

Abstract
For equimolar mixtures of ionic liquids with imidazolium‐based cations of very different electronic structure, we observe very pronounced surface enrichment effects by angle‐resolved X‐ray photoelectron spectroscopy (XPS). For a mixture with the same anion, that is, 1‐methyl‐3‐octylimidazolium hexafluorophosphate+1,3‐di(methoxy)imidazolium hexafluorophosphate ([C8C1Im][PF6]+[(MeO)2Im][PF6]), we find a strong enrichment of the octyl chain‐containing [C8C1Im]+ cation and a corresponding depletion of the [(MeO)2Im]+ cation in the topmost layer. For a mixture with different cations and anions, that is, [C8C1Im][Tf2N]+[(MeO)2Im][PF6], we find both surface enrichment of the [C8C1Im]+ cation and the [Tf2N]− (bis[(trifluoromethyl)sulfonyl]imide) anion, while [(MeO)2Im]+ and [PF6]− are depleted from the surface. We propose that the observed behavior in these mixtures is due to a lowering of the surface tension by the enriched components. Interestingly, we observe pronounced differences in the chemical shifts of the imidazolium ring signals of the [(MeO)2Im]+ cations as compared to the non‐functionalized cations. Calculations of the electronic structure and the intramolecular partial charge distribution of the cations contribute to interpreting these shifts for the two different cations.
Keywords: angle-resolved X-ray photoelectron spectroscopy (ARXPS), density functional theory, electronic structure, ionic liquid mixture, surface science
1. Introduction
Neat ionic liquids (ILs) have been studied extensively since they are promising candidates for different applications,1 e. g. in the fields of catalysis,2, 3, 4 fuel cells,5,6 lithium ion batteries,7, 8, 9 solvents,10,11 lubricants,12,13 sensors,14,15 and gas separation technologies,15, 16, 17 to name only a few. In many of these applications, the surface of the IL plays a decisive role, because it is its interface to the environment. Therefore, tailoring surface properties e. g. by choosing specific anion‐cation combinations being preferentially present at the surface is of high relevance. Recently, research started to focus also on IL mixtures, which enable an even higher number of potential anion and cation combinations.1,18 By gaining knowledge about the interface properties of various anion and cation combinations, we hope to get more insight into the understanding of the outermost surface layer of IL mixtures.
While a number of investigations exists for a variety of IL mixtures concerning bulk physical properties such as viscosity, density, structure, and conductivity,19,20 surface science studies on IL mixtures are less common. The surface composition of binary IL mixtures with non‐functionalized imidazolium‐based ILs was investigated using various surface science techniques such as time‐of‐flight secondary ion mass spectrometry (TOF‐SIMS),21,22 low‐energy ion scattering (LEIS),23,24 Rutherford backscattering (RBS),22,25, 26, 27 angle‐resolved X‐ray photoelectron spectroscopy (ARXPS),28, 29, 30 reactive atom scattering‐laser‐induced fluorescence (RAS‐LIF),20,31 and by simulations.20,26,32,33
Concerning equimolar IL mixtures with different anions, that is, [C4C1Im][Tf2N]1‐x[PF6]x TOF‐SIMS investigations by Souda indicate a strong surface enrichment of [Tf2N]−.21 By RBS, Nakajima et al.25 proposed [Tf2N]− surface enrichment also for [CnC1Im][Tf2N]0.5[PF6]0.5 (n=4, 6), but to a lesser degree. On the other hand, they did not observe a pronounced preferential surface enrichment of any of the anions for [C6C1Im][Tf2N]0.5Cl0.5 and [C6C1Im][PF6]0.5Cl0.5 mixtures.25 In a more recent study, the same group again investigated related IL mixtures with RBS and indeed found some surface enrichment;27 they estimated the mole fraction of the surface composition and explained the observed effects with a simple thermodynamic calculation. The authors concluded that larger ions are surface‐enriched compared to smaller ions. Furthermore, in a LEIS study on similar ILs, namely stoichiometric mixtures of [C4C1Im]+ with different anions, Cl− or I− with [Tf2N]−, and [HOSO3]− with [BF4]−, Lovelock and co‐workers24 found strong enrichment of [Tf2N]− and [BF4]−. They suggest that in a mixture with one common cation but different anions, the anion with the weaker cation‐anion intermolecular interaction is enriched in the outermost surface layer at the expense of the other anion.
Another series of studies addressed mixtures of ILs with the same anion, but imidazolium‐based cations with one shorter and one longer alkyl chain. A TOF‐SIMS study for [C2C1Im]0.5[C8C1Im]0.5[PF6] and [C2C1Im]1‐x[C8C1Im]x[BF4] reports the long alkyl chains to be located at the outer surface.21 In a comparative RBS and TOF‐SIMS study on [C2C1Im]1‐x [C10C1Im]x[Tf2N] mixtures (x=0.1, 0.5), Nakajima et al.22 also proposed surface segregation of the longer chain, but found a higher mole fraction of [C10C1Im]+ at the surface in TOF‐SIMS. In line with the observations for mixtures with different anions in the above mentioned studies, they attribute the lower degree of surface enrichment found in RBS to a lower surface sensitivity as compared to TOF‐SIMS and LEIS. In MD simulations for binary [C2C1Im]1‐x[C8C1Im]x[TfO] mixtures, the octyl chain of the [C8C1Im]+ cation was found to be preferentially located at the surface, with a decreasing ratio of the number density along with a higher mole fraction of this cation, in line with trends in the above mentioned experiments.33
In addition, there are some XPS studies available for IL mixtures with a common cation (anion) but with different anions (cations) of different coordination strength,28,29 which can be understood in terms of interionic partial charge transfer between cations and anions.34 Core level binding energy shifts with respect to the neat ILs indicate that the electronic structure on the ions in the bulk is modified when changing the mixture composition, as has been demonstrated for imidazolium‐29 as well as for pyrollidinium‐35 based IL mixtures. It was also shown that specific IL mixtures change the turnover frequency of a catalytic reaction by modifying the anion‐catalyst interaction.29 Recently, also a ternary mixture of [C8C1Im][Tf2N]1‐x‐y[SnCl3]xCly was studied by XPS, indicating a partial charge transfer from the Cl− anion to the [C8C1Im]+ cation.36 Concerning surface composition, a binary mixture of [C2C1Im]0.9[C12C1Im]0.1[Tf2N] was investigated by ARXPS in our group finding no pronounced enrichment of the cation with the longer C12 alkyl chain for this combination.30
The same [C2C1Im]1‐x[C12C1Im]x[Tf2N] mixture as well as a mixture with a fluorinated side chain were recently investigated by RAS‐LIF combined with MD simulations.31 It was shown that the component with the lower surface tension is preferentially located at the surface of the mixture.
Herein, we present for the first time a surface science study of an IL with methoxy (MeO) functionalization at the imidazolium ring, that is, 1,3‐di(methoxy)imidazolium hexafluorophosphate [(MeO)2Im][PF6]37,38 (see Table 1), and its equimolar mixtures with ILs containing a non‐functionalized [C8C1Im]+ cation and the same or a different anion; the two anions are the medium‐sized hexafluorophosphate ([PF6]−) and the large, less coordinating bis[(trifluoromethyl)sulfonyl]imide ([Tf2N]−). The tethering of methoxy groups at the imidazolium ring in [(MeO)2Im][PF6] is expected to have a strong influence on the electronic structure of the imidazolium ring compared to 1,3‐dialkylimidazolium systems,38 which can be accessed by XPS as has been previously shown for a thiouronium‐functionalized IL.39 Dialkoxyimidazolium compounds are interesting due to their ability to dissolve a reasonable amount of the greenhouse gases CO2 40 and N2O.41 They are also suitable in synthesis due to the fact that N‐heterocyclic carbenes (NHCs) are easily made from imidazolium‐based ILs which then can serve as catalyst or solvent in reactions. It was shown that similar compounds as the [(MeO)2Im][PF6], e. g. with a bromo‐substituent attached to the C2‐position or with more bulky substituents, form several metal‐NHC‐complexes.37,42 Furthermore, [(MeO)2Im][PF6] was demonstrated to be catalytically active in a Suzuki coupling reaction, but to a lesser extent than a standard 1,3‐bis(2,4,6‐trimethylphenyl)imidazolium (IMes) salt due to less bulky substituents at the imidazolium ring.38 Furthermore, di(methoxy)imidazolium‐based salts are used for enhancing reactions in water.43
Table 1.
