

Abstract
Lipid messengers exert their function on short time scales at distinct subcellular locations, yet most experimental approaches for perturbing their levels trigger cell‐wide concentration changes. Herein, we report on a coumarin‐based photocaging group that can be modified with organelle‐targeting moieties by click chemistry and thus enables photorelease of lipid messengers in distinct organelles. We show that caged arachidonic acid and sphingosine derivatives can be selectively delivered to mitochondria, the ER, lysosomes, and the plasma membrane. By comparing the cellular calcium transients induced by localized uncaging of arachidonic acid and sphingosine, we show that the precise intracellular localization of the released second messenger is crucial for the signaling outcome. Ultimately, we anticipate that this new class of caged compounds will greatly facilitate the study of cellular processes on the organelle level.
Keywords: arachidonic acid, caged compounds, calcium signaling, organelle-specific photorelease, sphingosine
Cellular signaling outcome is a function of messenger and compartment identity.1 Good examples are the function of IP3 as a trigger of calcium transients upon binding to its ER‐located receptor,2 the specific recruitment of cellular effector proteins such as PKC3 or Munc134 to the plasma membrane by diacylglycerol, or the role of PI3P and other lipids as identity‐defining entities on endosomes.5 Despite the obvious need for studies of these molecules in their natural cellular environment, our ability to perturb lipid levels in defined cellular compartments remains severely limited. Genetic perturbations of metabolizing enzymes appear to offer a straightforward approach, yet accurate prediction of the resulting lipid or metabolite profiles remains difficult owing to the existence of many parallel biosynthetic pathways and feedback loops. Highly promising optogenetic6 or chemical dimerizer based (CID) approaches7 typically involve light‐ or small‐molecule‐induced recruitment of an enzyme to generate the desired molecule at the intended subcellular localization, but the available applications for lipid messengers are still limited.6 In contrast, photochemical release of lipid messengers from inactive (caged) precursors offers a much broader substrate scope, but spatial control of the induced concentration bursts by optical means remains highly challenging.8, 9, 10
Ideally, caged compounds should be prelocalized to their target organelles prior to photorelease by dedicated chemical groups to ensure spatially controlled uncaging.11 This strategy has been realized in a limited number of examples by us and others.12, 13, 14, 15 However, its applicability is compromised by the fact that caged compound sets have to be generated by total synthesis for each messenger and the respective subcellular localizations. Addressing this issue, we streamlined the development of caged compounds for organelle‐specific photorelease by synthesizing a coumarin‐based caging group (“click cage”) that can be attached to bioactive molecules and subsequently modified by click chemistry with established organelle‐targeting moieties16 (Scheme 1).
Scheme 1.
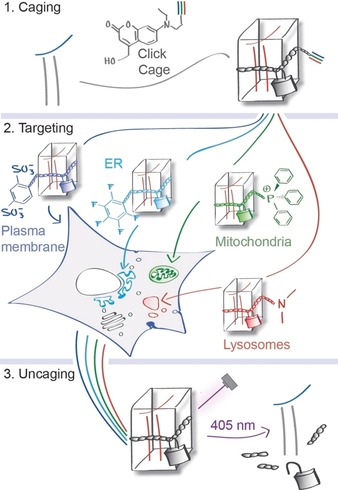
Schematic representation of organelle‐specific uncaging. Lipid messengers are equipped with an alkyne‐containing photocaging group and functionalized with targeting groups that localize them to specific compartments in living cells, where the active compounds can subsequently be photoreleased (uncaged) without initially affecting the messenger levels in neighboring compartments.
