

Abstract
Multidimensional single‐cell analysis requires approaches to visualize complex data in intuitive 2D graphs. In this regard, t‐distributed stochastic neighboring embedding (tSNE) is the most popular algorithm for single‐cell RNA sequencing and cytometry by time‐of‐flight (CyTOF), but its application to polychromatic flow cytometry, including the recently developed 30‐parameter platform, is still under investigation. We identified differential distribution of background values between samples, generated by either background calculation or spreading error (SE), as a major source of variability in polychromatic flow cytometry data representation by tSNE, ultimately resulting in the identification of erroneous heterogeneity among cell populations. Biexponential transformation of raw data and limiting SE during panel development dramatically improved data visualization. These aspects must be taken into consideration when using computational approaches as discovery tools in large sets of samples from independent experiments or immunomonitoring in clinical trials.
Keywords: polychromatic flow cytometry, single cell, high‐dimensional data, T cell, tSNE, CD8
The advent of powerful single‐cell technologies, such as single‐cell RNA‐seq (scRNAseq) and cytometry by time‐of‐flight (CyTOF), and the recent improvement of polychromatic flow cytometry to measure up to 30 parameters simultaneously, required the development of new tools to visualize complex multidimensional data in 2D space 1. t‐Distributed stochastic neighboring embedding (tSNE) is widely used in this regard and enables to define heterogeneity within a cell population as well as the rapid identification of similarities and differences between samples 2. Despite its capability to reduce dimensionality, tSNE maintains the original geometry of the high‐dimensional data and has the advantage to identify rare subpopulations if they are sufficiently distinct from the rest of the cells at the immunophenotypic or gene expression level 2. This is particularly useful in polychromatic flow cytometry, where virtually unlimited number of cells can be analyzed owing to reduced costs and increased throughput as compared to scRNAseq or CyTOF. These aspects render flow cytometry a technology of choice for the measurement of multiple parameters in large clinical trials where dozens of samples are to be analyzed.
In the last years, much effort has been dedicated to harmonize flow cytometry assays for basic science and clinical trials, especially in regard to machine standardization 3, 4, 5, 6, reagents 7, protocols 8, and data analysis 9, 10. The technology is extremely robust and reproducible in identifying rare cell populations in independent experiments performed by different operators 11, rather much variability is introduced when independent users analyze the same data due to subjective gating 9. As a consequence, unsupervised algorithms for the identification of cell populations in high‐dimensional data sets would provide an objective approach for data analysis 9. As these approaches make the use of every single cell in the input, technical variability between independent experiments or measurement errors may easily generate artefacts and lead to the misidentification of cell populations. Here, we tested the applicability of tSNE to 27‐parameter single‐cell flow cytometry data.
Materials and Methods
Sample Collection
All experiments using human buffy coats were approved by the Humanitas Research Hospital IRB. Peripheral blood mononuclear cells (PBMCs) were isolated from buffy coats by Ficoll separation and frozen in liquid nitrogen according to standard procedures, as described in Ref. 11.
Flow Cytometer Quality Control (QC)
Machine QC was performed according to the protocol developed by Perfetto et al. 3. Briefly, optimal voltage settings of PMTs were determined in two steps: first, by defining the separation of dimly stained Cytocal beads (Thermo Fisher Scientific, Waltham, MA, USA) over unstained compensation beads (Compbeads; BD Biosciences, San Jose, CA, USA) at 50 V intervals (range 350–800 V), and second, by defining the separation of quantum simply cellular beads (QSCB; previously stained with fluorescently conjugated monoclonal antibodies) over unstained Compbeads at ±25 V of the target voltage defined in the first step. Inter‐experiment QC was performed by running single‐peak Rainbow beads (Spherotec, Lake Forest, IL, USA) and unstained Compbeads and by adjusting PMT voltages to reach target values. Laser delays were adjusted manually.
Polychromatic Flow Cytometry for the Detection of Surface and Intracellular Antigens
All data were acquired between January and December 2017 on a FACS Symphony A5 flow cytometer (BD Biosciences) equipped with five lasers (UV, 350 nm; violet, 405 nm; blue, 488; yellow/green, 561 nm; red, 640 nm; all tuned at 100 mW, except UV tuned at 60 mW) or with a FACSAria cell sorter equipped with four lasers (violet, 405 nm; blue, 488; yellow/green, 561 nm; red, 640 nm; all tuned at 50 mW). Flow cytometry data were compensated in FlowJo by using single‐stained controls (Compbeads incubated with fluorescently conjugated antibodies), as described in Ref. 12.