Summary of ILs investigated in this study.
| Chemical formula | Structure | Name |
|---|---|---|
| IL1: [C8C1Im][Tf2N] |
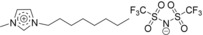
|
1‐methyl‐3‐octylimidazolium bis[(trifluoromethyl)sulfonyl]imide |
| IL2: [C8C1Im][PF6] |
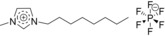
|
1‐methyl‐3‐octylimidazolium hexafluorophosphate |
| IL3: [(MeO)2Im][PF6] |
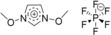
|
1,3‐di(methoxy)imidazolium hexafluorophosphate |
| IL4: [Me(EG)2C1Im][Tf2N] |
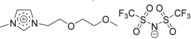
|
1‐[2‐(2‐methoxy‐ethoxy)ethyl]‐3‐methylimidazolium bis[(trifluoromethyl)sulfonyl]‐imide |
As experimental method, we use ARXPS for probing the surface layer of the IL samples. Our Dual Analyzer System for Surface Analysis (DASSA)44 setup allows for simultaneously recording spectra in a surface sensitive and a bulk sensitive mode, which directly permits to identify surface enrichment or depletion effects of all atoms (apart from hydrogen) being present in the ions. Compared to LEIS and TOF‐SIMS (and to a lower extent also RBS), XPS has the disadvantage that it is sensitive not only to the topmost layer, but depending on the emission angle, the signals stem from the first 7–9 nm (10–15 IL layers; at 0° emission) or from the first 1–1.5 nm (1–2 IL layers; at 80° emission). On the other hand, it is quantitative and allows us to differentiate elements in different chemical environments (i. e. the four inequivalent carbon atoms in [C8C1Im][Tf2N] and different O 1s species) by their chemical shift.
2. Results and Discussion
We investigated equimolar mixtures of ILs with different cations and anions, namely IL1: [C8C1Im][Tf2N], IL2: [C8C1Im][PF6], and IL3: [(MeO)2Im][PF6]; for an overview and the full names of these three ILs, see Table 1. In particular, we studied the mixture of IL2 and IL3, who share the same anion, and the mixture of IL1 and IL3 with different anions and cations. For all ILs and IL mixtures, we measured the core levels of all elements by angle‐resolved XPS at normal emission (0°, “bulk sensitive”) and at grazing emission (80°, “surface sensitive”), to elucidate possible surface enrichment effects. Due to the fact that the XPS peaks of the different elements show pronounced chemical shifts for the different cations and anions, we can base all conclusions on the consistent behavior of several signals.
The neat IL1 and IL2 have already been studied in detail in the past, and a pronounced protruding of the alkyl chains towards the vacuum has consistently been reported.44, 45, 46 On the other hand, we are not aware of any previous XPS studies of IL3, which contains the very specific [(MeO)2Im]+ cation. Therefore, we studied this neat IL in some detail and compare its spectra to those of IL2, and with another oxygen‐containing IL, that is, IL4: [Me(EG)2C1Im][Tf2N] (1‐[2‐(2‐methoxy‐ethoxy)ethyl]‐3‐methylimidazolium bis[(trifluoromethyl)sulfonyl]imide; see Table 1) as reference. This analysis, along with the charge on atoms calculations, will allow for a comparison of the electronic structure of these methoxy and polyethylene‐glycol (PEG) ILs. Notably, for none of the ILs Si 2p signals, indicative of surface‐active polysiloxane contaminations,47,48 are observed in the presented data, confirming the high purity of our ILs.
IL1, IL2, and IL4 are liquid at room temperature and IL3 has a melting point in the regime of 83–84 °C. The presented XP spectra of IL1–IL3 and the respective discussions are based on measurements at 90 °C to enable the comparison of the ILs in their liquid state; IL4 was measured at room temperature. The F 1s peaks of the respective anions are used as an internal reference: Spectra of ILs containing the [PF6]− anion are referenced to the FPF6 peak at 686.8 eV, whereas the FTf2N peak of ILs comprising the [Tf2N]− anion is set to 688.8 eV. Notably, XPS of IL3 in the solid state at room temperature show the presence of additional O 1s, N 1s, and C 1s signals. These additional signals increase with increasing irradiation time, indicating a decomposition of the [(MeO)2Im]+ cation under X‐radiation. By heating the sample to 90 °C, these signals almost disappear from the XPS detection volume, probably due to the evaporation of volatile decomposition products and/or dissolution of non‐volatile components into the bulk.
2.1. Normal Emission Spectra of the Neat ILs and Both IL Mixtures
In Figure 1, the 0° emission XP spectra of the four ILs and the investigated equimolar mixtures are depicted in the a) F 1s, b) O 1s, c) N 1s, d) C 1s, e) S 2p, and f) P 2p regions. The F 1s region (Figure 1a) shows the characteristic FPF6 peak of the [PF6]− anion at 686.8 eV for IL2, IL3, and the IL2+IL3 mixture, and the FTf2N peak of the [Tf2N]− anion at 688.8 eV for IL1 and IL4; for the mixture IL1+IL3 both F 1s peaks are observed.
Figure 1.
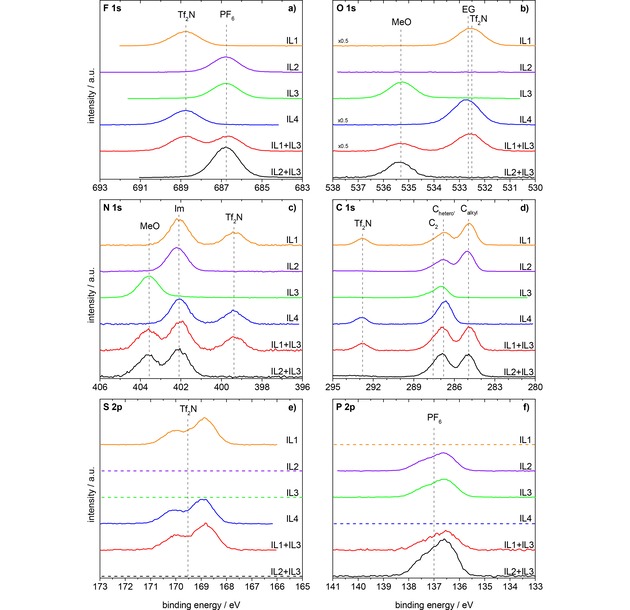
F 1s, O 1s, N 1s, C 1s, S 2p, and P 2p spectra of the investigated ILs and IL mixtures in 0° emission, measured with Al Kα radiation. Orange: IL1, [C8C1Im][Tf2N]; violet: IL2, [C8C1Im][PF6]; green: IL3, [(MeO)2Im][PF6]; blue: IL4, [Me(EG)2C1Im][Tf2N]; red: equimolar mixture of IL1+IL3, [C8C1Im][Tf2N]+ [(MeO)2Im][PF6]; black: equimolar mixture of IL2+IL3, [C8C1Im][PF6]+[(MeO)2Im][PF6]. All spectra are normalized to the fitted peak intensity of the F 1s spectrum of [(MeO)2Im][PF6]. Abbreviations: MeO for [(MeO)2Im]+, Im for N 1s of [C8C1Im]+ and [Me(EG)2C1Im]+, EG for O 1s of [Me(EG)2C1Im]+, PF6 for [PF6]−, Tf2N for [Tf2N]−.
In the O 1s region (Figure 1b), we observe no signal for IL2, the OTf2N signal at 532.6 eV for IL1, the OMeO signal at 535.3 eV for IL3, and the combined OTf2N+OEG signals at 532.7 eV for IL4. The mixture IL2+IL3 contains the OMeO signal and the mixture IL1+IL3 also the OTf2N signal.
In the N 1s region (Figure 1c) of IL1, IL2, IL4, and the mixtures IL1+IL3 and IL2+IL3, we find the imidazolium signal, NIm, at 402.1 eV and for IL1, IL4, and the IL1+IL3 mixture additionally the NTf2N signal at 399.4 eV; for the [(MeO)2Im]+ cation in IL3 and in the mixtures IL1+IL3 and IL2+IL3, the imidazolium nitrogen signal, NMeO, is found at the significantly higher binding energy of 403.6 eV.
In the C 1s region (Figure 1d), the signal of the carbon atoms, Chetero, next to N or O heteroatoms (see Scheme 1) of all neat ILs and IL mixtures are found around 287.3 eV (for a more detailed discussion see below); the carbon atoms, Calkyl, of the alkyl chain in IL1, IL2, and the mixtures IL1+IL3 and IL2+IL3 have binding energies around 285.0 eV, and the carbon atoms, CTf2N, of the [Tf2N]− anion of IL1, IL4, and the mixture IL1+IL3 are at 292.8 eV.
Scheme 1.
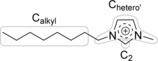
Definition of the carbon species: Calkyl, Chetero′, and C2.
In the S 2p and P 2p regions (Figure 1e, f), we observe the spin‐orbit peaks of the [Tf2N]− and the [PF6]− anions centered at 169.5 and 137.0 eV, respectively. Note that the binding energies of the anion and cation signals in the neat ILs and also in the equimolar mixtures, all agree to within 0.1 eV. One particular aspect to be mentioned is the fact that in all energy regions the signals of the different anions and cations are significantly separated from each other (see Figure 1), which allows for a detailed quantitative analysis, which is particularly relevant for the equimolar IL mixtures. Taking the atomic sensitivity factors (ASFs) into account, the quantitative analysis of the 0° spectra of all neat ILs is in accordance with the nominally expected atom ratios (see Table S1–S4).
In the following, we will compare the spectra of the four neat ILs in detail. While the spectra of IL1 have already been discussed in the past,44, 45, 46 the spectra of IL3, [(MeO)2Im][PF6], and to some extent also IL2 and IL4, [Me(EG)2C1Im][Tf2N], are presented in Figure 1 for the first time; in case of IL4, the very similar IL 1‐[2‐(2‐Ethoxyethoxy)ethyl]‐3‐methylimidazolium bis‐[(trifluoromethyl)sulfonyl]imide ([Et(EG)2C1Im][Tf2N]), that is, an IL with an ethyl instead of a methyl group as end group of the long side chain, has already been discussed in detail in ref [47].
2.1.1. Comparison of [C8C1Im]+ and [(MeO)2Im]+
As IL2 and IL3 have the same anion but different cations, we start by comparing the corresponding spectra. The most pronounced difference is observed for the imidazolium N 1s peak, which is shifted from 402.2 eV (NIm) for the [C8C1Im]+ cation in IL2 to 403.6 eV (NMeO) for the [(MeO)2Im]+ cation in IL3, that is, by 1.4 eV to higher binding energies. Smaller, but significant shifts towards higher binding energy are also found in the C 1s region: For IL2, [C8C1Im][PF6], two signals at 285.1 and 286.8 eV can be distinguished; they are assigned to carbon atoms bound only to hydrogen or carbon (Calkyl) and to carbon atoms bound to one hetero atom like nitrogen or oxygen (Chetero′), respectively. The small shoulder at 287.7 eV is assigned to the C2 carbon atom between the two nitrogen atoms. It carries a major part of the positive charge of the imidazolium ring,49,50 and thus, exhibits a higher binding energy than the other Chetero′ atoms. For [(MeO)2Im][PF6], the Chetero′ and C2 signals are shifted to 287.0 and 288.1 eV, that is, by 0.2–0.4 eV to higher binding energy, respectively. Due to the presence of methyl only, and thus, the absence of an alkyl chain, no corresponding signal is observed at 285.0 eV. The shift of the N 1s and C 1s signals of the [(MeO)2Im]+ cation towards higher binding energy by 1.4 and 0.2–0.4 eV, respectively, compared to the [C8C1Im]+ cation, is an indication that the imidazolium ring of [(MeO)2Im]+ has a lower electron density compared to [C8C1Im]+.
The O 1s spectrum of IL3 shows a very characteristic OMeO peak at 535.3 eV. Since IL2 does not contain an oxygen atom, we compare the spectrum to that of IL4, which also contains an oxygen atom in its functionalized PEG chain of the cation and also in the [Tf2N]− anion. The OEG and OTf2N levels have a very similar binding energy within our energy resolution and thus, only a single peak is seen for IL4, which is observed at a slightly higher binding energy of 532.7 eV, as compared to 532.6 eV found for the [Tf2N]− anion in IL1. Relative to this peak, the OMeO peak of IL3 is shifted towards higher binding energy by 2.6 eV resulting in a binding energy of 535.3 eV. This value is extraordinarily large and to the best of our knowledge oxygen with such a high binding energy is only described in polymers, e. g. 535.5 eV for peroxy‐oxygens in dibenzoyl peroxide.51
2.1.2. Calculations of Atomic Charges in [C2C2Im]+, [(MeO)2Im]+, and [Me(EG)1C1Im]+
To elucidate the observed large chemical shifts of IL3 in the O 1s and N 1s regions, atomic charges have been calculated for isolated cations in the gas phase: In an initial‐state picture, shifts to higher binding energies as observed for the uncommon [(MeO)2Im]+ cation of IL3 should be due to a decrease in electron density close to the probed atoms. For [(MeO)2Im]+, two low‐energy conformers (based on out‐of‐plane ether group orientations, see SI for detail) were identified. Atomic and group charges have been determined using both the NBO (natural bond orbital) and ChelpG (charges from electrostatic potential using a grid based method) methodologies. Net NBO charges of −0.35 and −0.02 electrons (e) were found at the OMeO and NMeO atoms, respectively (see Figure 2). The shorter 3‐(2‐methoxyethyl)‐1‐methylimidazolium ([Me(EG)1C1Im]+) is used as model for [Me(EG)2C1Im]+. Four stable conformers were obtained for [Me(EG)1C1Im]+ and charge analysis carried out for the highest and lowest energy structures, which also exhibit significant structural differences (see SI for details), yielding NBO charges of −0.60 to −0.57 e at the OEG atoms and −0.34 to −0.33 e at the NIm atoms. For comparison [C2C2Im]+, which is representative of the common [C8C1Im]+ cation of IL1 and IL2, has similar NBO values of −0.35 to −0.34 e for the NIm atoms. Apart from differences in absolute values between NBO and ChelpG analysis (due to the very different character of both methods), the relative changes in charge are similar to those identified for the NBO charges (see Table S9 for ChelpG charges).
Figure 2.
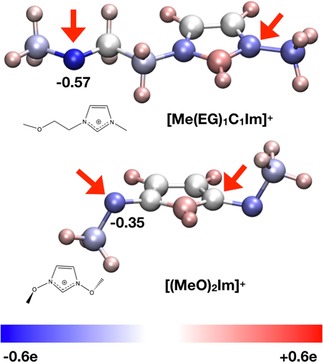
Pictorial representation of NBO charge distribution in [Me(EG)1C1Im]+ and [(MeO)2Im]+; blue is more negative, white neutral, and red more positive: For both cations, arrows mark the atoms of interest, nitrogen and oxygen.
The atomic charges found for NMeO in [(MeO)2Im]+ are considerably more positive (less negative) than those found for [Me(EG)1C1Im]+ and [C2C2Im]+. The more positive environment for NMeO is consistent with the higher XPS binding energies obtained for [(MeO)2Im]+. The NBO group charge for the imidazolium ring has also been determined; +0.93, +0.34, and +0.31 for [(MeO)2Im]+, [Me(EG)1C1Im]+, and [C2C2Im]+, respectively, clearly identifying a lower electron density on the imidazolium ring of [(MeO)2Im]+ compared to the latter, which is fully consistent with the XPS binding energies measured.
For oxygen, the NBO atomic charges for OMeO in [(MeO)2Im]+ are also considerably more positive than those found for [Me(EG)1C1Im]+. These values clearly indicate that electron density at the OMeO atoms is much lower for [(MeO)2Im]+, which explains the observed strong core level shift of more than +2.5 eV towards higher XPS binding energies. Notably, the examination of the molecular orbitals shows pπ‐orbitals on the OMeO atom interacting with the (aromatic) imidazolium ring pπ‐orbitals, linking them in a Möbius type way (see Figure 3). Thus, delocalization of the OMeO atom lone pair orbitals across the ring π‐system is occurring, reducing the overall charge on the OMeO atom.
Figure 3.
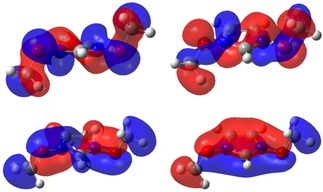
Molecular orbitals from [(MeO)2Im]+ showing the OMeO atom participating in aromatic delocalization of the imidazolium ring.
