGFB2, TGFB3, TGFBR1, and TGFBR2. Since October 2016, the BGN gene was added to the panel (analyzed in 166 patients), while SCARF2, which was not associated with TAD but had previously been selected in view of a possible differential diagnosis ‘Congenital contractural arachnodactyly’ and ‘Van den Ende–Gupta syndrome,’ was excluded from routine analysis. The previously described bioinformatics read‐depth‐based tool XHMM was used for CNV detection in the NGS sequencing data. CNV confirmation was performed using either a home‐made MLPA test, in combination with the P300 or the P200 MLPA kit of MRC Holland, or an SNP array. Detailed information on the analyzed genes and applied methodologies are available in the Supporting Materials and Methods.
2.2. Clinical data
Informed consent for NGS gene panel analysis was obtained from all 810 patients after genetic counseling by the referring physician. The main reasons for analysis of this gene panel include familial or early onset aortic aneurysms or dissections or signs of generalized connective tissue disorders. The majority of patients was referred by a clinical geneticist who frequently participated in a multidisciplinary team specialized in connective tissue disorders. A standardized survey was sent to the referring physicians in order to collect the medical data of patients carrying an identified genetic variant (including ophthalmologic and cardiologic findings, family history, and physical examination). Written informed consent was obtained from the patients and/or their parents with an aberration detected by XHMM, as more detailed medical data were published. Under Dutch law, assessment of the study protocol by our ethics committee was not indicated because only genetic and clinical data collected during regular patient care were used.
3. RESULTS
A pathogenic or likely pathogenic variant in an H‐TAD‐associated gene was identified in 66 of 810 index patients (8.1%). Of these, 60 (90.9%) were identified using routine NGS panel analysis (variant‐calling analysis). In the other six cases (9.1%), a pathogenic or likely pathogenic CNV was detected using XHMM. In 84 patients (10.4%), only variants of unknown significance (VUS) were identified. No pathogenic or likely pathogenic variants and/or VUS were identified in 660 patients (81.5%). The mean age at DNA diagnostics of index patients with a pathogenic or likely pathogenic variant was 35 years (median 36, range 0–77). The mean age of the remaining patients was 46 years (median 49, range 0–78). There was a male preponderance in index patients with a pathogenic or likely pathogenic variant, VUS, or without a VUS or pathogenic variant (68%, 64%, and 67%, respectively).
3.1. Genetic and clinical data in patients with variants identified by variant‐calling analysis
Table 1 provides an overview of the molecular data of the 60 pathogenic or likely pathogenic variants identified by variant‐calling analysis. Of these variants, 37 (62%) have not been described previously and all of them were unique. Heterozygous pathogenic or likely pathogenic variants were identified in FBN1 (N = 18, 30%), ACTA2 (N = 8, 13.3%), SMAD3 (N = 7, 11.7%), COL3A1 (N = 6, 10%), TGFB2 (N = 4, 6.7%), TGFBR1 (N = 3, 5%), TGFBR2 (N = 3, 5%), FBN2 (N = 3, 5%), MYH11 (N = 2, 3.3%), TGFB3 (N = 2, 3.3%), PRKG1 (N = 1, 1.7%), and NOTCH1 (N = 1, 1.7%). Homozygous pathogenic SLC2A10 variants were identified in two patients (3.3%). No (likely) pathogenic variants were found in BGN, EFEMP2, ELN, PLOD1, SKI, SMAD2, and SMAD4. In addition, 90 VUS were identified (patients 9, 52, 67–150; Table 1 and Supporting Information Table S1). In six patients (patients 9 and 52 in Table 1 and Supporting Information Table S1; and patients 69, 75, 90, and 127 in Supporting Information Table S1), two VUS (in different genes) were identified. An overview of the clinical data of all 60 patients with a pathogenic or likely pathogenic variant identified by variant‐calling analysis is provided in Table 2. The clinical data of patients 67–150 with a VUS are available in Supporting Information Table S2.
Table 1.
Summary of the genetic features of patients with a pathogenic or likely pathogenic variant detected by variant‐calling analysis of 21 H‐TAD genes
| Patient | Gene | Nucleotide change | Protein change | Effect | Domain | Conservation | SIFT/MutationTaster/Polyphen‐2/Grantham distance | MAF ExAC | Segregation analysisa | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | ACTA2 | c.115C > T | p.(Arg39Cys) | Missense | Actin | Baker's yeast b | + / + / − / 180 | Absent | yes | ( Hoffjan et al., 2011 ) |
| 2 | ACTA2 | c.116G > A | p.(Arg39His) | Missense | Actin | Baker's yeast b | + / + / − / 29 | Absent | yes | ( Guo et al., 2009 ) |
| 3 | ACTA2 | c.179C > A | p.(Ala60Glu) | Missense | Actin | Baker's yeastb | + / + / + / 107 | Absent | n.a. | Novel |
| 4 | ACTA2 | c.419C > T | p.(Ala140Val) | Missense | Actin | Baker's yeastb | + / + / ±/ 64 | Absent | yes | (Lerner‐Ellis et al., 2014) |
| 5c | ACTA2 | c.445C > T | p.(Arg149Cys) | Missense | Actin | Baker's yeast b | + / + / + / 180 | Absent | yes | ( Guo et al., 2007 ) |
| 6 | ACTA2 | c.835A > G | p.(Thr279Ala) | Missense | Actin | Baker's yeastb | + / + / − / 58 | Absent | n.a. | Novel |
| 7 | ACTA2 | c.854T > C | p.(Met285Thr) | Missense | Actin | Baker's yeastb | + / + / ± / 81 | Absent | n.a. | Novel |
| 8 | ACTA2 | c.1120C > T | p.(Arg374Cys) | Missense | Actin | C. elegans (FCUT Baker's yeast) | + / + / − / 180 | 1 / 121346 | n.a. | Novel |
| 9d | COL3A1 | c.318_325del | p.(Pro107Argfs*13) | Frameshift (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 10 | COL3A1 | c.555del | p.(Gly186Valfs*36) | Frameshift (NMD expected) | NA | NA | NA | Absent | yes, incomplete penetrance | ( Pepin et al., 2014 ; Schwarze et al., 2001 ) |
| 11 | COL3A1 | c.971G > A | p.(Gly324Asp) | Missense | Triple helix | Chicken b | + / + / ± / 94 | Absent | de novo e | Novel |
| 12 | COL3A1 | c.2050G > A | p.(Gly684Arg) | Missense | Triple helix | Chicken b | + / + / + / 125 | Absent | yes | Novel |
| 13 | COL3A1 | c.3219_3222dup | p.(Ala1075Trpfs*20) | Frameshift (NMD expected) | NA | NA | NA | Absent | Maternally inherited | Novel |
| 14 | COL3A1 | c.3446G > A | p.(Gly1149Asp) | Missense | Triple helix | Chicken b | + / + / + / 94 | Absent | n.a. | ( Frank et al., 2015 ) |
| 15 | FBN1 | c.32T > G | p.(Leu11Arg) | Missense | Signal peptide | Dogb | + / + / + / 102 | Absent | n.a. | (Baetens et al., 2011) |
| 16 | FBN1 | c.439C > T | p.(Gln147*) | Nonsense (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 17 | FBN1 | c.986dup | p.(Asp330Argfs*18) | Frameshift (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 18 | FBN1 | c.2177A > G | p.(Glu726Gly) | Missense | EGF‐like 11 | Tetraodon b | + / + / + / 98 | Absent | n.a. | ( Stheneur et al., 2009 ) |
| 19 | FBN1 | c.2645C > T | p.(Ala882Val) | Missense | TB 4 | Tetraodonb | + / + / + / 64 | Absent | n.a. | (Aragon‐Martin et al., 2010; Comeglio et al., 2007; Howarth, Yearwood, & Harvey, 2007; Hung et al., 2009; B. Loeys et al., 2004; Robinson et al., 2012) |
| 20 | FBN1 | c.2660G > A | p.(Cys887Tyr) | Missense | TB 4 | Tetraodon b | + / + / + / 194 | Absent | n.a. | Novel |
| 21 | FBN1 | c.2668T > C | p.(Cys890Arg) | Missense | TB 4 | Tetraodon b | + / + / + / 180 | Absent | n.a. | ( Collod‐Beroud et al., 2003 ; Kielty, Rantamaki, Child, Shuttleworth, & Peltonen, 1995 ) |