Cells previously frozen in liquid nitrogen were used in all experiments. Vials of PBMCs from three different donors were thawed in five independent, replicate experiments and stained with a 25‐color flow cytometry panel of antibodies as described below. Cells were added to RPMI1640 supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin, 1% l‐glutamine, and 20 μg/ml deoxyribonuclease I from bovine pancreas (Sigma‐Aldrich, St. Louis, MO, USA). Subsequently, cells were extensively washed and stained immediately with the Zombie Aqua Fixable Viability Kit (BioLegend, San Diego, CA, USA) in phosphate‐buffered saline for 15 min at room temperature (RT). Cells were then washed and stained with the combination of antibodies, purchased from BD Biosciences, BioLegend, or eBioscience, as indicated in Supporting Information Table S1. Antibodies were previously titrated to define optimal concentration, as described in Refs. 12, 13. Staining of CXCR5, CCR7, and CXCR3 was performed for 20 min at 37°C, while all other surface markers (except CD3) were stained for 20 min at RT. Ki‐67, CD3, and transcription factors were detected intracellularly following the fixation of cells with the FoxP3/transcription factor staining buffer set (eBioscience, Waltham, MA, USA) according to manufacturer's instructions and by incubating with specific antibodies for 30 min at 4 °C.
High‐Dimensional Single‐Cell Data Preprocessing and Analysis by tSNE
27‐Parameter Flow Cytometry Standard (FCS) 3.0 files were imported into FlowJo software version 9 or 10 and left untreated or biexponentially transformed (the same transformation for all files, performed in version 10) prior to tSNE analysis. Alternatively, background fluorescence events below the arbitrary threshold of 100, the maximum common value for all fluorescence parameters that is below the threshold of positive antigen expression in this dataset, were transformed by a process referred to as “background randomization.” Briefly, all fluorescence events comprised in the interval between the lowest value of the log distribution (as obtained from the machine) and 100 were randomly distributed evenly across all channels. Replicate samples are expected to contain a similar number of events in this interval, although with different distributions across fluorescence channels, and for this reason are forced to have the same distribution by background randomization.
Following transformation, 27‐parameter samples were assigned with a computational barcode for their unique identification, concatenated (3,000 CD8+ T cells per sample for a total of 45,000 CD8+ T cells) and visualized with tSNE (Barnes–Hut implementation) in FlowJo. The following parameters were tuned in preliminary experiments 14, then used at default values (unless otherwise specified) as we observed no substantial changes in the final distribution: iterations, 1000; perplexity, 40; initialization, deterministic; theta, 0.5; eta, 200. In some experiments, perplexity was changed to determine variability in tSNE cluster distribution and data display/representation (Figure 3a). All parameters except for lineage markers (CD3, CD4, and CD8), Aqua dead cell marker and Eomes transcription factor, were included in the analysis.
Figure 3.
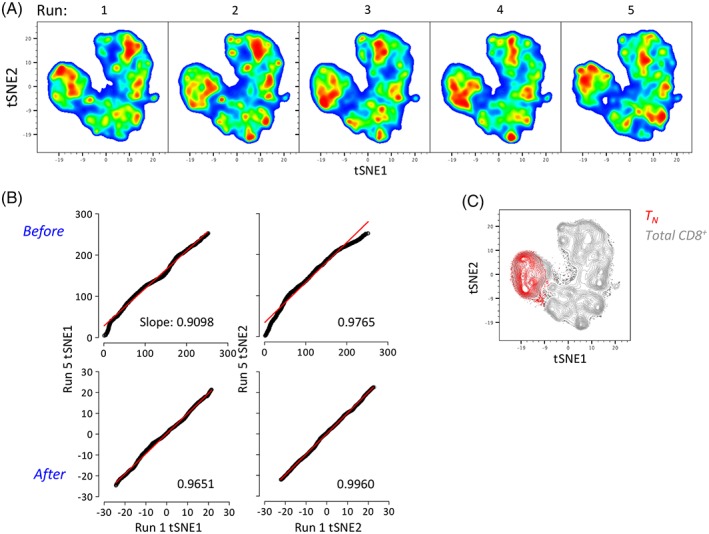
Biexponentlal transformation allows reproducibility of multidimensional data representation by tSNE. (a) tSNE representation (perplexity = 100) of 27‐parameter flow cytometry data across five independent replicate experiments following biexponential transformation in Flowlo version 10. Each run shows pooled CD8+ T cells from three different donors for simplicity (3,000 cells each). (b) Linear correlation of tSNE1 and tSNE2 axes values between Run 1 and Run 5 before and after biexponential transformation. Numbers in the plots indicate the slope of the line. (c) tSNE map of CD8+ TN cells (red) on top of total CD8+ T cells after biexponential transformation.
To test the influence of background fluorescence on the final tSNE representation independently of FlowJo software, PBMC samples from two different healthy donors were stained with a nine‐color panel (Supporting Information Table S1) within the same experiment and acquired on a FACSAria cell sorter. Markers were selected in a way that CD8+ TN cells are defined only by negative expression, that is, CD56−, HLA‐DR−, CD4−, and CD45RO−. Cells were further gated as CCR7+, although CCR7 was not included in the analysis. In this way, only background, and not positive fluorescence (i.e., protein abundance on the cell surface) defines CD8+ TN cells in the tSNE display. Compensated FCS 3.0 files (containing 20,000 cells/sample) were exported as .txt, concatenated in R analyzed using the Rtsne function (from the Rtsne package, using default options with the exception of PCA parameter that was set equal to “FALSE”) to calculate the coordinates for the 2D tSNE plot. Resulting data were further converted in FCS 3.0 format and analyzed with FlowJo 10.
Influence of Spreading Error (SE) on tSNE Map Calculation
To test the effect of SE 15 on the tSNE map calculation, we designed a polychromatic panel where SE is contributed only by CD57 BV605 spreading into the YG610 channel (in this experiment detecting anti‐CCR7 PE‐CF594; Figure 2a). A PBMC sample was stained as above with a backbone panel including Zombie Aqua and antibodies directed to recognizing the following antigens: CD3, CD4, CD8, CD27, CD45RA, CD45RO, CCR7, and CD56. After wash, cells were split into four replicate tubes, three of which were subsequently stained with increasing amounts of anti‐CD57 conjugated to BV605. The fourth tube was left unstained as a control. Then, 20,000 cells from each of the four samples (80,000 in total) were concatenated and visualized with tSNE. The following markers were given as input: CD27, CD45RA, CD45RO, CCR7, and CD56. To make sure that the final result was dependent only on SE and not by the abundance of CD57 expression, the CD57 parameter was excluded from tSNE calculation.
Figure 2.
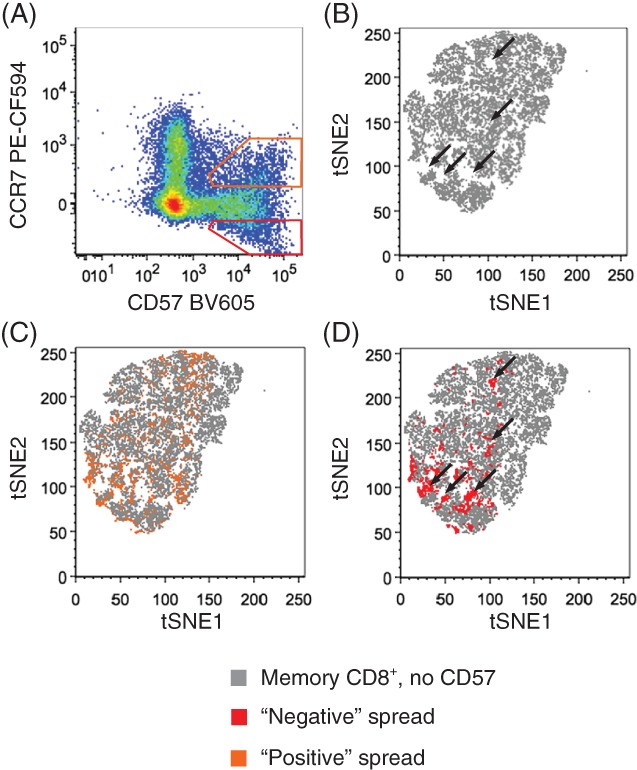
Contribution of SE to variability of multidimensional data visualization by tSNE. (a) Fluorescence spreading of anti‐CD57 BV605 into anti‐CCR7 PE‐CF594. Orange gate identifies cells spreading in the positive fraction while red gate identifies cells spreading in the negative fraction of the secondary detector. (b) tSNE plot of TMEM cells, gated as in Figure 1c, of a sample not contaning anti‐CD57 BV605. Overlay of positive‐ (c) and negative‐spreading (d) CD57+ cells, identified as in (a), on top of TMEM cells. In (b) and (d), black arrows identify holes in the tSNE map that are filled by negative‐spreading CD57+ cells.
Results
We evaluated the usefulness of tSNE to determine the heterogeneity of multidimensional single‐cell data generated by 27‐parameter polychromatic flow cytometry. For this purpose, PBMC samples from three healthy donors were thawed in five independent, replicate experiments and stained with 24 different fluorescently conjugated monoclonal antibodies directed to recognize antigens expressed by CD8+ T cells. A viability dye was used to exclude dead cells. The full gating strategy used to identify CD8+ T cells is depicted in Supporting Information Figure S1a. A single concatenated file containing 45,000 events from the different experiments was subjected to tSNE analysis (see “Methods” section for details on the procedure). Following debarcoding of replicate samples, we noticed that tSNE maps were substantially different in independent experiments, specifically runs 1–2, and runs 3–4 were mostly similar to each other (on the basis of the abundance of cells identified in the arbitrary gate) and that those from run 5 were the most different compared to runs 1–4 (Figure 1a). We thus aimed to investigate the reason at the basis of such variability, which characterized both tSNE1 and tSNE2 dimensions (Figure 1b). First of all, we tested whether samples from different runs harbored differential abundance of immune populations, for example due to the loss of certain subsets as a consequence of the freeze/thaw procedure. Therefore, we quantified the expression of single markers, one at a time, by standard gating and identified similar percentages of antigen expression (Supporting Information Figure S1b). These data also confirm the reproducibility of the flow cytometry technology. Overall, more variability between measurements could be observed for those markers not having a clear separation over negative cells, in our case T‐bet and Eomes. In particular, Eomes displayed the highest variability among replicate stainings and was thus excluded from further tSNE analysis.
Figure 1.
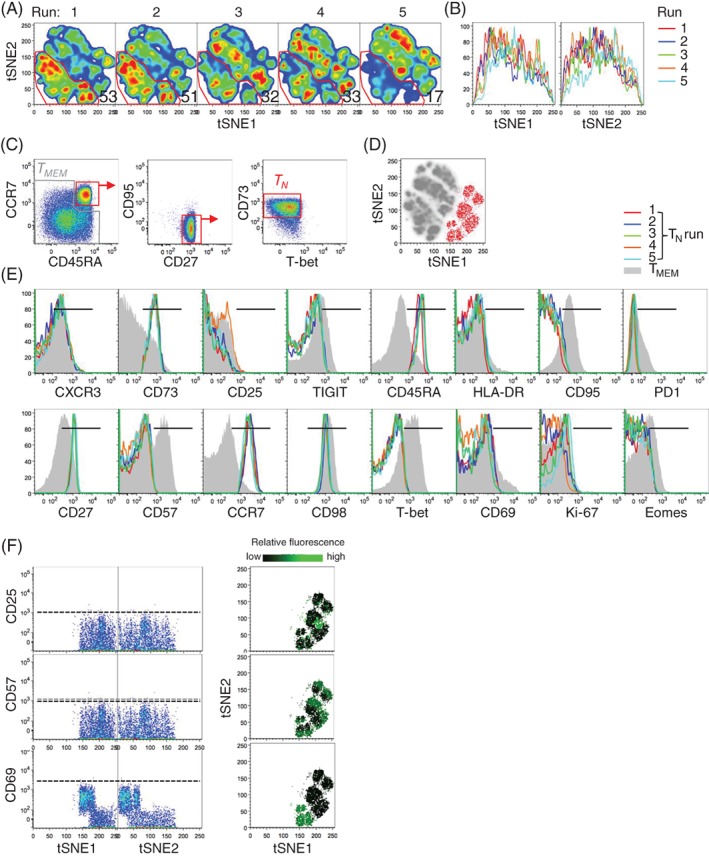
Contribution of background fluorescence to variability of multidimensional data visualization by tSNE. (a) tSNE map of 27‐parameter flow cytometry data from five replicate experiments. Numbers indicate the percentage of cells identified by the arbitrary gate. (b) Overlay of tSNE1 and tSNE2 axes obtained as in (a). (c) Gating strategy used for the identification of human CD8+ TN (red) and TMEM (grey) cells. (d) Overlay of gated CD8+ TN cells on top of the tSNE map obtained as in (a). Grey cells in the background are TMEM. (e) Histogram overlays of antigen expression by CD8+ TN across the five experiments. TMEM cells in grey are reported as a control. Black horizontal bar indicates positivity. Antigens poorly expressed by peripheral blood CD8+ T cells, i.e., IRF4, CD71, TIM3, CXCR5, and FoxP3, are not depicted. (f) Dot plots of tSNE1 and tSNE2 axes vs. antigens in gated CD8+ TN cells (left) and relative fluorescence levels of markers on TN cell tSNE clusters (right). Dotted horizontal bars indicate threshold of positivity. In all panels, each run shows pooled CD8+ T cells from three different donors for simplicity (3,000 cells each). TN: naive T cells; TMEM; memory T cells.
Differential distribution of events in the tSNE map among independent, replicate samples could be due to real biological heterogeneity (e.g., subsets composition) or to technical artifacts in the single‐cell data. To test this, we reasoned that a phenotypically homogeneous T‐cell population would be identified by a single island in the map. Therefore, we selected highly purified CD8+ naive T (TN) cells on the basis of a combination of six markers that are known to be associated with naivety (i.e., CD45RA+CCR7+CD27+CD95−Tbet−CD73+; Figure 1c) 16, 17 and plotted them on top of the tSNE map. Surprisingly, tSNE classified TN cells into multiple putative subpopulations (Figure 1d), despite the relative homogeneity of TN cell immunophenotype (either positive antigen expression or negative antigen expression; Figure 1e). Since data points (cells) are distributed in a stochastic way in the final tSNE map, we tested whether the final representation is dependent on the initial seed. To this end, we ran the algorithm several times with random seeds, and obtained very similar results (data not shown). We then thought of additional factors other than true biological heterogeneity that could drive the “clustering” of high‐dimensional flow cytometry events in the tSNE space, including differences in (1) compensation, (2) daily instrument performance, (3) positive signals, as a consequence of point 2 but also possibly caused by some sort of batch effect, as well as differences in background values (i.e., the biological negative fraction not containing fluorochrome‐conjugated antibodies), and (4) SE generated by errors in the measurement of photons at red/far red wavelengths 15.
The impact of fluorescence compensation on the final tSNE representation is intuitive, as incorrect compensation may result in newly generated populations of cells in the multidimensional space. Despite subtle differences in the final compensated data may occur between samples, correct compensation was generated by running experiment‐specific, single‐stained controls and by carefully examining single pairwise combinations 12. Matrix view of all possible combinations of antigen expression revealed comparable fluorescence distribution of CD8+ T cells from two representative experiments (run 1 and run 5; Supporting Information Figure S2), thus suggesting that compensation is not responsible for tSNE heterogeneity in this dataset.
Daily calibration according to Perfetto et al. 3 allowed adjusting voltages in order to obtain comparable positive and background fluorescence values in different experiments (Supporting Information Figure S3). We observed slight changes in background levels only in run 3, which can hardly explain differential tSNE distributions across the five runs. Positive signals from reference beads were highly comparable. As for beads, we observed reproducible measurements of antigens that are expressed on CD8+ TN cells, that is, CXCR3, CD73, CD45RA, CD27, CCR7, and CD98 (Figure 1e), indicating that a possible batch effect due to sample preparation is negligible. Instead, we noticed substantial variability in the distribution of the background for some parameters such as CD25, CD57, CD69, and Ki67 (Figure 1e), which ultimately affected the final tSNE output by erroneously identifying virtual subpopulations of cells (Figure 1f, left). Such putative heterogeneity identified by tSNE among CD8+ TN cells has no biological meaning (indeed CD8+ TN cells express little, if any CD69, CD25, or CD57; Figure 1e) and seems to be driven, at least in part, by cells with very low fluorescence values that are piled on the low end of the log scale, an effect generally referred to as the “log artifact” 18. By plotting the differential fluorescence intensity of background events on top of the tSNE map depicting TN cells, as identified from Figure 1d, it was possible to appreciate that TN “clusters” indeed harbor different background values of CD25, CD57, and CD69 (Figure 1f, right). Randomization of background, that is, the redistribution of negative events evenly across all channels below an arbitrary threshold (set at 100 for all parameters), improved the reproducibility of the tSNE plots (Supporting Information Figures S4a and S4b), thereby demonstrating that background values are indeed a source of variability.
Despite the reproducibility of the flow cytometry assay, a residual batch effect may still be present due to small variability in positive fluorescence across independent experiments. To overcome this, we analyzed two samples that were stained within the same experiment with the same analytical workflow. We also validated our results independently of the flow cytometer and the analytical platform. Also in this case, tSNE displays of CD8+ TN cells from the two samples were distinct (Supporting Information Figure S5a) and correlated with different levels of CD45RO, but not of CD56, HLA‐DR, and CD4 background levels (Supporting Information Figures S5b and S5c). Specifically, low background levels of CD45RO were associated with high tSNE1 axis and low tSNE2 axis values, and vice versa. These data confirm our previous observation that the influence of background fluorescence on tSNE display is a generalized phenomenon.
We next tested the contribution of SE to variability in tSNE representation. SE is directly proportional to the square root of the intensity of the staining, may vary between samples as a result of differential antigen abundance, and can potentially generate new cell clusters as a consequence of fluorescence spreading in the “negative” and “positive” regions of secondary dimensions (Figure 2a). Despite having exactly the same immunophenotypes and virtually the same biological functions, one could hypothesize that cells in the spread interval could be inaccurately mapped to different tSNE regions because defined by very different values in the multidimensional space. To test the individual contribution of SE, we mapped “negative” and “positive” SE on the top of a control sample that did not contain CD57 BV605 (Figure 2b). In this way, we noticed that while cells with positive SE values overlap to tSNE islands already occupied by other cells (Figure 2c; indeed, cells double positive for CD57 and CCR7 may exist in the sample), cells with negative SE values fill empty regions of the tSNE plot (Figure 2d), thus further contributing to generate new islands with irrelevant biological meaning. In conclusion, different background measurements and SE are major sources of variability in multidimensional single‐cell polychromatic flow cytomertry data representation by tSNE.
SE is an intrinsic physical characteristic of the dyes and cannot be corrected following data acquisition 4. However, SE can be minimized by mounting more powerful lasers so to increase photon emission, and by optimizing filter/mirror combinations or during panel development, if required in specific flow cytometry panels so to increase detection of dimly expressed antigens 12. In contrast, background fluorescence representation can be modified by using computational approaches, the most common of which is biexponential transformation 18. While positive data remain untouched, this approach circumvents the log artifact by normalizing data around zero, thus decreasing background variability between samples (Supporting Information Figure S6). Figure 3a shows that biexponential transformation dramatically improved tSNE representation of five replicate 27‐parameter flow cytometry analyses. Improvement could be also observed by correlating tSNE axes values from two experiments with the highest variability (i.e., run 1 and run 5; Figure 3b). Finally, CD8+ TN cells (identified as in Figure 1c) appeared relatively homogeneous following biexponential transformation when compared with nontransformed data (Figures 3c). Similar results were obtained with a different panel (Supporting Information Figure S7).
Discussion
Identification of trends in multidimensional single‐cell data that are associated with a certain pathology or immune status has been pursued since the development of flow cytometers capable of measuring more than 7–8 colors 19. The advent of new reagents and technologies such as CyTOF and scRNAseq extended this capability to dozens and thousands of parameters, respectively, thus making the development of new computational approaches for the visualization of such data a fundamental need 20. tSNE is currently the most popular approach in this regard as it displays single cells on an intuitive 2D graph and where relative antigen or gene expression can be visualized by the color tone 2. Despite limited in its analysis capability compared to the abovementioned technologies, polychromatic flow cytometry, now capable to measure up to 30 parameters simultaneously, is still the most popular and versatile single‐cell technology that is available in thousands of laboratories around the world. It is therefore anticipated that approaches such as tSNE will be largely utilized to display such data. While flow cytometry and CyTOF share multiple aspects in terms of protocols, reagents, and concept, the nature of their single‐cell data is different. Since flow cytometry relies on fluorescence measurements, scientists have to inevitably deal with background fluorescence, compensation, and SE that otherwise are poorly present in CyTOF.
Here, we have identified background fluorescence and SE to impact substantially the visualization of multidimensional single‐cell data by tSNE, specifically resulting in the generation of virtual “islands” that do not reflect real heterogeneity at the phenotypic or biological level. Differences in background, but not positive fluorescence, could be observed among replicate samples run in independent experiments. This was not due to machine performance, which confirmed to be very stable over time, rather variability in background seems to be sample dependent (Supporting Information Figure S4). These low (negative) fluorescence signals are irrelevant when analyzing data by standard gating, as no biological information beyond the number of cells contained in the negative fraction should be considered. Nevertheless, fluorescence “heterogeneity” in the negative interval may lead to substantial confusion when displaying multidimensional datasets with approaches such as tSNE, where every single fluorescence value, being it positive or negative, is considered.
Randomizing negative events below an arbitrary threshold evenly across all channels improved visualization but did not solve the issue in full, probably because of the lack of an algorithm capable to introduce channel‐specific thresholds (in fact, threshold of positivity is different in every single channel; Supporting Information Figure S4c), rather biexponential transformation, which normalizes negative data around zero without impacting the positive ones, substantially reduces the variability among independent experiments, thus enabling to obtain comparable tSNE plots of replicate, independent measurements. However, it was not possible to obtain complete overlap of tSNE plots between the biologically identical samples (Figure 3a). We attribute this to small variations in background or positive fluorescence, compensation or SE that ultimately affect multiple combinations in the multidimensional space and that are captured by the tSNE algorithm. Indeed, we further identified SE as a major contributor of variation in multidimensional flow data representation. This study describes a situation where a single pairwise fluorochrome combination that is dramatically affected by SE is sufficient to generate cell clusters with irrelevant biological meaning. As SE can affect multiple combinations in the 30‐parameter space 4 (Lugli and Roederer, unpublished observation), it is anticipated that data representation by tSNE would be progressively worsened when increasing the number of parameters.
A wrong assumption is that cells occupying different areas of the tSNE display harbor intrinsic biological heterogeneity; therefore, it is not rare to see the use of tSNE maps to identify putative subsets in a given population or set of samples. On the basis of our results, gating on the tSNE map followed by phenotypic analysis would lead to overfragmentation of immunophenotypes that complicates, rather than simplifies, multidimensional data analysis. Instead, clustering algorithms should be run in parallel to identify relevant cell subpopulations, as it is now done for scRNAseq. Along with this, it is of foremost importance to validate computational results with raw data, i.e., once the clusters are identified, to evaluate the relative abundance of antigen expression in that specific subpopulation either by percentage or MFI, if not both, followed by critical evaluation of immunophenotypes. Moreover, beyond machine, reagent, and computational standardization, immunophenotyping of replicate samples must be included in independent experiments, so to estimate the overall variability of the assay. Integration of classical gating and novel computational approaches will be pivotal to achieve the discovery of novel biological insights in high‐dimensional polychromatic flow cytometry data.
Conflict of Interest
The Laboratory of Translational Immunology receives reagents in kind from BD Biosciences, Italy, as a part of a collaborative research agreement. Other authors have no conflicts of interest to disclose.
Supporting information
Supplementary Figure 1. 27‐parameter FACS is reproducible across independent experiments. a) Gating strategy of the identification of CD8+ T cells in peripheral blood used in this study. b) Frequency of CD8+ T cells expressing a given antigen in 3 different healthy donors (HD). Each dot represents a single independent experiment. The grey bar in the background represents the interquartile range of the distribution.
Supplementary Figure 2. Similar distribution of compensated data between independent experiments. Matrix overlay of antigen expression between run 1 and run 5.
Supplementary Figure 3. Machine performance across independent experiments. a) Background and b) positive signals across independent experiments as revealed by unstained and single‐peak rainbow beads, respectively. Channel labels refer to the laser source (UV: ultraviolet; V: violet; B: blue; YG: yellow/green; R: red) and the median of wavelength detection by the bandpass filter.
Supplementary Figure 4. Background randomization of negative events improves visualization of high‐dimensional FACS data. a) tSNE map of 27‐parameter flow cytometry data from 5 replicate experiments following randomization of background at 100. Numbers in plots indicate the percentage of cells identified by the arbitrary gate. Each run shows pooled CD8+ T cells from 3 different donors for simplicity (3,000 cells each). b) Overlay of tSNE1 ans tSNE2 axes obtained as in a. c) Dot plots of tSNE1 and tSNE2 axes vs. antigens in gated CD8+ TN cells. Dotted horizontal bar indicates threshold of positivity of CD25, CD57 and CD69 expression (y axis). The black arrow indicates subpopulations identified following randomization of negative events below a threshold of 100.
Supplementary Figure 5. Different tSNE displays are associated with different background fluorescence values. PBMCs from two donors, stained in the same day with the same mix of multiple fluorescently‐conjugated monoclonal antibodies, were acquired on a FACSAria cell sorter, then concatenated for further analysis by tSNE in R. tSNE analysis included the following parameters: CD56 PE‐Cy5, HLA‐DR APC‐H7, CD4 APC and CD45RO PerCP‐Cy5.5. a) tSNE representation of total events and of TN‐gated CD8+ T cells (defined as CD4–CD45RO–CCR7+). b) Fluorescence levels of CD56, HLA‐DR, CD4 and CD45RO in CD8+ TN cells from the two samples included in the analysis. c) Dot plots of tSNE1 and tSNE2 axes vs. CD45RO in CD8+ TN‐gated cells from one representative donor. Dotted horizontal bar indicates threshold of positivity of CD45RO (y axis).
Supplementary Figure 6. Biexponential transformation normalizes background fluorescence across independent experiments. Histogram overlays of antigen expression by CD8+ TN across the 5 experiments following biexponential transformation. TMEM cells in grey are reported as a control. Black horizontal bar indicates positivity. Antigens poorly expressed by peripheral blood CD8+ T cells, i.e., IRF4, CD71, TIM3, CXCR5 and FoxP3, are not depicted. Each run shows pooled CD8+ T cells from 3 different donors for simplicity (3,000 cells each).
Supplementary Figure 7. Validation of the biexponential transformation approach. Overlay of gated CD8+ TN cells, defined as CD45RO– CCR7+ on top of the tSNE map before and after transformation. Grey cells in the background are TMEM cells. TN: naïve T cells; TMEM; memory T cells.
Acknowledgments
We wish to thank Pratip K. Chattopadhyay (NYU, New York, USA) and Stephen P. Perfetto (Vaccine Research Center, NIH, Bethesda, USA) for sharing information on the 30‐parameter flow cytometry platform, Gianluca Rotta and Jens Fleischer (BD Biosciences) for helping with instrument setup, and Eliver Ghosn (Emory University), Darya Orlova (Stanford University), and the members of the Laboratory of Translational Immunology for critical discussion of the article. EMCM is a recipient of the Fondazione Umberto Veronesi postdoctoral fellowship. JB is a recipient of the “Fondo di beneficenza Intesa San Paolo” fellowship from AIRC. EL is an International Society for the Advancement of Cytometry (ISAC) Marylou Ingram scholar. Purchase of the BD FACS Symphony A5 has been in part defrayed by a grant from Italian Ministry of Health (agreement no. 82/2015).
Literature Cited
- 1. Tanay A, Regev A. Scaling single‐cell genomics from phenomenology to mechanism. Nature. 2017;541:331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amir el‐AD, Davis KL, Tadmor MD, Simonds EF, Levine JH, Bendall SC, Shenfeld DK, Krishnaswamy S, Nolan GP, Pe'er D. viSNE enables visualization of high dimensional single‐cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31:545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perfetto SP, Ambrozak D, Nguyen R, Chattopadhyay PK, Roederer M. Quality assurance for polychromatic flow cytometry using a suite of calibration beads. Nat Protoc. 2012;7:2067–2079. [DOI] [PubMed] [Google Scholar]
- 4. Nguyen R, Perfetto S, Mahnke YD, Chattopadhyay P, Roederer M. Quantifying spillover spreading for comparing instrument performance and aiding in multicolor panel design. Cytometry Part A. 2013;83A:306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kalina T, Flores‐Montero J, Lecrevisse Q, Pedreira CE, van der Velden VH, Novakova M, Mejstrikova E, Hrusak O, Bottcher S, Karsch D, et al. Quality assessment program for EuroFlow protocols: summary results of four‐year (2010‐2013) quality assurance rounds. Cytometry Part A. 2015;87A:145–156. [DOI] [PubMed] [Google Scholar]
- 6. Westera L, van Viegen T, Jeyarajah J, Azad A, Bilsborough J, van den Brink GR, Cremer J, Danese S, D'Haens G, Eckmann L, et al. Centrally determined standardization of flow cytometry methods reduces interlaboratory variation in a prospective multicenter study. Clin Transl Gastroenterol. 2017;8:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maecker HT, McCoy JP, Nussenblatt R. Standardizing immunophenotyping for the Human Immunology Project. Nat Rev Immunol. 2012;12:191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McGowan I, Anton PA, Elliott J, Cranston RD, Duffill K, Althouse AD, Hawkins KL, De Rosa SC. Exploring the feasibility of multi‐site flow cytometric processing of gut associated lymphoid tissue with centralized data analysis for multi‐site clinical trials. PLoS One. 2015;10:e0126454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aghaeepour N, Finak G, Flow CAPC, Consortium D, Hoos H, Mosmann TR, Brinkman R, Gottardo R, Scheuermann RH. Critical assessment of automated flow cytometry data analysis techniques. Nat Methods. 2013;10:228–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Finak G, Langweiler M, Jaimes M, Malek M, Taghiyar J, Korin Y, Raddassi K, Devine L, Obermoser G, Pekalski ML, et al. Standardizing Flow Cytometry Immunophenotyping Analysis from the Human ImmunoPhenotyping Consortium. Sci Rep. 2016;6:20686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lugli E, Gattinoni L, Roberto A, Mavilio D, Price DA, Restifo NP, Roederer M. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat Protoc. 2013;8:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lugli E, Zanon V, Mavilio D, Roberto A. FACS analysis of memory T lymphocytes. Methods Mol Biol. 2017;1514:31–47. [DOI] [PubMed] [Google Scholar]
- 13. Cossarizza A, Chang HD, Radbruch A, Akdis M, Andra I, Annunziato F, Bacher P, Barnaba V, Battistini L, Bauer WM, et al. Guidelines for the use of flow cytometry and cell sorting in immunological studies. Eur J Immunol. 2017;47:1584–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van der Maaten LJP, Hinton GE. Visualizing high‐dimensional data using t‐SNE. J Mach Learn Res. 2008;9:2579–2605. [Google Scholar]
- 15. Roederer M. Spectral compensation for flow cytometry: visualization artifacts, limitations, and caveats. Cytometry. 2001;45:194–205. [DOI] [PubMed] [Google Scholar]
- 16. Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, et al. A human memory T cell subset with stem cell‐like properties. Nat Med. 2011;17:1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mahnke YD, Brodie TM, Sallusto F, Roederer M, Lugli E. The who's who of T‐cell differentiation: human memory T‐cell subsets. Eur J Immunol. 2013;43:2797–2809. [DOI] [PubMed] [Google Scholar]
- 18. Parks DR, Roederer M, Moore WA. A new "Logicle" display method avoids deceptive effects of logarithmic scaling for low signals and compensated data. Cytometry Part A. 2006;69A:541–551. [DOI] [PubMed] [Google Scholar]
- 19. Lugli E, Pinti M, Nasi M, Troiano L, Ferraresi R, Mussi C, Salvioli G, Patsekin V, Robinson JP, Durante C, et al. Subject classification obtained by cluster analysis and principal component analysis applied to flow cytometric data. Cytometry Part A. 2007;71A:334–344. [DOI] [PubMed] [Google Scholar]
- 20. Spitzer MH, Nolan GP. Mass cytometry: single cells, many features. Cell. 2016;165:780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. 27‐parameter FACS is reproducible across independent experiments. a) Gating strategy of the identification of CD8+ T cells in peripheral blood used in this study. b) Frequency of CD8+ T cells expressing a given antigen in 3 different healthy donors (HD). Each dot represents a single independent experiment. The grey bar in the background represents the interquartile range of the distribution.
Supplementary Figure 2. Similar distribution of compensated data between independent experiments. Matrix overlay of antigen expression between run 1 and run 5.
Supplementary Figure 3. Machine performance across independent experiments. a) Background and b) positive signals across independent experiments as revealed by unstained and single‐peak rainbow beads, respectively. Channel labels refer to the laser source (UV: ultraviolet; V: violet; B: blue; YG: yellow/green; R: red) and the median of wavelength detection by the bandpass filter.
Supplementary Figure 4. Background randomization of negative events improves visualization of high‐dimensional FACS data. a) tSNE map of 27‐parameter flow cytometry data from 5 replicate experiments following randomization of background at 100. Numbers in plots indicate the percentage of cells identified by the arbitrary gate. Each run shows pooled CD8+ T cells from 3 different donors for simplicity (3,000 cells each). b) Overlay of tSNE1 ans tSNE2 axes obtained as in a. c) Dot plots of tSNE1 and tSNE2 axes vs. antigens in gated CD8+ TN cells. Dotted horizontal bar indicates threshold of positivity of CD25, CD57 and CD69 expression (y axis). The black arrow indicates subpopulations identified following randomization of negative events below a threshold of 100.
Supplementary Figure 5. Different tSNE displays are associated with different background fluorescence values. PBMCs from two donors, stained in the same day with the same mix of multiple fluorescently‐conjugated monoclonal antibodies, were acquired on a FACSAria cell sorter, then concatenated for further analysis by tSNE in R. tSNE analysis included the following parameters: CD56 PE‐Cy5, HLA‐DR APC‐H7, CD4 APC and CD45RO PerCP‐Cy5.5. a) tSNE representation of total events and of TN‐gated CD8+ T cells (defined as CD4–CD45RO–CCR7+). b) Fluorescence levels of CD56, HLA‐DR, CD4 and CD45RO in CD8+ TN cells from the two samples included in the analysis. c) Dot plots of tSNE1 and tSNE2 axes vs. CD45RO in CD8+ TN‐gated cells from one representative donor. Dotted horizontal bar indicates threshold of positivity of CD45RO (y axis).
Supplementary Figure 6. Biexponential transformation normalizes background fluorescence across independent experiments. Histogram overlays of antigen expression by CD8+ TN across the 5 experiments following biexponential transformation. TMEM cells in grey are reported as a control. Black horizontal bar indicates positivity. Antigens poorly expressed by peripheral blood CD8+ T cells, i.e., IRF4, CD71, TIM3, CXCR5 and FoxP3, are not depicted. Each run shows pooled CD8+ T cells from 3 different donors for simplicity (3,000 cells each).
Supplementary Figure 7. Validation of the biexponential transformation approach. Overlay of gated CD8+ TN cells, defined as CD45RO– CCR7+ on top of the tSNE map before and after transformation. Grey cells in the background are TMEM cells. TN: naïve T cells; TMEM; memory T cells.