2.1.3. Comparison of the Anion Signals of IL1, IL2, and IL3
To complete the comparison of the spectra of IL3 to the other ILs, we address the anion. Notably, the binding energies for IL2 and IL3 are identical. In the P 2p region, we observe one asymmetric peak, centered at 137.0 eV, due to the non‐resolved spin‐orbit‐splitting of the 2p3/2 and 2p1/2 levels. In the F 1s spectra, one single peak (FPF6) is found at 686.8 eV. If we compare the anion signals of IL3 with those of IL1, we find the spin‐orbit‐split S 2p doublet centered at 169.5 eV in the S 2p region instead of the P 2p signals. Furthermore, the FTf2N peak at 688.8 eV is clearly separated from the FPF6 peak, which enables accurate quantification in the mixtures (see below); the anion‐related NTf2N and CTf2N peaks have already been described above.
The analysis of IL1–IL4 shows that the differences of the F 1s, O 1s, N 1s, C 1s, S 2p, and P 2p levels are a clear signature of the respective cations and anions and will serve as extremely helpful, multiple fingerprint in analyzing the IL mixtures below.
2.1.4. Equimolar Mixture of IL2 and IL3
Next we address the equimolar mixture of IL2 and IL3 in Figure 1, that is, a mixture of ILs with the same anion [PF6]−. Overall, all signals are found at identical binding energies as for the neat ILs. For the F 1s, P 2p, and O 1s regions, in each case only one signal is found, which is due to the anion (F 1s and P 2p) and the [(MeO)2Im]+ cation (O 1s). In the N 1s and C 1s region, the signals of the two cations are superimposed. The NIm peak is found at 402.1 eV and the NMeO peak at 403.6 eV. The Chetero′ and C2 signals of [C8C1Im]+ and [(MeO)2Im]+ in the neat ILs are only slightly shifted (0.2–0.4 eV) relative to each other, which leads to a combined peak at 286.9 eV with a high energy shoulder at 287.9 eV, and the well separated Calkyl peak at 285.0 eV. Within the margin of error, the binding energies of the F 1s, O 1s, N 1s, C 1s, and P 2p peaks are coinciding with the binding energies of the previously presented IL2 and IL3.
The comparison of the intensity of the two N 1s peaks and the two dominating C 1s peaks yields to an at first sight stunning result: From the IL2:IL3=[C8C1Im]+ : [(MeO)2Im]+=1 : 1 stoichiometry (50 % : 50 %) of the equimolar IL mixture, one would expect an intensity ratio of 1 : 1 for the NIm : NMeO signals. Inspection of Figure 1c (and Table S6), however, shows a ratio of 1 : 0.85 (54 % : 46 %), that is, a reduced amount of [(MeO)2Im]+ in the near‐surface region probed by XPS. Since weighting errors can be ruled out due to our accuracy being below 1 %, we propose that the observed deviation is due to a pronounced surface enrichment of the [C8C1Im]+ cations in the top most layer, which goes along with a depletion of the [(MeO)2Im]+ cation. Considering an inelastic mean free path of the C 1s and N 1s electrons of 3 nm (at kinetic energies of 1150±60 eV), the contribution of the topmost layer to the total signal at normal emission is 20–25 %. Assuming a simplified model with the topmost layer containing only [C8C1Im]+ cations and all layers underneath being mixed stoichiometrically (1 : 1), a signal ratio of 1.2 : 0.8 (60 % : 40 %) is expected in XPS, which would yield an even stronger increase of the nitrogen signals of [C8C1Im]+, on expense of the [(MeO)2Im]+ signals, than observed by us.
2.1.5. Equimolar Mixture of IL1 and IL3
Finally, we discuss the spectra of the equimolar mixture of IL1 and IL3 in Figure 1, that is, the mixture of ILs with different anions and different cations to search for possible selective enrichment of anions or cations. For this IL mixture, we find contributions from different cations and anions in nearly all regions; only the S 2p and P 2p spectra show solely contributions from one of the anions each. In the F 1s region, we find the FTf2N and the FPF6 peaks of the two anions at 688.8 and 686.7 eV, respectively. The O 1s region displays the OTf2N and OMeO peaks at 532.6 and 535.3 eV, respectively. In the N 1s region, we find three peaks, the NTf2N peak at 399.4 eV, the NMeO peak at 403.6 eV, and the NIm peak at 402.0 eV. The carbon region shows the CTf2N peak at 292.8 eV, the Calkyl peak at 284.9 eV, and the superposition of the Chetero′+C2 peaks of the [C8C1Im]+ and [(MeO)2Im]+ cations at 286.8 and 287.8 eV, respectively. Within the margin of error, the binding energies of the F 1s, O 1s, N 1s, C 1s, S 2p, and P 2p peaks are coinciding with the binding energies of the previously presented neat IL1 and IL3.
When comparing the intensities of the different anion and cation signals, we again find apparent deviation from the nominal stoichiometry. From the 50 % : 50 % stoichiometry of the equimolar IL mixture, [C8C1Im][Tf2N]+[(MeO)2Im][PF6] (IL1+IL3), one would expect an intensity ratio of 2 : 2 : 1 for the NIm : NMeO : NTf2N signals and of 7 : 10 : 2 for the Calkyl : Chetero′+C2 : CTf2N signals. As for the IL2+IL3 mixture, the [(MeO)2Im]+ signals are lower than expected from the stoichiometry, which indicates again that this cation is depleted from the surface and that [C8C1Im]+ is enriched at the surface. Information on possible enrichment effects of the anions can be derived also from the F 1s spectra; the ∼14 % larger peak area of the FTf2N as compared to the FPF6 peak (Table S5) indicates a depletion of the [PF6]− anions in the topmost layer.
2.2. Angle‐Resolved XPS Results
To obtain more detailed information of surface enrichment and depletion effects, we will present and analyze angle‐resolved XP spectra of the neat IL2 and IL3, and in particular the IL mixtures in the following. IL1 has already been discussed in literature.44, 45, 46 For this IL, a significant increase of the Calkyl is observed at 80°, that is, the signal at 80° increases by about 22 % (see SI, Figure S1). This behavior is a clear indication of the surface enrichment of the alkyl chains by preferential orientation of the cations. Concomitantly, most of the other signals decrease by a factor up to 13 %. For IL4, only minor changes with emission angle are seen (Figure S2), indicating the absence of major enrichment/orientation effects at the outer surface. This is in line with our earlier47 observations for other PEG ILs. For the signals of the [Tf2N]− anion, we find a minor surface enrichment of the CF3 groups and a depletion of the corresponding SO2 groups.21,22,47,52
2.2.1. Neat IL2 and IL3
We start with the analysis of the spectra of the neat IL2, [C8C1Im][PF6]. In Figure 4, the F 1s, O 1s, N 1s, C 1s, and P 2p spectra are depicted for 0° (black) and 80° (red) emission. We observe a strong enhancement of the Calkyl signal (increase in peak intensity of 31 %) at 80° as compared to 0°, which indicates that the alkyl chain of the [C8C1Im]+ cation is enriched at the surface, pointing away from the surface towards vacuum. For the FPF6, NIm, Chetero′+C2, and PPF6 signals, we find a damping at 80° due to the surface‐enriched alkyl groups on top. This damping is most pronounced for the F 1s level and least pronounced for the P 2p level, which is attributed to the increasingly lower kinetic energy of the emitted photoelectron, ∼800 eV for F 1s vs. ∼1350 eV for P 2p.
Figure 4.
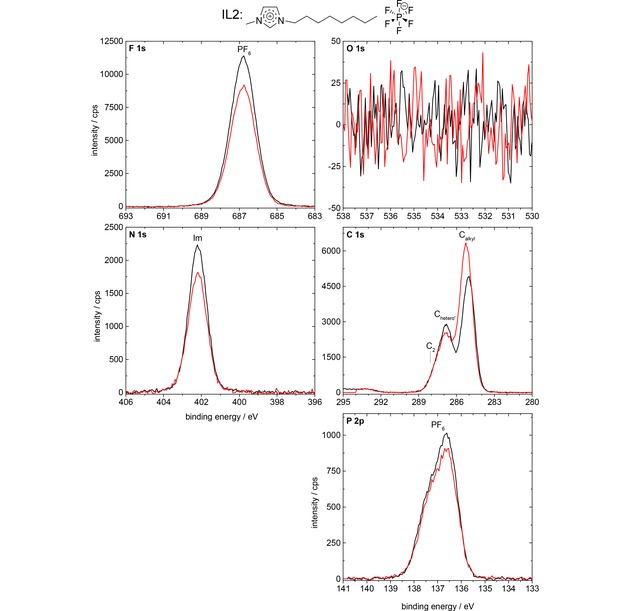
F 1s, O 1s, N 1s, C 1s, and P 2p spectra of IL2, [C8C1Im][PF6], in 0° (black) and 80° (red) emission, measured with Al Kα radiation.
The F 1s, O 1s, N 1s, C 1s, and P 2p spectra of the neat IL3, [(MeO)2Im][PF6], are depicted in Figure 5 for 0° (black) and 80° (red) emission. We observe no pronounced intensity changes (peak area changes) with emission angle; the apparent lower peak height at 80° for most core levels (0–2 %) goes along with a slight peak shift and broadening of the peaks; the latter is probably due to the inhomogeneous final state screening in the topmost layer, which contributes to 80 % of the signals at 80°, but only to 25 % of the signal at 0°. The absence of significant signal intensity differences for the various core levels in 80° and 0° emission indicates a homogeneous distribution of anions and cations at the surface and in the bulk of the IL. This is in line with previous experience that surface enrichment effects are typically found for systems with functionalized and non‐functionalized alkyl chains with a chain length of at least 4 carbon units.52,53 Notably, the small peak at 285.0 eV, is due to a minor contamination of the IL (see Experimental Section).
Figure 5.
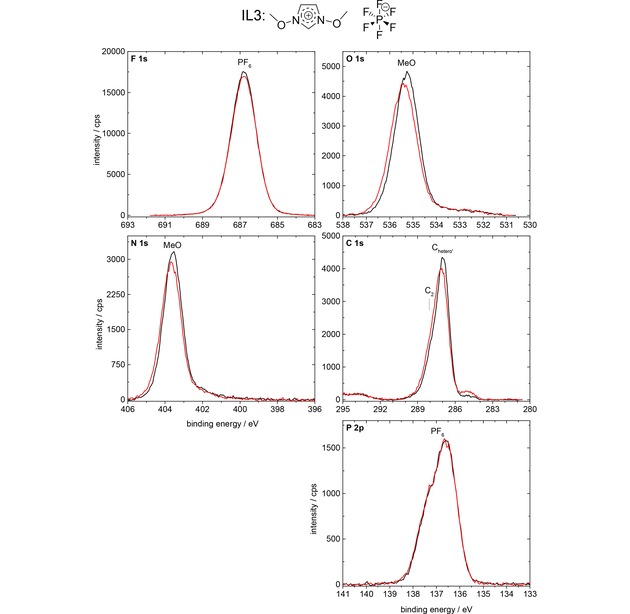
F 1s, O 1s, N 1s, C 1s, and P 2p spectra of IL3, [(MeO)2Im][PF6], in 0° (black) and 80° (red) emission, measured with Al Kα radiation.
2.2.2. Equimolar Mixture of IL2 and IL3
Next, we analyze the angle‐resolved XP spectra of the equimolar mixture of IL2 and IL3 in Figure 6, that is, the IL mixture of ILs with the identical [PF6]− anion. The Calkyl signal shows a very strong enhancement (increase in peak height of ∼69 %) at 80° (red) as compared to 0° (black) emission, which indicates that the [C8C1Im]+ cation is enriched at the surface with the alkyl chain pointing away from the surface towards vacuum. At the same time, the NMeO and OMeO signals of the [(MeO)2Im]+ cation show only ∼45 % intensity at 80° as compared to 0° emission, indicating the depletion of this cation from the topmost layer. For the FPF6 and PPF6 signals, a moderate decrease of the 80° signals as compared to 0° signals is observed, very similar to that of the neat IL2, which is again due to the damping of the anions by the surface‐enriched octyl chain of the [C8C1Im]+ cation. Interestingly, the damping of the NIm signal at 402.1 eV is weaker than for the neat IL2. This behavior is attributed to the increased surface sensitivity in 80°, where the contribution from the [C8C1Im]+‐enriched topmost layer dominates (∼80 % of total signal intensity originates from this layer), whereas at 0° this enriched layer has a much smaller contribution (25 %), while the majority of the signal (75 %) stems from the bulk underneath with a 1 : 1 composition. The same argument also explains the comparably weak damping of the combined CIm+CMeO signal at ∼287 eV. Taking all these results together, we find a pronounced surface enrichment of the [C8C1Im]+ cation, which goes along with a surface depletion of the [(MeO)2Im]+ cation. From the 80° emission data a concentration of ∼69 mol% of [C8C1Im]+ is deduced for the first 1–1.5 nm. This conclusion is perfectly in line with the conclusions already drawn above from the comparison of the 0° intensities of the C 1s and N 1s core levels of the different cations discussed in the context of Figure 1. The observed enrichment of [C8C1Im]+ on expense of [(MeO)2Im]+ in the top‐most layer is attributed to the thermodynamic driving force to obtain the lowest possible surface tension, that is, surface free energy. Despite the fact that no experimental surface tension values are available for the neat IL3 or for the IL2+IL3 mixture, it appears plausible that the weakly coordinating [C8C1Im]+ with its long alkyl chain exhibits a considerable lower surface tension than the more polar [(MeO)2Im]+, and is thus enriched at the surface.54
Figure 6.
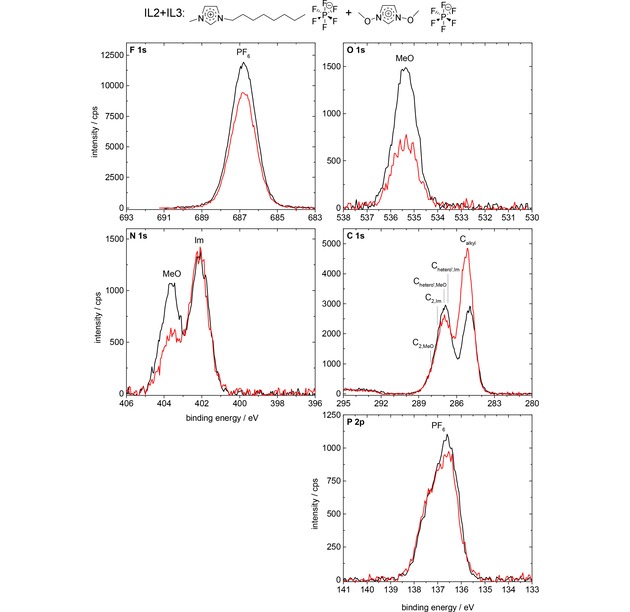
F 1s, O 1s, N 1s, C 1s, and P 2p spectra of the equimolar IL2+IL3 mixture, [C8C1Im][PF6]+[(MeO)2Im][PF6], in 0° (black) and 80° (red) emission, measured with Al Kα radiation.
2.2.3. Equimolar Mixture of IL1 and IL3
Finally, we address the surface composition of the equimolar mixture of IL1 and IL3 (see Figure 7), that is, the mixture of ILs with different cations and anions. The Calkyl signal is considerably enhanced (by ∼47 %) at 80° (red) as compared to 0° (black), which again indicates that the [C8C1Im]+ cation is enriched at the surface with the alkyl chain preferentially pointing towards the vacuum. Again, also the N 1s and O 1s signals of the [(MeO)2Im]+ cation show a much lower intensity (only ∼62 %) at 80° emission, due to the depletion of this cation from the topmost layer. These signal changes result in a concentration of ∼69 mol% of [C8C1Im]+ probed in 80° emission for the first 1–1.5 nm. Concerning the anions, we find a pronounced decrease of the FPF6 (by 39 %) and PPF6 signals (by 27 %), indicating the depletion of [PF6]− from the topmost layer. On the other hand, we find a moderate increase (up to 14 %) of the FTf2N, OTf2N, NTf2N, CTf2N, and STf2N signals at 80° as compared to 0°. This increase is in contrast to the neat IL1, where all anion signals decreased by 2–13 % in intensities at 80°, and indicates the enrichment of [Tf2N]− in the topmost layer of the IL1+IL3 mixture indicating a concentration of ∼68 mol% of [Tf2N]− in the first 1–1.5 nm. In sum, our ARXPS results clearly show that pronounced surface enrichment of [C8C1Im]+ and [Tf2N]− and depletion of [(MeO)2Im]+ and [PF6]− also occurs in equimolar IL mixtures with different anions and cations. Concerning the cations, again the [C8C1Im]+ cation is enriched at the surface, due to its presumed lower contribution to the surface tension. For the anion, we find [Tf2N]− enriched at the surface while [PF6]− is depleted. We attribute this to the lower contribution to the surface tension of the former; indeed, surface tension measurements for [C8C1Im][Tf2N] and [C8C1Im][PF6] yield values of 29.5 mN/m for the former and 32.5 mN/m for the latter.55
Figure 7.
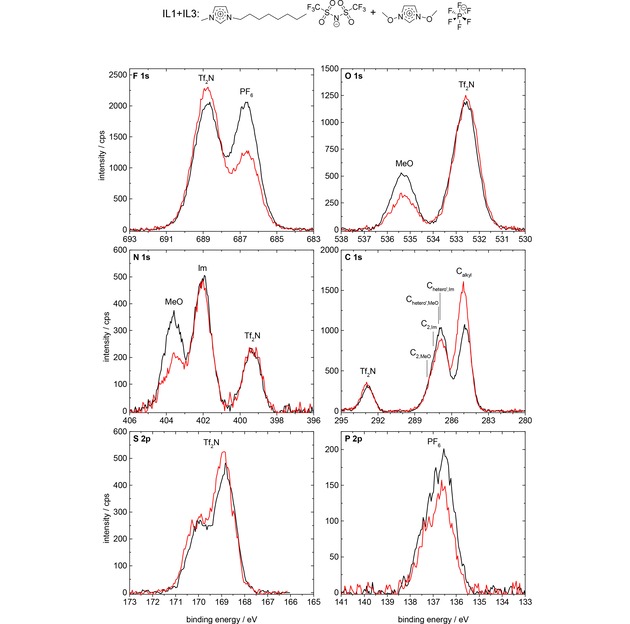
F 1s, O 1s, N 1s, C 1s, S 2p, and P 2p spectra of the equimolar IL1+IL3 mixture, [C8C1Im][Tf2N]+[(MeO)2Im][PF6], in 0° (black) and 80° (red) emission, measured with Al Kα radiation.
3. Conclusions
We have studied ILs with a methoxy functionalized imidazolium‐based cation, that is, [(MeO)2Im][PF6], and equimolar mixtures of this IL by angle‐resolved X‐ray photoelectron spectroscopy. In the [(MeO)2Im][PF6]+[C8C1Im][PF6] mixture, the second IL has the same anion, but the non‐functionalized [C8C1Im]+ as cation. In the [(MeO)2Im][PF6]+[C8C1Im][Tf2N] mixture, the second IL consists of different anions and cations. For the neat [(MeO)2Im][PF6], we observe that the tethering of methoxy groups at the imidazolium ring leads to pronounced changes in the electronic structure of the imidazolium rings as compared to the situation for the non‐functionalized ring; these changes are reflected by pronounced differential shifts towards higher binding energies of the imidazolium nitrogen and carbon atoms as well as for the oxygen atoms linked to the ring. DFT calculations for the [(MeO)2Im]+ cation combined with an atomic charge analysis are fully in line with the observed XPS shifts in an initial state picture: In contrast to the situation for PEG‐ or alkyl‐chains, the electron density is considerably reduced in the imidazolium ring and also at the oxygen of the methoxy group, when oxygen is directly linked to the nitrogen atoms. For the two equimolar mixtures, we found no additional changes in the electronic structure, that is, the binding energies are unchanged as compared to the neat ILs within the margin of error. For both equimolar mixtures of ILs with different cations ([(MeO)2Im]+ or [C8C1Im]+) and anions ([PF6]− or [Tf2N]−), we always observed pronounced surface enrichment effects by angle‐resolved XPS. For the mixture with the same [PF6]− anion, we find a pronounced enrichment of the octyl chain‐containing [C8C1Im]+ cation and a corresponding depletion of the [(MeO)2Im]+ cation in the topmost layer. For a mixture of these cations with different anions, we find surface enrichment of the [C8C1Im]+ cation and the [Tf2N]− anion, while [(MeO)2Im]+ and [PF6]− are depleted from the surface. The observed behavior is proposed to be due to a lowering of surface tension by the enriched components.
Experimental Section
Materials
[C8C1Im][PF6] was purchased from Sigma‐Aldrich (purity>95 %). [(MeO)2Im][PF6] was synthesized with a purity of >99 %.37,38 [Me(EG)2C1Im][Tf2N] and [C8C1Im][Tf2N] were synthesized as was published earlier;45,47 the PEG‐functionalized IL [Me(EG)2C1Im] [Tf2N] was used for comparison of binding energy positions. [(MeO)2Im][PF6] shows a minor contamination in its solid state at room temperature, which almost disappears by heating the sample to 90 °C. All neat ILs were used without further purification. For ARXPS, a thin layer (thickness about 0.1 mm) of the neat ILs or the equimolar mixtures was placed onto a molybdenum sample holder and transferred to the vacuum chamber via a load‐lock, where they remained at least twelve hours for degassing. The equimolar mixtures of [(MeO)2Im][PF6] with [C8C1Im][Tf2N] or [C8C1Im][PF6] were prepared with acetonitrile (Sigma‐Aldrich, purity 99.8 %) as a co‐solvent to ensure proper mixing of the respective ILs. They were dripped on the molybdenum sample holder with a disposable glass pipette outside and put into the load‐lock after slow evaporation of the co‐solvent acetonitrile.
Angle‐Resolved X‐Ray Photoelectron Spectroscopy (ARXPS)
All angle‐resolved XP spectra were measured with our DASSA44 apparatus, which is a modified Multiprobe® XP system (OMICRON NanoTechnology GmbH). To our knowledge, this ARXPS system is the first setup to be equipped with two hemispherical energy analyzers (ARGUS‐type). The analyzers are mounted at emission angles ϑ of 0° and 80° with respect to the surface normal of a horizontally aligned sample. This enables the simultaneous measurement of 0° and 80° emission spectra of liquids, which would drip off the sample holder upon sample rotation away from the horizontal position. Measuring simultaneously at both angles has the additional advantage that the measuring time for the two spectra is reduced by a factor of two and possible time‐ (or radiation‐) induced changes can be ruled out (or minimized). As X‐ray source, we use monochromated Al Kα radiation, impinging onto the sample under the magic angle of 54.7°, with respect to both analyzers. The surface sensitivity of XPS increases with increasing emission angle, proportional to 1/cos(ϑ). Considering the inelastic mean free path of electrons in organic matter of 2.6 (F 1s)–3.3 nm (P 2p)56 at the given kinetic energies of the photoelectrons (∼800 (F 1s)–1300 eV (P 2p)), the information depth at 0° amounts to 7–9 nm, while at 80° only the topmost 1–1.5 nm are probed. All 80° spectra are scaled up by a so‐called geometry factor obtained for each data set according to ref [44]. Consequently, a larger (smaller) signal at 80° as compared to 0° unambiguously indicates that the corresponding element has a higher (lower) concentration at the surface than in the bulk. Therefore, conclusions about surface enrichment and surface orientation effects are possible.
The X‐ray source (XM 1000) was operated at a power of 238 W, with a spot size of the monochromated Al Kα radiation (hν=1486.6 eV) of 1.0 mm×2.0 mm on the sample. For both analyzers, we used a curved slit of 1.0 mm×10.0 mm and the high magnification mode. These settings led to an acceptance angle of ±3.1° and a full‐width‐at‐half‐maximum (FWHM) of the analysis area of 0.31 mm×3.2 mm in 0° emission. The pass energy was set to 35 eV resulting in an overall instrumental energy resolution of 0.4 eV.
All spectra were referenced to the F 1s signals of the [PF6]− and [Tf2N]− anions at 686.8 and 688.8 eV, respectively, to compensate for charging effects and different sample work functions. With this binding energy referencing, the Calkyl of [C8C1Im]+ (see Scheme 1 for peak assignment) is located at 285.0±0.1 eV.
The peaks in the different regions were analyzed for quantification. Quantitative analysis was possible by using the intensity of the fitted peaks and taking into account the ASF of each signal.44 For ILs containing the [Tf2N]− anion, a three‐point linear background was subtracted in C 1s spectra; for all other spectra, a two‐point linear background subtraction was used. The signals were fitted with a Gauss‐Lorentz function with 30 % Lorentz contribution. The XP signals of P 2p and S 2p are composed of two components, namely the spin‐orbit‐split 2p1/2 and 2p3/2 levels. The two components always have the same FWHM and binding energy separation (0.90 eV for P 2p and 1.21 eV for S 2p), and an area ratio of 1 : 2. When fitting the C 1s spectra constraints were employed (for peak assignment see Scheme 1): The area ratios of the C2, Chetero′, and Calkyl peaks measured at 0° were set to their nominal ratios; at 80°, no area constraints were applied for Calkyl. For IL1 and IL2 (see Table 1), the separation of C2 and Chetero′ is set to 0.90 eV, with the FWHM of Chetero′ 1.10 times wider than that of C2 and Calkyl. For IL4, the FWHM of Chetero′ is 1.19 times that of C2. For IL3, the Chetero′ and C2 are separated by 1.10 eV, with the FWHM of Chetero′ 1.02 times wider than that of C2. All fit parameters are extracted by fitting several C 1s spectra as mentioned in ref [44]. Note that in earlier publications C2 and Chetero′ were combined in one Chetero peak due to a lower resolution than for the DASSA setup used here.
Calculation of Atomic Charges
In order to correlate XPS core level binding energies with the electron density, density functional theory (DFT) calculations were carried out and the atomic partial charges on individual atoms were compared via two charge analysis methods: NBO and ChelpG.57,58 The calculations are performed for [(MeO)2Im]+, [C2C2Im]+, and [Me(EG)1C1Im]+. Thereby, the [C2C2Im]+ cation is taken as a model cation for the larger [C8C1Im]+ cation studied by ARXPS as well as for direct comparison with [(MeO)2Im]+ without oxygen being implemented in the cation.
DFT calculations were performed at the B3LYP−D3(BJ)/6‐311G(d,p) level using Becke's three‐parameter exchange functional in combination with the Lee, Yang, and Parr correlation functional (B3LYP).59,60 Grimme's D3 dispersion correction with Becke‐Johnson (BJ) damping was used to account for dispersion.61, 62, 63, 64 The 6‐311G(d,p) basis set was employed for all calculations. All calculations were carried out employing the Gaussian 09 (revision D.01) suite of programs.65 Structures were fully optimized under no symmetry constraints and maxima and minima have been confirmed by vibrational analysis (one or no imaginary frequencies). NBO analysis employed version 6.0 (not the default version within G09).66,67 All molecular orbitals have been generated using the Gaussview software at the 0.02 au isosurface.68
The NBO charge analysis is based on atomic orbitals, and as such provides a “localized” view. The electron density is decomposed into a basis of natural atomic orbitals, which can be described as the effective “natural orbitals of an atom” in a particular molecular environment. The negative charge on the atoms is calculated by assigning electron density to atoms on which the basis atomic orbitals are centered; the balance with the positive nucleus charge then provides the NBO charge on this atom.57 The ChelpG charge analysis is based on the electrostatic potential (ESP) created by the DFT‐derived electron density. Atomic partial charges are fitted in order to reproduce the ESP (beyond the van‐der‐Waals radii of all the atoms in the molecule); in this sense the charges produce an “external” view of the ion or molecule. It has been shown that both NBO and ChelpG charge analysis can be well correlated with experimental XPS binding energies and near edge X‐ray absorption fine structure (NEXAFS) spectroscopy.69,70 While fitted trends may show a good correlation with both NBO and ChelpG there is a more robust physical basis for the correlation of NBO charges with the experimental data presented here, which leads us to focus on the NBO charges. The ChelpG charges and a discussion of the associated limitations are presented in the SI.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We thank N. Taccardi for providing us with the IL [C8C1Im][Tf2N]. B.S.J.H. and H.‐P.S. thank the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation programme for financial support, in the context of the Advanced Investigator Grant “ILID” to H.‐P.S. (Grant Agreement No. 693398‐ILID).
B. S. J. Heller, C. Kolbeck, I. Niedermaier, S. Dommer, J. Schatz, P. Hunt, F. Maier, H.-P. Steinrück, ChemPhysChem 2018, 19, 1733.
References
- 1. Plechkova N. V., Seddon K. R., Chem. Soc. Rev. 2008, 37, 123–150. [DOI] [PubMed] [Google Scholar]
- 2. Steinrück H.-P., Phys. Chem. Chem. Phys. 2012, 14, 5010–5029. [DOI] [PubMed] [Google Scholar]
- 3. Kernchen U., Etzold B., Korth W., Jess A., Chem. Eng. Technol. 2007, 30, 985–994. [Google Scholar]
- 4. Steinrück H.-P., Libuda J., Wasserscheid P., Cremer T., Kolbeck C., Laurin M., Maier F., Sobota M., Schulz P. S., Stark M., Adv. Mater. 2011, 23, 2571–2587. [DOI] [PubMed] [Google Scholar]
- 5. Yasuda T., Watanabe M., MRS Bull. 2013, 38, 560–566. [Google Scholar]
- 6. Díaz M., Ortiz A., Ortiz I., J. Membr. Sci. 2014, 469, 379–396. [Google Scholar]
- 7. Forgie J. C., Khakani S. El, MacNeil D. D., Rochefort D., Phys. Chem. Chem. Phys. 2013, 15, 7713–7721. [DOI] [PubMed] [Google Scholar]
- 8. Bayley P. M., Best A. S., MacFarlane D. R., Forsyth M., Phys. Chem. Chem. Phys. 2011, 13, 4632–4640. [DOI] [PubMed] [Google Scholar]
- 9. Ferrari S., Quartarone E., Tomasi C., Ravelli D., Protti S., Fagnoni M., Mustarelli P., J. Power Sources 2013, 235, 142–147. [Google Scholar]
- 10. Capello C., Fischer U., Hungerbuhler K., Green Chem. 2007, 9, 927–934. [Google Scholar]
- 11. Canales R. I., Brennecke J. F., J. Chem. Eng. Data 2016, 61, 1685–1699. [Google Scholar]
- 12. Steinrück H.-P., Wasserscheid P., Catal. Lett. 2015, 145, 380–397. [Google Scholar]
- 13. Xiao H., Tribol. Trans. 2017, 60, 20–30. [Google Scholar]
- 14. Silvester D. S., Analyst 2011, 136, 4871–4882. [DOI] [PubMed] [Google Scholar]
- 15. Sun P., Armstrong D. W., Anal. Chim. Acta 2010, 661, 1–16. [DOI] [PubMed] [Google Scholar]
- 16. Baltazar Q. Q., Leininger S. K., Anderson J. L., J. Chromatogr. A 2008, 1182, 119–127. [DOI] [PubMed] [Google Scholar]
- 17. Poole C. F., Poole S. K., J. Sep. Sci. 2011, 34, 888–900. [DOI] [PubMed] [Google Scholar]
- 18. Lopes J. N. Canongia, Cordeiro T. C., Esperança J. M. S. S., Guedes H. J. R., Huq S., Rebelo L. P. N., Seddon K. R., J. Phys. Chem. B 2005, 109, 3519–3525. [DOI] [PubMed] [Google Scholar]
- 19. Clough M. T., Crick C. R., Grasvik J., Hunt P. A., Niedermeyer H., Welton T., Whitaker O. P., Chem. Sci. 2015, 6, 1101–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bruce D. W., Cabry C. P., Lopes J. N. C., Costen M. L., D'Andrea L., Grillo I., Marshall B. C., McKendrick K. G., Minton T. K., Purcell S. M., Rogers S., Slattery J. M., Shimizu K., Smoll E., Tesa-Serrate M. A., J. Phys. Chem. B 2017, 121, 6002–6020. [DOI] [PubMed] [Google Scholar]
- 21. Souda R., Surf. Sci. 2010, 604, 1694–1697. [Google Scholar]
- 22. Nakajima K., Miyashita M., Suzuki M., Kimura K., J. Chem. Phys. 2013, 139, 224701. [DOI] [PubMed] [Google Scholar]
- 23. Garcia I. J. Villar, Fearn S., Gregorio G. F. De, Ismail N. L., Gschwend F. J. V., McIntosh A. J. S., Lovelock K. R. J., Chem. Sci. 2014, 5, 4404–4418. [Google Scholar]
- 24. Villar-Garcia I. J., Fearn S., Ismail N. L., McIntosh A. J. S., Lovelock K. R. J., Chem. Commun. 2015, 51, 5367–5370. [DOI] [PubMed] [Google Scholar]
- 25. Nakajima K., Oshima S., Suzuki M., Kimura K., Surf. Sci. 2012, 606, 1693–1699. [Google Scholar]
- 26. Nakajima K., Nakanishi S., Chval Z., Lísal M., Kimura K., J. Chem. Phys. 2016, 145, 184704. [DOI] [PubMed] [Google Scholar]
- 27. Nakajima K., Nakanishi S., Lísal M., Kimura K., J. Mol. Liq. 2017, 230, 542–549. [Google Scholar]
- 28. Men S., Licence P., Chem. Phys. Lett. 2017, 681, 40–43. [Google Scholar]
- 29. Villar-Garcia I. J., Lovelock K. R. J., Men S., Licence P., Chem. Sci. 2014, 5, 2573–2579. [Google Scholar]
- 30. Maier F., Cremer T., Kolbeck C., Lovelock K. R. J., Paape N., Schulz P. S., Wasserscheid P., Steinrück H.-P., Phys. Chem. Chem. Phys. 2010, 12, 1905–1915. [DOI] [PubMed] [Google Scholar]
- 31. Smoll E. J. Jr., Tesa-Serrate M. A., Purcell S. M., D′Andrea L., Bruce D. W., Slattery J. M., Costen M. L., Minton T. K., McKendrick K. G., Faraday Discuss. 2018, 206, 497–522. [DOI] [PubMed] [Google Scholar]
- 32. Palchowdhury S., Bhargava B. L., Phys. Chem. Chem. Phys. 2015, 17, 19919–19928. [DOI] [PubMed] [Google Scholar]
- 33. Palchowdhury S., Bhargava B. L., J. Phys. Chem. B 2016, 120, 5430–5441. [Google Scholar]
- 34. Cremer T., Kolbeck C., Lovelock K. R. J., Paape N., Wölfel R., Schulz P. S., Wasserscheid P., Weber H., Thar J., Kirchner B., Maier F., Steinrück H.-P., Chem. Eur. J. 2010, 16, 9018–9033. [DOI] [PubMed] [Google Scholar]
- 35. Men S., Lovelock K. R. J., Licence P., Phys. Chem. Chem. Phys. 2011, 13, 15244–15255. [DOI] [PubMed] [Google Scholar]
- 36. Men S., Licence P., Chem. Phys. Lett. 2017, 686, 74–77. [Google Scholar]
- 37. Laus G., Schwärzler A., Schuster P., Bentivoglio G., Hummel M., Wurst K., Kahlenberg V., Lörting T., Schütz J., Peringer P., Bonn G., Nauer G., Schottenberger H., Z. Naturforsch. 2007, 62b, 295–308. [Google Scholar]
- 38. Bartz S., Blumenröder B., Kern A., Fleckenstein J., Frohnapfel S., Schatz J., Wagner A., Z. Naturforsch. 2009, 64b, 629–638. [Google Scholar]
- 39. Taccardi N., Niedermaier I., Maier F., Steinrück H.-P., Wasserscheid P., Chem. Eur. J. 2012, 18, 8288–8291. [DOI] [PubMed] [Google Scholar]
- 40. Revelli A.-L., Mutelet F., Jaubert J.-N., J. Phys. Chem. B 2010, 114, 12908–12913. [DOI] [PubMed] [Google Scholar]
- 41. Revelli A.-L., Mutelet F., Jaubert J.-N., J. Phys. Chem. B 2010, 114, 8199–8206. [DOI] [PubMed] [Google Scholar]
- 42. Laus G., Wurst K., Kahlenberg V., Kopacka H., Kreutz C., Schottenberger H., Z. Naturforsch. 2010, 65b, 776–782. [Google Scholar]
- 43. Brendgen T., Fahlbusch T., Frank M., Schühle D. T., Seßler M., Schatz J., Adv. Synth. Catal. 2009, 351, 303–307. [Google Scholar]
- 44. Niedermaier I., Kolbeck C., Steinrück H.-P., Maier F., Rev. Sci. Instrum. 2016, 87, 045105. [DOI] [PubMed] [Google Scholar]
- 45. Kolbeck C., Cremer T., Lovelock K. R. J., Paape N., Schulz P. S., Wasserscheid P., Maier F., Steinrück H.-P., J. Phys. Chem. B 2009, 113, 8682–8688. [DOI] [PubMed] [Google Scholar]
- 46. Hammer T., Reichelt M., Morgner H., Phys. Chem. Chem. Phys. 2010, 12, 11070–11080. [DOI] [PubMed] [Google Scholar]
- 47. Kolbeck C., Killian M., Maier F., Paape N., Wasserscheid P., Steinrück H.-P., Langmuir 2008, 24, 9500–9507. [DOI] [PubMed] [Google Scholar]
- 48. Gottfried J. M., Maier F., Rossa J., Gerhard D., Schulz P. S., Wasser-scheid P., Steinrück H.-P., Z. Phys. Chem. 2006, 220, 1439–1453. [Google Scholar]
- 49. Iuga C., Solís C., Alvarez-Idaboy J. R., Martínez M. Á., Mondragón M. A., Vivier-Bunge A., J. Mol. Model. 2014, 20, 2186–2194. [DOI] [PubMed] [Google Scholar]
- 50. Yamada T., Tominari Y., Tanaka S., Mizuno M., Fukunaga K., Materials 2014, 7, 7409–7422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Dilks A., J. Polym. Sci. Polym. Chem. Ed. 1981, 19, 1319–1327. [Google Scholar]
- 52. Kolbeck C., Niedermaier I., Deyko A., Lovelock K. R. J., Taccardi N., Wei W., Wasserscheid P., Maier F., Steinrück H.-P., Chem. Eur. J. 2014, 20, 3954–3965. [DOI] [PubMed] [Google Scholar]
- 53. Lovelock K. R. J., Kolbeck C., Cremer T., Paape N., Schulz P. S., Wasserscheid P., Maier F., Steinrück H.-P., J. Phys. Chem. B 2009, 113, 2854–2864. [DOI] [PubMed] [Google Scholar]
- 54. Oliveira M. B., Domínguez-Pérez M., Cabeza O., Lopes-da-Silva J. A., Freire M. G., Coutinho J. A. P., J. Chem. Thermodyn. 2013, 64, 22–27. [Google Scholar]
- 55. Kolbeck C., Lehmann J., Lovelock K. R. J., Cremer T., Paape N., Wasserscheid P., Fröba A. P., Maier F., Steinrück H.-P., J. Phys. Chem. B 2010, 114, 17025–17036. [DOI] [PubMed] [Google Scholar]
- 56. Roberts R. F., Allara D. L., Pryde C. A., Buchanan D. N. E., Hobbins N. D., Surf. Interface Anal. 1980, 2, 5–10. [Google Scholar]
- 57. Reed A. E., Curtiss L. A., Weinhold F., Chem. Rev. 1988, 88, 899–926. [Google Scholar]
- 58. Breneman C. M., Wiberg K. B., J. Comput. Chem. 1990, 11, 361–373. [Google Scholar]
- 59. Becke A. D., Phys. Rev. A 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- 60. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789. [DOI] [PubMed] [Google Scholar]
- 61. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 62. Grimme S., Ehrlich S., Goerigk L., J. Comput. Chem. 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 63. Becke A. D., Johnson E. R., J. Chem. Phys. 2005, 123, 154101. [DOI] [PubMed] [Google Scholar]
- 64. Becke A. D., Johnson E. R., J. Chem. Phys. 2006, 124, 221101. [DOI] [PubMed] [Google Scholar]
- 65.G. W. T. M. J. Frisch, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. Montgomery, J. A., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, j. Cioslowski, D. J. Fox, Gaussian 09 2009.
- 66.E. D. Glendening, J. K. Badenhoop, A. E. Reed, J. E. Carpenter, J. A. Bohmann, C. M. Morales, C. R. Landis, F. Weinhold, Theoretical Chemistry Institute, University of Wisconsin, Madison, WI, 2013; http://nbo6.chem.wisc.edu/.
- 67. Glendening E. D., Landis C. R., Weinhold F., J. Comput. Chem. 2013, 34, 1429–1437. [DOI] [PubMed] [Google Scholar]
- 68.GaussView, Version 6, R. Dennington, T. A. Keith, J. M. Millam, Semichem Inc., Shawnee Mission, KS, 2016.
- 69. Fogarty R. M., Matthews R. P., Clough M. T., Ashworth C. R., Brandt-Talbot A., Corbett P. J., Palgrave R. G., Bourne R. A., Chamberlain T. W., Hoogerstraete T. Vander, Thompson P. B. J., Hunt P. A., Besley N. A., Lovelock K. R. J., Phys. Chem. Chem. Phys. 2017, 19, 31156–31167. [DOI] [PubMed] [Google Scholar]
- 70. Fogarty R. M., Rowe R., Matthews R. P., Clough M. T., Ashworth C. R., Brandt A., Corbett P. J., Palgrave R. G., Smith E. F., Bourne R. A., Chamberlain T. W., Thompson P. B. J., Hunt P. A., Lovelock K. R. J., Faraday Discuss. 2018, 206, 183–201. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary