

ABSTRACT
Background: The clinical demarcation of the syndrome progressive myoclonus ataxia is unclear, leading to a lack of recognition and difficult differentiation from other neurological syndromes.
Objectives: The objective of this study was to apply a refined definition of progressive myoclonus ataxia and describe the clinical characteristics in patients with progressive myoclonus ataxia and with isolated cortical myoclonus.
Methods: A retro‐ and prospective analysis was performed in our tertiary referral center between 1994 and 2014. Inclusion criteria for progressive myoclonus ataxia patients were the presence of myoclonus and ataxia with or without infrequent (all types, treatment responsive) epileptic seizures. Inclusion criteria for isolated cortical myoclonus was the presence of isolated cortical myoclonus. Clinical and electrophysiological characteristics data were systematically scored.
Results: A total of 14 progressive myoclonus ataxia patients (males, 7; females, 7), median age 14.5 years, and 8 isolated cortical myoclonus patients (males, 2; females, 6), median age 23.5 years, were identified. In 93% of the progressive myoclonus ataxia patients, ataxia started first (median 2 years) followed by myoclonus (4 years) and finally infrequent epilepsy (9.3 years), with a progressive course in 93%. In 64% of the progressive myoclonus ataxia patients, a genetic underlying etiology was identified, including 3 not earlier reported causative progressive myoclonus ataxia genes. In isolated cortical myoclonus patients, myoclonus started at (median) 12 years with progression over time in 63% and a single epileptic seizure in 1 patient. No genetic causes were identified.
Conclusion: Using a refined definition, we could create a rather homogenous progressive myoclonus ataxia group. Patients with isolated cortical myoclonus have a different course and do not appear to evolve in progressive myoclonus ataxia. The refined progressive myoclonus ataxia definition is a successful first step toward creating a separate syndrome for both clinical practice and future genetic research. © 2018 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: ataxia, isolated cortical myoclonus, myoclonus, progressive myoclonus ataxia, Ramsay Hunt
Progressive myoclonus ataxia (PMA) is a rare disorder and defined in the literature as progressive ataxia and myoclonus, without prominent decline in cognitive functions, and with or without infrequent epileptic seizures.1 PMA was previously known as the Ramsay Hunt syndrome, named after sir James Ramsay Hunt who first described this triad of symptoms in 1921.2 PMA can be acquired as in celiac disease but is mainly caused by genetic disorders, including inborn errors of metabolism and neurodegenerative disorders, such as myoclonic epilepsy with ragged red fibers syndrome and North Sea progressive myoclonus epilepsy.3 Despite the suspicion of a genetic cause, only in less than 50% a causative genetic defect can be identified.4 Identification of an etiological cause is important as some disorders require specific treatment and also with regard to the patient's prognosis and appropriate genetic counseling.
As a clear demarcation of clinical features of PMA is absent, problems arise regarding the recognition and differentiation of PMA from other neurological syndromes. PMA can especially be hard to distinguish from the related but more extensively studied progressive myoclonus epilepsy (PME) syndrome. PMA and PME syndromes are characterized by the combination of myoclonus, ataxia, and epilepsy and mainly differ in the severity of specific features. By definition, PME patients compared to PMA present with more prominent (frequent) epilepsy and have in addition, severe cognitive deterioration.1 It is unclear where to draw the line between infrequent and frequent epilepsy and if (mild) cognitive decline should be part of the PMA criteria. In the past, the difficulties in discriminating PME and PMA led to a discussion between experts from the epilepsy field arguing for lumping, whereas movement disorder experts favored splitting.5, 6 Recently, this discussion restarted in the disorder North Sea PME caused by a mutation in the Golgi SNAP receptor complex 2 gene (GOSR2) gene, which is described both as PMA and PME in the literature.7, 8
The aim of the present study is to apply refined clinical criteria for PMA in a Dutch cohort of patients presented in a tertiary movement disorder clinic. The criteria include symptoms of ataxia and myoclonus and a clear definition of infrequent epilepsy, comprising of treatment‐responsive epilepsy enfolding all seizure types. We do not define the decline in cognitive functions. PMEs are known to progress into refractory epilepsy (frequent epilepsy) and can even result in death as a result of status epilepticus; therefore, the response on appropriate antiepileptic therapy can be used as feature of infrequent epilepsy and hence PMA.9 Compared to previous criteria of PMA in which only tonic‐clonic seizures were taken into account, we included all seizure types because difference in type is not associated with difference in health‐related quality of life (HRQoL) and should not determine severity of epilepsy.10, 11
In this study, we focus on the clinical and electrophysiological features and the detected etiology and expect that this will lead to a homogenous group of PMA patients with different genetic causes. At the onset of symptoms, the severity and course of PMA are not always clear in an individual patient. We therefore also include patients with isolated cortical myoclonus (ICM) as little is known in the literature of this group of patients. By including ICM patients we can study if the course of ICM and PMA is similar or different and determine if ICM can be a potential predecessor of the devastating PMA syndrome. The clustering of rare patients facilitates future genetic research with the ultimate goal of insight in the pathology and increase the diagnostic yield in patients with suspected genetic causes of PMA.
Methods
A retrospective analysis was performed of PMA and ICM patients who visited the University Medical Center of Groningen (Groningen, The Netherlands) in the period 1994 to 2014. In addition, we prospectively included PMA and ICM patients visiting the outpatient clinic between June 2014 and January 2017. The medical ethical committee of the University Medical Center of Groningen approved this study (no. 2014.256).
To identify patients presenting with PMA, the terms “myoclonus” AND “ataxia” were used when searching the hospital's electronic database. Patients of all ages were included. Inclusion criteria for PMA syndrome were based on the following criteria: (1) the presence of myoclonus, (2) the presence of ataxia, and (3) no or infrequent epilepsy (treatment‐responsive epilepsy including all types of epileptic seizures). These criteria were partly derived from the most recent definition stated by the Marseille Consensus Group in 1990. We changed the criteria in three ways. We left out progression of core features and deterioration of cognitive function as essential criteria. The third difference is the specification of infrequent epilepsy (Table 1).1
Table 1.
A short overview of criteria included in the refined definition we proposed in this study compared to the definition stated by the Marseille Consensus Group
| Criteria | Refined definition, 2017 | Marseille Group definition, 1990 |
|---|---|---|
| Myoclonus | ✓ | ✓ |
| Ataxia | ✓ | ✓ |
| Infrequent epilepsy | Treatment‐responsive All types of seizures | ✓ |
| Mild cognitive decline | ✗ | ✓ |
| Progressive course | ✗ | ✓ |
To identify ICM patients, a retrospective analysis was performed using the Clinical Neurophysiological database for those in whom video polymyography was part of the routine diagnostic workup. The electrophysiological diagnosis of cortical myoclonus required a burst duration below 100 ms and is supported by (1) the presence of action‐induced or stimulus‐sensitive myoclonic jerks, (2) a (multi)focal distribution predominantly affecting the face and distal limbs, (3) the presence of negative myoclonus, (4) confirmation of the cortical origin with back averaging, coherence analysis, and/or somatosensory evoked potential.12
We systematically scored the following clinical characteristics: age, gender, age at onset, age at examination, family history, progression of motor symptoms, onset of myoclonic/ataxic symptoms, distribution of myoclonus/ataxia, stimulus sensitivity and provoking factors of myoclonus, frequency of epilepsy, severity of cognitive decline, psychiatric symptoms, oculomotor abnormalities, and underlying etiology. The severity of cognitive decline was based on the expert opinion stated by the neurologists in clinical notes. The following features of polymyography, if applied, were also systematically scored: burst duration, presence of negative myoclonus, and, if present, back averaging, coherence analysis, and somatosensory evoked potential.
Statistical Analysis
Using IBM SPSS Statistics version 23 (Armonk, New York, USA), descriptive statistics were used to study discriminative features between patients presenting with PMA and patients with ICM.
Results
Patients
A total of 38 patients were identified suffering from myoclonus and ataxia, of which 14 patients were considered to adhere to the refined definition of PMA. A total of 30 patients were identified with cortical myoclonus, 8 of which with ICM. Patients were excluded in case insufficient information was available (n = 5), another movement disorder (dystonia, chorea, tics, tremor and parkinsonian signs) was present (n = 19), or an acquired etiology was causative of the movement disorder (n = 8). Following the diagnostic algorithm of myoclonus, an acquired etiology was ruled out if the use of medication or toxic agents, laboratory test, and MRI were not explanatory for the myoclonic jerks.13 Furthermore, the presence of prominent epilepsy was reason for exclusion in case of refractory epilepsy (frequent epilepsy; n = 4) or in case of epilepsy being reason for initial referral (n = 10), as this study is from the perspective of the movement disorder clinician (Supplemental Figure 1).
Table 2 gives an overview of the clinical characteristics. An extensive summary of the individual patients is given in Supplemental Table 1.
Table 2.
Clinical characteristics of progressive myoclonus ataxia and isolated cortical myoclonus
| Clinical characteristics | Progressive myoclonus ataxia | Isolated cortical myoclonus |
|---|---|---|
| Patients, n, F/M | 14, (7/7) | 8, (6/2) |
| Age,a IQR | 14.5 (7.3‐31) | 23.5 (18.8‐25) |
| Age of onset disease,a IQR | 2 (0.9‐2.3) | 12 (7‐18) |
| Progressive course | 13 | 5 |
| Duration of disease,a IQR | 13.5 (6.3‐27) | 10.5 (5.3‐16) |
| Duration of follow‐up in years,a IQR | 8.8 (2.3‐18.2) | 3.1 (0.7‐4.2) |
| Myoclonus | ||
| Onset acute/gradual/unknown | 0/13/1 | 2/5/1 |
| Distribution (F/A/UL/LL) | 7/7/14/8 | 3/2/8/4 |
| Action induced | 11 | 5 |
| Ataxia | ||
| Onset acute/gradual | 0/14 | ‐ |
| Distribution (A/UL/LL) | 8/13/11 | ‐ |
| Ataxic gait | 14 | ‐ |
| Dysarthria | 10 | ‐ |
| Infrequent epilepsy | 4 | 1 |
| Cognitive decline (mild/severe) | 8 (8/0) | 2 (2/0) |
| Psychiatric symptoms | 7 | 1 |
| Oculomotor abnormalities | 9 | 3 |
| Positive family history | 2 | 1 |
| Etiology | ||
| North Sea PME | 4 | 0 |
| SCA 13 | 2 | 0 |
| SCA 5 | 1 | 0 |
| HADDS | 1 | 0 |
| Nonprogressive congenital cerebellar ataxia | 1 | 0 |
| Unknown | 5 | 8 |
A, axial area; F, face; HADDS, hypotonia ataxia and delayed development syndrome; IQR, interquartile range; LL, lower limbs; PME, progressive myoclonus epilepsy; SCA, spinocerebellar ataxia; UL, upper limbs.
Median.
PMA
The 14 PMA patients (males, 7; females, 7) had a median age of 14.5 years (interquartile range [IQR] 7.3‐31). In 13 patients, the presenting symptom was reported, of whom 11 patients started with ataxic symptoms, 1 patient with myoclonic jerks, and 1 with simultaneously ataxic and myoclonic symptoms. The ataxic symptoms started at the median age of 2 years (IQR 1‐2.3). The onset was gradual, with both upper and lower limb involvement in 11, an ataxic gait in 14, and dysarthria in 10 cases. At the median age of 4 years (IQR 2.3‐6) myoclonus started with a gradual onset, worsening during action and most commonly affecting the upper limbs. Myoclonus was stimulus sensitive (somatosensory and auditory) in 4 cases, and negative myoclonus was seen in 6 cases. In 4 patients, infrequent epilepsy was observed starting at a median age of 9.3 years (IQR 6‐11.4). All 4 patients with seizures were therapy responsive and showed a decrease in frequency, resulting in 0 to 1 seizures per year, after the optimal dose of antiepileptic therapy was initiated. Figure 1 illustrates the clinical course of the 14 patients based on our definition of PMA.
Figure 1.
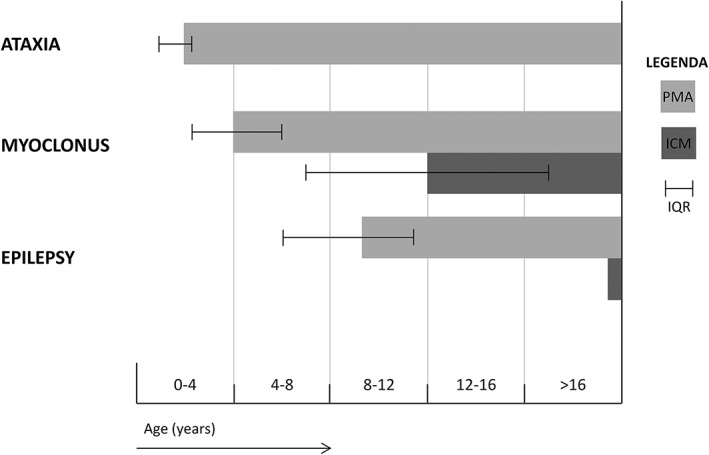
The clinical course of progressive myoclonus ataxia (PMA) and isolated cortical myoclonus (ICM). The median age of onset, with the interquartile range (IQR), is displayed for ataxia, myoclonus, and epilepsy, represented by the start of the bar. The length of the bar represents duration of disease.
A decline of cognitive function was seen in 8 of 14 PMA patients, all of whom showed mild deterioration. Associated psychiatric problems were present in 7 cases: an autism spectrum disorder in 3, attention deficit hyperactivity disorder in 2, and behavioral problems without a diagnosis in 2 cases. In 9 cases, saccadic oculomotor abnormalities were described. A positive family history was present in 2 unrelated patients. The motor symptoms deteriorated over time in 13 patients, reported by patients as worsening of daily activity performance, during a median follow‐up period of 8.8 years (IQR 2.3‐18.2). During this period, 2 patients became wheelchair bound and 3 patients used walking aids in daily life.
Polymyography was performed in 8 patients showing an electromyography (EMG) burst duration of <100 milliseconds, compatible with a cortical origin of the myoclonus. Additional electrophysiological tests were obtained in a limited number of these patients, cortico‐coherence analysis was positive in 2/3 cases, and giant somatosensory evoked potential (SEP) was present in 0/1 cases, supporting the cortical origin. EEG was performed in 11 patients. In 4 cases, epileptic abnormalities (ie, multifocal spikes and sharp waves, with generalization) were observed, and in 5 a slow background rhythm was observed. In reaction to a visual stimulation, a photoparoxysmal response was present in 1 patient, combined with cortical myoclonus. These findings are compatible with cortical reflex myoclonus. The anatomical origin of myoclonus in the remaining cases was not further specified and could not be established based on the retrospective data.
In 9 patients (64%) with PMA, an underlying etiological cause was identified (see Supplemental Table 1). The diagnoses included North Sea PME (n = 4), spinocerebellar ataxia 5 (n = 1) and spinocerebellar ataxia 13 (n = 2), hypotonia ataxia and delayed development syndrome (n = 1), and nonprogressive congenital cerebellar ataxia (n = 1). In the 5 remaining cases, no cause could be identified despite the application of the novel diagnostic myoclonus algorithm, including the performance of next generation sequencing (NGS).13 The 2 deceased patients had died before the myoclonus panel was introduced.
ICM
A total of 8 patients (males, 2; females, 6) with a median age of 23.5 years (IQR 18.8‐25) were diagnosed with ICM. Myoclonus was first observed at a median age of 12 years (IQR 7‐18; Figure 1), of which 5 cases showed a gradual onset and 2 (sub)acute; in 1 patient onset was unknown. The myoclonic jerks were most commonly present in the upper limbs. In 5 cases the jerks worsened during action. Two patients showed somatosensory stimulus sensitivity, and in 5 cases negative myoclonus was observed. One patient had a generalized tonic‐clonic seizure at age 40 years.
Mild cognitive decline was observed in 2 patients, and 1 patient reported depressive symptoms. In 3 patients, abnormal vertical eye movements were seen: an “around‐the‐house” sign in 2 cases and saccadic abnormalities in 1 case. A positive family history was present in 2 patients. Five patients showed progression of myoclonus during a median follow‐up of 3.1 years (IQR 0.7‐4.2). None of the patients showed deterioration in walking abilities.
Polymyography in all 8 cases showed a burst duration of <100 milliseconds. Coherence analysis showed a cortical origin in 1/4 cases, back averaging in 0/3, and giant SEP was present in 1/6 cases. EEG showed both mild epileptic abnormalities (ie, focal spikes) and mild slow background rhythm in 1 patient; solely epileptic abnormalities (ie, generalized spike and wave complexes) in 2 patients; and solely a slow background rhythm in another patient.
No underlying genetic cause could be identified in any of the patients presenting with ICM despite thorough diagnostic workup. The diagnostic algorithm for myoclonus has been applied in all cases including NGS. In 6 cases, NGS was negative, and in 2 it is pending.
Discussion
In this study, we studied a group of 14 patients adhering to our refined definition of PMA and detected a homogenous phenotype with a fixed order of symptoms. PMA in patients started with ataxia at a young age (2 years; IQR 1‐2.3), with subsequent occurrence of mainly cortical myoclonus (4 years; IQR 2.3‐6), followed by infrequent (treatment responsive) epilepsy (9.3 years; IQR 6‐11.4). The motor symptoms were progressive in the majority during a median follow‐up period of 8.8 years (IQR 2.3‐18.2). Mild cognitive deterioration was detected in 8 (57%) patients. In the PMA group, a genetic diagnose was made in 64% of the patients. In the patients with ICM, the age at onset of myoclonus is later in early puberty (12 years; IQR 7‐18). In the ICM patients, no genetic cause was detected.
With the help of our suggested refined definition, we were able to create a homogenous group of PMA patients. The underlying etiology in 64% of the PMA patients is remarkably higher than the previously reported 50%.4 This high percentage of identified causes is likely as result of the recent increase of known genes and the application of NGS. An additional determinant could be the strict and less elaborate PMA definition by the specification of infrequent epilepsy and, however less probable, omission of cognitive decline. The increased percentage of identified underlying etiologies does not preclude the discovery of new causal molecular defects as 36% of our PMA cases remain without an etiological diagnosis despite excessive diagnostic investigations. The approach of clustering patients with homogenous clinical phenotypes to discover new genes has proven to be successful in the past by finding a mutation in GOSR2 causal of North Sea PME.7 The high impact of identifying such a molecular defect can be observed as the mutation in GOSR2 accounts for 29% of our PMA cases. Only by clustering homozygous clinical phenotypes in our expertise center have we already identified 3 etiologies and 4 de novo mutations that broaden the differential diagnosis of PMA for they had not been previously associated with this syndrome. These include spinocerebellar ataxia 5, hypotonia ataxia and delayed development syndrome, and nonprogressive congenital cerebellar ataxia.
Knowledge regarding the clinical course of PMA patients is essential to guide clinicians in daily practice. In patients with childhood‐onset ataxia, physicians should consider the syndrome of PMA in their differential diagnosis. In the application of childhood‐onset ataxia molecular diagnostic tests, disorders associated with PMA should be included in sequencing panels or diagnostic filters. In addition, clinicians should actively search for the myoclonic jerks in these patients. The jerks are easily overlooked and sometimes hard to distinguish from ataxic symptoms, whereas myoclonus can be medically treated.14 Hopefully this insight into the clinical course will result in the syndrome of PMA to be better recognized and treated and the diagnostic delay for these patients shortened.
Associated symptoms were frequently observed in PMA patients and mainly included psychiatric symptoms and oculomotor abnormalities. Psychiatric comorbidities have never been scored systematically in a cohort of PMA patients but are increasingly recognized as clinical features of many genetically determined movement disorders and generalized epilepsies.15, 16 Furthermore, saccadic eye movement abnormalities have been described probably related to the cerebellar ataxia.17 Associated features of underlying etiologies, which were previously described in literature, were confirmed in our patients as well. The 4 patients identified with GOSR2 presented with areflexia and deformities of the feet, although no scoliosis was seen in our cohort.18 Hypotonia and cognitive decline were observed as associated features of PMA caused by a mutation in early B cell factor 3 gene (EBF3), and speech delay was observed in the patient with a mutation in the nonprogressive congenital cerebellar ataxia gene.19, 20
From our data it appears unlikely that ICM is the predecessor of PMA as only 1 of the PMA patients started with myoclonic jerks. Furthermore, the age at onset is later (12 years; IQR 7‐18) with a rather benign course in the ICM group when compared with the PMA group. The age of onset in the ICM patients resembled that of the most common form of PME, Unverricht‐Lundborg disease. A recent study describing 77 Unverricht‐Lundborg disease patients showed a mean age of onset of 11.4 ± 3.2 years.21 Epilepsy is usually ground for initial referral.22 We focused on patients referred for movement disorders and excluded patients with epilepsy as initial reason for referral. Therefore, it cannot be excluded that the 8 ICM patients in our study are predecessors of PME. However, it appears highly unlikely because in only 1 of the ICM patients, 1 epileptic seizure occurred during the follow‐up of 3.2 years (IQR 0.6‐4.5).
Our study has certainly some limitations. To validate the new proposed criteria for PMA it is important to compare the novel criteria to the former criteria and appraise their value in distinguishing PMA from other neurological syndromes, focusing primarily on PME. In the current study, PME patients were not included as our cohort was assembled from the perspective of the movement disorder specialist. Epilepsy is usually the initial reason for referral in PME patients,22 and therefore, including a cohort of patients from an epilepsy clinic is important for future validation studies of PMA criteria. Furthermore, the number of patients in our study was small, with the majority retrospectively identified at a single tertiary referral center. This warrants a larger prospective multinational study to increase the sample size to further delineate the clinical syndrome of PMA with standardized evaluation, phenotyping the movement disorders in detail and evaluation of the diagnostic yield of molecular testing.
In conclusion, our proposed refined definition of PMA creates a homogenous group of patients with a high number of genetic causes, including 3 causes of PMA not previously reported. Patients with ICM have a different course and do not appear to evolve in the PMA syndrome. The suggested definition of PMA needs further validation, but appears to enable clinicians to recognize PMA and make a diagnosis earlier in life and is also a tool to facilitate future genetic research.
Author Roles
1) Research project: A. Conception, B. Organization, C. Execution; 2) Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3) Manuscript: A. Writing of the first draft, B. Review and Critique.
S.V.: 1B, 1C, 3A, 3B
R.Z.: 1A, 1B, 1C, 3B
J.W.J.E.: 1C, 3B
C.E.B.: 1B, 1C, 3B
T.J.K.: 1A, 1B, 1C, 3B
M.A.J.T.: 1A, 1B, 1C, 3B
Full financial disclosure for the previous 12 months
T.J.K. declares no conflict of interests. He received grants from Metabolic Power Fundation and Metakids foundation and Ride4Kids foundation (all nonprofit) for studying movement disorders in metabolic diseases. He received research grants from Actelion pharmaceuticals (profit) for studying movement disorders in Niemann‐Pick‐C disease and received an honorarium for presenting at a sponsored meeting on Niemann‐Pick‐C. M.A.J.T. is funded by Fonds Nuts‐Ohra, Prinses Beatrix Fonds, Gossweiler Foundation, Fonds Psychische gezondheid, Phelps Stichting, Hersenstichting, Stichting Beatrix kinderziekenhuis, Stichting Wetenschapsfonds Dystonie Vereniging, the Parkinson patienten vereniging and unrestricted educational grants from Ipsen, Allergan, Merz, Acthelion, and Medtronic. S.V., R.Z., J.W.J.E, and C.E.B. have nothing to disclose.
Supporting information
Supplement Figure 1
Supplement table 1. Extensive description of clinical characteristics and mutation type.
Relevant conflicts of interests/financial disclosures: Nothing to report.
References
- 1. Aicardi J, Andermann E, Andermann F, et al. Classification of progressive myoclonus epilepsies and related disorders. Ann Neurol 1990;28:113‐116. [DOI] [PubMed] [Google Scholar]
- 2. Hunt JR. Dyssynergia cerebellaris myoclonica—primary atrophy of the dentate system: a contribution to the pathology and symptomatology of the cerebellum. Brain 1921;44:490‐538. [Google Scholar]
- 3. Zutt R, Tijssen MAJ, Elting JW. Myoclonus In: Wolters E, ed. Parkinson Disease and Other Movement Disorders. 1st ed. Amsterdam, The Netherlands: VU University Press; 2014:513‐533. [Google Scholar]
- 4. Borg M. Symptomatic myoclonus. Neurophysiol Clin 2006;36:309‐318. [DOI] [PubMed] [Google Scholar]
- 5. Andermann F, Berkovic S, Carpenter S, Andermann E. The Ramsay Hunt syndrome is no longer a useful diagnostic category. Mov Disord 1989;4:13‐17. [DOI] [PubMed] [Google Scholar]
- 6. Marsden CD, Obeso JA. Viewpoints on the Ramsay Hunt syndrome. The Ramsay Hunt syndrome is a useful clinical entity. Mov Disord 1989;4:6‐12. [DOI] [PubMed] [Google Scholar]
- 7. Corbett MA, Schwake M, Bahlo M, et al. A mutation in the Golgi Qb‐SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am J Hum Genet 2011;88:657‐663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Egmond ME, Verschuuren‐Bemelmans CC, Nibbeling EA, et al. Ramsay Hunt syndrome: clinical characterization of progressive myoclonus ataxia caused by GOSR2 mutation. Mov Disord 2014;29:139‐143. [DOI] [PubMed] [Google Scholar]
- 9. Andermann F, Rouleau G, Lehesjoki E. Progressive myoclonus epilepsy: the gene‐empowered era. Epilepsia 2016;18:1‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Taylor RS, Sander JW, Taylor RJ, Baker GA. Predictors of health‐related quality of life and costs in adults with epilepsy: a systematic review. Epilepsia 2011;52:2168‐2180. [DOI] [PubMed] [Google Scholar]
- 11. Marsden CD, Harding AE, Obeso JA, Lu C‐S. Progressive myoclonic ataxia (the Ramsay Hunt syndrome). Arch Neurol 1990;47:1121‐1125. [DOI] [PubMed] [Google Scholar]
- 12. Zutt R, Elting JW, van der Hoeven JH, Lange F, Tijssen MAJ. Myoclonus subtypes in tertiary referral center. Cortical myoclonus and functional jerks are common. Clin Neurophysiol 2016;128:253‐259. [DOI] [PubMed] [Google Scholar]
- 13. Zutt R, van Egmond ME, Elting JW, et al. A novel diagnostic approach to patients with myoclonus. Nat Rev Neurol 2015;11:687‐697. [DOI] [PubMed] [Google Scholar]
- 14. Van Egmond ME, Elting JWJ, Kuiper A, et al. Myoclonus in childhood‐onset neurogenetic disorders: the importance of early identification and treatment. Eur J Paediatr Neurol 2015;19:726‐729. [DOI] [PubMed] [Google Scholar]
- 15. Peall KJ, Lorentzos MS, Heyman I, et al. A review of psychiatric co‐morbidity described in genetic and immune mediated movement disorders. Neurosci Biobehav Rev 2017;80:23‐35. [DOI] [PubMed] [Google Scholar]
- 16. Loughman A, Bendrups NA, D'Souza WJ. A systematic review of psychiatric and psychosocial comorbidities of genetic generalised epilepsies (GGE). Neuropsychol Rev 2016;26:364‐375. [DOI] [PubMed] [Google Scholar]
- 17. Winchester S, Singh PK, Mikati MA. Ataxia. Handb Clin Neurol 2013;112:1213‐1217. [DOI] [PubMed] [Google Scholar]
- 18. Dibbens LM, Rubboli G. GOSR2: a progressive myoclonus epilepsy gene. Epileptic Disord 2016;18:111‐114. [DOI] [PubMed] [Google Scholar]
- 19. Chao HT, Davids M, Burke E, et al. A syndromic neurodevelopmental disorder caused by de novo variants in EBF3. Am J Hum Genet 2017;100:128‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thevenon J, Lopez E, Keren B, et al. Intragenic CAMTA1 rearrangements cause non‐progressive congenital ataxia with or without intellectual disability. J Med Genet 2012;49:400‐408. [DOI] [PubMed] [Google Scholar]
- 21. Franceschetti S, Michelucci R, Canafoglia L, et al. Progressive myoclonic epilepsies Definitive and still undetermined causes. Neurology 2014;82:405‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Crespel A, Ferlazzo E, Franceschetti S, et al. Unverricht‐Lundborg disease. Epileptic Disord 2016;18:28‐37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement Figure 1
Supplement table 1. Extensive description of clinical characteristics and mutation type.