| 22 | FBN1 | c.2953G > A | p.(Gly985Arg) | Missense | TB 5 | Tetraodon b | + / + / + / 125 | Absent | n.a. | ( Faivre et al., 2009 ; Howarth et al., 2007 ; B. Loeys, Nuytinck, Delvaux, De Bie, & De Paepe, 2001 ; Rommel et al., 2005 ; Turner et al., 2009 ; Yoo et al., 2010 ) |
| 23 | FBN1 | c.3152T > G | p.(Phe1051Cys) | Missense | EGF‐like 15 | Tetraodon b | + / + / + / 205 | Absent | n.a. | Novel |
| 24 | FBN1 | c.3373C > T | p.(Arg1125*) | Nonsense (NMD expected) | NA | NA | NA | Absent | yes | ( Attanasio et al., 2008 ; Comeglio et al., 2007 ; Hung et al., 2009 ; Magyar et al., 2009 ; Rommel et al., 2005 ; Sheikhzadeh et al., 2012 ; Stheneur et al., 2009 ) |
| 25 | FBN1 | c.4987T > C | p.(Cys1663Arg) | Missense | EGF‐like 28 | Zebrafish b | + / + / + / 180 | Absent | n.a. | ( Dietz, Saraiva, Pyeritz, Cutting, & Francomano, 1992 ; Stheneur et al., 2009 ; Yoo et al., 2010 ) |
| 26 | FBN1 | c.5015del | p.(Cys1672Leufs*10) | Frameshift (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 27 | FBN1 | c.5699G > C | p.(Cys1900Ser) | Missense | EGF‐like 32 | Zebrafish b | + / + / + / 112 | Absent | n.a. | ( Stheneur et al., 2009 ) |
| 28 | FBN1 | c.6031T > C | p.(Cys2011Arg) | Missense | EGF‐like 34 | Zebrafish b | + / + / + / 180 | Absent | de novo e | Novel |
| 29 | FBN1 | c.6942C > G | p.(Tyr2314*) | Nonsense (NMD expected) | NA | NA | NA | Absent | de novo e | Novel |
| 30 | FBN1 | c.7708G > A | p.(Glu2570Lys) | Missense | EGF‐like 45 | Tetraodonb | + / + / + / 56 | Absent | n.a. | (Arbustini et al., 2005; Attanasio et al., 2008; Soylen et al., 2009) |
| 31 | FBN1 | c.8188C > T | p.(Arg2730Trp) | Missense | C‐terminal domain | Tetraodonb | + / + / + / 101 | Absent | n.a. | Novel |
| 32 | FBN1 | c.8578_8579dup | p.(Asp2860Glufs*4) | Frameshift (NMD not expected) | Asprosin chain | NA | NA | Absent | n.a. | Novel |
| 33 | FBN2 | c.3812G > C | p.(Gly1271Ala) | Missense | EGF‐like 19 | Chickenb | + / + / + / 60 | Absent | n.a. | (Buchan et al., 2014) |
| 34 | FBN2 | c.3889G > A | p.(Gly1297Ser) | Missense | EGF‐like 20 | Chickenb | + / + / + / 56 | 2 / 121372 | Paternally inherited | Novel |
| 35 | FBN2 | c.7526_7527del | p.0 | Frameshift (NMD confirmed) | NA | NA | NA | Absent | n.a. | Novel |
| 36 | MYH11 | c.3315‐5G > A | p.? | Splice (NMD not expected) | Coiled coil region | NA | NA | Absent | n.a. | Novel |
| 37 | MYH11 | c.5293C > T | p.(Arg1765Trp) | Missense | Coiled coil region | Zebrafishb | + / + / + / 101 | 1 / 115948 | n.a. | Novel |
| 38 | NOTCH1 |
|
p.(Tyr708Cys) | Missense | EGF‐like 18 | Tetraodon (FCUT Fruitfly) | + / + / + / 194 | Absent | de novo (inferred) | Novel |
| 39 | PRKG1 | c.530G > A | p.(Arg177Gln) | Missense | cGMP‐binding, high affinity | C. elegans b | − / + / + / 43 | Absent | n.a. | ( Guo et al., 2013 ) |
| 40 | SLC2A10 | c.510G > A f | p.(Trp170*) | Nonsense (NMD expected) | NA | NA | NA | Absent | n.a. (consaguineous parents) | ( Coucke et al., 2006 ; Moceri et al., 2013 ) |
| 41 | SLC2A10 | c.1276G > T f | p.(Gly426Trp) | Missense | Transmembrane helical region 10 | Tetraodon b | + / + / + / 184 | 3 / 116638 | confirmed parental carriership | ( Callewaert et al., 2008 ) |
| 42 | SMAD3 | c.1A > T | p.(Met1?) | Loss of initiation codon | Initiator methionine | C. elegans b | NA | Absent | n.a. | Novel |
| 43 | SMAD3 | c.391_394dup | p.(Thr132Argfs*35) | Frameshift (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 44 | SMAD3 | c.492dup | p.(Asn165*) | Frameshift (NMD expected) | NA | NA | NA | Absent | Yes | Novel |
| 45 | SMAD3 | c.802C > T | p.(Arg268Cys) | Missense | MH2 | C. elegans b | + / + / + / 180 | Absent | Yes | Novel |
| 46 | SMAD3 | c.893A > G | p.(Tyr298Cys) | Missense | MH2 | Fruitfly | − / + / + / 194 | Absent | Yes | Novel |
| 47 | SMAD3 | c.1010‐2A > G | p.? |
|
MH2 | NA | NA | Absent | n.a. | Novel |
| 48 | SMAD3 | c.1179dup | p.(Cys394Leufs*4) | Frameshift (NMD not expected) | MH2 | NA | NA | Absent | Yes | ( Aubart et al., 2014 ) |
| 49 | TGFB2 | c.709G > T | p.(Glu237*) | Nonsense (NMD expected) | NA | NA | NA | Absent | n.a. | Novel |
| 50 | TGFB2 | c.979C > T | p.(Arg327Trp) | Missense | Transforming growth factor beta‐2 chain | Frog | + / + / + / 101 | Absent | n.a. | (Lindsay et al., 2012; Schubert, Landis, Shikany, Hinton, & Ware, 2016) |
| 51 | TGFB2 | c.989G > A | p.(Arg330His) | Missense | Transforming growth factor beta‐2 chain | Tetraodon | + / + / + / 29 | Absent | Incomplete penetrance? | Novel |
| 52 d | TGFB2 | c.1017‐1G > T | p.? | Splice (NMD possible) | Transforming growth factor beta‐2 chain | NA | NA | Absent | de novo | Novel |
| 53 | TGFB3 | c.899G > A | p.(Arg300Gln) | Missense | Latency‐associated peptide chain | Fruitfly b | + / + / + / 43 | Absent | Yes | (Matyas, Naef, Tollens, & Oexle, 2014 ) |
| 54 | TGFB3 | c.1075A > C | p.(Ser359Arg) | Missense | Transforming growth factor beta‐3 chain | Fruitflyb | + / + / + / 110 | Absent | n.a. | Novel |
| 55 | TGFBR1 | c.790G > A | p.(Ala264Thr) | Missense | Protein kinase | Fruitflyb | + / + / + / 58 | Absent | yes (incomplete penetrance) | Novel |
| 56 | TGFBR1 | c.1255+2T > C | p.[Tyr378Asnfs*3, 0] | Splice (exon 7 skipping partially stable at RNA level) | Protein kinase | NA | NA | Absent | yes | Novel |
| 57 | TGFBR1 | c.1460G > A | p.(Arg487Gln) | Missense | Protein kinase | Fruitfly b | + / + / + / 43 | Absent | de novo e | (Akutsu et al., 2007 ; Jondeau et al., 2016 ; B. L. Loeys et al., 2006 ; Matyas et al., 2006 ; Melenovsky et al., 2008 ; Yang et al., 2012 ) |
| 58 | TGFBR2 | c.1565G > A | p.(Arg522Gln) | Missense | Protein kinase | Zebrafishb | + / + / + / 43 | 1 / 121046 | Paternally inherited | Novel |
| 59 | TGFBR2 | c.1630G > T | p.(Glu544*) | Nonsense (NMD not expected) | Protein kinase | NA | NA | Absent | n.a. | Novel |
| 60 | TGFBR2 | c.1669C > T | p.(Gln557*) | Nonsense (NMD not expected) | Not in functional domain/region | NA | NA | Absent | n.a. | Novel |
Used RefSeq transcripts (based on Genome build: GRCh37/hg19): ACTA2: NC_000010.10(NM_001141945.2), COL3A1: NC_000002.11(NM_000090.3), FBN1: NC_000015.9(NM_000138.4), FBN2: NC_000005.9(NM_001999.3), MYH11: NC_000016.9(NM_001040113.1), NOTCH1: NC_000009.11(NM_017617.3), PRKG1: NC_000010.10(NM_001098512.2), SLC2A10: NC_000020.10(NM_030777.3), SMAD3: NC_000015.9(NM_005902.3), TGFB2: NC_000001.10(NM_001135599.2), TGFB3: NC_000014.8(NM_003239.4), TGFBR1: NC_000009.11(NM_004612.2), TGFBR2: NC_000003.11(NM_001024847.2).
Pathogenic variants (class 5) are depicted in bold.
FCUT, functionally conserved up to; n.a., not available; NA, not applicable; NMD, nonsense mediated mRNA decay
Yes, segregation analysis performed in (at least) one family member, variant segregated accordingly.
No further alignment available.
This family is recently described in literature(Overwater & Houweling, 2017).
A variant of unknown significance was identified in these patients as well (Supporting Information Table S1).
Paternity and maternity not confirmed.
Homozygous variant.
‐ Tolerated (SIFT), polymorphism (MutationTaster), and benign (Polyphen‐2) predictions.
±Possibly damaging (Polyphen‐2) prediction.
+Deleterious (SIFT), Disease‐causing (MutationTaster), probably damaging (Polyphen‐2) predictions.
Alignment, SIFT, MutationTaster, Polyphen‐2, Grantham distance: Alamut GRCh37 accessed July 2017.
Table 2.
Summary of the clinical features of patient with a pathogenic or likely pathogenic variant detected by variant‐calling analysis of 21 H‐TAD genes
| Family history | |||||||
|---|---|---|---|---|---|---|---|
| Patient | Involved gene | Sex, agea | Cardiovascular feature(s) | Systemic feature(s) | Genotype | Relative | Phenotype |
| 1 | ACTA2 | ♀, 16 | PDA | None |
|
|
|
| 2 | ACTA2 | ♂, 28 | Dis (A and B, 26 y), BAV | None |
|
|
|
| 3 | ACTA2 | ♂, 46 | Dis (A, 45 y) | None | ? | No relatives clinically affected | |
| 4 | ACTA2 | ♀, 69 | Dis (B, 61 y; A, 65 y) | None |
|
|
|
| 5c | ACTA2 | ♂, 36 | Dis (B, 36 y) | Iris flocculi, livedo reticularis |
|
|
|
| 6 | ACTA2 | ♂, 73 | An (AoR, 52 mm, 69 y) | None | ? | No relatives clinically affected | |
| 7 | ACTA2 | ♂, 22 | Dis (A, 21 y), BAV | PP, SS, Myopia −5/−5 dpt | ? | No relatives clinically affected | |
| 8 | ACTA2 | ♂, 57 | Dis (B, 57 y), An (AoR 41 mm, 57 y) | Myopia −4 dpt, pneumothorax | ? | B | SUD (58 y) |
| 9d | COL3A1 | ♂, 59 | Rup (AoA, 54 y), An (AA, 59 y) | None |
|
|
|
| 10 | COL3A1 | ♂, 52 | Dis (A, 47 y), An (subclavian and vertebral artery, 52 y) | Increased AHR | ? | No relatives clinically affected | |
| 11 | COL3A1 | ♀, 44 | Dis (B, 44 y) | NA | ‐ | Si |
|
| 12 | COL3A1 | ♀, 31 | An (renal and carotid artery), Dis (mammary‐, subclavian‐ and iliac artery), occlusion (brachial artery) | None |
|
|
|
| 13 | COL3A1 | ♂, 42 | Dis (A, 38 y) | Hyperkyphosis, hypermobile fingers |
|
|
|
| 14 | COL3A1 | ♂, 45 | Dis (coronary artery, 42 y), An (AAo, 47 mm, 45 y) | Soft skin | ? | No relatives clinically affected | |
| 15 | FBN1 | ♂, 66 | Dis (B, 49 y), An (subclavian artery, AA, 54 y) | NA | ? | So | Clinical features of MFS |
| 16 | FBN1 | ♀, 27 | An (AoR, 41 mm, 27 y), MVP | Arachnodactyly |
|
|
|
| 17 | FBN1 | ♂, 35 | An (AoR, 50 mm, 35 y), ASD, atrial flutter (23 y) |
|
|
|
|
| 18 | FBN1 | ♂, 5 | An (AAo, 27 mm, Z‐score +2.7, 5 y), VSD | PP, hyperkyphosis, wrist sign +, dolichocephaly, malar hypoplasia, EL, BS 8/9 | ? | No relatives clinically affected | |
| 19 | FBN1 | ♂, 53 | An (thoracic aorta, 80 mm, 53 y) | Wrist and thumb sign +, IH | ? | PF | Multiple relatives with An and/or Dis |
| 20 | FBN1 | ♀, 36 | An (AoR, severe, 35 y), MVP | Scoliosis, PC, Myopia −6.5 dpt, SS | ? | No relatives clinically affected | |
| 21 | FBN1 | ♂, 11 | NA | Increased AHR, PD, clinical features of MFS | NA | ||
| 22 | FBN1 | ♂, 32 | Dis (A, 15 y), MVP | Marfanoid habitus, PP, reduced elbow extension, arachnodactyly, HAP, crowding, myopia ‐5/−3 dpt, SS | ? | No relatives clinically affected | |
| 23 | FBN1 | ♀, 0 | An (AoR,0 y), MI, TI | PC, joint contractures, arachnodactyly, dysmorphic facial features | ? | No relatives clinically affected | |
| 24 | FBN1 | ♂, 3 | None | Height +3.4 SD, arachnodactyly, HAP, ptosis, epicanthal folds, delayed speech |
|
|
|
| 25 | FBN1 | ♀, 29 | An (AoR, 41 mm, 29 y), MI | Arachnodactyly, HAP, dolichocephaly, EL, RD | ? | F | SD (42 y), myocardial infarction |
| 26 | FBN1 | ♀, 11 | MVP | Marfanoid habitus, PP, wrist and thumb sign +, joint luxations, SS, recurrent hematomas | ? | Clinically not affected | |
| 27 | FBN1 | ♀, 9 | None | Increased AHR, PC, club foot, PP, thumb sign +, downslanting, malar hypoplasia, myopia, recurrent hematomas | ? | No relatives clinically affected | |
| 28 | FBN1 | ♂, 5 | None | Tall stature, arachnodactyly, PP, PC, wrist sign +, HAP, hypermobility, macular degeneration | de novo e | ||
| 29 | FBN1 | ♀, 10 | An (AAo, 31 mm, Z‐score +2.7, 10 y) | PD, PP, arachnodactyly, HAP, dolichocephaly, myopia | de novo e | ||
| 30 | FBN1 | ♂, 54 | Dis (A, 54 y) | Pneumothorax, NA | + | So(2) | Clinically not affected |
| 31 | FBN1 | ♀, 46 | An (AAo, 46 mm, 46 y), cerebral infarction (33 y), stenosis (axillary‐, brachial‐ and subclavian artery, 36 y) | Hypermobile fingers | ? | No relatives clinically affected | |
| 32 | FBN1 | ♂, 0 | MI, TI | PC, PP, dolichocephaly, downslanting, enophthalmos, floppy ears | ? | No relatives clinically affected | |
| 33 | FBN2 | ♂, 10 | TI | Tall stature, PE, HAP, crowding | ? | MF | An (aorta), hypermobility |
| 34 | FBN2 | ♂, 55 | Borderline An (AAo, 39 mm, 54 y) | PE, hyperkyphosis, hammer toes, downslanting, myopia | + | F | Clinically not affected |
| 35 | FBN2 | ♂, 65 | An (AAo, 45 mm, 64 y) | Hammer toes, HAP, enophthalmos, prominent eyes, and nose, malar hypoplasia |
|
|
|
| 36 | MYH11 | ♂, 71 | Dis (A and B, 70 y), An (AA, 54 mm, 71 y) | None | ? | M | Rup (aorta, deceased) |
| 37 | MYH11 | ♂, 59 | An (AAo, 46 mm, 58 y), BAV, PFO | PP, malar hypoplasia, cutaneous hyperextensibility | ? | No relatives clinically affected | |
| 38 | NOTCH1 | ♂, 77 | An (AAo and AoA, 85 mm, 77 y) | None | ? |
|
|
| 39 | PRKG1 | ♂, 52 | Dis (subclavian‐, iliac‐ and brachiocephalic artery, 42 y), borderline an (AAo, 40 mm, 52 y) | SS | ? | No relatives clinically affected | |
| 40 | SLC2A10 | ♀, 15 | Arterial tortuosity (aorta, pulmonary artery, carotid arteries), MI, ASD | PP, hypermobile fingers, hypermobility, thumb sign +, clinodactyly, hypertelorism, periorbital fullness, | ? | No relatives clinically affected | |
| 41 | SLC2A10 | ♂, 0 | An (AoR, 17 mm, Z‐score +3.3, 5 months), PFO, abnormal course AoA, and pulmonary vessels | Arachnodactyly, abnormal thumb position, downslanting, hypertelorism, HAP, retrognathia diaphragmatic hernia |
|
|
|
| 42 | SMAD3 | ♀, 62 | Dis (A, 60 y), MI | PP, early onset arthrosis, myopia −2.5/−4 dpt | ? | F | An (AA, deceased, 67 y) |
| 43 | SMAD3 | ♂, 68 | An (thoracic aorta) | Tall stature, PE, scoliosis, early onset arthrosis, mild myopia | + | D | Tall stature, arachnodactyly |
| 44 | SMAD3 | ♀, 37 | Dis (coronary artery, 32 y), VSD | Brachydactyly type E, hypertelorism, prominent venous pattern, varicose veins, recurrent hematomas, myopia −6 dpt, IH, UH |
|
|
|
| 45 | SMAD3 | ♀, 76 | Dis (B, 63 y), An (AoA, 60 mm, 70 y) | Arthralgia, genu valgum, hypermobility, IH |
|
|
|
| 46 | SMAD3 | ♂, 17 | None | Scoliosis, PE, flat cornea |
|
|
|
| 47 | SMAD3 | ♀, 51 | Dis (A, B, 51 y) | Scoliosis, arthralgia, early onset arthrosis | + | So | Clinically not affected |
| 48 | SMAD3 | ♀, 40 | Borderline an (AoR, 40 y), MVP, MI | Wrist and thumb sign +, SS |
|
|
|
| 49 | TGFB2 | ♀, 19 | None |
|
|
|
|
| 50 | TGFB2 | ♂, 39 | An (AoR, 55 mm, 25 y), MVP | Scoliosis, PD, wrist and thumb sign +, hypermobility, recurrent hematomas in iliopsoas muscle, dural ectasia | ? | No relatives clinically affected | |
| 51 | TGFB2 | ♂, 0 | None | Arachnodactyly, joint contractures, retrognathia |
|
|
|
| 52d | TGFB2 | ♂, 32 | An (AoR, 44 mm, 32 y) | PC, PP, arachnodactyly, HAP, dolichocephaly, enophthalmos, malar hypoplasia, crowding, myopia −6.5 dpt, pneumothorax |
|
|
|
| 53 | TGFB3 | ♂, 43 | None | Increased AHR, PD, thumb sign +, BS 6/9 |
|
|
|
| 54 | TGFB3 | ♂, 59 | AVI (25 y), An (AoR, 46 mm, 25 y; AoR, 55 mm, AAo 48 mm, 57 y) | PP, HAP, downslanting, UH | ‐ | So | Clinically not affected |
| 55 | TGFBR1 | ♂, 56 | Dis (A and B, 56 y) | Scoliosis, PE, dolichocephaly, enophthalmos, malar hypoplasia | + | M | Clinically not affected |
| 56 | TGFBR1 | ♂, 33 | An (AoR, 43 mm, 31 y) | SS, dural ectasia |
|
|
|
| 57 | TGFBR1 | ♂, 16 | Dis (thoracic aorta, deceased, 16 y) | PE, tall stature, scoliosis, arachnodactyly | de novo | ||
| 58 | TGFBR2 | ♂, 14 | An (AoR, 40 mm, Z‐score +4.3, 14 y), VSD, DCRV | None | + | F | An (AoR, 42 mm, 52 y) |
| 59 | TGFBR2 | ♂, 15 | None | PD, hyperkyphosis, arthralgia, myopia ‐3 dpt | NA | ||
| 60 | TGFBR2 | ♀, 16 | An (AoR, 44 mm, 16 y), MVP | PP, arachnodactyly, hypermobility, luxations of hips and knees, bifid uvula, hypertelorism, blue sclerae | NA | ||
AA, abdominal aortic; AAo, ascending aorta; AHR, arm / height ratio; An, aneurysm; AoA, aortic arch; AoR, aortic root; ASD, atrial septal defect; AVI, aortic valve insufficiency; B, brother; BAV, bicuspid aortic valve; BS, Beighton score; CVD, cardiovascular disease; D, daughter; DCRV, double chambered right ventricle; Dis, dissection; dpt, dioptre; EL, ectopia lentis; F, father; GDa, granddaughter; GSo, grandson; HAP, highly arched palate; HE, heterozygous carrier; HT, hypertension; IH, inguinal hernia; M, mother; MF, maternal family; MFS, Marfan syndrome; MGF, maternal grandfather; MI, mitral valve insufficiency; MU, maternal uncle; MVP, mitral valve prolapse; N, nephew; NA, no further information available; PA, paternal aunt; PC, pectus carinatum; PCo, paternal cousin; PD, pectus deformity; PDA, patent ductus arteriosus; PE, pectus excavatum; PF, paternal family; PFO, patent foramen ovale; PGF, paternal grandfather; PGM, paternal grandmother; PP, pes plani; PU, paternal uncle; RD, retinal detachment; Rup, rupture; SD, standard deviation; Si, sister; Sib, sibling; So, son; SS, skin striae; SUD, sudden death; TI, tricuspid valve insufficiency; UH, umbilical hernia; VSD, ventricular septal defect
Age (in years) at DNA diagnostics.
Low‐grade mosaicism detected by NGS analysis in the father of the index patient.
This family is recently described in literature (Overwater & Houweling, 2017).
A variant of unknown significance was identified in these patients as well (Supporting Information Table S1).
Paternity and maternity not confirmed.
+ variant present
− variant absent
? unknown
3.2. Genetic and clinical data in patients with a CNV identified by XHMM analysis
The results of the XHMM analysis in the six patients with a CNV (patients 61–66) are depicted in Figure 1 and are summarized in Table 3.
Figure 1.
Genomic copy‐number variants in H‐TAD patients based on XHMM analysis. PCA: principal‐component analysis; XHMM: eXome hidden Markov model. A, MYLK gene; deletion of exons 17 and 18. B, PRKG1 gene; deletion of exon 3. C, SMAD3; deletion of exon 6. D, TGFB2; deletion of exons 4, 5, 6, and 7. E, NOTCH1 gene; whole gene duplication. F, SCARF2 gene; whole gene deletion. Graphic representation of the copy‐number variants in each gene based on XHMM analysis. Horizontal axis indicates physical position of the CNVs. Vertical axis indicates sample Z‐score of PCA‐normalized read depth. Deletions are colored in red, and duplications are colored in green
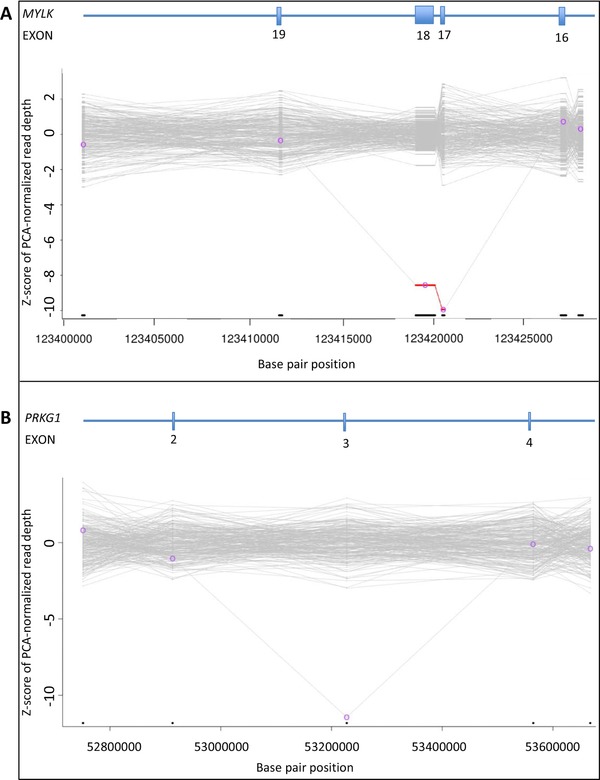
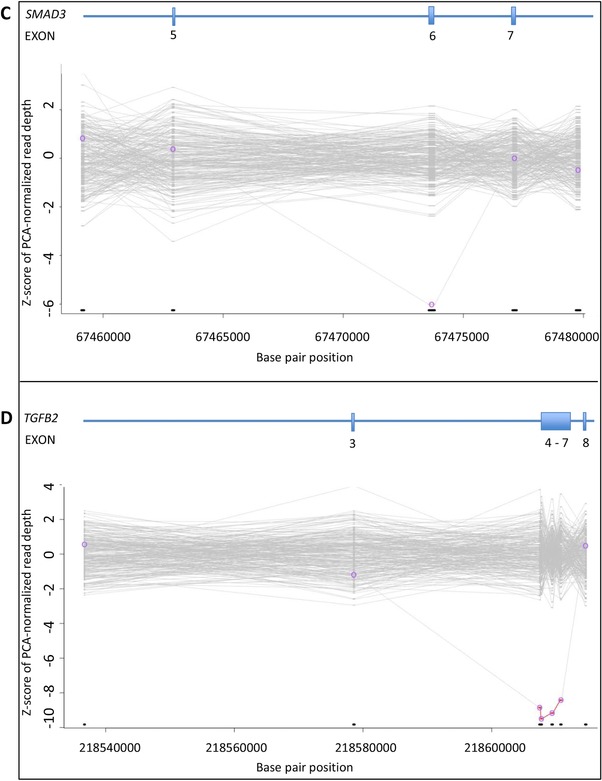
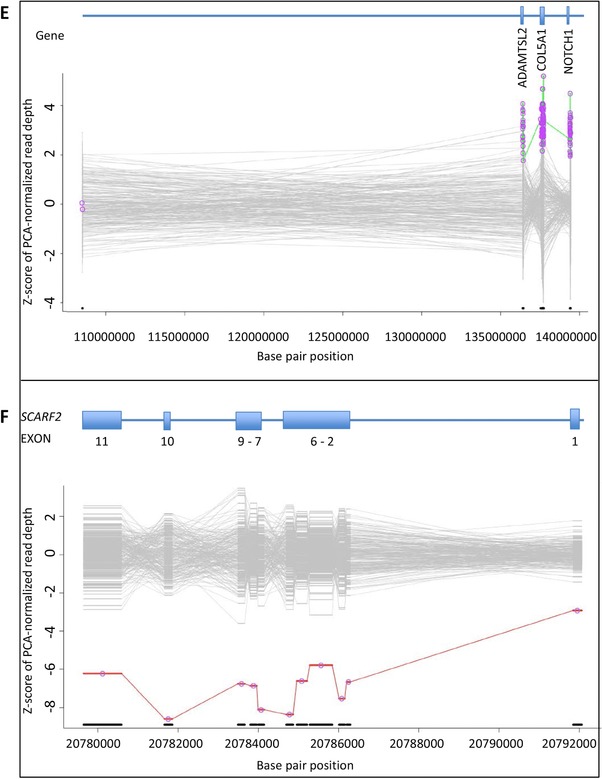
Table 3.
Summary of the genetic features of six patients with a pathogenic or likely pathogenic CNV
| Patient | Gender, agea | Involved gene, exon(s) based on XHMM analysis | Loss/gain | Protein change | Effect | Confirmed CNV | Validation technique | Variant classification |
|---|---|---|---|---|---|---|---|---|
| 61 | M, 66 | MYLK; exon 17 and 18 | Loss |
|
|
MYLK, deletion exon 17 en 18b | MLPA | 5 |
| 62 | M, 36 | PRKG1; exon 3c | Loss | p.(Asp145_Thr183delinsAla) | in‐frame deletion‐insertion | PRKG1, deletion exon 3c | MLPA | 4 |
| 63 | M, 31 | SMAD3; exon 6d | Loss | p.(Asp220_Ile290del) | in‐frame deletion | SMAD3, deletion exon 6d | MLPA | 5 |
| 64 | M, 17 | TGFB2; exons 4–7e | Loss | p.(Ile199_Arg390del) | in‐frame deletion | TGFB2, deletion exons 4–7e | MLPA | 5 |
| 65 | F, 0 | Duplication NOTCH1; whole genef | Gain | NA | NA | unbalanced translocation: 46,XX,der(7)t(7;9)(p22.3;q33.3)f | SNP array and karyotyping | 5 |
| 66 | M, 0 | Deletion SCARF2; whole geneg | Loss | NA | NA |
|
SNP array | 5 |
CNV, copy‐number variation; MLPA, multiplex ligation‐dependent probe; NA, not applicable; NMD, nonsense mediated mRNA decay; XHMM, eXome hidden Markov model.
Age (in years) at DNA diagnostics.
HGVS nomenclature: NC_000003.11(NM_053025.3)(MYLK): c.(2390+1_2391‐1)_(3448+1_3449‐1)del.
HGVS nomenclature: NC_000010.10(NssssssssM_001098512.2)(PRKG1): c.(433+1_434‐1)_(547+1_548‐1)del.
HGVS nomenclature: NC_000015.9(NM_005902.3)(SMAD3): c.(658+1_659‐1)_(871+1_872‐1)del.
HGVS nomenclature: NC_000001.10(NM_001135599.2)(TGFB2): c.(594+1_595‐1)_(1170+1_1171‐1)del.
ISCN nomenclature after additional SNP array and karyotyping.
ISCN nomenclature after additional SNP array.
In patient 61, a deletion of two exons in the MYLK gene was identified (NM_053025.3: c.(2390+1_2391‐1)_(3448+1_3449‐1)del). This deletion is predicted to generate an out‐of‐frame deletion in the long transcript of the MYLK gene (NM_053025.3) and a loss of the first 682 coding nucleotides, including the alternative translation initiation codon in the smooth‐muscle cell‐specific transcript encoding isoform 5 (Uniprot Q15746‐7). This male patient was diagnosed with a type B dissection at the age of 60 years and developed a type A dissection at the age of 65 years. He was treated surgically (Bentall procedure). Medical history and physical examination did not reveal any other signs of a connective tissue disorder. Pedigree analysis revealed that his sister suddenly died at the age of 53 years. No medical records, autopsy, or DNA were available. The 35‐year‐old son of the index patient did not carry the two‐exon deletion of MYLK. Until now, no other relatives opted for genetic testing.
In patient 62, a deletion of one exon of PRKG1 was detected (NM_001098512.2: c.(433+1_434‐1)_(547+1_548‐1)del). This deletion is predicted to lead to an in‐frame deletion of 39 amino acids and the insertion of an Alanine residue and encompasses a large part of the high‐affinity cGMP‐binding domain of the PRKG1 protein including Arginine177. A recurrent substitution of this arginine for glutamine has been reported in patients with H‐TAD and shown to have a gain‐of‐function effect (Guo et al., 2013). At the age of 35 years, this male patient was diagnosed with an aortic root dilatation, a type A dissection, aortic valve insufficiency, and dilated cardiomyopathy. He was treated surgically (Bentall procedure). His skin showed stretch marks on the shoulders and chest. Medical history, ophthalmological evaluation, and physical examination did not reveal any other features of a connective tissue disorder. A cardiomyopathy gene panel analysis (50 genes) did not result in the identification of a genetic cause for his dilated cardiomyopathy. Family history showed no clinically affected relatives. No relatives were available for cardiologic evaluation and DNA diagnostics.
In patient 63, a deletion of one exon in SMAD3, predicted to result in an in‐frame deletion of part of the MH2 domain, was found (NM_005902.3: c.(658+1_659‐1)_(871+1_872‐1)del). This male patient was followed up from the age of eight years, after his father, who was diagnosed with a chronic dissection of the ascending aorta at the age of 33 years, suddenly died at the age of 37 years. The paternal grandmother died at the age of 39 years, possibly caused by an aortic dissection as well. The patient was diagnosed with an aortic root dilatation with a maximal diameter of 48 mm and a dilated left coronary artery at the age of 30 years. He was treated surgically (David procedure). Physical examination revealed pes plani, a prominent venous pattern on the chest and arms, and several dysmorphic facial features including dolichocephaly, hypertelorism, and retrognathia. He had no signs of early onset osteoarthritis.
In patient 64, a four‐exon deletion was detected in the TGFB2 gene (NM_001135599.2: c.(594+1_595‐1)_(1170+1_1171‐1)del). This deletion is predicted to result in an in‐frame deletion of a large part of the TGFB2 protein. This 17‐year‐old male patient was under regular cardiologic surveillance because of TAAD in his father and paternal grandfather. At the age of 17 years cardiologic evaluation revealed an aortic root dilatation of 39 mm (Z‐score +3.28). Moreover, he had inguinal hernia repair at the age of one year, recurrent patellar dislocation, an asymmetric pectus deformity, and mild dysmorphic facial features including a long face, downslanting palpebral fissures, and a highly arched palate. The intragenic TGFB2 deletion was also present in his clinically affected father (clinical features include aortic root aneurysm requiring surgery at age 31 and aortic dissection at age 46) and his 11‐year‐old sister (features consisted of pectus deformity and highly arched palate and mild myopia). The phenotypes of all family members will be described in more detail elsewhere (Vliegenthart et al., manuscript in preparation). All intragenic deletions were confirmed by MLPA analysis (Supporting Information Figure S1).
In patients 65 and 66, XHMM findings were suggestive of a larger chromosomal abnormality. In patient 65, a duplication of the entire NOTCH1 gene was detected. COL5A1 and ADAMTSL2, which are located in the same chromosomal region (9q) and are present in our NGS platform, were also duplicated in this newborn female patient who presented after birth with several dysmorphic features. Facial features included frontal bossing, deep‐set eyes, low set ears with overfolded helices, and a crumpled left ear with a preauricular tag, micrognathia, and a small mouth. In addition, flexion contractures of elbows, wrists, and knees and striking arachnodactyly were noticed. Based on these features, she was initially suspected to have neonatal Marfan syndrome or Beals syndrome. Because XHMM analysis indicated a large 9q duplication, an SNP array was performed. A copy‐number gain at 9q33.3–q34.43 (11.8Mb; hg19; chr9:129172353–141020389) and a copy‐number loss at 7p22.3 (2Mb; hg19; chr7:43360–2067625) were found. Subsequent karyotyping revealed an unbalanced translocation 46,XX,der(7)t(7;9)(p22.3;q33.3). Parental cytogenetic studies showed that her father carried a balanced reciprocal translocation; 46,XY,t(7;9)(p22.3;q33.3). Results of the array and karyotyping are shown in Figure 2A. In the literature, overlapping phenotypic manifestations such as similar craniofacial features, joint contractures, and arachnodactyly have been described in the 9q duplication syndrome (Amarillo, O'Connor, Lee, Willing, & Wambach, 2015). During follow‐up, she was treated for bleeding esophageal varices probably caused by portal vein thrombosis, which have not been described in patients with a 9q duplication syndrome and/or 7p22.3 deletion previously.
Figure 2.
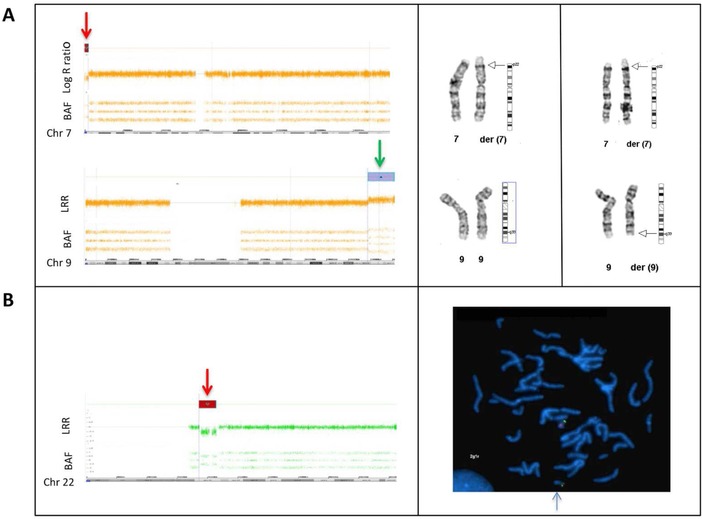
Further characterization of XHMM results by additional (cyto‐) genetic testing. BAF, B allele frequency; Chr, chromosome; der, derivate chromosome; LLR, log R ratio; FISH, fluorescence in situ hybridization. A, SNP array profile of chromosomes 7 and 9 are shown on the left. The top plot of each image shows the LRR, which provides an estimation of the copy number for each marker aligned to its chromosomal position. The bottom plot of each image shows the BAF for each SNP aligned to its chromosomal position. SNP array analysis revealed a terminal copy‐number loss at 7p22.3 (2Mb; GRCh37; chr7:43360‐2067625) indicated with a red arrow and a terminal copy‐number gain at 9q33.3–q34.43 (11.8Mb; GRCh37; chr9:129172353–141020389) indicated with a green arrow. Chromosomes 7 and 9 from the index (left) with the unbalanced translocation and the father (right) carrying the balanced translocation are shown on the right. The breakpoints of the reciprocal translocation are indicated with an arrow. The index has the derivative chromosome 7 lacking a short segment from the short arm of chromosome 7 that is replaced by an extra copy of a terminal segment of chromosome 9q. The father has two derivative chromosomes 7 and 9, each carrying a segment of the other chromosome. B, SNP array profile of chromosome 22 is shown on the left. SNP array analysis revealed a copy‐number loss at 22q11.2 (3.2Mb; GRCh37; chr22:20779645_20792061) indicated with a red arrow. The results of metaphase FISH on blood from the mother is presented on the right. The 22q11.2 region is recognized by the HIRA probe, producing a red signal. The green signal is from the ARSA probe hybridizing with the ARSA gene on chromosome band 22q13.33. The 22q11.2 deletion is indicated by a blue arrow. Metaphase FISH analysis revealed that the mother is also a carrier of the 22q11.2 deletion (ish del(22)(q11.2q11.2)(HIRA‐))
Finally, a deletion of the entire SCARF2 gene, located at 22q11, was detected in patient 66. This newborn male patient presented with severe perinatal problems, including asphyxia and the need for resuscitation, after an uncomplicated pregnancy. Furthermore, initially a connective tissue disorder was suspected based on the presence of a relative dilatation of the aortic root in relation to the body surface area (16 mm, Z‐score +3) and a strangulated inguinal hernia. Physical examination revealed unilateral postaxial polydactyly without any other dysmorphic features. Simultaneous analysis of the NGS H‐TAD gene panel and SNP array revealed that the heterozygous deletion of SCARF2 was part of a 22q11.2 deletion (i.e., DiGeorge syndrome) (3.2Mb; hg19; chr22:20779645_20792061). A normal male karyotype (46,XY) was seen. Parental fluorescence in situ hybridization (FISH) revealed that his mother also carried the 22q11.2 deletion (ish del(22)(q11.2q11.2)(HIRA‐)). Results of array and FISH are shown in Figure 2B. Except for delayed motor and speech development at childhood and complaints of fatigue and recurrent infections, his mother had no medical problems. Cardiac ultrasound showed no abnormalities. Most clinical features of the index patient, including inguinal hernia and postaxial polydactyly, were consistent with the established diagnosis. During follow‐up the relative dilatation of the aortic diameter was normalized.
4. DISCUSSION
This study provides the results of the molecular and clinical findings in the largest cohort of patients suspected of H‐TAD reported in the literature to date. In addition, this is the first report describing CNV analyses of 21 H‐TAD‐associated genes using variant‐calling analysis combined with XHMM analysis. In this cohort of 810 patients, a pathogenic or likely pathogenic variant was identified in 66 patients (8.1%). Overall, we identified a relatively low number of pathogenic or likely pathogenic variants in our H‐TAD cohort compared to previous studies that identified mutations in 10.3% to 35.5% (Campens et al., 2015; Lerner‐Ellis et al., 2014; Poninska et al., 2016; Proost et al., 2015; Wooderchak‐Donahue et al., 2015; Ziganshin et al., 2015). This wide range is likely to be explained by differences in clinical and demographic characteristics of the study populations and different inclusion criteria used for genetic testing. In general, DNA testing in the Netherlands is increasingly offered at a lower threshold to TAAD patients (e.g., not only to very young patients or patients with a positive family history for H‐TAD), which may explain the relatively low mutation detection yield.
Using routine NGS analysis (variant‐calling analysis) pathogenic or likely pathogenic variants were identified in FBN1, ACTA2, SMAD3, COL3A1, TGFB2, TGFBR1, TGFBR2, FBN2, MYH11, TGFB3, SLC2A10, PRKG1, and NOTCH1. As expected, most of the pathogenic and likely pathogenic variants were detected in FBN1 (N = 18, 30%). Of these, at least 14 (78%) fulfilled the revised Marfan criteria. However, the proportion of pathogenic FBN1 and COL3A1 variants in this cohort is biased because single‐gene analysis of these two genes is still offered in our institute and variants in these genes detected using single‐gene analysis were not included in this study. Therefore, it is likely that in patients with a highly suggestive phenotype of vascular Ehlers–Danlos syndrome, single‐gene analysis of COL3A1 was requested instead of NGS panel analysis. This might explain the high proportion of COL3A1 variants predicted to result in haploinsufficiency detected in this study (3 of 6 = 50%, compared with approximately 4% of nonsense/frameshift variants currently reported in the COL3A1 LOVD database; https://eds.gene.le.ac.uk/home.php?select_db=COL3A1), as the phenotype in patients with COL3A1 haploinsufficiency is often confined to vascular events (Leistritz, Pepin, Schwarze, & Byers, 2011).
Of the pathogenic and likely pathogenic variants identified, 37 (67%) have not been described previously. None of these variants were identified more than once in our patient cohort. This emphasizes the extreme allelic heterogeneity of H‐TAD‐related disorders. Young age at diagnosis, a positive family history, and presence of syndromic features were shown to be the strongest predictors for the identification of a disease‐causing variant in the literature (P = 0.001–0.01) (Campens et al., 2015). The observation that the mean age at DNA testing in the group of patients with a pathogenic or likely pathogenic variant was 11 years lower than the mean age in the groups without a pathogenic or likely pathogenic variant is in line with this. However, 10 of the 66 patients with a pathogenic or likely pathogenic variant were over the age of 60 years at the time of DNA testing (15.2%). Of these, three patients (30%) had a negative family history for aortic disease, sudden death < 45 years, or systemic features of a connective tissue disorder. These observations underscore the reduced and age‐dependent penetrance with a high degree of clinical heterogeneity in H‐TAD. In five patients with an identified pathogenic or likely pathogenic variant, DNA testing of both parents suggested a de novo occurrence, while in one case a de novo occurrence was inferred as the variant was detected in mosaic status. This was in line with the negative family history for aortic disease in these families.
Of the 66 pathogenic or likely pathogenic variants, six were CNVs detected by XHMM analysis. These aberrations account for an incremental yield of 9.1% of the identified pathogenic or likely pathogenic variants, underscoring the relevance of adding a technique to identify CNVs in TAAD patients. The CNVs included (multi‐)exon deletions in MYLK, PRKG1, SMAD3, and TGFB2. To the best of our knowledge, intragenic (multi‐)exon deletions have not been reported in these genes before. The clinical features of the patients with these (multi‐)exon deletions did not differ notably from the known phenotypic manifestations related to variants in these genes. Moreover, a large duplication including the whole NOTCH1 gene and a large deletion encompassing SCARF2 were detected by XHMM analysis. These aberrations were part of an unbalanced translocation (46,XX,der(7)t(7;9)(p22.3;q33.3)) and a 22q11.2 deletion (22q11.2(20779645_20792061)x1), respectively, and were classified as the cause of the clinical features of the patients.
The results of this study underline the importance of CNV analysis using a bioinformatics tool such as XHMM in the clinical diagnostic care for TAAD patients. As CNV analysis is often not routinely performed for most genes included in this NGS platform, these CNVs would not have been detected by regular genetic analysis. Four of the six detected CNVs in this study were small intragenic deletions (two single‐exon deletions, one 2‐exon, and one 4‐exon deletion). These are generally not detected by routine CGH or SNP array analysis. This highlights the importance of using a CNV detection tool, which allows detection of CNVs with (small) single‐exon resolution. Based on the results of this study, single‐exon‐sensitive deletion/duplication analysis on a routine basis should be recommended in patients suspected of H‐TAD.
5. CONCLUSION
In 66 of 810 (8.1%) patients suspected of H‐TAD, a pathogenic or likely pathogenic variant was identified using our NGS gene panel in combination with XHMM analysis. Six of these 66 pathogenic or likely pathogenic variants (9.1%) were a CNV, not detectable by routine NGS analysis. This study is the first to describe the incremental yield of CNV analysis in patients suspected of H‐TAD. Our study underscores the importance of CNV analysis using a bioinformatics tool such as XHMM in the clinical diagnostic care for H‐TAD patients.
Supporting information
Supplementary data: materials and methods
Figure S1. Confirmation of identified intragenic deletions with MLPA analysis
Table S1. Summary of the genetic features of patients with a variant of unknown significance detected by variant‐calling analysis of 21 H‐TAD genes
Table S2. Summary of the clinical features of the patient with a variant of unknown significance detected by variant‐calling analysis of 21 H‐TAD genes
Table S3. Overview of the genes analysed in this study
Table S4. Tools used to classify variants.
DECLARATIONS
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Under Dutch law, assessment of the study protocol by our ethics committee was not indicated because only genetic and clinical data collected during regular patient care were used.
CONSENT FOR PUBLICATION
Written informed consent was obtained from the patients and/or their parents with an aberration detected by XHMM, as more detailed medical data were published. Informed consent for DNA diagnostics was obtained from all 810 patients after genetic counseling by the referring physician.
AVAILABILITY OF DATA AND MATERIAL
All data and protocols used for this study are either included in the article (or in its supporting files) or are available upon request.
COMPETING INTERESTS
The authors declare that they have no competing interests.
FUNDING
Not applicable
AUTHORS' CONTRIBUTIONS
EO, JPT, ACH, and AM initiated the project. The genetic tests were validated and/or supervised by LM, PR, MMW, EV, and AM. The clinical data of the patients were collected by EO, MJHB, AFB, IB, ED, JMH, YHH, MK, IPK, LAM, JMAV, KKY, PJGZ, MG, JPT, and ACH. The first draft of the manuscript was written by EO and LM. This was supervised by JPT, ACH, and AM. The manuscript was read and approved by all authors.
Overwater E, Marsili L, Baars MJH, et al. Results of next‐generation sequencing gene panel diagnostics including copy‐number variation analysis in 810 patients suspected of heritable thoracic aortic disorders. Human Mutation. 2018;39:1173–1192. 10.1002/humu.23565
ACH and AM contributed equally to this work.
Communicated by Reed E. Pyeritz
REFERENCES
- Ackerman, M. J. , Priori, S. G. , Willems, S. , Berul, C. , Brugada, R. , Calkins, H. , … European Heart Rhythm, A. (2011). HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: This document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA). Europace, 13(8), 1077–1109. 10.1093/europace/eur245 [DOI] [PubMed] [Google Scholar]
- Akutsu, K. , Morisaki, H. , Takeshita, S. , Sakamoto, S. , Tamori, Y. , Yoshimuta, T. , … Morisaki, T. (2007). Phenotypic heterogeneity of Marfan‐like connective tissue disorders associated with mutations in the transforming growth factor‐beta receptor genes. Circulation Journal, 71(8), 1305–1309. [DOI] [PubMed] [Google Scholar]
- Amarillo, I. E. , O'Connor, S. , Lee, C. K. , Willing, M. , & Wambach, J. A. (2015). De novo 9q gain in an infant with tetralogy of Fallot with absent pulmonary valve: Patient report and review of congenital heart disease in 9q duplication syndrome. American Journal of Medical Genetics. Part A, 167A(12), 2966–2974. 10.1002/ajmg.a.37296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragon‐Martin, J. A. , Ahnood, D. , Charteris, D. G. , Saggar, A. , Nischal, K. K. , Comeglio, P. , … Arno, G. (2010). Role of ADAMTSL4 mutations in FBN1 mutation‐negative ectopia lentis patients. Human Mutation, 31(8), E1622–E1631. 10.1002/humu.21305 [DOI] [PubMed] [Google Scholar]
- Arbustini, E. , Grasso, M. , Ansaldi, S. , Malattia, C. , Pilotto, A. , Porcu, E. , … Tavazzi, L. (2005). Identification of sixty‐two novel and twelve known FBN1 mutations in eighty‐one unrelated probands with Marfan syndrome and other fibrillinopathies. Human Mutation, 26(5), 494 10.1002/humu.9377 [DOI] [PubMed] [Google Scholar]
- Attanasio, M. , Lapini, I. , Evangelisti, L. , Lucarini, L. , Giusti, B. , Porciani, M. , … Pepe, G. (2008). FBN1 mutation screening of patients with Marfan syndrome and related disorders: Detection of 46 novel FBN1 mutations. Clinical Genetics, 74(1), 39–46. 10.1111/j.1399-0004.2008.01007.x [DOI] [PubMed] [Google Scholar]
- Aubart, M. , Gobert, D. , Aubart‐Cohen, F. , Detaint, D. , Hanna, N. , d'Indya, H. , … Jondeau, G. (2014). Early‐onset osteoarthritis, Charcot‐Marie tooth like neuropathy, autoimmune features, multiple arterial aneurysms and dissections: An unrecognized and life threatening condition. Plos One, 9(5), e96387 10.1371/journal.pone.0096387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baetens, M. , Van Laer, L. , De Leeneer, K. , Hellemans, J. , De Schrijver, J. , Van De Voorde, H. , … Coucke, P. J. (2011). Applying massive parallel sequencing to molecular diagnosis of Marfan and Loeys‐Dietz syndromes. Human Mutation, 32(9), 1053–1062. 10.1002/humu.21525 [DOI] [PubMed] [Google Scholar]
- Biddinger, A. , Rocklin, M. , Coselli, J. , & Milewicz, D. M. (1997). Familial thoracic aortic dilatations and dissections: A case control study. Journal of Vascular Surgery, 25(3), 506–511. [DOI] [PubMed] [Google Scholar]
- Buchan, J. G. , Alvarado, D. M. , Haller, G. E. , Cruchaga, C. , Harms, M. B. , Zhang, T. , … Gurnett, C. A. (2014). Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Human Molecular Genetics, 23(19), 5271–5282. 10.1093/hmg/ddu224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert, B. L. , Willaert, A. , Kerstjens‐Frederikse, W. S. , De Backer, J. , Devriendt, K. , Albrecht, B. , … De Paepe, A. M. (2008). Arterial tortuosity syndrome: Clinical and molecular findings in 12 newly identified families. Human Mutation, 29(1), 150–158. 10.1002/humu.20623 [DOI] [PubMed] [Google Scholar]
- Campens, L. , Callewaert, B. , Mosquera, Muino , L., Renard , M., Symoens , S., De Paepe , … De Backer, J. (2015). Gene panel sequencing in heritable thoracic aortic disorders and related entities ‐ results of comprehensive testing in a cohort of 264 patients. Orphanet Journal of Rare Diseases, 10, 9 10.1186/s13023-014-0221-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coady, M. A. , Davies, R. R. , Roberts, M. , Goldstein, L. J. , Rogalski, M. J. , Rizzo, J. A. , … Elefteriades, J. A. (1999). Familial patterns of thoracic aortic aneurysms. Archives of Surgery, 134(4), 361–367. [DOI] [PubMed] [Google Scholar]
- Collod‐Beroud, G. , Le Bourdelles, S. , Ades, L. , Ala‐Kokko, L. , Booms, P. , Boxer, M. , … Boileau, C. (2003). Update of the UMD‐FBN1 mutation database and creation of an FBN1 polymorphism database. Human Mutation, 22(3), 199–208. 10.1002/humu.10249 [DOI] [PubMed] [Google Scholar]
- Comeglio, P. , Johnson, P. , Arno, G. , Brice, G. , Evans, A. , Aragon‐Martin, J. , … Child, A. (2007). The importance of mutation detection in Marfan syndrome and Marfan‐related disorders: Report of 193 FBN1 mutations. Human Mutation, 28(9), 928 10.1002/humu.9505 [DOI] [PubMed] [Google Scholar]
- Coucke, P. J. , Willaert, A. , Wessels, M. W. , Callewaert, B. , Zoppi, N. , De Backer, J. , … De Paepe, A. (2006). Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nature Genetics, 38(4), 452–457. 10.1038/ng1764 [DOI] [PubMed] [Google Scholar]
- den Hartog, A. W. , Franken, R. , van den Berg, M. P. , Zwinderman, A. H. , Timmermans, J. , Scholte, A. J. , … Groenink, M. (2016). The effect of losartan therapy on ventricular function in Marfan patients with haploinsufficient or dominant negative FBN1 mutations. Netherlands Heart Journal, 24(11), 675–681. 10.1007/s12471-016-0905-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietz, H. C. , Saraiva, J. M. , Pyeritz, R. E. , Cutting, G. R. , & Francomano, C. A. (1992). Clustering of fibrillin (FBN1) missense mutations in Marfan syndrome patients at cysteine residues in EGF‐like domains. Human Mutation, 1(5), 366–374. 10.1002/humu.1380010504 [DOI] [PubMed] [Google Scholar]
- Eccles, D. M. , Balmana, J. , Clune, J. , Ehlken, B. , Gohlke, A. , Hirst, C. , … Gomez Garcia, E. B. (2016). Selecting patients with ovarian cancer for germline BRCA mutation testing: Findings from guidelines and a systematic literature review. Advances in Therapy, 33(2), 129–150. 10.1007/s12325-016-0281-1 [DOI] [PubMed] [Google Scholar]
- Faivre, L. , Collod‐Beroud, G. , Callewaert, B. , Child, A. , Binquet, C. , Gautier, E. , … Boileau, C. (2009). Clinical and mutation‐type analysis from an international series of 198 probands with a pathogenic FBN1 exons 24–32 mutation. European Journal of Human Genetics, 17(4), 491–501. 10.1038/ejhg.2008.207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank, M. , Albuisson, J. , Ranque, B. , Golmard, L. , Mazzella, J. M. , Bal‐Theoleyre, L. , … Jeunemaitre, X. (2015). The type of variants at the COL3A1 gene associates with the phenotype and severity of vascular Ehlers‐Danlos syndrome. European Journal of Human Genetics, 23(12), 1657–1664. 10.1038/ejhg.2015.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken, R. , den Hartog, A. W. , Radonic, T. , Micha, D. , Maugeri, A. , van Dijk, F. S. , … Pals, G. (2015). Beneficial outcome of losartan therapy depends on type of FBN1 Mutation in Marfan syndrome. Circulation. Cardiovascular Genetics, 8(2), 383–388. 10.1161/CIRCGENETICS.114.000950 [DOI] [PubMed] [Google Scholar]
- Fromer, M. , Moran, J. L. , Chambert, K. , Banks, E. , Bergen, S. E. , Ruderfer, D. M. , … Purcell, S. M. (2012). Discovery and statistical genotyping of copy‐number variation from whole‐exome sequencing depth. American Journal of Human Genetics, 91(4), 597–607. 10.1016/j.ajhg.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fromer, M. , & Purcell, S. M. (2014). Using XHMM software to detect copy number variation in whole‐exome sequencing data. Current Protocols in Human Genetics, 81, 7 23 21–21. 10.1002/0471142905.hg0723s81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gago‐Diaz, M. , Blanco‐Verea, A. , Teixido‐Tura, G. , Valenzuela, I. , Campo, Del , M., Borregan , … Brion, M. (2014). Whole exome sequencing for the identification of a new mutation in TGFB2 involved in a familial case of non‐syndromic aortisc disease. Clinica Chimica Acta, 437, 88–92. 10.1016/j.cca.2014.07.016 [DOI] [PubMed] [Google Scholar]
- Guo, D. C. , Pannu, H. , Tran‐Fadulu, V. , Papke, C. L. , Yu, R. K. , Avidan, N. , … Milewicz, D. M. (2007). Mutations in smooth muscle alpha‐actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genetics, 39(12), 1488–1493. 10.1038/ng.2007.6 [DOI] [PubMed] [Google Scholar]
- Guo, D. C. , Papke, C. L. , Tran‐Fadulu, V. , Regalado, E. S. , Avidan, N. , Johnson, R. J. , … Milewicz, D. M. (2009). Mutations in smooth muscle alpha‐actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. American Journal of Human Genetics, 84(5), 617–627. 10.1016/j.ajhg.2009.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, D. C. , Regalado, E. , Casteel, D. E. , Santos‐Cortez, R. L. , Gong, L. , Kim, J. J. , … Milewicz, D. M. (2013). Recurrent gain‐of‐function mutation in PRKG1 causes thoracic aortic aneurysms and acute aortic dissections. American Journal of Human Genetics, 93(2), 398–404. 10.1016/j.ajhg.2013.06.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiratzka, L. F. , Bakris, G. L. , Beckman, J. A. , Bersin, R. M. , Carr, V. F. , Casey, D. E., Jr. , … Society for Vascular, M. (2010). 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM guidelines for the diagnosis and management of patients with thoracic aortic disease: Executive summary. A report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology, American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons, and Society for Vascular Medicine. Catheterization and Cardiovascular Interventions, 76(2), E43–E86. [DOI] [PubMed] [Google Scholar]
- Hoffjan, S. , Waldmuller, S. , Blankenfeldt, W. , Kotting, J. , Gehle, P. , Binner, P. , … Scheffold, T. (2011). Three novel mutations in the ACTA2 gene in German patients with thoracic aortic aneurysms and dissections. European Journal of Human Genetics, 19(5), 520–524. 10.1038/ejhg.2010.239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth, R. , Yearwood, C. , & Harvey, J. F. (2007). Application of dHPLC for mutation detection of the fibrillin‐1 gene for the diagnosis of Marfan syndrome in a National Health Service Laboratory. Genetic Testing, 11(2), 146–152. 10.1089/gte.2006.0514 [DOI] [PubMed] [Google Scholar]
- Hoyert, D. L. , Arias, E. , Smith, B. L. , Murphy, S. L. , & Kochanek, K. D. (2001). Deaths: Final data for 1999. National Vital Statistics Reports, 49(8), 1–113. [PubMed] [Google Scholar]
- Hung, C. C. , Lin, S. Y. , Lee, C. N. , Cheng, H. Y. , Lin, S. P. , Chen, M. R. , … Su, Y. N. (2009). Mutation spectrum of the fibrillin‐1 (FBN1) gene in Taiwanese patients with Marfan syndrome. Annals of Human Genetics, 73(Pt 6), 559–567. 10.1111/j.1469-1809.2009.00545.x [DOI] [PubMed] [Google Scholar]
- Jondeau, G. , Ropers, J. , Regalado, E. , Braverman, A. , Evangelista, A. , Teixedo, G. , … Montalcino Aortic, C. (2016). International registry of patients carrying TGFBR1 or TGFBR2 mutations: Results of the MAC (Montalcino aortic consortium). Circulation. Cardiovascular Genetics, 9(6), 548–558. 10.1161/CIRCGENETICS.116.001485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelsen, J. R. , Dawany, N. , Martinez, A. , Grochowski, C. M. , Maurer, K. , Rappaport, E. , … Devoto, M. (2015). A de novo whole gene deletion of XIAP detected by exome sequencing analysis in very early onset inflammatory bowel disease: A case report. BMC Gastroenterology [Electronic Resource], 15, 160 10.1186/s12876-015-0394-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalique, Z. , Lyons, O. T. , Clough, R. E. , Bell, R. E. , Reidy, J. F. , Schwarze, U. , … Taylor, P. R. (2009). Successful endovascular repair of acute type B aortic dissection in undiagnosed Ehlers‐Danlos syndrome type IV. European Journal of Vascular and Endovascular Surgery, 38(5), 608–609. 10.1016/j.ejvs.2009.07.009 [DOI] [PubMed] [Google Scholar]
- Kielty, C. M. , Rantamaki, T. , Child, A. H. , Shuttleworth, C. A. , & Peltonen, L. (1995). Cysteine‐to‐arginine point mutation in a 'hybrid' eight‐cysteine domain of FBN1: Consequences for fibrillin aggregation and microfibril assembly. Journal of Cell Science, 108(Pt 3), 1317–1323. [DOI] [PubMed] [Google Scholar]
- Koyama, S. , Sato, H. , Wada, M. , Kawanami, T. , Emi, M. , & Kato, T. (2017). Whole‐exome sequencing and digital PCR identified a novel compound heterozygous mutation in the NPHP1 gene in a case of Joubert syndrome and related disorders. BMC Medical Genetics [Electronic Resource], 18(1), 37 10.1186/s12881-017-0399-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leistritz, D. F. , Pepin, M. G. , Schwarze, U. , & Byers, P. H. (2011). COL3A1 haploinsufficiency results in a variety of Ehlers‐Danlos syndrome type IV with delayed onset of complications and longer life expectancy. Genetics in Medicine, 13(8), 717–722. 10.1097/GIM.0b013e3182180c89 [DOI] [PubMed] [Google Scholar]
- Lerner‐Ellis, J. P. , Aldubayan, S. H. , Hernandez, A. L. , Kelly, M. A. , Stuenkel, A. J. , Walsh, J. , & Joshi, V. A. (2014). The spectrum of FBN1, TGFbetaR1, TGFbetaR2 and ACTA2 variants in 594 individuals with suspected Marfan syndrome, Loeys‐Dietz syndrome or thoracic aortic aneurysms and dissections (TAAD). Molecular Genetics and Metabolism, 112(2), 171–176. 10.1016/j.ymgme.2014.03.011 [DOI] [PubMed] [Google Scholar]
- Lindsay, M. E. , Schepers, D. , Bolar, N. A. , Doyle, J. J. , Gallo, E. , Fert‐Bober, J. , … Loeys, B. L. (2012). Loss‐of‐function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nature Genetics, 44(8), 922–927. 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys, B. , De Backer, J. , Van Acker, P. , Wettinck, K. , Pals, G. , Nuytinck, L. , … De Paepe, A. (2004). Comprehensive molecular screening of the FBN1 gene favors locus homogeneity of classical Marfan syndrome. Human Mutation, 24(2), 140–146. 10.1002/humu.20070 [DOI] [PubMed] [Google Scholar]
- Loeys, B. , Nuytinck, L. , Delvaux, I. , De Bie, S. , & De Paepe, A. (2001). Genotype and phenotype analysis of 171 patients referred for molecular study of the fibrillin‐1 gene FBN1 because of suspected Marfan syndrome. Archives of Internal Medicine, 161(20), 2447–2454. [DOI] [PubMed] [Google Scholar]
- Loeys, B. L. , Schwarze, U. , Holm, T. , Callewaert, B. L. , Thomas, G. H. , Pannu, H. , … Dietz, H. C. (2006). Aneurysm syndromes caused by mutations in the TGF‐beta receptor. New England Journal of Medicine, 355(8), 788–798. 10.1056/NEJMoa055695 [DOI] [PubMed] [Google Scholar]
- Magyar, I. , Colman, D. , Arnold, E. , Baumgartner, D. , Bottani, A. , Fokstuen, S. , … Matyas, G. (2009). Quantitative sequence analysis of FBN1 premature termination codons provides evidence for incomplete NMD in leukocytes. Human Mutation, 30(9), 1355–1364. 10.1002/humu.21058 [DOI] [PubMed] [Google Scholar]
- Matyas, G. , Arnold, E. , Carrel, T. , Baumgartner, D. , Boileau, C. , Berger, W. , & Steinmann, B. (2006). Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome‐related disorders. Human Mutation, 27(8), 760–769. 10.1002/humu.20353 [DOI] [PubMed] [Google Scholar]
- Matyas, G. , Naef, P. , Tollens, M. , & Oexle, K. (2014). De novo mutation of the latency‐associated peptide domain of TGFB3 in a patient with overgrowth and Loeys‐Dietz syndrome features. American Journal of Medical Genetics. Part A, 164A(8), 2141–2143. 10.1002/ajmg.a.36593 [DOI] [PubMed] [Google Scholar]
- Melenovsky, V. , Adamira, M. , Kautznerova, D. , Voska, L. , Weichet, J. , Loeys, B. , & Pirk, J. (2008). Aortic dissection in a young man with Loeys‐Dietz syndrome. Journal of Thoracic and Cardiovascular Surgery, 135(5), 1174–1175, 1175 e1171. 10.1016/j.jtcvs.2008.01.003 [DOI] [PubMed] [Google Scholar]
- Milewicz, D. , Hostetler, E. , Wallace, S. , Mellor‐Crummey, L. , Gong, L. , Pannu, H. , … Regalado, E. (2016). Precision medical and surgical management for thoracic aortic aneurysms and acute aortic dissections based on the causative mutant gene. Journal of Cardiovascular Surgery, 57(2), 172–177. [PubMed] [Google Scholar]
- Milewicz, D. M. , Regalado, E. S. , Shendure, J. , Nickerson, D. A. , & Guo, D. C. (2014). Successes and challenges of using whole exome sequencing to identify novel genes underlying an inherited predisposition for thoracic aortic aneurysms and acute aortic dissections. Trends in Cardiovascular Medicine, 24(2), 53–60. 10.1016/j.tcm.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moceri, P. , Albuisson, J. , Saint‐Faust, M. , Casagrande, F. , Giuliano, F. , Devos, C. , … Jeunemaitre, X. (2013). Arterial tortuosity syndrome: Early diagnosis and association with venous tortuosity. Journal of the American College of Cardiology, 61(7), 783 10.1016/j.jacc.2012.06.070 [DOI] [PubMed] [Google Scholar]
- Olsson, C. , Thelin, S. , Stahle, E. , Ekbom, A. , & Granath, F. (2006). Thoracic aortic aneurysm and dissection: Increasing prevalence and improved outcomes reported in a nationwide population‐based study of more than 14,000 cases from 1987 to 2002. Circulation, 114(24), 2611–2618. 10.1161/CIRCULATIONAHA.106.630400 [DOI] [PubMed] [Google Scholar]
- Overwater, E. , & Houweling, A. C. (2017). Iris flocculi and type B aortic dissection. Ophthalmology, 124(11), 1711 10.1016/j.ophtha.2017.05.011 [DOI] [PubMed] [Google Scholar]
- Pepin, M. G. , Schwarze, U. , Rice, K. M. , Liu, M. , Leistritz, D. , & Byers, P. H. (2014). Survival is affected by mutation type and molecular mechanism in vascular Ehlers‐Danlos syndrome (EDS type IV). Genetics in Medicine, 16(12), 881–888. 10.1038/gim.2014.72 [DOI] [PubMed] [Google Scholar]
- Pollack, J. R. , Sorlie, T. , Perou, C. M. , Rees, C. A. , Jeffrey, S. S. , Lonning, P. E. , … Brown, P. O. (2002). Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proceedings of the National Academy of Sciences of the United States of America, 99(20), 12963–12968. 10.1073/pnas.162471999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poninska, J. K. , Bilinska, Z. T. , Franaszczyk, M. , Michalak, E. , Rydzanicz, M. , Szpakowski, E. , … Ploski, R. (2016). Next‐generation sequencing for diagnosis of thoracic aortic aneurysms and dissections: Diagnostic yield, novel mutations and genotype phenotype correlations. Journal of Translational Medicine, 14(1), 115 10.1186/s12967-016-0870-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poultney, C. S. , Goldberg, A. P. , Drapeau, E. , Kou, Y. , Harony‐Nicolas, H. , Kajiwara, Y. , … Buxbaum, J. D. (2013). Identification of small exonic CNV from whole‐exome sequence data and application to autism spectrum disorder. American Journal of Human Genetics, 93(4), 607–619. 10.1016/j.ajhg.2013.09.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proost, D. , Vandeweyer, G. , Meester, J. A. , Salemink, S. , Kempers, M. , Ingram, C. , … Van Laer, L. (2015). Performant mutation identification using targeted next‐generation sequencing of 14 thoracic aortic aneurysm genes. Human Mutation, 36(8), 808–814. 10.1002/humu.22802 [DOI] [PubMed] [Google Scholar]
- Regalado, E. S. , Guo, D. C. , Santos‐Cortez, R. L. , Hostetler, E. , Bensend, T. A. , Pannu, H. , … Milewicz, D. M. (2016). Pathogenic FBN1 variants in familial thoracic aortic aneurysms and dissections. Clinical Genetics, 89(6), 719–723. 10.1111/cge.12702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regalado, E. S. , Guo, D. C. , Villamizar, C. , Avidan, N. , Gilchrist, D. , McGillivray, B. , … Milewicz, D. M. (2011). Exome sequencing identifies SMAD3 mutations as a cause of familial thoracic aortic aneurysm and dissection with intracranial and other arterial aneurysms. Circulation Research, 109(6), 680–686. 10.1161/CIRCRESAHA.111.248161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson, E. N. , van der Linde, D. , Sherrah, A. G. , Vallely, M. P. , Wilson, M. , Bannon, P. G. , & Jeremy, R. W. (2016). Familial non‐syndromal thoracic aortic aneurysms and dissections ‐ Incidence and family screening outcomes. International Journal of Cardiology, 220, 43–51. 10.1016/j.ijcard.2016.06.086 [DOI] [PubMed] [Google Scholar]
- Robinson, D. O. , Lin, F. , Lyon, M. , Raponi, M. , Cross, E. , White, H. E. , … Baralle, D. (2012). Systematic screening of FBN1 gene unclassified missense variants for splice abnormalities. Clinical Genetics, 82(3), 223–231. 10.1111/j.1399-0004.2011.01781.x [DOI] [PubMed] [Google Scholar]
- Rommel, K. , Karck, M. , Haverich, A. , von Kodolitsch, Y. , Rybczynski, M. , Muller, G. , … Arslan‐Kirchner, M. (2005). Identification of 29 novel and nine recurrent fibrillin‐1 (FBN1) mutations and genotype‐phenotype correlations in 76 patients with Marfan syndrome. Human Mutation, 26(6), 529–539. 10.1002/humu.20239 [DOI] [PubMed] [Google Scholar]
- Schubert, J. A. , Landis, B. J. , Shikany, A. R. , Hinton, R. B. , & Ware, S. M. (2016). Clinically relevant variants identified in thoracic aortic aneurysm patients by research exome sequencing. American Journal of Medical Genetics. Part A, 170A(5), 1288–1294. 10.1002/ajmg.a.37568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze, U. , Schievink, W. I. , Petty, E. , Jaff, M. R. , Babovic‐Vuksanovic, D. , Cherry, K. J. , … Byers, P. H. (2001). Haploinsufficiency for one COL3A1 allele of type III procollagen results in a phenotype similar to the vascular form of Ehlers‐Danlos syndrome, Ehlers‐Danlos syndrome type IV. American Journal of Human Genetics, 69(5), 989–1001. 10.1086/324123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheikhzadeh, S. , Kade, C. , Keyser, B. , Stuhrmann, M. , Arslan‐Kirchner, M. , Rybczynski, M. , … von Kodolitsch, Y. (2012). Analysis of phenotype and genotype information for the diagnosis of Marfan syndrome. Clinical Genetics, 82(3), 240–247. 10.1111/j.1399-0004.2011.01771.x [DOI] [PubMed] [Google Scholar]
- Shlien, A. , & Malkin, D. (2010). Copy number variations and cancer susceptibility. Current Opinion in Oncology, 22(1), 55–63. 10.1097/CCO.0b013e328333dca4 [DOI] [PubMed] [Google Scholar]
- Soylen, B. , Singh, K. K. , Abuzainin, A. , Rommel, K. , Becker, H. , Arslan‐Kirchner, M. , & Schmidtke, J. (2009). Prevalence of dural ectasia in 63 gene‐mutation‐positive patients with features of Marfan syndrome type 1 and Loeys‐Dietz syndrome and report of 22 novel FBN1 mutations. Clinical Genetics, 75(3), 265–270. 10.1111/j.1399-0004.2008.01126.x [DOI] [PubMed] [Google Scholar]
- Spataro, N. , Roca‐Umbert, A. , Cervera‐Carles, L. , Valles, M. , Anglada, R. , Pagonabarraga, J. , … Bosch, E. (2017). Detection of genomic rearrangements from targeted resequencing data in Parkinson's disease patients. Movement Disorders, 32(1), 165–169. 10.1002/mds.26845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stheneur, C. , Collod‐Beroud, G. , Faivre, L. , Buyck, J. F. , Gouya, L. , Le Parc, J. M. , … Boileau, C. (2009). Identification of the minimal combination of clinical features in probands for efficient mutation detection in the FBN1 gene. European Journal of Human Genetics, 17(9), 1121–1128. 10.1038/ejhg.2009.36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapar, A. , & Cooper, M. (2013). Copy number variation: What is it and what has it told us about child psychiatric disorders? Journal of the American Academy of Child and Adolescent Psychiatry, 52(8), 772–774. 10.1016/j.jaac.2013.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner, C. L. , Emery, H. , Collins, A. L. , Howarth, R. J. , Yearwood, C. M. , Cross, E. , … Foulds, N. C. (2009). Detection of 53 FBN1 mutations (41 novel and 12 recurrent) and genotype‐phenotype correlations in 113 unrelated probands referred with Marfan syndrome, or a related fibrillinopathy. American Journal of Medical Genetics. Part A, 149A(2), 161–170. 10.1002/ajmg.a.32593 [DOI] [PubMed] [Google Scholar]
- Wooderchak‐Donahue, W. , VanSant‐Webb, C. , Tvrdik, T. , Plant, P. , Lewis, T. , Stocks, J. , … Bayrak‐Toydemir, P. (2015). Clinical utility of a next generation sequencing panel assay for Marfan and Marfan‐like syndromes featuring aortopathy. American Journal of Medical Genetics. Part A, 167A(8), 1747–1757. 10.1002/ajmg.a.37085 [DOI] [PubMed] [Google Scholar]
- Yang, J. H. , Ki, C. S. , Han, H. , Song, B. G. , Jang, S. Y. , Chung, T. Y. , … Kim, D. K. (2012). Clinical features and genetic analysis of Korean patients with Loeys‐Dietz syndrome. Journal of Human Genetics, 57(1), 52–56. 10.1038/jhg.2011.130 [DOI] [PubMed] [Google Scholar]
- Yoo, E. H. , Woo, H. , Ki, C. S. , Lee, H. J. , Kim, D. K. , Kang, I. S. , … Kim, J. W. (2010). Clinical and genetic analysis of Korean patients with Marfan syndrome: Possible ethnic differences in clinical manifestation. Clinical Genetics, 77(2), 177–182. 10.1111/j.1399-0004.2009.01287.x [DOI] [PubMed] [Google Scholar]
- Ziganshin, B. A. , Bailey, A. E. , Coons, C. , Dykas, D. , Charilaou, P. , Tanriverdi, L. H. , … Elefteriades, J. A. (2015). Routine genetic testing for thoracic aortic aneurysm and dissection in a clinical setting. Annals of Thoracic Surgery, 100(5), 1604–1611. 10.1016/j.athoracsur.2015.04.106 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data: materials and methods
Figure S1. Confirmation of identified intragenic deletions with MLPA analysis
Table S1. Summary of the genetic features of patients with a variant of unknown significance detected by variant‐calling analysis of 21 H‐TAD genes
Table S2. Summary of the clinical features of the patient with a variant of unknown significance detected by variant‐calling analysis of 21 H‐TAD genes
Table S3. Overview of the genes analysed in this study
Table S4. Tools used to classify variants.