We chose to use a dialkylaminocoumarin scaffold as the caging group as this group can be cleaved with 405 nm laser light, which is available on most confocal microscopes. This relatively short wavelength offers the possibility to combine live‐cell uncaging experiments with fluorescence imaging of green, red, and far‐red fluorescent proteins. Furthermore, the alkyl residues of the dialkylamino group can be readily replaced with functional moieties without affecting the photocleavage reaction.17
We generated arachidonic acid and sphingosine derivatives equipped with the new caging group as these cellular messengers have been reported to perform widely varying functions depending on their subcellular localization.12, 15 The click‐cage coumarin 1 was synthesized starting from coumarin intermediate 2. Reductive amination of the aromatic amino group yielded the ethylated coumarin species 3, which was subsequently equipped with an alkyne group using 3‐butynyl tosylate. Removal of the acetate protection group yielded the desired alkyne‐functionalized coumarin alcohol 1. Click‐caged arachidonic acid 6 was obtained by EDC‐mediated esterification, and the organelle‐targeted derivatives were generated by CuI‐mediated cycloaddition (click chemistry) using the cationic triphenylphosphonium azide 7 for the mitochondrial probe 11, the tertiary amino azide 8 for the lysosomal probe 12, the sulfonated azide 9 for the plasma‐membrane‐specific probe 13, and the perfluorinated azide 10 for the ER probe 14. Similarly, click‐caged sphingosine 17 was synthesized by attaching the click‐cage chloroformate 16 to the primary amino group of sphingosine. The mitochondria‐, lysosome‐, plasma‐membrane‐, and ER‐targeted caged sphingosine derivatives 18–21 were generated in a similar fashion as the corresponding arachidonic acid derivatives (Scheme 2).
Scheme 2.
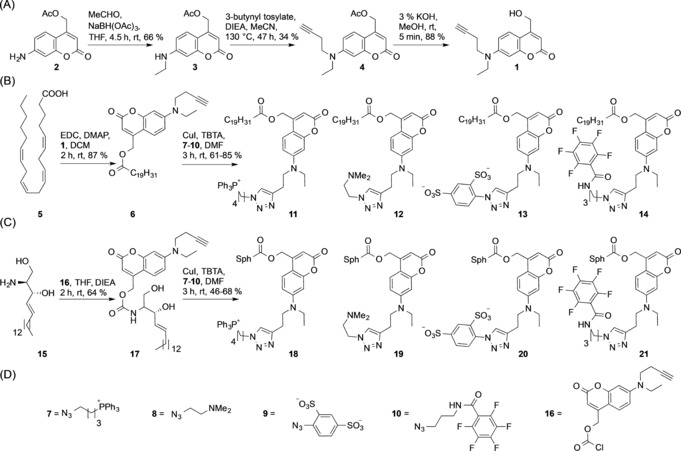
Synthesis of organelle‐targeted caged arachidonic acid and sphingosine derivatives. A) Synthesis of the alkyne‐containing click‐cage photocleavable group 1. B) Synthesis of caged arachidonic acid derivatives for mitochondrial (11), lysosomal (12), plasma membrane (13), and ER photorelease (14). C) Synthesis of caged sphingosine derivatives for mitochondrial (18), lysosomal (19), plasma membrane (20), and ER (21) photorelease. D) Functionalized azido compounds utilized for click reactions in (B) and (C) and click‐cage chloroformate 16 utilized in (C).
We next investigated the photorelease efficiencies of compounds 6, 11–14, and 17–21 in vitro by comparing their performance to the respective published diethylaminocoumarin‐caged arachidonic acid20 and sphingosine21 derivatives 22 and 23 and found that the respective organelle‐targeting caging groups could be cleaved by UV light with similar efficiencies compared to the parent coumarin compounds (Figure S2 and Table S2; see the Supporting Information for details). These findings are in line with the fact that the detected bands in the absorption and fluorescence spectra appear largely unchanged in shape, despite varying intensities for individual compounds (Figure S1).
In order to assess the pre‐uncaging localization of the functionalized caged compounds, we loaded HeLa cells with suitable amounts of caged messengers and the corresponding organelle markers (see the Supporting Information for details), and analyzed the subcellular localization by confocal fluorescence microcopy while controlling for bleed‐through (Figure S3). Both the caged sphingosine (18–21; Figure 1 A) and arachidonic acid (11–14) derivatives (Figure S4 A) did indeed localize to their target organelles. The subcellular localizations of click‐caged sphingosine (17) and arachidonic acid (6) derivatives were found to be very similar to the localization observed for the respective untargeted diethylaminocoumarin‐caged arachidonic acid and sphingosine derivatives 22 and 23 (Figure 1 B and Figure S4 B).
Figure 1.
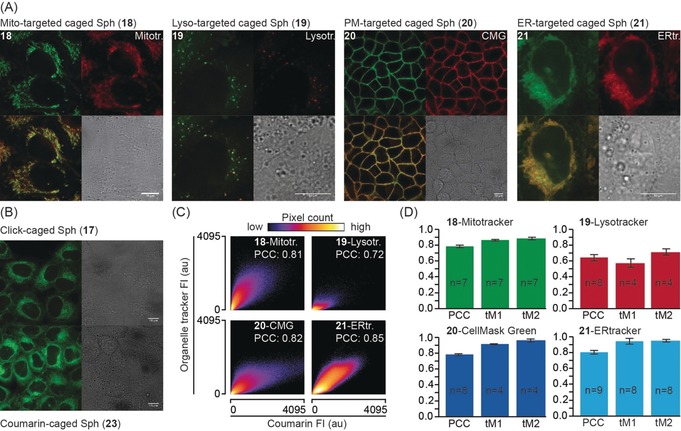
A) Cellular localization of organelle‐targeted caged sphingosine derivatives. The upper left panels depict the localization of the sphingosine probes 18–21 (left to right for mitochondria‐ (18), lysosome‐ (19), plasma‐membrane‐ (20), and ER‐targeted (21) probes). The upper right panels show the respective organelle markers (left to right: mitotracker green, lysotracker green, cell mask green, ER tracker). The lower left panels show overlays of probe and organelle marker fluorescence while the lower right panels depict the corresponding bright‐field images. B) Cellular localization of click‐caged sphingosine 17 and untargeted diethylaminocoumarin‐caged sphingosine 23. Scale bars: 10 μm in all images. C, D) Quantification of the colocalization between caged compounds and organelle markers. 2D histograms of representative images show correlation between pixel intensities in both channels. Source images for the 2D histograms are displayed in Figure S5. Bar graphs show Pearson correlation coefficients (PCC) of non‐thresholded images and Manders correlation coefficients above the autothreshold for both channels, in every case derived from a minimum of four images. Only images that could be thresholded using a standardized automated routine were included for the determination of Manders overlap coefficients to avoid human bias. Error bars indicate standard deviation.
Colocalization analysis using both the Pearson correlation coefficient (PCC) as well as thresholded Manders overlap coefficients confirmed a high degree of colocalization between organelle‐targeted caged lipids and the corresponding organelle markers (Figure 1 C, D and Figure S4C, D) in most cases, indicated by PCC values of 0.72 or higher. Only two probes exhibited slightly lower PCC values, namely the lysosome‐targeted sphingosine probe 19 and the ER‐targeted arachidonic acid probe 14. These effects are likely caused by higher background staining in the case of 19 and the occurrence of insoluble aggregates at the high concentrations necessary for loading cells with 14. Interestingly, these trends were not observed for the lysosome‐targeted arachidonic acid probe 12 and the ER‐targeted sphingosine probe 21, indicating that commonly used organelle‐targeting groups might display varying efficiencies for driving chemically different parent molecules to their respective target compartments, which might cause artifacts in biological experiments if not properly controlled for.
Arachidonic acid as well as sphingosine are prominently involved in cellular signaling cascades, particularly in eliciting intracellular calcium responses.21, 22 We therefore tested whether uncaging of these messengers in distinct organelles would lead to differential modulation of intracellular calcium levels. First, we established compound loading procedures that ensured comparable incorporation into the target organelle membranes. While suitable conditions were readily determined for the plasma‐membrane‐, mitochondria‐, and lysosome‐targeted probes (11–13, 18–20), we found that the ER‐targeted probes were incorporated to a significantly lower extent, likely owing to the reduced solubility caused by the presence of the perfluorinated C6F5 group (Figure S6; see the Supporting Information for details). As this effect was much more pronounced for the arachidonic acid derivative 14 than for the sphingosine probe 21, we did not include 14 in the subsequent calcium experiments. To monitor calcium responses, HeLa cells were loaded with the calcium indicator Fluo‐4‐AM and the respective caged compounds (see the Supporting Information for details) and imaged at room temperature using a Zeiss LSM880 confocal microscope. Uncaging was carried out by a 1.5 s flash illumination of the entire field of view using a 385 nm LED. We detected strikingly different patterns of calcium transients in response to uncaging of arachidonate and sphingosine in distinct cellular membranes. Sphingosine uncaging at the plasma membrane did not trigger calcium transients, whereas uncaging in lysosomes, the ER, or mitochondria elicited robust transients (Figure 2 B). On the other hand, arachidonate uncaging at the plasma membrane and in mitochondria triggered calcium increases, whereas photorelease in lysosomes triggered significantly smaller effects (Figure 2 C). These findings are in line with the localization of the main known intracellular targets of sphingosine (TPC1, in endosomes and lysosomes) and arachidonic acid (GPR40, at the plasma membrane), which are both known to be involved in calcium signaling.21, 23 The reasons for the occurrence of calcium transients after photorelease in mitochondria are less evident and could either be attributed to calcium release from mitochondrial stores in response to the photoinduced changes of mitochondrial lipid composition or to rapid transport of arachidonic acid and sphingosine to their respective sites of action. In contrast to a recently published result,15 we did not find differential calcium signaling patterns for mitochondrial and untargeted sphingosine photorelease. This might be due to differences in the utilized cell lines and/or final concentrations after photoactivation.
Figure 2.
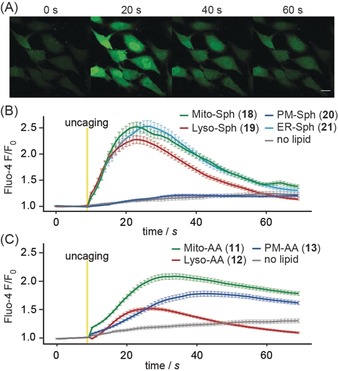
A) Time lapse montage of Fluo‐4 responses after uncaging of sphingosine from the mitochondria‐targeted probe 18. Scale bar: 20 μm. B, C) Mean normalized Fluo‐4 intensities after uncaging of sphingosine (B) and arachidonic acid (C) from probes 11–13 and 18–21. Error bars represent SEM, yellow bars indicate uncaging events.
Crucially, using whole‐field‐of‐view flash illumination uncaging allowed us to analyze single‐cell Ca2+ responses on large numbers of cells (Figure S7). This enabled us to account for non‐normal distributions of cellular responses, which would complicate analyses of calcium signaling solely based on averaged traces. In the current dataset, such distributions are not observed. For example, the lower average calcium signals after photorelease of arachidonic acid in lysosomes corresponded to a global shift of the observed distribution towards smaller single‐cell responses compared to photorelease in mitochondria or at the plasma membrane (Figure S7 B).
Taken together, we have reported a method for the photorelease of lipid messengers in an organelle‐specific manner based on the novel “click cage” concept. The underlying modular design allowed us to rapidly generate multiple photocaged messengers for applications in different organelles by click‐chemistry‐mediated functionalization with organelle‐targeting groups. Using calcium signaling as an example, we demonstrated that photorelease of sphingosine and arachidonic acid in various organelles caused strikingly different signaling patterns. Organelle‐targeted caged lipid messengers will enable studies of cellular signaling with much improved spatial precision, and we anticipate that photochemical probes of this type will play an important role as discovery tools in cell‐biological studies.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank the Mass Spectrometry Facility and Light Microscopy Facility of MPI‐CBG for their support. Furthermore, we acknowledge Hanna Schmid, Nils Krüger, Cristina Jiménez López, and Annett Lohmann for support during the project. A.N. gratefully acknowledges financial support by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (758334 ASYMMEM) and the Deutsche Forschungsgemeinschaft (DFG) as a member of the TRR83 consortium. D.H. gratefully acknowledges financial support by the Deutsche Forschungsgemeinschaft (DFG) as a member of the TRR83 and TRR186 consortia.
N. Wagner, M. Stephan, D. Höglinger, A. Nadler, Angew. Chem. Int. Ed. 2018, 57, 13339.
Contributor Information
Dr. Doris Höglinger, Email: doris.hoeglinger@bzh.uni-heidelberg.de.
Dr. André Nadler, Email: nadler@mpi-cbg.de.
References
- 1. Groves J. T., Kuriyan J., Nat. Publ. Gr. 2010, 17, 659–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mikoshiba K., J. Neurochem. 2007, 102, 1426–1446. [DOI] [PubMed] [Google Scholar]
- 3. Newton A. C., Am. J. Physiol. Endocrinol. Metab. 2010, 298, E395–E402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bauer C. S., Woolley R. J., Teschemacher A. G., Seward E. P., J. Neurosci. 2007, 27, 212–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gruenberg J., Curr. Opin. Cell Biol. 2003, 15, 382–388. [DOI] [PubMed] [Google Scholar]
- 6. Spangler S. M., Bruchas M. R., Curr. Opin. Pharmacol. 2017, 32, 56–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Voß Stephanie, Klewer L., Wu Y. W., Curr. Opin. Chem. Biol. 2015, 28, 194–201. [DOI] [PubMed] [Google Scholar]
- 8. Höglinger D., Nadler A., Schultz C., Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2014, 1841, 1085–1096. [DOI] [PubMed] [Google Scholar]
- 9. Ellis-Davies G. C. R., Nat. Methods 2007, 4, 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Nadler A., Reither G., Feng S., Stein F., Reither S., Müller R., Schultz C., Angew. Chem. Int. Ed. 2013, 52, 6330–6334; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 6455–6459. [Google Scholar]
- 11. Muro E., Atilla-Gokcumen G. E., Eggert U. S., Mol. Biol. Cell 2014, 25, 1819–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Nadler A., Yushchenko D. A., Müller R., Stein F., Feng S., Mulle C., Carta M., Schultz C., Nat. Commun. 2015, 6, 10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Horinouchi T., Nakagawa H., Suzuki T., Fukuhara K., Miyata N., Bioorg. Med. Chem. Lett. 2011, 21, 2000–2002. [DOI] [PubMed] [Google Scholar]
- 14. Leonidova A., Pierroz V., Rubbiani R., Lan Y., Schmitz A. G., Kaech A., Sigel R. K. O., Ferrari S., Gasser G., Chem. Sci. 2014, 5, 4044–4056. [Google Scholar]
- 15. Feng S., Harayama T., Montessuit S., David F. P. A., Winssinger N., Martinou J. C., Riezman H., eLife 2018, 7, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Xu W., Zeng Z., Jiang J. H., Chang Y. T., Yuan L., Angew. Chem. Int. Ed. 2016, 55, 13658–13699; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 13858–13902. [Google Scholar]
- 17. Lin Q., Bao C., Fan G., Cheng S., Liu H., Liu Z., Zhu L., J. Mater. Chem. 2012, 22, 6680–6688. [Google Scholar]
- 18. Rostovtsev V. V., Green L. G., Fokin V. V., Sharpless K. B., Angew. Chem. Int. Ed. 2002, 41, 2596–2599; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2002, 114, 2708–2711. [Google Scholar]
- 19. Tornøe C. W., Christensen C., Meldal M., J. Org. Chem. 2002, 67, 3057–3064. [DOI] [PubMed] [Google Scholar]
- 20. Carta M. et al., Neuron 2014, 81, 787–799. [DOI] [PubMed] [Google Scholar]
- 21. Höglinger D., Haberkant P., Aguilera-Romero A., Riezman H., Porter F. D., Platt F. M., Galione A., Schultz C., eLife 2015, 4, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Meves H., Br. J. Pharmacol. 2008, 155, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Itoh Y., Kawamata Y., Harada M., Kobayashi M., Fujii R., Fukusumi S., Ogi K., Hosoya M., Tanaka Y., Uejima H., Tanaka H., Maruyama M., Satoh R., Okubo S., Kizawa H., Komatsu H., Matsumura F., Noguchi Y., Shinohara T., Hinuma S., Fujisawa Y., Fujino M., Nature 2003, 422, 173–176. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary